

Abstract
The central nervous system (CNS) constitutively expresses complement (C) membrane receptors and complement proteins, including the component C5a. This is a crucial terminal effector of the C cascade, mostly involved in pain and neuroinflammatory conditions. Aberrant activation of C5a protein and its receptor C5aR has been reported to play a critical role in neurodegenerative diseases, with important clinical consequences. Here we have investigated the effects of DF3016A, a novel selective C5aR antagonist, able to penetrate the blood-brain barrier (BBB), on cortical neurons exposed to oxygen-glucose deprivation-reoxygenation (OGD/R), a neuroinflammation-related process. We demonstrated that a mild ischemic insult induces an early upregulation of C5aR associated with the over-production of pro-inflammatory cytokines and the over-expression of the transcriptional regulatory factor miR-181. Furthermore, we report the first experimental evidence of the effect of DF3016A, modulating complement component C5a, on neurons in a model of injury. Interestingly, DF3016A protects neuronal viability by restoring intracellular calcium levels, thus opposing the increase in pro-inflammatory cytokine levels and miR-181 expression. Based on our results, we suggest that DF3016A is a novel C5aR antagonist promoting protective effects against OGD/R-induced damage that could be a new therapeutic approach to controlling CNS neuroinflammatory conditions.
Keywords: Neuroinflammation, Complement, C5a, Cortical neurons, Cytokines, Pain
Introduction
Neuroinflammation and neurodegenerative processes have been strongly related to chronic pain (Alexander et al. 2008; Varrassi et al. 2015). They may be consequent to the activation of complement in the central nervous system (CNS) (Van Beek et al. 2003). More recently, the importance of neurodegenerative processes has been related to concomitant pathological conditions connected to pain such as paraneoplastic neuropathies, rheumatic, and joint diseases (Fusco et al. 2017; Zis et al. 2017; Zis et al. 2017a). Despite being immunologically separate from the circulating plasma, and in the absence of blood-brain barrier (BBB) damage, cells of the CNS express many components of immunity including all complement (C) factors (Woodruff et al. 2010; O'Barr et al. 2001). The C system, composed of a large number of serum proteins and membrane-bound receptors, among others, protects from infection through innate and adaptive immune mechanisms (Carroll 1998; Song et al. 2000). In the CNS, complement component 5a (C5a) and its cell membrane receptor (C5aR) are constitutively expressed not only in astrocytes and microglia (Müller-Ladner et al. 1996) but also in neurons (Davoust et al. 1999; O'Barr et al. 2001; Pavlovski et al. 2012). The C cascade pathway is involved in pain mechanisms (Gasque et al. 1997), and the inappropriate activation of complement produces a local inflammatory reaction. Much evidence indicates that C5a and C5aR play a critical role in neurodegenerative diseases (Yanamadala and Friedlander 2010; Zhou et al. 2009; Quadros and Cunha 2016; Barnum 2002; Farkas et al. 2003), and the C5a has a number of effects on neurons in vitro (Mukherjee and Pasinetti 2001; Farkas et al. 1998; Pavlovski et al. 2012). For more than 30 years, researchers have actively investigated the role of the immune system (IS) and specifically, C proteins, in human neurodegenerative diseases and mouse models (Alexander et al. 2008). C production in the CNS not only protects from invading pathogens but also may have a regulatory role in neuronal processes (Farkas et al. 2012; Farkas et al. 2008). However, there is compelling evidence that it can exert a dual role in the CNS, depending on the pathophysiological context (van Beek et al. 2003). Stevens et al. (2007) reported a C involvement during brain development in physiological homeostasis and pruning of unwanted synapses, suggesting that the aberrant reactivation of complement-mediated synapse elimination may take place in neurodegenerative conditions (Mukherjee and Pasinetti 2000). After ischemic stroke, the progression of brain injury also involves complement mediated changes (Alawieh et al. 2015).
Acute ischemic stroke is responsible for almost 90% of neuronal injuries following deprivation of oxygen and nutrients. During the ischemic cerebrovascular events, there are two zones: the “ischemic core” and the “ischemic penumbra.” Focal ischemic stroke induces neuronal death in the “core” and molecular and cellular alterations in the “penumbra,” changes that are temporal/spatial-dependent. The term ischemic penumbra is generally used to define the tissue surrounding the severe ischemic core where blood flow is reduced to cause hypoxia, but not sufficient to result in irreversible failure of energy metabolism and cellular necrosis (Astrup et al. 1981). Ischemic penumbra is a diagnostic and biochemical target for brain plasticity, neuroprotection, and neurorepair. Moreover, a sublethal preconditioning injury strongly increases the neuronal resistance to subsequent injury by stimulating protective processes (Badaut et al. 2005; Kawahara et al. 2004; Narayanan and Perez-Pinzon 2017).
Oxygen-glucose deprivation/reoxygenation (OGD/R) procedure provides an in vitro model of neuroinflammation-related processes (Goldberg and Choi 1993) that are crucial elements in the injury onset and progression. To better understand the relevance of C5a modulation in this context, we ran a medicinal chemistry program (Moriconi et al. 2014) that led to the identification of DF3016A, a novel potent and selective C5aR inhibitor with tailored pharmacological properties. Herein, we report and evaluate the in vitro characterization and the experimental effects of DF3016A, on primary rat cortical neurons exposed to sublethal OGD/R.
Materials and Methods
Cell Isolation and Culture
Polymorphonuclear cells (PMNs) were obtained from buffy coats of heparinized peripheral blood from adult healthy volunteers, as previously described (Bertini et al. 2004). Ethical clearance was obtained by local ethical review committees and conformed to Italian regulations. PMNs were separated by dextran sedimentation followed by hypotonic lysis of contaminating red blood cells as previously described (Bertini et al. 2004). Cell viability, as measured by trypan blue dye exclusion, was greater than 98%.
Murine and rat PMNs were isolated from peritoneal cavities injected with 1.5 ml of 3% thioglycolate in saline to male Balb/c mice (20–25 g and 7–9 weeks of age, provided by Charles River, Calco, Italy) or with 10 ml of 1.5 ml of 3% thioglycolate in saline to SD rat (370–450 g and 3–4 months of age provided by Charles River, UK). Four hours after injection, the animals were sacrificed by decapitation and peritoneal cavities were washed with saline. PMNs were recovered and centrifuged at 600×g for 10 min. The pellet was resuspended in Hank’s Balanced Salt Solution (HBSS); then the cells were counted using Türk’s solution and diluted at 3 × 106 cells/ml.
Migration Assay
Migration of human, rat, and mouse PMNs was evaluated using a 48-well micro-chemotaxis chamber, as previously described (Bertini et al. 2004). Briefly, 25 μl of control medium HBSS for human and rodent PMNs, or chemoattractant solution 10 nM chemokine (CXC motif) ligand 1 (CXCL1), 1 nM CXCL8 or CXCL1/KC for rodent PMNs, 10 nM C5a or 10 nM mC5a or rC5a for rodent PMNs, 10 nM CXCL12, and 10 nM n-formylmethionyl-leucyl-phenylalanine (fMLP) were seeded in the lower compartment of the chemotaxis chamber. Fifty microliters of cell suspension (1.5 × 106/ml for human PMNs, 3.0 × 106/ml for rat and mouse PMNs) pre-incubated at 37 °C for 15 min in the presence or absence of different concentrations of DF3016A or vehicle was seeded in the upper compartment. The chamber was incubated at 37 °C in air with 5% CO2 for 45 min. At the end of incubation, filters containing migrated cells were removed, fixed, and stained with Diff-Quik, and five oil immersion fields at high magnification (×100; Zeiss microscope) were counted after sample coding.
Peritoneal Murine Macrophage Preparation and Lipopolysaccharide-Induced Prostaglandin E2 Production
Peritoneal exudate cells were collected from peritoneal washings of male mice Balb/c mice (20–25 g and 7–9 weeks of age, provided by Charles River, Calco, Italy), 5 days after i.p. injection of 3% thioglycollate in saline (1.5 ml per mouse), as previously reported (Mascagni et al. 2000). Cells were placed at 1 × 106 ml−1 in 96-well plates and non-adherent cells removed by gentle washing 2 h later. DF3016A was then added to adherent macrophages 20 min before adding lipopolysaccharide (LPS) (1 μg ml−1). Control cells received vehicle at the appropriate dilution. Total prostaglandin E2 (PGE2) production was determined in the supernatant 24 h after LPS stimulation. PGE2 levels were measured by Enzyme Immunoassay (EIA) Kit (sensitivity 2.5 pg per well).
In Vitro Selectivity
DF3016A was tested in the SafetyScreen44 Panel performed at Eurofins Cerep SA (France) by radio ligand binding assays to assess the off-target activities towards a panel of GPCRs, enzymes, ion channels, transporters, and nuclear receptors. All selected targets are recommended by four major pharmaceutical companies (Bowes et al. 2012). The compound was tested at a single concentration of 10 μM in triplicate.
DF3016A was dissolved in dimethyl sulfoxide (DMSO) to achieve 10 mM stock solution that was diluted with water/HBSS to a final concentration of 10 μM. Cell membrane homogenates (48 μg protein) were incubated for 60 min at 22 °C with the respective reference compound in the absence or presence of the test compound in a buffer containing 50 mM Tris-HCl (pH 7.4), 2 mM MgCl2, and 1 mM ethylene-diamine-tetra-acetic acid (EDTA). After incubation, the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard Instruments, Meriden, CT, USA) presoaked with 0.3% polyethyleneimine (PEI), and rinsed several times with ice-cold 50 mM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard Instruments). The filters were dried, then counted for radioactivity in a scintillation counter (Topcount, Packard Instruments) using a scintillation cocktail (Microscint-O, Packard Instruments).
Brain Penetration in the Rat
DF3016A was studied in male Sprague Dawley rats (n = 3) to investigate the BBB penetration after oral administration. SD rats (370–450 g and 3–4 months of age at the time of testing) were purchased from Charles River, UK. All animals were housed in groups of three per room under controlled conditions of temperature (19 to 21 °C) and humidity (50%), and maintained on a 12-h light/dark cycle (lights on at 7 and lights off at 19 including a 30-min dawn/dusk lights increasing/dimming period, respectively). Animals had free access to rat chow pellets CRM (P) and water except during the experimental procedures. All animals were weighed immediately before the test. The test drug was administered orally at 30 mg/kg using saline solution (0.9% NaCl) as vehicle. The animals were sacrificed at Tmax (2 h postdose) with sodium pentobarbital administered intraperitoneally. Cardiac blood samples, 0.6 ml, were obtained and placed in EDTA-coated tubes. The tubes were spun at 13,000 rpm for 4 min and 100 μl of supernatant taken and immediately stored at − 80 °C prior to analysis. Plasma samples were analyzed by liquid chromatography–mass spectrometry (LC-MS/MS) following extraction by protein precipitation with internal standard in acetonitrile, and levels of parent measured against an extracted calibration curve of plasma samples spiked with test compound. The whole brain was harvested then placed in a plastic tube and immediately frozen at − 80 °C until required. Brain tissue were homogenized and analyzed by LC-MS/MS following extraction by protein precipitation, and levels of parent measured against an extracted calibration curve of brain homogenate samples spiked with the test compound. Total exposure levels of the test compound in brain and plasma at 2 h postdose were determined.
Primary Cortical Cultures
Neuronal cultures were prepared from cortices of 17–18-day-old Sprague-Dawley rat fetuses. Two pregnant rats, provided from Envigo RMS S.r.l. (Z.I. Azzida, 57–33,049 S. Pietro al Natisone, Udine Italy), were euthanized by exposition to a gradually rising concentration of CO2 and then decapitated. Rat brains were isolated using standard procedures, and after the elimination of meninges, the cortical tissues were dissociated by trituration and digestion with 20 U/ml papain (Invitrogen, Karlsruhe, Germany) for 30 min at 37 °C. Approximately 1 × 106 cells in 1 ml DMEM/F12 medium containing 0.2 mM Glutamax, 1% penicillin/streptomycin (Pen/Strep), 5% fetal bovine serum, and 5% horse serum were seeded into poly-D-lysine pretreated plates. After 4–6 h, the medium was changed with Neurobasal containing the serum-free B27 supplement (2%), 1% Pen/Strep, and 0.2 mM Glutamax that selectively inhibits glial proliferation. Neurons were maintained in a humidified atmosphere (5% CO2/95% air) at 37 °C and used for experiments after 8–10 days in vitro. All experimental procedures were performed according to Italian law 116/92, authorization no. 104–2013-A.
Drug Treatment
DF3016A was dissolved in DMSO. The final concentration of DMSO in the medium was below 0.1%. To characterize toxicity, primary cultures were treated for 24 h with different DF3016A concentrations (10 nM, 50 nM, 100 nM, 500 nM, 1 μM, 5 μM, 10 μM, 100 μM, 0.5 mM, 1 mM), finally treated with suitable concentration, and exposed to OGD/R (1 h first, and then 3 h). Control conditions underwent neither drug treatment nor OGD/R. After treatments, cultures were used for in vitro experiments. Different tests were carried out as described below.
Experimental Model
We performed an experimental model in vitro reproducing neuroinflammation stress condition. Less than 50% mortality was induced to mimic the damage occurred in the area surrounding the ischemic core where neurons still have capacity for recovery. Previously time–response experiments conducted for more than 1 h (2 h) of OGD induced more than 50% mortality (data not shown). On this basis, we selected only 1-h OGD that induces about 20% mortality in primary neuronal cultures and allows resilience of alive neurons.
OGD/R
DF3016A (500 nM) was added to the cortical neurons which were exposed to O2 and glucose deprivation with glucose-free Neurobasal in Hypoxia Chamber for Cell Culture (BioSpherix) at 5% CO2 atmosphere-85% N2. O2 concentration was monitored by using a ProOx sensor controller, and the 0.03% O2 value was considered acceptable. Cells were incubated in this anaerobic chamber for 1 h and then removed from the anaerobic environment. Hence, neurobasal media were substituted and cultures placed in 95% air and 5% CO2 conditions, for 3 h of reoxygenation. Cells of control group were treated identically except that they were not exposed to OGD/R.
Cell Viability Evaluation
Quantifications of neuronal viability were assessed by MTS method using Cell Titer One Solution Cell Proliferation Assay (Promega Corporation Madison, WI, USA) based on 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenil)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (dilution 1:10). Viable cells with active metabolism convert MTT into formazan, detected by absorbance measurements at 490 nm in a BioTeek Elx800 microplate reader. Acridine Orange (AO) and Propidium Iodide (PI) double staining have been used to evaluate viable and dead cells in controls and exposed neurons. Images have been acquired through a fluorescent microscope combined with a Digital Color Camera.
Isolation of RNA and qRT-PCR
The total RNA, including microRNA (miRNA), from cells in cultures were extracted with Ribospin Kit (GenaAll) followed by Riboclear Plus Kit (GenaAll) to remove residual DNA according to the manufacturer’s protocols.
Total mRNA transcription (2μg/20 μl) was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and reverse transcription for MicroRNA (10 ng/15 μl) was performed using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems).
The qPCR 20 μl/reactions for miR-181a (assay ID: hsa-miR-181a) and C5aR (assay ID: Rn02134203_s1) were conducted in triplicate using TaqMan® MicroRNA Assay Kit and TaqMan® Gene Expression Assays protocol (Applied Biosystems), respectively, following the manufacturer’s instructions. Reactions were performed using a Step One Plus Applied Biosystems thermocycler (Life Technologies) with the default protocol. Briefly, for each reaction 40 cycles of amplification with the following profile were performed: 95 °C for 10 min for the first cycle, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min.
Relative intensity of PCR-specific amplicons was calculated using the 2−ΔΔCt method (Livak and Schmittgen 2001). Beta-actin (assay ID: Rn00667869_m1) and U6 (assay ID: 001973) small nuclear RNA (snRNA) (Han et al. 2015) were used as internal control to normalize the level of each transcript.
Fluo-4 NW Calcium Assay
Neuronal cells, grown on 96-well plates, were exposed to OGD/R and treated with 500 nM of DF3016A; growth media were removed, and 100 μl of the dye loading solution (F36206 Fluo-4 NW Calcium Assay Kit, Invitrogen) was added carefully to each well. Plates were incubated at 37 °C for 30 min and then at room temperature for an additional 30 min. Fluorescence was revealed at 516 nm with Victor 3 Model 1420-012 Multi-label Microplate Reader. Data were presented as the ratio between Ca++ influx and neuronal viability.
ELISAs
Quantitative measurements of cellular proteins (C5a, TNF-α, IL-1β, and IL-6) in cell culture supernatants were determined by ELISA methods using Rat IL-6, Mouse TNFα, Rat IL-1β PicoKine™ ELISA Kits (Boster Biological Technology) and Rat C5a ELISA Kit (Elabscience Biotechnology Inc.) according to the manufacturer’s instructions. All tests were performed in triplicate.
Data Analysis
Data are expressed as mean ± standard error of mean (SE). Mean group differences were evaluated by one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. P values less than 0.05 were considered statistically significant. All statistical analyses were performed using SPSS 19.0 software.
Results
In Vitro Characterization
In vitro pharmacological characterization showed that DF3016A did not inhibit spontaneous cell migration per se yet potently inhibited the C5a-induced human PMN migration with an IC50 of 50 nM and cross-reactions with rat and mouse C5aR (IC50 = 45 nM and 37 nM, respectively). The compound exhibited a high degree of selectivity with no effect on PMN migration induced by other leukocyte activators such as CXCL8, CXCL1, CCL3, fMLP, CXCL12, and on LPS-induced PGE2 accumulation up to 10 μM concentration.
In Vitro Selectivity
DF3016A was tested in order to assess the off-target activities towards a panel of GPCRs, enzymes, ion channels, transporters, and nuclear receptors (44 targets in total, according to the SafetyScreen44 assay provided by Eurofins Cerep). Radioligand binding assays were carried out at the test concentration of 10 μM in triplicate. We found that DF3016A did not show any inhibition of the assayed receptors, confirming the high selectivity towards the C5aR.
Brain Penetration in Rats
With the aim of assessing the therapeutic potential of DF3016A in CNS disorders, brain penetration of the molecule was tested in SD rats after oral administration at 30 mg/kg. At 2 h postdose (Tmax), we found that the levels of DF3016A were 104,683 ng/ml in plasma and 4172 ng/g in brain, with a brain to plasma ratio of 0.04. Once adjusted for the free plasma and brain fractions (0.7% and 7.7%, respectively), this increases to an unbound brain to plasma ratio (Kpuu) of 0.44 (Table 1). This means that oral DF3016A is able to sufficiently penetrate the brain.
Table 1.
Concentrations of DF3016A in plasma and brain of rats following oral dosing (30 mg/kg)
| DF3016A | |||||
|---|---|---|---|---|---|
| rPPB Fu | rBTB Fu | PK (30 mg/kg, PO) | |||
| Plasma Cmax (ng/mL) | Brain Cmax (ng/g) | Kp | Kpuu | ||
| 0.7% | 7.7% | 104,683 ± 8867 | 4172 ± 323 | 0.04 | 0.44 |
The values of plasma and brain concentrations are the mean ± S.D. (n = 3)
rPPB rat plasma protein binding, rBTB rat brain tissue binding, Kp brain to plasma ratio, Kpuu unbound brain to plasma ratio
DF3016A Evaluation on Neuronal Viability
To evaluate the effects of DF3016A on neuronal viability we performed dose-response experiments with different DF3016A concentrations for 24 h. Data were obtained using the MTS assay from three different cultures; neurons exhibited dose-dependent changes in cellular viability (Fig. 1a). From 10 nM to 5 μM of DF3016A concentration, no significant mortality was observed compared to the control. On the basis of these results, the 500 nM concentration was chosen for performing the subsequent experiments, and cortical neurons were treated for 24, 48, and 72 h without significant viability changes (Fig. 1b).
Fig. 1.

a Effect of different concentration of DF3016A on neuronal cortical cells. Values in the columns are means ± SEM of three different cultures. The line + symbol shows the same data as a percentage of vitality. b Viability of neurons treated with 500 nM of DF3016A for 24, 48, and 72 h
Cellular Viability After OGD-R Injury
To examine the effect of DF3016A during OGD and at different time points of re-oxygenation, 500 nM of DF3016A was added to the culture medium. As estimated by the MTS assay, the viability of OGD/R cultures was reduced by about 20% compared to the control. Following 3 to 48 h of reperfusion, the treatment with 500 nM DF3016A significantly attenuated OGD/R-induced cell death (Fig. 2). As described, neuronal viability was also assessed using Acridine Orange/Ethidium Bromide (AO/ET) staining. Viable cells appear evenly green, in contrast to dead cells that appear as red spots due to chromatinic deposits. OGD/R exposure induced an increase in neuronal loss (Fig. 3c, d) while in ischemic neurons, treated with DF3016A, the neuronal death caused by OGD/R exposure was close to that of control (Fig. 3e, f).
Fig. 2.

Cortical neurons treated with DF3016A 500 nM and exposed to OGD and different reoxygenation time points (3, 12, 24, 48 h). Data are means ± SEM of three different cultures. *P < 0.05, **P < 0.01, ***P < 0.001 (Bonferroni’s post hoc test). CTR-UT control-untreated, OGD/R-UT neurons exposed to OGD/R, CTR-DF control treated with 500 nM DF3016A, OGD/R-DF neurons exposed to OGD/R and treated with 500 nM DF3016A
Fig. 3.

Acridine Orange/Ethidium Bromide (AO/ET) staining for discrimination of live and dead cells. a, b represent control neurons; c, d are neurons exposed to OGD/R 3 h; and e, f are neurons treated with 500 nM DF3016A and exposed to OGD/R 3 h
Effects of DF3016A Treatment on Neuronal Calcium Influx
C5aR has been suggested to also act as a modulator of Ca2+ influx via L-type voltage-gated Ca2+ channels (L-VGCCs) in neurons (Farkas et al. 2012). As shown in Fig. 4, OGD/R-UT exposure induced an abnormal Ca2+ influx (P < 0.001 vs non-exposed cells) related to neuronal loss (shown in Fig. 2) probably due to excitotoxicity events. Notably, the 500-nM C5a antagonist treatment triggered a significant reduction of Ca2+ influx in OGD/R neurons (P < 0.001 OGD/R-DF vs OGD/R-UT cells).
Fig. 4.

Calcium influx measured in control and DF-treated cultures exposed to OGD/R
DF3016A as an Antagonist on C5a And C5aR Expression
From the results obtained after OGD/R exposure, regarding the expression of C5aR and C5a protein release, we have observed a significant decrease of C5a released protein (Fig. 5a), tightly related to the C5aR over-expression (Fig. 5b). DF3016A treatment reverted C5aR gene expression to the control levels (Fig. 5b). Likewise, DF3016A restored C5a protein level to not significant values compared to control (Fig. 5a).
Fig. 5.
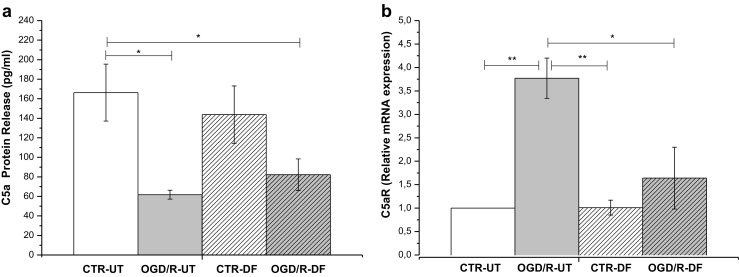
a OGD/R-UT condition dramatically reduced the C5a protein level but induced a significant increase of C5aR gene expression. b DF3016A C5aR antagonist notably reduced C5aR transcription (mean ± SEM six independent experiments). *P < 0.05, **P < 0.01, ***P < 0.000 (Bonferroni’s post hoc test). CTR/R-UT control-untreated, OGD/R-UT OGD/R exposed neurons, CTR/R-DF control treated with 500 nM DF3016A, OGD/R-DF OGD/R exposed neurons treated with 500 nM DF3016A
DF3016A Effects on Pro-Inflammatory Cytokines
Considering that C5aR activation largely contributes to inflammatory responses in several diseases leading to neurodegenerative conditions (Choudhry et al. 2016; Yanamadala and Friedlander 2010), we analyzed the major pro-inflammatory cytokine release after OGD/R exposure in neuronal cultures with and without 500-nM DF3016A treatment. ELISA analysis showed a significant upregulation of TNF-α (Fig. 6a) and IL-1β (Fig. 6b) during ischemic neuronal injury. The IL-6 release was higher in OGD/R, even if data were not significant (Fig. 6c). The effect on IL-6 was surprising. We presume that our sublethal stimulation was unable to elicit enough IL-6 expression in cortical neurons. Indeed, recent data demonstrated that high IL-6 levels are released in dorsal root ganglion cultures with high satellite glial cell levels (80%) and is stimulated by lipopolysaccharide release (Leisengang et al. 2018). C5aR antagonist treatment lead to a restoration of control conditions of ischemic/hypoxic neurons (Fig. 6a, b).
Fig. 6.
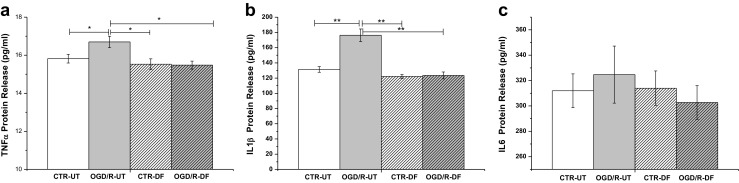
Graph showing pro-inflammatory cytokine release after OGD/R exposure and DF3016A treatment (mean ± SEM of four independent experiments; *P < 0.05, **P < 0.01) (Bonferroni’s post hoc test). CTR-UT control-untreated, OGD/R-UT OGD/R exposed neurons, CTR-DF control treated with 500 nM DF3016A, OGD/R-DF OGD/R exposed neurons treated with 500 nM DF3016A
Effects of DF3016A on miR-181a Expression
MicroRNAs, small non-coding RNA molecules, play an essential role in the regulation of gene expression through the degradation/inhibition of mRNA targets. In particular, miR-181a seems to be actively involved in the response to inflammatory stimuli (Xie et al. 2013). OGD/R induced a significant upregulation of miR-181 expression (P < 0.01 vs non-exposed cells), but DF3016A treatment completely attenuated the miR-181a over-expression (P < 0.001 OGD/R-DF vs OGD/R-UT) (Fig. 7).
Fig. 7.

Graph showing the miR-181a relative gene expression levels under OGD/R condition and DF3016A treatment. Data from four independent experiments (mean ± SEM). **P < 0.01, ***P < 0.000 (Bonferroni’s post hoc test). CTR-UT control-untreated, OGD/R-UT OGD/R exposed neurons, CTR-DF control treated with 500 nM DF3016A, OGD/R-DF OGD/R exposed neurons treated with 500 nM DF3016A
Discussion
Neuroinflammation can involve activation of the brain immune system in response to an inflammatory challenge (Alexander et al. 2008). It is characterized by cellular and molecular changes within the brain that involve many immunological mediators such as the complement system (Van Beek et al. 2003). Neuroinflammation and the subsequent neurodegenerative processes have clear clinical consequences (Paladini et al. 2015; Paladini et al. 2016; Fusco et al. 2017; Zis et al. 2017; Zis et al. 2017a; Varrassi et al. 2018) Uncontrolled complement activation can lead to excess tissue inflammation and damage; moreover, neuroinflammation induces sensitization and synaptic hyperactivity in the peripheral and central nervous system (Varrassi et al. 2015).
Here we evaluated the effects of DF3016A, a new molecule that acts as a selective C5aR antagonist. C5a is a crucial terminal component of the complement cascade supporting nociceptive sensitization and inflammation. The results reported here show that DF3016A is a potent and specific inhibitor of C5aR (IC50 in the nanomolar range) with a high selectivity based on a panel of GPCRs. Furthermore, DF3016A is characterized as an orally bioavailable C5aR antagonist (rat bioavailability = 69%, oral t1/2 = 6.1 h) with good brain penetration (Kpuu = 0.44) upon oral administration. Based on this evidence, we further characterized the effects of the compound on primary rat cortical neurons exposed to sublethal OGD/R, an in vitro model of neuroinflammation-related processes. Firstly, we showed that treatment with DF3016A for 24 h in the concentration range 10 nM–5 μM did not affect neuronal viability and the toxic concentration of DF3016A (TC50 = dose that kills 50% of the cells) was between 0.5 and 1 mM. Additionally, a longer treatment for up to 72 h with 500 nM DF3016A did not affect significantly neuronal viability. We then tested the effect of the drug using the OGD/R model that produces deficits in synaptic function, which has been widely used as an in vitro model to mimic the effects of ischemia-reperfusion injury and neuroinflammation in neurodegenerative diseases. Our experimental protocol was based on the exposure of primary neuronal cultures to 1 h of OGD and 3 h of reoxygenation. The protocol also allowed us to study the early neuronal response, based on the finding of no significant differences in the viability of cultures kept in the reoxygenation state for up to 48 h. This insult induces a mild degree of neuronal death and allows investigation of the functional activity of viable neurons. During ischemia, an excess of calcium influx is believed to be one of the first events triggering excitotoxic cell death (Mukherjee and Pasinetti 2001; Farkas et al. 1998). The increase in intracellular calcium level is an important element in the signal-transduction network activated after the binding of C5a fragment complement to cell-surface receptors (Monk and Partridge 1993; Triantafilou et al. 2013). We observed that DF3016A treatment nullified the large increase of Ca2+ influx induced by OGD/R and restored calcium concentrations to control levels. Previous work has demonstrated that the increased levels of released C5a in neuronal cultures exposed to OGD are tightly linked to apoptosis (Pavlovski et al. 2012). In the present study, we did not observe any increase in release of C5a protein in OGD/R neurons. On the contrary, C5aR gene expression was upregulated after OGD/R treatment. This upregulation is an interesting and may suggest that our OGD/R model induces an early upregulation of C5aR expression, and subsequent binding of C5a to its receptor, explaining the lower levels of C5a protein observed. It is noteworthy that the complement pathway can be activated immediately after neuronal injury, and the increased receptor expression is one of the first steps in this response (Woodruff et al. 2010). Moreover, our results may not be in contrast to those previously observed after 12 h of OGD (Pavlovski et al. 2012), as the increase in C5a protein could be a subsequent process activated by more detrimental injuries, leading neuronal cells to apoptosis. Indeed, production of a proteolytic fragment of C5a is a late event in the complement cascade, regulated by many complement-regulatory molecules which are subject to control by the expression of several receptors such as inflammatory mediators (Woodruff et al. 2010; Van Beek et al. 2000). However, DF3016A treatment restores the control situation, with the expression of C5a maintaining the unchanged levels of the protein in OGD/R-treated neurons. As shown by different authors (Sayah et al. 1999; Song et al. 2018), C5a protein binding to its receptor can lead to the production of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 which in turn affect complement expression (Busch et al. 2013). In keeping with these pieces of evidence, in our experimental model, we observed overexpression of the major pro-inflammatory cytokines in OGD/R-exposed cultures. Remarkably, DF3016A treatment abolished the increase. The complex cytokine network in CNS is involved not only in the immune response but also in a variety of physiological and pathological processes (Szelényi 2001). Nevertheless, even though there are reports that low concentrations of cytokines can exhibit beneficial effects on neuronal viability and neurological function (Di Loreto et al. 2000; Vezzani and Viviani 2015), an injury-induced overexpression promotes neuronal death.
Increasing evidence supports a significant role for miRNAs in response to cerebral ischemia (Ouyang and Giffard 2013; Dharap et al. 2009). MiRNAs are small non-coding RNA molecules that control gene expression at the post-transcriptional level, and are abundantly expressed during brain development and in the adult mammalian brain (Kos et al. 2016). MiRNAs target messenger RNAs (mRNAs). MiRNAs can induce mRNA degradation or the repression of the process of translation to modulate gene expression, and a single miRNA can bind and regulate several mRNA targets. The miR-181 family, especially miR-181a and miR-181b, are enriched in the brain (Miska et al. 2004), and their aberrant expression has been associated with brain diseases. The knockdown of miR-181a enhanced the production of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, IL-8) (Hutchison et al. 2013), and increased levels of the anti-inflammatory cytokine IL-10 result from miR-181 over-expression (Murray 2005; de Vries 1995) in a murine stroke model. In a model of cerebral ischemia, the expression of miR-181 increases in the area where cell death occurs, whereas expression is lower in the penumbra zone where cells can survive (Ouyang et al. 2012). Altogether, these data indicate that miR-181 is involved in general transcriptional profiles of cellular stress response and cellular survival-related changes. Therefore, we focused on the investigation of miR-181a in our model. Our data revealed increases in miR-181a expression after OGD/R exposure that could represent an early neuronal response to ischemic injury. DF3016A treatment completely abolished this over-expression, suggesting that this C5aR antagonist could affect immune-mediator expression also through interactions with transcriptional factors such as miR-181a. The overall profile of DF3016A made it an excellent candidate for additional in vitro and in vivo studies to assess the pharmacological effects of a C5aR antagonist with good brain penetration.
Conclusion
Altogether, these results support for the first time the hypothesis that DF3016A, a potent and selective C5aR inhibitor, might exhibit neuroprotective effects against neuroinflammatory processes induced by OGD/R in primary cortical neurons. This could have important clinical consequences. Our experimental model allowed the investigation of CNS molecular and biological mechanisms in a controlled environment in vitro and on individual cell types. We did not study potential interactions with different cellular phenotypes in the brain. In conclusion, this study suggests that DF3016A might be a candidate molecule for a novel therapeutic approach to neuroinflammation-related diseases. Further researches are in progress to evaluate the possible cellular and molecular mechanisms underlying the observed neuroprotection.
Acknowledgements
The authors are indebted to Professor Anthony Dickenson for the critical review of the final manuscript.
Funding
This work was supported by the Paolo Procacci Foundation (PPF) and Dompé Farmaceutici SpA funds.
Compliance with Ethical Standards
Conflicts of Interest
Laura Brandolini, Gianluca Bianchini, and Marcello Allegretti are employees of Dompé Farmaceutici SpA, Italy. The company has interests in the development of C5aR antagonists for the treatment of pain conditions. The other authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice where the studies were conducted.
Footnotes
The original version of this article was revised due to a retrospective Open Access order.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Laura Brandolini and Marta Grannonico contributed equally to this work.
Change history
5/6/2019
The article The Novel C5aR Antagonist DF3016A Protects Neurons Against Ischemic Neuroinflammatory Injury, written by Laura Brandolini, Marta Grannonico, Gianluca Bianchini, Alessia Colanardi, Pierluigi Sebastiani, Antonella Paladini, Alba Piroli, Marcello Allegretti, and Giustino Varrassi.
References
- Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, Tomlinson S. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J Neuroinflammation. 2015;12:247. doi: 10.1186/s12974-015-0464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin-Yang in neuroinflammation—neuro-protection and -degeneration. J Neurochem. 2008;107(5):1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12(6):723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- Badaut J, Hirt L, Price M, de Castro Ribeiro M, Magistretti PJ, Regli L. Hypoxia/hypoglycemia preconditioning prevents the loss of functional electrical activity in organotypic slice cultures. Brain Res. 2005;1051(1–2):117–122. doi: 10.1016/j.brainres.2005.05.063. [DOI] [PubMed] [Google Scholar]
- Barnum SR. Complement in central nervous system inflammation. Immunol Res. 2002;26(1–3):7–13. doi: 10.1385/IR:26:1-3:007. [DOI] [PubMed] [Google Scholar]
- Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V, Cesta MC, Galliera E, Martinez FO, Di Bitondo R, Troiani G, Sabbatini V, D'Anniballe G, Anacardio R, Cutrin JC, Cavalieri B, Mainiero F, Strippoli R, Villa P, Di Girolamo M, Martin F, Gentile M, Santoni A, Corda D, Poli G, Mantovani A, Ghezzi P, Colotta F. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci U S A. 2004;101(32):11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov. 2012;11(12):909–922. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Busch C, Girke G, Kohl B, Stoll C, Lemke M, Krasnici S, Ertel W, Silawal S, John T, Schulze-Tanzil G. Complement gene expression is regulated by pro-inflammatory cytokines and the anaphylatoxin C3a in human tenocytes. Mol Immunol. 2013;53(4):363–373. doi: 10.1016/j.molimm.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- Choudhry N, Li K, Zhang T, Wu KY, Song Y, Farrar CA, Wang N, Liu CF, Peng Q, Wu W, Sacks SH, Zhou W. The complement factor 5a receptor 1 has a pathogenic role in chronic inflammation and renal fibrosis in a murine model of chronic pyelonephritis. Kidney Int. 2016;90(3):540–554. doi: 10.1016/j.kint.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- de Vries JE. Immunosuppressive and anti-inflammatory properties of interleukin 10. Ann Med. 1995;27(5):537–541. doi: 10.3109/07853899509002465. [DOI] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29(4):675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Loreto S, Corvetti L, Maccarone R, Piancatelli D, Adorno D. Interleukin 1-beta modulates the effects of hypoxia in neuronal culture. J Neuroimmunol. 2000;106(1–2):32–42. doi: 10.1016/s0165-5728(00)00209-5. [DOI] [PubMed] [Google Scholar]
- Farkas I, Baranyi L, Takahashi M, Fukuda A, Liposits Z, Yamamoto T, Okada H. A neuronal C5a receptor and an associated apoptotic signal transduction pathway. J Physiol. 1998;507(Pt 3):679–687. doi: 10.1111/j.1469-7793.1998.679bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas I, Takahashi M, Fukuda A, Yamamoto N, Akatsu H, Baranyi L, Tateyama H, YamamotoT ON, Okada H. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer’s disease. J Immunol. 2003;170:5764–5771. doi: 10.4049/jimmunol.170.11.5764. [DOI] [PubMed] [Google Scholar]
- Farkas I, Varju P, Szabo E, Hrabovszky E, Okada N, Okada H, Liposits Z. Estrogen enhances expression of the complement C5a receptor and the C5a-agonist evoked calcium influx in hormone secreting neurons of the hypothalamus. Neurochem Int. 2008;52(4–5):846–856. doi: 10.1016/j.neuint.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Farkas I, Sárvári M, Aller M, Okada N, Okada H, Likó I, Liposits Z. Estrogen receptor α and β differentially mediate C5aR agonist evoked Ca2+-influx in neurons through L-type voltage-gated Ca2+ channels. Neurochem Int. 2012;60(6):631–639. doi: 10.1016/j.neuint.2012.02.024. [DOI] [PubMed] [Google Scholar]
- Fusco M, Skaper S, Coaccioli S, Paladini A, Varrassi G. Degenerative joint diseases and neuroinflammation. Pain Pract. 2017;17:522–532. doi: 10.1111/papr.12551. [DOI] [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Götze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. 1997;150(1):31–41. [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;3(8):3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Ge X, Tan J, Chen F, Gao H, Lei P, Zhang J. Establishment of lipofection protocol for efficient miR-21 transfection into cortical neurons in vitro. DNA Cell Biol. 2015;34(12):703–709. doi: 10.1089/dna.2015.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison ER, Kawamoto EM, Taub DD, Lal A, Abdelmohsen K, Zhang Y, Wood WH, 3rd, Lehrmann E, Camandola S, Becker KG, Gorospe M, Mattson MP. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61(7):1018–1028. doi: 10.1002/glia.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara K, Yanoma J, Tanaka M, Nakajima T, Kosugi T. Nitric oxide produced during ischemia is toxic but crucial to preconditioning-induced ischemic tolerance of neurons in culture. Neurochem Res. 2004;29(4):797–804. doi: 10.1023/b:nere.0000018853.30131.4d. [DOI] [PubMed] [Google Scholar]
- Kos A, Olde Loohuis N, Meinhardt J, van Bokhoven H, Kaplan BB, Martens GJ, Aschrafi A. MicroRNA-181 promotes synaptogenesis and attenuates axonal outgrowth in cortical neurons. Cell Mol Life Sci. 2016;73(18):3555–3567. doi: 10.1007/s00018-016-2179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisengang S, Ott D, Murgott J, Gerstberger R, Rummel C, Roth J. Primary cultures from rat dorsal root ganglia: responses of neurons and glial cells to somatosensory or inflammatory stimulation. Neuroscience. 2018;394:1–13. doi: 10.1016/j.neuroscience.2018.10.018. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(− delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mascagni P, Sabbatini V, Biordi L, Martinotti S, Allegretti M, Marullo A, Caselli G, Bertini R. R- and S-isomers of nonsteroidal anti-inflammatory drugs differentially regulate cytokine production. Eur Cytokine Netw. 2000;11(2):185–192. [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5:R68. doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk PN, Partridge LJ. Characterization of a complement-fragment-C5a-stimulated calcium-influx mechanism in U937 monocytic cells. Biochem J. 1993;295(Pt 3):679–684. doi: 10.1042/bj2950679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriconi A, Cunha TM, Souza GR, Lopes AH, Cunha FQ, Carneiro VL, Pinto LG, Brandolini L, Aramini A, Bizzarri C, Bianchini G, Beccari AR, Fanton M, Bruno A, Costantino G, Bertini R, Galliera E, Locati M, Ferreira SH, Teixeira MM, Allegretti M. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc Natl Acad Sci U S A. 2014;111:16937–16942. doi: 10.1073/pnas.1417365111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Pasinetti GM. The role of complement anaphylatoxin C5a in neurodegeneration: implications in Alzheimer’s disease. J Neuroimmunol. 2000;105(2):124–130. doi: 10.1016/s0165-5728(99)00261-1. [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem. 2001;77(1):43–49. doi: 10.1046/j.1471-4159.2001.00167.x. [DOI] [PubMed] [Google Scholar]
- Müller-Ladner U, Jones JL, Wetsel RA, Gay S, Raine CS, Barnum SR (1996) Enhanced expression of chemotactic receptors in multiple sclerosis lesions. J Neurol Sci 144(1–2):135–41 [DOI] [PubMed]
- Murray PJ. The primary mechanism of the IL-10-regulated anti-inflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102(24):8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SV, Perez-Pinzon MA. Ischemic preconditioning treatment of astrocytes transfers ischemic tolerance to neurons. Cond Med. 2017;1(1):2–8. [PMC free article] [PubMed] [Google Scholar]
- O'Barr SA, Caguioa J, Gruol D, Perkins G, Ember JA, Hugli T, Cooper NR. Neuronal expression of a functional receptor for the C5a complement activation fragment. J Immunol. 2001;166(6):4154–4162. doi: 10.4049/jimmunol.166.6.4154. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Giffard RG. MicroRNAs regulate the chaperone network in cerebral ischemia. Transl Stroke Res. 2013;4(6):693–703. doi: 10.1007/s12975-013-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE, Sun X, Giffard RG. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis. 2012;45(1):555–563. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini A, Fusco M, Coaccioli S, Skaper SD, Varrassi G. Chronic pain in the elderly: the case for new therapeutic strategies. Pain Physician. 2015;18:E863–E876. [PubMed] [Google Scholar]
- Paladini A, Fusco M, Cenacchi T, Schievano C, Piroli A, Varrassi G. Palmitoylethanolamide, a special food for medical purposes, in the treatment of chronic pain: a pooled data meta-analysis. Pain Physician. 2016;19:11–24. [PubMed] [Google Scholar]
- Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;6(9):3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- Quadros AU, Cunha TM. C5a and pain development: an old molecule, a new target. Pharmacol Res. 2016;112:58–67. doi: 10.1016/j.phrs.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Sayah S, Ischenko AM, Zhakhov A, Bonnard AS, Fontaine M. Expression of cytokines by human astrocytomas following stimulation by C3a and C5a anaphylatoxins: specific increase in interleukin-6 mRNA expression. J Neurochem. 1999;72(6):2426–2436. doi: 10.1046/j.1471-4159.1999.0722426.x. [DOI] [PubMed] [Google Scholar]
- Song WC, Sarrias MR, Lambris JD. Complement and innate immunity. Immunopharmacology. 2000;49:187–198. doi: 10.1016/s0162-3109(00)80303-3. [DOI] [PubMed] [Google Scholar]
- Song Y, Wu KY, Wu W, Duan ZY, Gao YF, Zhang LD, Chong T, Garstka MA, Zhou W, Li K. Epithelial C5aR1 signaling enhances uropathogenic Escherichia coli adhesion to human renal tubular epithelial cells. Front Immunol. 2018;9:949. doi: 10.3389/fimmu.2018.00949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Szelényi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54(4):329–338. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126(Pt 13):2903–2913. doi: 10.1242/jcs.124388. [DOI] [PubMed] [Google Scholar]
- Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, Fontaine M. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. 2000;161(1):373–382. doi: 10.1006/exnr.1999.7273. [DOI] [PubMed] [Google Scholar]
- Van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Varrassi G, Fusco M, Coaccioli S, Paladini A. Chronic pain and neurodegenerative processes in elderly people. Pain Pract. 2015;15(1):1–3. doi: 10.1111/papr.12254. [DOI] [PubMed] [Google Scholar]
- Varrassi G, Fusco M, Skaper SD, Battelli D, Zis P, Coaccioli S, Pace MC, Paladini A. A pharmacological rationale to reduce the incidence of opioid induced tolerance and hyperalgesia: a review. Pain Ther. 2018;7:59–75. doi: 10.1007/s40122-018-0094-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96(Pt a):70–82. doi: 10.1016/j.neuropharm.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. NeuroMolecular Med. 2010;12(2):179–192. doi: 10.1007/s12017-009-8085-y. [DOI] [PubMed] [Google Scholar]
- Xie W, Li Z, Li M, Xu N, Zhang Y. miR-181a and inflammation: miRNA homeostasis response to inflammatory stimuli in vivo. Biochem Biophys Res Commun. 2013;430(2):647–652. doi: 10.1016/j.bbrc.2012.11.097. [DOI] [PubMed] [Google Scholar]
- Yanamadala V, Friedlander RM. Complement factors and their G protein-coupled receptors in neuroprotection and neurodegeneration. Trends Mol Med. 2010;16(2):69–76. doi: 10.1016/j.molmed.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50(3):1392–1399. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zis P, Daskalaki A, Bountouni I, Sykioti P, Varrassi G, Paladini A. Depression and chronic pain in the elderly: links and management challenges. Clin Interv Aging. 2017;12:709–720. doi: 10.2147/CIA.S113576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zis P, Paladini A, Piroli A, McHugh PC, Varrassi G, Hadjivassiliou Pain as first manifestation of paraneoplastic neuropathies: a systematic review and meta-analysis. Pain Ther. 2017;6:143–151. doi: 10.1007/s40122-017-0076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]