

Abstract
Reaction of the mononuclear nonheme complex [FeII(CH3CN)(N3PyS)]BF4 (1) with an HNO donor, Piloty’s acid (PhSO2NHOH, P.A.), at low temperature affords a high-spin (S = 2) FeII-P.A. intermediate (2), characterized by 57Fe Mössbauer and Fe K- edge X-ray absorption (XAS) spectroscopies, with interpretation of both supported by DFT calculations. The combined methods indicate that P.A. anion binds as the N-deprotonated tautomer (PhSO2NOH−) to [FeII(N3PyS)]+, leading to 2. Complex 2 is the first spectroscopically characterized example, to our knowledge, of P.A. anion bound to a redox-active metal center. Warming of 2 above −60 °C yields the stable {FeNO}7 complex [Fe(NO)(N3PyS)]BF4 (4), as evidenced by 1H NMR, ATR-IR, and Mössbauer spectroscopies. Isotope labeling experiments with 15N-labeled P.A. confirm that the nitrosyl ligand in 4 derives from P.A. In contrast, addition of a second equivalent of a strong base leads to S-N cleavage and production of an {FeNO}8 species, the deprotonated analog of an Fe-HNO complex. This work has implications for the targeted delivery of HNO/ NO−/NO‧ to nonheme Fe centers in biological and synthetic applications, and suggests a new role for nonheme FeII complexes in the assisted degradation of HNO donor molecules.
Graphical Abstract

INTRODUCTION
Nitroxyl (HNO/NO−, also known as azanone or nitrosyl hydride) is a critically important biomolecule whose endogenous production and pharmacology have yet to be resolved.1–6 The HNO molecule is a positive cardiac inotrope used in the treatment of heart failure,7 and is also a potent inhibitor of aldehyde dehydrogenase,8–10 showing promise as a therapeutic for alcohol addiction. It is currently thought that HNO participates in signal transduction in biology by coordinating to the heme center in soluble guanylate cyclase.11–12 Metal-bound HNO species (Mn+(HNO)) have been implicated as reactive intermediates in a number of catalytic cycles that process NOx,13–15 but only a few examples of Mn+(HNO) complexes with first row metals have been reported to date.16–26 HNO has also been used as a surrogate for O2; for example substitution of O2 by HNO in reactions with the enzyme manganese quercetin 2,3-dioxygenase led to substrate nitroxygenation (i.e. HNO incorporation).27–28 Furthermore, there is considerable interest in the reduction/protonation of metal nitrosyl species (M(NO‧)) to produce viable HNO-releasing agents, as well as to examine the feasibility of HNO production from metal centers and NO‧ in vivo.24–25, 29–38
In contrast to NO‧, which is relatively stable under anaerobic conditions, HNO has an intrinsically short lifetime due to rapid (8.0 × 106 M−1 s−1)39 dimerization to give hyponitrous acid, HON=NOH, which further decays to yield N2O and H2O (8.0 × 10−4 s−1).40 To deliver the short-lived HNO in situ for biochemical studies or medical applications, donor molecules that release HNO in response to external stimuli (e.g. pH, temperature, light) must be employed. Sulfohydroxamic acids (RSO2NHOH, R = -alkyl, aryl) constitute one important class of HNO donors that release HNO mainly in response to pH,41 and in some cases, through photoactivation.42–43 Piloty’s acid (N-hydroxybenzenesulfonamide; P.A.), first reported in 1896,44 is the oldest member of this class, and releases HNO under basic conditions according to the mechanism shown in Scheme 1. However, donation of HNO by P.A. is slow (kmax ~ 10−3 to 10−4 s−1 at 25 °C).41
Scheme 1.

Mechanism of HNO Release from Piloty’s Acid
Given that basic conditions are necessary to produce HNO, the P.A.− anion is presumed to be the immediate precursor to HNO release. There are two possible tautomers of monodeprotonated P.A.−, either N- or O-deprotonated PHSO2NOH or PHSO2NHO, respectively. Given the relatively slow rates of HNO release from P.A., we hypothesized that the P.A.− anion could be an important player in the reactivity with a cationic metal center. Precedent for this hypothesis comes from a high-resolution crystal structure of zinc carbonic anhydrase (human isoform II) inhibited by P.A.45 This structure shows that P.A.− anion binds to the active site by coordinating to the zinc(II) center through the sulfonamido N atom, i.e. the N-deprotonated tautomer. Alkali metal salts of P.A.− anion (e.g. Li+, Na+) exist as the N-deprotonated tautomer in both the solid state and in dioxane solution, 46–47 and there is computational evidence to suggest that this tautomer is energetically favored. 48 Other data suggest that it is the O-deprotonated tautomer that leads to HNO release.49
The interactions of redox-active, biologically relevant metal ions such as Fe, Mn, or Co, with HNO donors such as P.A. have been examined previously, and in some cases, were shown to accelerate the decomposition of P.A. and related RSO2NHOH compounds.48, 50–51 However, there is still relatively little known about the mechanism of action between redox-active metal ions and RSO2NHOH compounds. In particular, direct structural or spectroscopic evidence for metal-P.A. adducts is lacking. We have set out to examine nonheme iron complexes and their interaction with P.A. and other RSO2NHOH donors in order to characterize the potential binding modes, mechanisms of action, and HNO-releasing properties of Fe/P.A. systems.
We previously described the reaction of an FeII-solvento complex, [FeII(CH3CN)(N3PyS)BF4 (1) with P.A. in the presence of KOtBu/18-crown-6 (1 equiv) at −40 °C. This reaction gave a new, thermally unstable species 2, which was characterized by UV-vis spectroscopy (λmax = 418, 501 nm).29 Reaction of 2 with a further equivalent of base at −40 °C afforded the {FeNO}8 complex [Fe(NO)(N3PyS)] (3) (λmax = 520, 720 nm), which was also obtained by one-electron reduction of an {FeNO}7 precursor, [Fe(NO)(N3PyS)]BF4 (4), with decamethylcobaltocene (CoCp*2). The {FeNO}8 complex 3 exhibited a rare S = 1 ground state,29,52 and was shown to produce N2O in 54% yield upon standing in solution at 23 °C. We reasoned that complex 2, the immediate precursor to the {FeNO}8 species, could be an Fe(HNO) or Fe-P.A. adduct, but no further spectroscopic characterization was obtained.
Herein we provide new characterization of 2 by 57Fe Mössbauer and Fe K-edge X-ray absorption spectroscopies, which show that 2 is in fact a high-spin (S = 2) FeII-P.A. adduct. Density functional theory (DFT) calculations, including predicted Mössbauer parameters for N-bound and O-bound Fe-P.A. adducts, support the spectroscopic results and suggest that the P.A. ligand in 2 is coordinated through nitrogen. Using a combination of Mössbauer, paramagnetic 1H NMR, and attenuated total reflectance infrared (ATR-IR) spectroscopies, we show that the thermal decay of 2 under strictly anaerobic conditions yields the {FeNO}7 complex 4. In contrast, the {FeNO}8 complex 3 is produced from adding more base, consistent with 2 being a precursor to Fe(HNO).
RESULTS AND DISCUSSION
Synthesis and solution state properties of [57FeII(CH3CN)(N3PyS)]BF4 (l-57Fe).
The FeII-thiolate starting material, [57FeII(CH3CN)(N3PyS)]BF4 (1-57Fe), was prepared from the isotopically enriched (57Fe, 95%) FeII-thioether precursor, [57FeII(CH3CN)(N3PySEtCN)](BF4)2 (5-57Fe) by using the method previously described for the normal abundance analogue (Scheme 2).29 Treatment of 5-57Fe with KOtBu (1 equiv) in dry, deoxygenated CH3CN at 23 °C produces an instantaneous color change from orange-red to wine-red, corresponding to removal of the base-labile 2-cyanoethyl protecting group from the sulfur donor. Pure 1-57Fe, a dark red-brown solid, is isolated in 65% crystalline yield after addition of excess diethyl ether to a concentrated CH3CN/toluene (1:1 v/v) solution of the product.
Scheme 2.

Preparation of Isotopically Labeled [FeII(CH3CN)(N3PyS)]BF4 (1-57Fe)
The zero-field 57Fe Mössbauer spectra of 5-57Fe and 1-57Fe, recorded at 80 K in frozen CH3CN, are shown in Figure 1. Thioetherligated 5-57Fe produces a sharp quadrupole doublet with δ = 0.44 and |ΔEQ| = 0.55 mm s−1, consistent with a low-spin (is, S = 0) FeII ground state. Thiolate-ligated 1-57Fe exhibits parameters (δ = 0.43, |ΔEQ| = 0.31 mm s−1) that are also consistent with ls-FeII and accord well with the data previously collected at 5.4 K for crystalline 1 (δ = 0.41 and |ΔEQ| = 0.26 mm s−1).53 This result provides strong evidence that 1 retains its mononuclear, six-coordinate, solid state structure upon dissolution in CH3CN. The nearly identical isomer shifts observed for 5-57Fe and 1-57Fe (0.44 vs. 0.43 mm s−1) make good sense in light of the fact that these complexes possess very similar first coordination sphere metrics by single crystal X-ray diffraction. In the solid state, complexes 1 and 5 exhibit average Fe-Npy distances of 1.9594(13) Å and 1.9659(13) and Fe-S distances of 2.2848(4) and 2.3018 (4) Å, respectively.53–54 Despite their similar overall coordination environments, 5-57Fe and 1-57Fe have different |ΔEQ| values (0.55 vs. 0.31 mm s·1)1 which likely reflects the difference in thioether versus thiolate ligation.
Figure 1.

57Fe Mössbauer spectra (80 K in frozen CH3CN) showing the conversion of 5-57Fe and 1-57Fe according to Scheme 2. Experimental data are plotted as black dots while best fits are overlaid as solid lines. (a) Spectrum of 5-57Fe fit to a single doublet with δ = 0.44, |ΔEQ| = 0.55, ΓL=R = 0.29 mm s−1. (b) Spectrum of 1-57Fe fit to a single doublet with δ = 0.43, |ΔEQ| = 0.31, ΓL=R = 0.25 mm s−1.
Reactivity of 1 with Piloty’s acid.
It was shown previously that complex 1 reacted with Piloty’s acid (P.A.) and KOtBu/18-crown-6 in CH3CN at −40 °C to give a new, thermally unstable intermediate 2, which was characterized by UV-vis spectroscopy (λmax = 418, 501 nm).29 This species is not observed when the reaction is run at 23 °C (vide infra). We hypothesized that this intermediate might be either an Fe(HNO) or Fe(P.A.) adduct, but no further spectroscopic characterization was obtained. With 1-57Fe in hand, intermediate 2-57Fe could be examined by Mössbauer spectroscopy. To increase the stability of 2-57Fe, we changed to a mixed solvent system of n-butyronitrile/THF, which allowed for lower temperatures to be employed and for the solubility of KOtBu/ 18-crown-6 to be maintained.
The generation of 2-57Fe at ca. −60 °C in CiH1CN/THF is outlined in Scheme 3. Anaerobic, dropwise addition of a THF solution of KOtBu/18-crown-6 (1 equiv) to a pre-cooled mixture of 1-57Fe (5 mM) and P.A. (1 equiv) in C3H7CN produces a gradual color change from deep wine red to bright pink red over 2 h. This color change is due to loss of the intense charge-transfer bands for diamagnetic 1 (ε ~ 4000–5000 M−1 cm−1)53 as intermediate 2 (ε ~ 1000 M−1 cm−1) is formed, as shown previously. An aliquot of the cold, deoxygenated reaction mixture can be quickly transferred to a Mössbauer sample holder using air-free techniques. Alternatively, the reaction mixture can be rapidly frozen by pouring into N2(I), giving frozen pellets which then can be further pulverized to give a fine powder that is loaded into the Mössbauer sample holder.
Scheme 3.

Reaction of [FeII(CFFsCN)(N3PyS)]BF4 and Piloty’s Acid at −60 °C
A representative Miissbauer spectrum for the reaction mixture is shown in Figure 2. The major species, which accounts for 75% of the fitted area, has parameters consistent with a hs-FeII species (δ = 1.05, |ΔEQ| = 2.93 mm s−1; dashed blue line). We assign this doublet to 2-57Fe. A minor component corresponds to starting 1-57Fe (δ = 0.42, |ΔEQ| = 0.33 mm s−1; solid orange line, 7% offit). A third, broad component with δ = 0.36 and |ΔEQ| = 0.61 mm s−1 (18%) is required to fit the remaining area and corresponds to an unidentified product. Longer reaction times (3–6 h), or lower temperature (−80 °C), did not significantly increase the spectroscopic yield of 2-57Fe.
Figure 2.

57Fe Mössbauer spectrum (80 K, C3H7CN/THF, 9:1 v/v) of 1-57Fe + PA. (1 equiv) + KOtBu/18-crown-6 (1 equiv) reacted at −60 °C for 2 h according to Scheme 3. Experimental data (black dots); best fit (red line); majority hs FeII species 2-57Fe (dashed blue line, 75%; Voigt line shape); minor Is FeII species (solid orange line, 7%; Lorentzian line shape) corresponding to 1-57Fe; minor species (solid gray hne, 18%) corresponding to an unidentified product.
Our group has shown that an analogous change from low-spin to high-spin FeII occurs when I is dissolved in methanol.53, 55 This hs FeII species was assigned as [FeII(CH3OH)(N3PyS)]+ (1-CH3OH). The 80 K Mössbauer spectrum for 1-57Fe dissolved in methanol contains two quadrupole doublets, one arising from the acetonitrilebound species (60%), and the other arising from the proposed methanol adduct (δ = 1.05, |ΔEQ| = 2.77 mm s−1; 40%) (Figure S1). At room temperature, the CH3CN in 1 is completely displaced by CH3OH as evidenced by Evans method measurements,53 and the Mössbauer data show that upon freezing the re-binding of CH3CN is greatly favored. In contrast, the P.A.-derived ligand readily outcompetes either CH3CN or the related C3H7CN solvent for the Fe binding site. Notably, the isomer shifts for 1-57Fe in CH3OH and 2-57Fe are identical, which indicates that their overall coordination environments are similar. The combined observations suggest that binding of the reactive P.A. species to Fe is favored over CH3CN, and maintains the [FeII(N3PyS)]+ core structure.
As shown in Scheme 1, HNO release from Piloty’s acid is accompanied by generation of benzenesulfinate anion (PhSO2−), a possible ligand for [FeII(N3PyS)]+. To test whether 2-57Fe is an FeII-PhSO2−·adduct rather than a P.A. or HNO adduct, 1-57Fe was reacted with Na+PhSO2− according to Scheme 4. This reaction yielded a new, thermally stable hs FeII species (6-57Fe) with δ = 1.07 and |ΔEQ| = 3.11 mm s−1, which we assign to a coordinated FeII(O2SPh) adduct. Importantly, these parameters are significantly different from those seen for 2-57Fe. Complex 6-57Fe was also produced in good yield as seen by Möissbauer spectroscopy (84%) when 1-57Fe was treated with a previously reacted 1:1 mixture of P.A. and KOtBu/ 18-crown-6 at 23 °C (Scheme 4 and Figure 3). The latter result confirms that a 1:1 mixture of P.A. and strong base in organic solvent produces PhSO2−, as it does in water.41 Taken together, these experiments show that metastable 2 is not an FeII(PhSO2−) adduct.
Scheme 4.

Reaction of [FeII(CH3CN)(N3PyS)]BF4 and Benzenesulfinate Anion (PhSO2−) Sources at 23 °C
Figure 3.

57Fe Mössbauer spectmm (80 K, C3F7CN/THF, 9:1 v/v) for the reaction of 1-57Fe with a previously reacted 1:1 mixture of PA and KOtBu/18-crown-6 at 23 °C. Experimental data (black dots); best fit (red line); FeII(PhSO2−) adduct 6-57Fe (dashed green line, 84%); 1-57Fe (solid orange line, 12%); minor species (solid gray line, 4%) corresponding to an unidentified product.
Next, we considered the possibility that 2 may be an Fe-HNO adduct ({Fe(HNO)}8 in the Enemark-Feltham notation). The only Mössbauer data on a proposed {Fe(HNO)}8 species comes from cryoreduction of the {FeNO}7 adduct of taurine dioxygenase (TauD).56 This species could only be generated in low yield (17%), but gave Mössbauer parameters of δ = 0.80 and |ΔEQ| = 1.64 mm s−1 that were consistent with DFT calculations for a quintet (S = 2) {Fe(HNO)}8 structure. We turned to DFT methods to predict the Mössbauer parameters for [Fe(HNO)(N3PyS)]+. Optimized geometries of the S = 0, 1, and 2 spin states were obtained and employed for the Mössbauer calculations (see Supporting Information). The calculated isomer shifts range from 0.27 to 0.55 mm s−1 (estimated maximum errors of ±0.1 mm s−1),57 which are clearly inconsistent with the experimental data for 2-57Fe (δ = 1.05 mm s−1). The calculated quadrupole splittings (estimated maximum errors of ±0.5 mm s−1)57 are also in poor agreement with 2-57Fe. We conclude that 2 is not an Fe-HNO adduct.
Fe K-Edge X-ray Absorption Spectroscopy (XAS).
The normalized X-ray absorption near-edge structure (XANES) of complex 2 is shown in Figure 4, overlaid with the spectrum of starting 1 for comparison. The energies of the rising edge inflection points increase upon going from 1 (7120.7 eV) to 2 (7121.5 eV), consistent with back-donation of electron density from FeII to the N–O π* of P.A.
Figure 4.

Normalized Fe K-edge XANES of the hs FeII-P A. complex 2 (solid red line) and the FeII-solvento precursor 1 (solid blue line) (frozen C3H7CN/THF, 9:1 v/v).
The extended X-ray absorption fine structure (EXAFS) of 2 is shown in Figure 5a (solid green trace), along with an overlaid line of best fit (dashed black trace). The corresponding Fourier transforms of the EXAFS data and best fit are shown in Figure 5b. Relevant fitting parameters are provided in Table 1, and result from sequentially improved fits to the data (see Table S1). The best fit for complex 2 includes four N/O scatterers at 2.17 Å and one S scatterer at 2.66 Å, which correspond to the Fe(N3PyS) core. A fifth N/O scatterer at 1.95 Å, an Fe-O scatterer at 2.51 Å, and a second S scatterer at 3.25 can be isolated from the data. These structural features strongly support the assignment of 2 as a hs FeII-Piloty’s acid adduct.
Figure 5.

(a) Fe K-edge EXAFS of 2 plotted from k = 2 to 12 Å−1 (solid green line) overlaid with the corresponding best fit (dashed black line), (b) Fourier transforms of the EXAFS data, fit, and residual (data minus fit; solid gray line).
Table 1.
| CN | R (Å) | ± | σ2 | ± | |
|---|---|---|---|---|---|
| Fe-S | 1 | 2.66 | 0.01 | 0.005 | 0.001 |
| Fe-N | 4 | 2.17 | 0.01 | 0.010 | 0.001 |
| Fe-N(O) | 1 | 1.95 | 0.01 | 0.005 | 0.001 |
| Fe-S | 1 | 3.25 | 0.01 | 0.005 | 0.001 |
| Fe-C | 8 | 4.74 | 0.01 | 0.004 | 0.001 |
| Fe-C | 16 | 3.84 | 0.01 | 0.002 | 0.001 |
| Fe-0 | 1 | 2.51 | 0.02 | 0.001 | 0.002 |
See Table SI for a list of sequentially improved fits.
’EXAFS data were fit with EXAFSPAK using paths calculated by FEFF7. Coordination numbers were held constant, whereas distances (R) and Debye-Waller factors (σ2) were allowed to float. Goodness of fit was measured with F, which was defined as . E0 for the best fit was +3.64 eV.
Tabulated errors (±) correspond to fitting errors. Expected errors for Rare ± 0.02 Å; expected errors in CN are ca. 20–25% (reference 58).
DFT-calcuIated structures for the FeII-Piloty’s acid adduct (2).
Given the availability of EXAFS data for 2, a constrained geometry optimization approach was employed. Our computational studies commenced with quintet [FeII(κ1-N-PhSO2NOH)(N3PyS)] (5A, Figure 6 and Table S3) which contains an N-bound, monodeprotonated P.A. ligand in the sixth coordination site. From EXAFS, the Fe-S(N3PyS), Fe-S(P.A.), Fe-N(O), and Fe▪▪▪O(N) distances were fixed at 2.66,3.25,1.95, and 2.51 Å, respectively. The four Fe-N(N3PyS) distances, which typically have a large degree of asymmetry, were chosen such that their average would equal 2.17 A (i.e., the EXAFS value for four Fe-N scatterers), while all remaining distances were allowed to vary. Using this spectroscopically constrained structure led to the optimized geometry shown in Figure 6. Calculation of the 57Fe Mӧssbauer parameters for this geometry gave δ = 0.96 and |ΔEQ| = 2.81 mm s−1, both of which accord well with the experimental data for 2-57Fe (6 = 1.05 and |ΔEQ| = 2.93 mm s−1). Similarly, when the four Fe-N(N3PyS) distances were allowed to optimize without constraints (structure 5B, Table S3), the calculated Mössbauer parameters were δ = 1.06 and |ΔEQ| = 2.84 mm s−1. An increase in the average Fe-N(N3PyS) distance for 5B (2.28 Å) translates to an increase in isomer shift of ~0.1 mm s−1, but has no impact on the calculated quadrupole splitting. Thus, EXAFS-derived structures for 2 lead to calculated Mössbauer parameters that are fully consistent with the experimental data.
Figure 6.

DFT structure of 5[FeII(κ1-N-PhSO2NOH)(N3PySH)]] (5A) obtained by optimizing with constraints from the EXAFS data for 2 (Table 1). Calculated 57Fe Mӧssbauer parameters are shown in green. C and H atoms are depicted as brown and white spheres, respectively.
Unconstrained geometry optimizations for quintet FeII(κ1-N-PhSO2NOH)(N3PyS)] produce shorter Fe-S(N3PyS) distances of 2.4–2.5 Å, depending on the choice of functional or basis set. These distances are not consistent with the EXAFS results. We considered the possibility that the N3PyS sulfur donor might be protonated, which could lead to elongation of the Fe-S bond. Accordingly, we investigated [FeII(κ1-N-PhSO2NOH)(N3PySH)]; a tautomer of [FeII(κ1-N-PhSO2NOH)(N3PySH)] that could be obtained by internal proton transfer from P.A. to N3PyS. Although this structure exhibits an Fe-S(N3PySH) distance of 2.60 Å, which is similar to the EXAFS-derived distance, it is ~20 kcal mol−1 higher in energy than S-deprotonated structures obtained through fully relaxed optimizations. Thus, we conclude that 5A is the most reasonable structure for 2. However, we cannot exclude the possibility that hydrogen bonding or other electrostatic effects (such as an NSPyS▪▪▪K+ interaction) could contribute to elongation of the Fe-S bond in solution.
Generation of an {FeNO}7 complex from 2.
A rapid color change from bright red to dark brown is observed when samples of 2 are warmed to 23 °C. These changes correspond to loss of the UVvis bands for 2 and the appearance of a species with λmax = 350, 450,and 550 nm (Figure 7). This spectrum is consistent with Fe(NO)(N3PyS)]BF4 (4), an {FeNO}7 complex previously synthesized.59 A representative 1H NMR spectrum of final reaction mixtures in CD3CN is shown in Figure 8 (top), and contains a number of well-separated, paramagnetically shifted peaks between 35 and −5ppm. These signals match the spectrum seen for 4 (Figure 8, bottom). No other paramagnetically shifted 1H resonances are observed across the acquisition window (+130 to −50 ppm).
Figure 7.

UV-vis spectra (CH3CN, 0.1 mM Fe, −40 °C) comparing FeII-solvento starting material 1 (solid black line), FeII-P.A. complex 2 (dashed red line), and the thermal decay product(s) of 2 (solid brown line).
Figure 8.

Paramagnetic 1H-NMR spectra (CD3CN, 23 °C) showing the thermal decay product of 2 (top) and an authentic sample of the {FeNO}7 complex 4 (bottom).
The generation of 4 is further evidenced by attenuated total reflectance infrared (ATR-IR) spectroscopy with 15N-labeled Piloty’s acid, which was synthesized according to Scheme 5. Samples of 2-57Fe were prepared with 14N-P.A. and 15N-P.A. according to the standard protocol, yielding identical Mӧssbauer spectra which established the same purity level for both samples (Figures S3). Warming of these samples to 23 °C similarly yielded identical 1H NMR spectra consistent with the {FeNO}7 complex 4. These samples gave the ATR-IR spectra shown in Figure 9 (14N: black line; 15N: red line). The 15N minus 14N difference spectrum (blue line) contains a prominent band at 1642 cm−1 (positive intensity) that shifts to 1613 cm−1 (negative intensity) upon 15N substitution. This band is assigned to the v(N-O) mode of 4 and exhibits a downshift of 29 cm−1 that is in excellent agreement with the harmonic oscillator prediction for an isolated N-O stretch (−30 cm−1). A second isotopically sensitive band is observed at 1735 cm−1 (15N: 1703 cm−1), also arising from 4. These two v(N-O) modes belong to the S = ½ and S = 3/2 spin states of 4, which are both populated at 23 °C (Figure 9, green line).55, 59 No other 14N/15N shifts are evident in the ATR-IR data. Importantly, these results establish that the nitrosyl ligand in 4 derives from Piloty’s acid.
Scheme 5.

Synthesis of 15N-Labeled Piloty’s Acid
Figure 9.
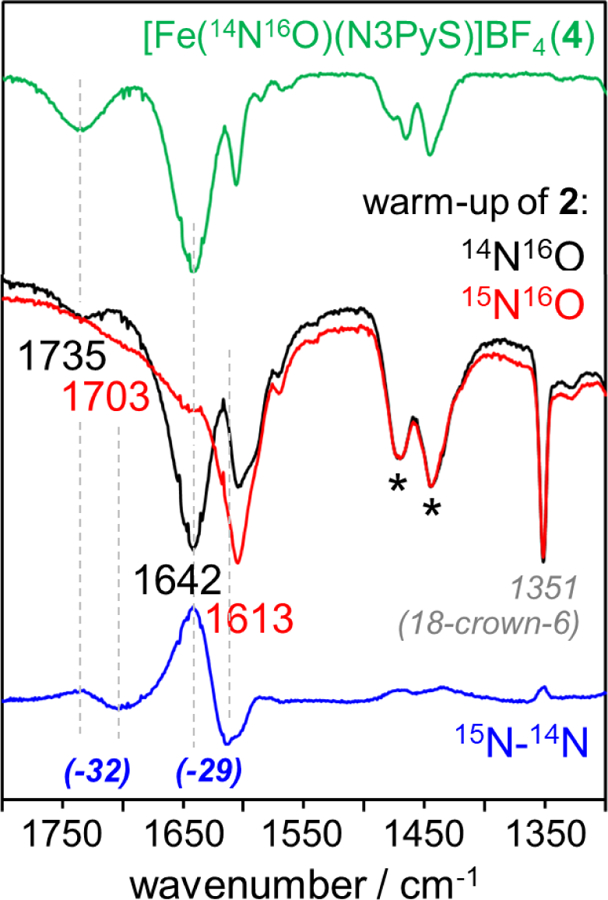
ATR-IR spectra following the thermal decay of 2. Top: authentic 4-14N (green line). Middle: thermal decay of 2-14N (black line) and 2-15N (red line), overlaid and normalized relative to the 1467 and 1444 cm−1 N3PyS ligand vibrations (denoted by*). The 1351 cm−1 mode originates from 18-crown-6. Bottom: difference spectrum (15N minus 14N, blue line) showing isotopically sensitive modes at 1735 and 1642 cm−1 corresponding to 4.
Mӧssbauer data for the thermal decay of 2-57Fe. is presented in Figure 10. Given that the spectrum of 4-57Fe. is well-resolved at 80 K in CH3CN (but not in mixtures of C3H7CN/THF), final reaction mixtures in C3H7CN/THF (9:1 v/v) were concentrated to dryness under high vacuum before adding CH3CN. The major species with δ = 0.31 and |ΔEQ| = 0.57 mm s−1 (78% offit) corresponds to 4-57Fe. A minor species with δ = 1.09 and |ΔEQ| = 3.12 mm s−1 (22% of fit) is also observed, which we tentatively attribute to coordination of the PhSO2−, by-product at Fe. Thus, the combined data (Mӧssbauer, NMR, ATR-IR, and UV-vis) confirm that the major product obtained from warming 2 at −60 °C to 23 °C is the {FeNO}7 complex 4.
Figure 10.

57Fe Mössbauer spectrum (80 Kin frozen CH3CN) showing the thermal decay of 2-57Fe. Experimental data (black dots); best fit (red line); majority {FeNO}7 product 4-57Fe (dashed purple line, 78%); minor hs FeII co-product (solid green line, 22%).
Mechanism.
A mechanism for the formation and decay of Fe-P.A. complex 2 is shown in Scheme 6. Deprotonation of P.A with KOtBu/18-crown-6 (1 equiv) at-60 °C affords the P.A. monoanion (PhSO2NOH) Our data suggest that this species displaces CH3CN from 1, yielding P.A.-bound 2, before HNO release can occur. The combined Mӧssbauer and XAS data, supported by DFT calculations, indicate that the P.A ligand in 2 is coordinated via nitrogen. Addition of a second equivalent of KOtBu/l8-crown-6 leads to the {FeNO}8 complex 3. In the absence of excess base, thermal decay of 2 from −60 °C to 23 °C induces S-N bond cleavage, leading to the {FeNO}7 complex 4 (78%). There is precedent for the production of {FeNO}7 complexes from Piloty’s acid and related RSO2NHOH compounds;60,61 however, these complexes were obtained from iron(III) precursors via HNO /NO− transfer. The production of an {FeNO}7 complex from Piloty’s acid, base, and an iron(II) precursor, as described here, is consistent with formation of a highly reactive {Fe(HNO)}8 species, which is then oxidized by an as yet unidentified pathway to give a stable {FeNO}7 complex. Efforts are ongoing to trap the elusive nonheme {Fe(HNO)}8 species.
Scheme 6.

Possible Mechanism of Formation of the FeII-PiIoty’s Acid Adduct and Reaction Pathways Leading to {FeNO}7 and {FeNO}8 Products
CONCLUSIONS
This work describes the characterization of a rare mononuclear, nonheme FeII-Piloty’s acid adduct by UV-vis and 57Fe Mӧssbauer spectroscopy, Fe K-edge XAS, and DFT. This adduct can be formed at low temperatures from an Fe(II) precursor, P.A., and one equivalent of a strong alkoxide base. Piloty’s acid and related RSO2NHOH compounds are synthetic precursors to HNO/NO−, and are known to react with Fe(III) centers to give {FeNO}7 species.60–61 Much less is known about the reactions of RSO2NHOH donors with Fe(II) complexes, where the anticipated products are less stable, one-electron reduced {FeNO}8 or {Fe(HNO)}8 species. In this regard, we have provided strong evidence for an unprecedented reaction where the P.A. donor, rather than free HNO, binds to a nonheme Fe(II) complex, which then ultimately facilitates nitroxyl production on the metal. Our results indicate that binding of monoanionic P.A. to the Fe(II) center is favored over spontaneous S-N bond cleavage to give HNO and benzenesulfinate anion (PhSO2−)
Our findings also have implications for the chemistry of HNO with nonheme iron-thiolate metalloenzymes. The FeII precursor used in this study, [FeII(CH3CN)(N3PyS]BF4, was previously shown to be a structural and functional mimic of the thiol dioxygenases.53, 59 It is well known that HNO reacts with free thiols to give sulfonamide (RS(O)NH2) and (RSSR) products, 12 but considerably less is known about the reaction of HNO with metal-bound thiols/thiolates. In the present example, we have shown that coordination of P.A. to Fe can lead to either sulfur-ligated {FeNO}8 or {FeNO}7 species, while the sulfur donor remains unmodified. Thus, formation of a metal-bound adduct might protect sulfur sites from rreversible modification by preventing formation of free HNO from P.A. or similar HNO donors.
EXPERIMENTAL SECTION
General Procedures.
All syntheses and manipulations were conducted in an N2-filled glovebox (Vacuum Atmospheres, O2 < 0.2 ppm, H2O < 0.5 ppm) or using standard Schlenk techniques under an atmosphere of dry Ar unless otherwise noted. 57Fe powder (95.5 atom %; 98% purity) was purchased from Cambridge Isotope Laboratories. Piloty’s acid (N-hydroxybenzenesulfonamide) and 15N-labeled Piloty’s acid were synthesized according to a literature procedure.62 Potassium tert-butoxide (KOtBu, sublimed grade, 99.99% trace metals basis) was purchased from Sigma-Aldrich and stored in the glovebox. 18-crown-6 was purchased from Sigma-Aldrich and purified by recrystallization from hot acetonitrile. The 18-crown-6-acetonitrile solvate thus obtained was dried under highvacuum at 35 °C for several hours to afford pure 18-crown-6. Acetonitrile, acetonitrile-d3, and toluene were distilled from CaH2. Tetrahydrofuran was distilled from a purple sodium/benzophenone ketyl solution. Butyronitrile was distilled from Na2CO3/KMnO4 according to a literature procedure.63 Diethyl ether was obtained from a PureSolv solvent purification system (SPS) and used without further purification. All solvents were degassed by a minimum of three freeze-pump-thaw cycles and stored over freshly activated 3 Å molecular sieves in the glove box.
Instrumentation.
UV-visible spectra were recorded on a Varian Cary 50 Bio spectrophotometer integrated with a Unisoku USP-203 cryostat. 1H NMR spectra were recorded on a Bruker Avance 400 MHz FT-NMR spectrometer at 298 K and referenced against residual solvent proton signals. Attenuated total reflectance (ATR) infrared spectra were obtained with a Golden Gate Reflectance diamond cell in a Nexus 670 Thermo-Nicolet FTIR spectrometer. 57Fe Mӧssbauer spectra were collected on a spectrometer from SEE Co. (Science Engineering & Education Co., MN) integrated with a Janis SVT-400-MOSS LHe/LN2 cryostat. The spectrometer was operated in the constant acceleration mode in a transmission geometry.
[57FeII(CH3CN)(N3PySEtCN)](BF4)2 (5-57Fe).
The title compound was prepared with a 57Fe enrichment level of approximately 80%. A three-neck, 100 mL round-bottom flask equipped with a stir bar was fitted with a reflux condenser. Amounts of 57Fe powder (33 mg, 0.58 mmol) and 56Fe powder (11 mg, 0.20 mmol) were added. A solution of HBF4 (48 wt. % in H2O, 0.3 mL, 2.9 equiv) was added via syringe, and the mixture was refluxed for 45 min until no Fe powder remained. The resulting aqueous solution of 57Fe-enriched 57Fe(BF4)2 was cooled to 23 °C and then diluted with CH3CN (5 mL). The N3PySEtCN ligand (355.1 mg, 0.786 mmol) was dissolved in CH3CN (10 mL), and the 57Fe(BF4)2 solution was added at 23 °C, producing an immediate color change from pale orange to dark red-orange. After 2 h, excess Et2O (20 mL) was added, resulting in precipitation of a brown residue. The supernatant was decanted by cannula, and the residue was dried in vacuo over P4O10. Slow vapor diffusion of Et2O into a concentrated CH3CN solution of the product furnished large red crystals of 5-57Fe after 24 h (413 mg, 73% yield).
[57FeII(CH3CN)(N3PyS)]BF4 (1-57Fe).
An amount of crystal-line 5-57Fe (81 mg, 0.11 mmol) was dissolved with vigorous stirring in CH3CN mL). A slurry of KOtBu (13 mg, 0.11 mmol, 1 equiv) in CH3CN (4 mL) was prepared and added dropwise at 23 °C to the solution of 5-57Fe, resulting in an immediate color change from dark red-orange to dark wine red. The mixture was stirred for 2.5 h, filtered through a short plug of Celite, and concentrated under reduced pressure. The dark red-brown residue was triturated with Et2O (3×5 mL) and dried to yield crude 1-57Fe. The crude product was dissolved in a mixture of CH3CN and toluene (1:1 v/v, 4 mL) and filtered through Celite. Excess Et2O (12 mL) was added, and the turbid mixture was placed in a −35 °C freezer overnight, resulting in precipitation of a red-brown microcrystalline solid. Removal of the yellow-brown supernatant and further drying under reduced pressure yielded pure 1-57Fe (42 mg, 65% yield).
57Fe Mössbauer Spectroscopy.
57Fe Mössbauer spectra were collected on frozen solution samples at 80 K in the absence of an applied magnetic field. 57Fe-enriched samples were prepared in custom Delrin cups (10.0 mm x 12.4 mm OD) equipped with tight-fitting Delrin inserts (5.0 mm x 11.0 mm). All samples were stored under LN2 prior to analysis. The zero velocity of each Mössbauer spectrum refers to the centroid of a 25 μm, metallic iron (α-Fe) foil collected at room temperature. The WMOSS program (SEE Co., formerly WEB Research Co., Edina, MN) was used to analyze the Mӧssbauer data. Data were fit to quadrupole doublets with Lorentzian or Voigt line shapes as specified.
Fe K-Edge X-ray Absorption Spectroscopy.
Fe K-edge XAS and extended X-ray absorption fine structure (EXAFS) data were collected at the 16-pole, 2-T wiggler Beamline 9–3 at the Stanford Synchrotron Radiation Lightsource (SSRL) under ring conditions of 500 mA and 3 GeV. All samples were 57Fe-enriched in order to assess purity by Mӧssbauer spectroscopy, and were prepared in custom screw-top Delrin Mössbauer cups. The base of each cup contained a slit (1 × 4 mm) that was covered in 38 μM Kapton to provide an X-ray transparent window, thus allowing for sequential Mӧssbauer and XAS measurements. Samples were maintained at 10 K in an Oxford liquid He flow cryostat. A Si(220) double-crystal monochromator was used for energy selection. A Rh-coated mirror (set to an energy cutoff of 9 key) was used for harmonic rejection. Internal incident energy calibration was achieved by assigning the first inflection points of a Fe foil placed downstream of the sample to 7111.2 eV. Data were collected in fluorescence mode using a Canberra 30-element Ge array detector windowed on Fe Kα emission. Elastic scatter into the detector was attenuated using a Soller slit with an upstream Co filter. Multiple scans were measured and averaged with SIXPACK64 software package. No spectral changes due to photo-damage were observed after multiple scans for these complexes. Data were normalized to post-edge jumps of 1.0 at 7130 eV in SIXPACK by applying a Gaussian normalization for the pre-edge and a three-region cubic spline to model the smooth background above the edge. EXAFS data were fitted using the OPT module of EXAFSPAK65 using initial scattering paths calculated using FEFF766–67.
Generation of 2-57Fe.
A dark red solution of 1-57Fe (5 mg, 9 μmol) in C3H7CN (1.6 mL) was combined with a stir bar in a 10 mL Schlenk flask. The flask was cooled to −60 °C in a slurry of dry ice and chloroform. A stock solution of Piloty’s acid (9 mg, 53 μmol in C3H7CN (1.15 mL) was prepared, and an aliquot (200 μL, 9 μmol, 1 equiv per Fe) was loaded into a gas-tight syringe. A stock solution of KOtBu (6 mg, 50 timol) and 18-crown-6 (13 mg, 50 μmol in THF (1.1 mL) was prepared, and an aliquot (200 μL, 9 μmol, 1 equiv per Fe) was loaded into a separate gas-tight syringe. The Piloty’s acid solution was added dropwise over 10 min to the solution of 1-57Fe at −60 °C, and stirring was continued for an additional 5 min. Next, the KOtBu/18-crown-6 solution was added dropwise over 10 min. Stirring was continued for 2 h, over which time the color of the reaction mixture turned from dark wine red to bright pink red. For Mӧssbauer spectroscopy, the reaction mixture was poured into N2(1), and the resulting frozen powder was packed into a sample cup under N2(1). Alternatively, the reaction mixture was further cooled to −98°C, and transferred by pre-cooled syringe to a pre-cooled sample cup. Samples for Fe K-edge X-ray absorption spectroscopy were prepared in an identical manner.
Reaction of 1-57Fe with Previously Reacted 1:1 Mixture of Piloty’s Acid and KOtBu/18-crown-6 at 23 °C.
Piloty’s acid (15 mg, 87 μmol) was dissolved in C3H7CN (1.0 mL). Amounts of KOtBu (11 mg, 94 μmol) and 18-crown-6 (24 mg, 90 μmol) were combined and dissolved in THF (1.0 mL). The solution of KOtBu/18-crown-6 was added to the solution of Piloty’s acid with stirring, resulting in a dark yellow mixture. The color of the reaction mixture rapidly bleached within ⁓5 s. Stirring was continued for 48 h at 23 °C, at which point the mixture appeared colorless and slightly turbid.
An amount of 1-57Fe (2 mg, 3 μmol) was dissolved in C3H7CN (600 μL). An aliquot of the previously reacted P.A./KOtBu/18-crown-6 mixture (80 μL; 3 μmol, 1 equiv per Fe based on initial P.A.) was added dropwise to 1-57Fe, resulting in an immediate color change from dark wine-red to bright pink-red. For Mössbauer spectroscopy, an aliquot of the reaction mixture (400μL) was transferred to a sample cup and frozen in N2(1).
Reaction of 1-57Fe with NaPhSO2. at 23 °C
An amount of 1-57Fe (2 mg, 3 Etmol) was dissolved in a mixture of C3H7CN (500 μL) and THF (50 μL). Sodium benzenesulfinate (11 mg, 66 μmol) and 18-crown-6 (18 mg, 70 μmol) were combined with C3H7CN (1.0 mL), and the resulting slurry was vigorously stirred for 1 h. An amount of the slurry (60 μL; 4 μmol Na+PhSO2−) was added dropwise to 1-57Fe with continued stirring, producing an immediate color change from dark wine-red to bright pink-red. For Mӧssbauer spectroscopy, an aliquot of the reaction mixture (400 μL) was transferred to a sample cup and frozen in N2(1).
Supplementary Material
ACKNOWLEDGMENTS
The NSF (CHE1566007 to D.P.G.) and NIH (R35GM124908 to K.M.L.) are gratefully acknowledged for financial support A.M.C. thanks the Harry and Cleio Greer Fellowship for support. XAS data were obtained at the Stanford Synchrotron Radiation Lightsoure (SSRL). Use of SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-ACO2–765F00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The Kirin cluster at JHU KSAS is thanked for CPU time to A.M.C
Footnotes
ASSOCIATED CONTENT
Supporting information
This material is available free of charge via the internet at http://pubs.acs.org. 57Fe Mӧssbauer data/fits, Fe K-edge EXAFS data/fits, and DFT calculations
The authors declare no competing financial interests.
REFERENCES
- 1.Suarez SA; Munoz M; Alvarez L; Venancio MF; Rocha WR; Bikiel DE; Marti MA; Doctorovich F HNO Is Produced by the Reaction of NO with Thiols. J. Am. Chem. Soc 2017,139,14483–14487. [DOI] [PubMed] [Google Scholar]
- 2.Kumar MR; Clover T; Olaitan AD; Becker C; Solouki T; Farmer PJ The reaction between GSNO and H2S: On the generation of NO, HNO and N20. Nitric Oxide 2018,77,96–105. [DOI] [PubMed] [Google Scholar]
- 3.Suarez SA; Neuman NI; Munoz M; Alvarez L; Bikiel DE; Brondino CD; Ivanovic-Burmazovic I; Miljkovic J; Filipovic MR; Marti MA; Doctorovich F Nitric oxide is reduced to HNO by proton- coupled nucleophilic attack by ascorbate, tyrosine, and other alcohols. A new route to HNO in biological media? J.Am. Chem. Soc 2015,137,4720–4727. [DOI] [PubMed] [Google Scholar]
- 4.Hamer M; Suarez SA; Neuman NI; Alvarez L; Munoz M; Marti MA; Doctorovich F Discussing endogenous NO●/HNO interconversion aided by phenolic drugs and vitamins. Inorg. Chem 2015,54,9342–9350. [DOI] [PubMed] [Google Scholar]
- 5.Irvine JC; Ritchie RH; Favaloro JL; Andrews KL; Widdop RE; Kemp-Harper BK Nitroxyl (HNO): the Cinderella of the nitric oxide story. Trends Pharmacol. Sci 2008,29,601–608. [DOI] [PubMed] [Google Scholar]
- 6.Paolocci N; Jackson MI; Lopez BE; Miranda K; Tocchetti CG; Wink DA; Hobbs AJ; Fukuto JM The pharmacology of nitroxyl (HNO) and its therapeutic potential: not just the Janus face of NO. Pharmacol Ther 2007,113,442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tocchetti CG; Wang W; Froehlich JP; Huke S; Aon MA; Wilson GM; Di Benedetto G; O’Rourke B; Gao WD; Wink DA; Toscano JP; Zaccolo M; Bers DM; Valdivia HH; Cheng H; Kass DA; Paolocci N Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ. Res 2007, 100, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagasawa HT; DeMaster EG; Redfern B; Shirota FN; Goon DJ Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide. J. Med. Chem 1990, 33, 3120–3122. [DOI] [PubMed] [Google Scholar]
- 9.DeMaster EG; Redfern B; Nagasawa HT Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem. Pharmacol 1998, 55, 2007–2015. [DOI] [PubMed] [Google Scholar]
- 10.Shoeman DW; Shirota FN; DeMaster EG; Nagasawa HT Reaction of nitroxyl, an aldehyde dehydrogenase inhibitor, with N-acetyl-L-cysteine. Alcohol 2000, 20, 55–59. [DOI] [PubMed] [Google Scholar]
- 11.Miller TW; Cherney MM; Lee AJ; Francoleon NE; Farmer PJ; King SB; Hobbs AJ; Miranda KM; Burstyn JN; Fukuto JM The effects of nitroxyl (HNO) on soluble guanylate cyclase activity: interactions at ferrous heme and cysteine thiols. J. Biol. Chem 2009, 284, 21788–21796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianco CL; Toscano JP; Bartberger MD; Fukuto JM The chemical biology of HNO signaling. Arch. Biochem. Biophys 2017, 617, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Einsle O; Messerschmidt A; Huber R; Kroneck PM; Neese F Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J. Am. Chem. Soc 2002, 124, 11737–11745. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y; Stroka JR; Kandemir B; Dickerson CE; Bren KL A Cobalt Metallopeptide Electrocatalyst for the Selective Reduction of Nitrite to Ammonium. J. Am. Chem. Soc 2018, 140, 16888–16892. [DOI] [PubMed] [Google Scholar]
- 15.Anderson JH The metabolism of hydroxylamine to nitrite by Nitrosomonas. Biochem. J 1964, 91, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin R; Farmer PJ The HNO adduct of myoglobin: Synthesis and characterization. J. Am. Chem. Soc 2000, 122, 2393–2394. [Google Scholar]
- 17.Sulc F; Fleischer E; Farmer PJ; Ma D; La Mar GN 1H NMR structure of the heme pocket of HNO-myoglobin. J. Biol. Inorg. Chem 2003, 8, 348–352. [DOI] [PubMed] [Google Scholar]
- 18.Sulc F; Immoos CE; Pervitsky D; Farmer PJ Efficient trapping of HNO by deoxymyoglobin. J. Am. Chem. Soc 2004, 126, 1096–1101. [DOI] [PubMed] [Google Scholar]
- 19.Immoos CE; Sulc F; Farmer PJ; Czarnecki K; Bocian DF; Levina A; Aitken JB; Armstrong R; Lay PA Bonding in HNO-myoglobin as characterized by X-ray absorption and resonance raman spectroscopies. J. Am. Chem. Soc 2005, 127, 814–815. [DOI] [PubMed] [Google Scholar]
- 20.Pervitsky D; Immoos C; Veer W; Farmer PJ Photolysis of the HNO adduct of myoglobin: transient generation of the aminoxyl radical. J. Am. Chem. Soc 2007, 129, 9590–9591. [DOI] [PubMed] [Google Scholar]
- 21.Kumar MR; Pervitsky D; Chen L; Poulos T; Kundu S; Hargrove MS; Rivera EJ; Diaz A; Colon JL; Farmer PJ Nitrosyl hydride (HNO) as an O2 analogue: long-lived HNO adducts of ferrous globins. Biochemistry 2009, 48, 5018–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montenegro AC; Amorebieta VT; Slep LD; Martin DF; Roncaroli F; Murgida DH; Bari SE; Olabe JA Three redox states of nitrosyl: NO+, NO●, and NO−/HNO interconvert reversibly on the same pentacyanoferrate(II) platform. Angew. Chem. Int. Ed. Engl 2009, 48, 4213–4216. [DOI] [PubMed] [Google Scholar]
- 23.Montenegro AC; Bari SE; Olabe JA Reactivity of iron(II)-bound nitrosyl hydride (HNO, nitroxyl) in aqueous solution. J. Inorg. Biochem 2013. 118, 108–114. [DOI] [PubMed] [Google Scholar]
- 24.Walter MIL; Dzul SP; Rodrigues AV; Stemmler TL; Telser J; Conradie J; Ghosh A; Harrop TC Synthesis of Con-NO- Complexes and Their Reactivity as a Source of Nitroxyl. J. Am. Chem. Soc 2016, 138, 12459–12471. [DOI] [PubMed] [Google Scholar]
- 25.Goodrich LE; Roy S; Alp EE; Zhao J; Hu MY; Lehnert N Electronic structure and biologically relevant reactivity oflow-spin {FeNO}8 porphyrin model complexes: new insight from a bis-picket fence porphyrin. Inorg. Chem 2013, 52, 7766–7780. [DOI] [PubMed] [Google Scholar]
- 26.Abucayon EG; Khade RL; Powell DR; Zhang Y; Richter-Addo GB Hydride Attack on a Coordinated Ferric Nitrosyl: Experimental and DFT Evidence for the Formation of a Heme Model-HNO Derivative. J. Am. Chern. Soc 2016, 138, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar MR; Zapata A; Ramirez AJ; Bowen SIC; Francisco WA; Farmer PJ Nitrosyl hydride (HNO) replaces dioxygen in nitroxygenase activity of manganese quercetin dioxygenase. Proc. Natl. Acad. Sci. USA 2011, 108, 18926–18931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han XZ; Kumar MR; Farmer PJ Nitroxygenation of quercetin by HNO. Tetrahedron Lett 2016, 57, 399–402. [Google Scholar]
- 29.Confer AM; McQ\lilken AC; Matsumura H.; Moenne-Loccoz P; Goldberg DPA Nonheme, High-Spin {FeNO}8 Complex that Spontaneously Generates N,O. J. Am. Chern. Soc 2017, 139, 10621–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patra AK; Di1be KS; Sanders BC; Papaefthymiou GC; Conradie J; Ghosh A; Harrop T C.A thermally stable {FeNO}8 complex: properties and biological reactivity of reduced MNO systems. Chem. Sci 2012, 3, 36069. [Google Scholar]
- 31.Sanders BC; Patra AIC; Harrop TC Synthesis, properties, and reactivity of a series of non-heme {FeNO}7/8 complexes: implications for Fenitroxyl coordination. J. Inorg. Biochem 2013, 118, 115–127. [DOI] [PubMed] [Google Scholar]
- 32.Rhine MA; Rodrigues AV; Bieber Urbauer RJ; Urbaner JL; Stemmler TL; Harrop TC Proton-induced reactivity of NO from a {CoNO}8 complex. J. Am. Chem. Soc 2014, 136, 12560–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhine MA; Sanders BC; Patra AK; Harrop TC Overview and new insights into the thiol reactivity of coordinated NO in {MNO}6/7/8 (M = Fe, Co) complexes. Tnorg. Chem 2015, 54, 9351–9366. [DOI] [PubMed] [Google Scholar]
- 34.Speelman AL; White CJ; Zhang B; Alp EE; Zhao J; Hu M; Krebs C; Penner-Hahn J; Lehnert N Non-heme High-Spin {FeNO}6−8 Complexes: One Ligand Platform Can Do lt All. J. Am. Chern. Soc 2018, 140, 1 1 341–11359. [DOI] [PubMed] [Google Scholar]
- 35.Kupper C; Schober A; Demeshko S; Bergner M; Meyer F An exclusively organometallic {FeNO}7 complex with tetracarbene ligation and a linear Fe NO unit. Tnorg. Chem 2015, 54, 3096–3098. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrino J; Bari SE; Bikiel DE; Doctorovich F Successful stabilization of the elusive species {FeNO}8 in a heme model. J. Am. Chem. Soc 2010, l32, 989–995. [DOI] [PubMed] [Google Scholar]
- 37.Carcia-Serres R; Crapperhaus CA; Bothe E; Bill E; Weyhermuller T; Neese F; Wieghardt K Structural, spectroscopic, and computational study of a11 octahedral, non-heme [Fe-NO]6−8 Series: [Fe(NO)(cyclam-ac)]2+/+/0. J. Am. Chem. Soc 2004, 126, 5138–5153. [DOI] [PubMed] [Google Scholar]
- 38.Li F; Meyer RL; Carpenter SH; Van Gelder LE; Nichols AW; Machan CW; Neidig ML; Matson EM Nitric oxide activation facilitated by cooperative multimetallic electron transfer within an ironfunctionalized polyoxovanadate-alkoxide cluster. Chern. Sci 2018, 9, 6379–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shafirovich V; Lymar SV Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc Natl. Acad. Sci. U.S.A 2002, 99, 7340–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchholz JR; Powell RE The Decomposition of Hyponitrous Acid. I. The Non-chain Reaction. J. Am. Chem. Soc 1963, 85, 509–511. [Google Scholar]
- 41.DuMond JF; King SB The chemistry of nitroxyl-relcasing compounds. Antioxid. Redox Signal 2011, 14, 1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y; Cink RB; Fejedelem ZA; Simpson MC; Seed AJ; Sampson P; Brasch NE Development of Photoactivatable Nitroxyl (HNO) Donors Incorporating the (3-Hydroxy-2-naphthalenyl)methyl Phototrigger. Eur.J. Org. Chem 2018, 1745–1755.
- 43.Carrone G; Pellegrino J; Doctorovich F Rapid generation of HNO induced by visible light. Chem. Commun 2017, 53, 5314–5317. [DOI] [PubMed] [Google Scholar]
- 44.Piloty O Ueber eine Oxydation des Hydroxylamins durch Benzolsulfochlorid. Ber. Dtsch. Chem. Ge, 1896, 29, 1559–1567. [Google Scholar]
- 45.Di Fiore A; Maresca A; Alterio V; Supuran CT; De Simone G Carbonic anhydrase inhibitors: X-ray crystallographic studies for the binding ofN-substituted benzenesulfonamides to human isoform IL Chem. Commun 2011, 47, 11636–11638. [DOI] [PubMed] [Google Scholar]
- 46.Exner O Acyl derivatives of hydroxylamine. IX. A spectroscopic study of tautomcrism of sulphohydroxamic acids. Collect. Czech. Chem. Commun 1964, 29, 1337–1343. [Google Scholar]
- 47.Exner O; Juskab T X-ray photoelectron spectroscopic study of sulphonamides: Charge distribution and tautomerism. Collect. Czech. Chem. Commun 1984, 49,51–57. [Google Scholar]
- 48.Alvarez L; Suarez SA; Bikiel DE; Reboucas JS; Batinic-Haberle I; Marti MA; Doctorovich F Redox potential determines the reaction mechanism ofHNO donors with Mn and Fe porphyrins: defining the better traps. lnorg. Chern 2014, 53, 7351–7360. [DOI] [PubMed] [Google Scholar]
- 49.Bonner FT; Ko Y Kinetic, isotopic, and nitrogen-15 NMR study of N-hydroxybenzenesulfonamide decomposition: an nitrosyl hydride (HNO) source reaction. Inorg. Chem 1992, 31, 2514–2519. [Google Scholar]
- 50.Marti MA; Bari SE; Estrin DA; Doctorovich F Discrimination of nitroxyl and nitric oxide by water-sofoble Mn(III) porphyrins. J. Am. Chem. Soc 2005, 127, 4680–4684 [DOI] [PubMed] [Google Scholar]
- 51.Subedi H; Brasch NE Mecha11istic studies of the reactions of the reduced, Vitamin B12 derivatives with the HNO donor Piloty’s acid: further evidence for oxidation of cob(I)alamin by (H)NO. Dalton Trans 2016, 45, 352–60. [DOI] [PubMed] [Google Scholar]
- 52.Dey A; Confer AM; Vilbert AC; Moenne-Loccoz P; Lancaster KM; Goldberg DP A Nonheme Sulfur-Ligated {FeNO}6 Complex and Comparison with Redox-lnterconvertible {FeNO}7 and {FeNO}8 Analogues. Angew. Chem. Tnt. Ed. Eng1 2018, 57, 13465–13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McQuilken AC; Jiang Y; Siegler MA; Goldberg DP Addition ofdioxygen to an N4S(thiolate) iron(II) cysteine dioxygenase model gives a structurally characterized sulfinato-iron(II) complex. J. Am. Chem. Soc 2012, 134, 8758–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Widger LR; Jiang Y; McQuilken AC; Yang T; Siegler MA; Matsumura H; Moenne-Loccoz P; Kumar D; de Visser SP; Goldberg DP Thioether-ligated iron(II) and iron(III)-hydroperoxo/alkylperoxo complexes with an H-bond donor in the second coordination sphere. Dalt.on Trans 2014, 43, 7522–7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McQuilken AC; Matsumura H; Durr M; Confer AM; Sheckelton JP; Siegler MA; McQuilken TM; Ivanovic-Burmazovic I; Moenne-Loccoz P; Goldberg DP Photoinitiated Reactivity of a ThiolateLigated, Spin-Crossover Nonheme {FeNO}7 Complex with Dioxygen. J.Am. Chem. Soc 2016, 138, 3107–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye S; Price JC; Barr EW; Green MT; Bollinger JM Jr.; Krebs C; Neese F Cryoreduction of the NO-adduct of taurine:alphaketoglutarate dioxygenase (TauD) yields an elusive {FeNO}8 species. J. Am. Chem. Soc 2010, 132, 4739–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papai M; Vanko G On Predicting Mössbauer Parameters of Iron-Containing Moleniles with Density-Fimctional Theory. J. Chem. Theory Comput 2013, 9, 5004–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Riggs-Gelasco PJ; Stemmler TL; Penner-Hahn JE XAFS of dinuclear metal sites in proteins and model compounds. Coard. Chern. Rev 1995, 144, 245–286. [Google Scholar]
- 59.McQuilken AC; Ha Y; Sutherlin KD; Siegler MA; Hodgson KO; Hedman B; Solomon EI; Jameson GN; Goldberg DP Preparation of non-heme {FeNO}7 models of cystcine dioxygenase: sulfur versus nitrogen ligation and photorelease of nitric oxide. J. Am. Chern. Soc 2013, 135, 14024–14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davies SC; Evans DJ; Hughes DL; Konkol M; Richards RL; Sanders JR; Sobota P Mononuclear, binuclear, trinuclear and tetranuclear iron complexes of the N(CH2CH2S)33− (NS3) ligand with nitrosyl co-ligands. J. Chem. Soc. Dalton Trans 2002, 2473–2482.
- 61.Suárez SA; Martí MA; De Biase PM; Estrin DA; Bari SE; Doctorovich F HNO trapping and assisted decomposition of nitroxyl donors by ferric hemes. Polyhedron 2007, 26, 4673–4679. [Google Scholar]
- 62.Aizawa K; Nakagawa H; Matsuo K; Kawai K; Ieda N; Suzuki T; Miyata N Piloty’s acid derivative with improved nitroxyl-releasing characteristics. Bioorganic Med. Chem. Lett 2013, 23,2340–2343. [DOI] [PubMed] [Google Scholar]
- 63.Armarego WLF; Perrin DD, Purification of Laboratory Chemicals, 4th ed. Butterworth-Heineman: Oxford, 1997. [Google Scholar]
- 64.Webb S SIXPACK, Stanford Synchrotron Lightsource, Stanford Linear Accelerator Center, Stanford University: Stanford, CA, 2002. [Google Scholar]
- 65.George GN EXAFSPAK, Stanford Synchrotron Radiation Lightsource, S. L. A. C., Stanford University. [Google Scholar]
- 66.Mustre de Leon J; Rehr JJ; Zabinsky SI; Albers RC Ab initio curved-wave x-ray-absorption fine structure. Phys. Rev. B 1991, 44, 4146–4156. [DOI] [PubMed] [Google Scholar]
- 67.Rehr JJ; Mustre de Leon J; Zabinsky SI; Albers RC Theoretical x-ray absorption fine structure standards. J. Am. Chem. Soc 1991,113,5135–5140. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.