


Abstract
In eukaryotes and archaea, tRNA genes frequently contain introns, which are removed during maturation. However, biological roles of tRNA introns remain elusive. Here, we constructed a complete set of Saccharomyces cerevisiae strains in which the introns were removed from all the synonymous genes encoding 10 different tRNA species. All the intronless strains were viable, but the tRNAPheGAA and tRNATyrGUA intronless strains displayed slow growth, cold sensitivity and defective growth under respiratory conditions, indicating physiological importance of certain tRNA introns. Northern analyses revealed that removal of the introns from genes encoding three tRNAs reduced the amounts of the corresponding mature tRNAs, while it did not affect aminoacylation. Unexpectedly, the tRNALeuCAA intronless strain showed reduced 5.8S rRNA levels and abnormal nucleolar morphology. Because pseudouridine (Ψ) occurs at position 34 of the tRNAIleUAU anticodon in an intron-dependent manner, tRNAIleUAU in the intronless strain lost Ψ34. However, in a portion of tRNAIleUAU population, position 34 was converted into 5-carbamoylmethyluridine (ncm5U), which could reduce decoding fidelity. In summary, our results demonstrate that, while introns are dispensable for cell viability, some introns have diverse roles, such as ensuring proper growth under various conditions and controlling the appropriate anticodon modifications for accurate pairing with the codon.
INTRODUCTION
tRNAs, well-known as central adapter molecules involved in protein translation, are a highly expressed class of small noncoding RNAs that constitute as much as 4–10% of total cellular RNAs (1). Despite their small body, tRNAs receive extensive processing, including terminal trimming, CCA addition, nucleotide modifications and splicing (see below). For example, ∼3 million cytosolic tRNA molecules are transcribed per generation from 275 tRNA genes dispersed throughout all the 16 chromosomes in the yeast Saccharomyces cerevisiae. The resulting primary transcripts must be properly matured in the same duration (2,3). Initially, tRNAs are synthesized in their precursor form with the 5′-leader and 3′-trailer sequences. Some pre-tRNAs in the eukaryotes and archaebacteria contain one or a few introns. The number of eukaryotic tRNA species (defined by their anticodon sequence) that are produced from intron-containing precursors ranges from 2 to 52, and each of the 46 tRNA species in the mushroom, Laccaria bicolor, has at least one isodecoder with an intron (3). In most cases, such tRNA genes are interrupted by a single intron inserted at one nucleotide 3′ to the anticodon, the so-called canonical position, whereas some tRNAs in archaebacteria and certain eukaryotic algae harbor more diverse types of introns (4–6). Eubacterial tRNA genes seldom have introns, and such rare introns are Group I or Group II self-splicing introns, which are different from those of eukaryotes and archaebacteria.
In S. cerevisiae, ∼20% of the total tRNA genes encoding 10 different tRNA families contain a single intron with 14–60 nt length at the canonical position. These extra sequences must be removed before final maturation. Because the intron interferes with formation of the universally conserved structure of the anti-codon stem loop, splicing machinery, which mainly consists of tRNA-splicing endonuclease (Sen) and tRNA ligases (also 2′-phosphotransferase in fungi and plants), is essential for viability in various eukaryotes (7–11).
It has been a long mystery the reason why some tRNA genes have the introns. As described above, pre-tRNA splicing is a prerequisite for the function of tRNAs in translation, and the resulting introns are efficiently degraded by the sequential actions of tRNA ligase and Xrn1p, at least in S. cerevisiae (12). Unlike for mRNAs, alternative splicing of pre-tRNAs is very rare. The archaebacterium, Caldivirga maquilingensis, utilizes splicing to produce three different isoacceptors of tRNAGly from five split tRNA fragments (13). In this case, one common 3′-fragment (38–3′ end) is joined with one or two fragments consisting of the 5′ halves. However, in the eukaryotes, interruption of tRNA genes with the intron does not enable production of more diverse transcripts from the limited number of tRNA genes. Some introns contribute to nucleotide modifications of the mature part of tRNAs. In another archaebacterium Haloferax volcanii, a box C/D small RNA is nested in the intron of tRNATrpCCA, and the intronic part of pre-tRNATrp or the excised intron is used to select nucleotides C34 and U39 on tRNATrp for 2′-O-methylation (14). In vitro modification assays using cell extracts demonstrated that yeast tRNA-pseudouridine synthase Pus1p only accepts the intron-containing pre-tRNAIleUAU for its pseudouridylation of U34 and U36 (15,16). Notably, the same enzyme can catalyze pseudouridylation of U27 on both the pre- and mature tRNAs. In addition, tRNA methyltransferase Trm4p in yeast and humans catalyzes intron-dependent m5C formation at C34 on tRNALeuCAA and that at C40 on tRNAPheGAA in yeast (17,18). Indeed, the position of 34 on the tRNAs base-pairs with the wobble position of the codon, and some modifications of position 34 affect accuracy and/or efficiency of decoding (19,20). However, deletion of the PUS1 gene or the TRM4 gene is not lethal in S. cerevisiae, indicating that these modifications are not essential for yeast growth or decoding by these tRNAs.
Another interesting point is that the presence or absence of an intron seems to be common among all synonymous genes encoding an isoacceptor or an isodecoder tRNA. For example, in S. cerevisiae and Schizosaccharomyces pombe, there is no isoacceptor consisting of mixed isodecoders with and without the intron on their genomes. Among the organisms with more complex genome structures, certain tRNA species are encoded by both intron-containing and intronless genes, although these two types of genes tend to derive from different origins, as occurs for tRNATyrGUA in Arabidopsis thaliana and tRNALeuCAA in Homo sapiens. Thus, there might be an unknown system or selective pressure to maintain or exclude the introns in whole synonymous tRNA genes encoding certain isoacceptor/isodecoders on the genome.
Not like the in vitro analyses described above, essentiality of the tRNA intron for viability and tRNA function has been examined only in a limited number of tRNA species in vivo. Requirement of the tRNA intron was first examined in vivo in tRNASerCGA of S. cerevisiae, which is encoded by a single gene, and revealed that the intron is dispensable for yeast viability (21). Similarly, the tRNATrp intron nesting the box C/D small RNA could be removed from its gene without affecting the growth of H. volcanii, and again the tRNA is encoded by the single gene (22). Our previous study found that removal of the intron from S. cerevisiae tRNATrp, which is encoded by six genes with the same sequence including the intron part, has no significant growth and protein synthesis defects (23). However, because only a few tRNA species have been analyzed to date, a tRNA species that harbors an intron essential for growth of the organism may indeed exist.
In this report, to extend our analyses of the physiological roles of the tRNA introns, we used the sequential deletion technique to remove the introns from all the synonymous genes encoding one of the 10 intron-containing tRNA species. Using this method, we obtained a complete set of the total 10 mutants of S. cerevisiae, each harboring deletions of all the introns of a single tRNA species, and examined the impact of intron removal on tRNA production and their functions on protein synthesis. Among the 10 intronless mutants, the tRNAPhe and tRNATyr intronless mutants showed impaired cell growth especially at low temperatures, and tRNAIleUAU, tRNALysUUU, tRNAPhe and tRNATyr mutants exhibited various extent of growth defects on non-fermentable carbon sources. None of the 10 intronless mutants showed strong translation defects on ribosome profiles. The intronless mutants of tRNALeuCAA and tRNAPhe had lower amounts of 5.8S rRNA than the wild-type strain, and the former showed an abnormal nucleolar morphology. We also confirmed in vivo requirement of the tRNA intron for pseudouridylation of positions 34 and 36 of tRNAIleUAU, and unexpectedly found that the intron protects tRNAIleUAU against an aberrant modification. Overall, our results show that all the tRNA introns in S. cerevisiae are dispensable for yeast growth while the existence of a canonical intron in certain tRNA genes and/or precursors has impacts on various physiological aspects to maximize yeast viability.
MATERIALS AND METHODS
Constructions of intronless tRNA plasmids
We analyzed the tRNA genes on the S. cerevisiae genome annotated in Saccharomyces Genome Database (https://www.yeastgenome.org/) and Genomic tRNA Database (http://lowelab.ucsc.edu/GtRNAdb/). Intronless tRNA plasmids for gene integration were constructed essentially as described by Mori et al. (23) and are summarized in Supplementary Table S1A. Briefly, a DNA fragment comprising an intronless tRNA gene and its flanking fragments were amplified by megaprimer polymerase chain reaction (PCR) and normal PCR, respectively, with appropriate primer sets listed in Supplementary Table S2. Then, the two fragments were sequentially introduced into a vector, pTYE374, using restriction sites fused to the ends of the PCR products. To introduce a dual marker cassette into the resulting plasmid, a unique restriction site between the two PCR fragments on the pTYE374 derivative was digested, blunted by T4 DNA polymerase and ligated with an appropriate DNA fragment with one of the dual marker cassettes (i.e. hisG::ADE2::GAL1p-GIN11M86::camr::hisG), which was released from the host plasmids by NotI digestion and blunted by T4 DNA polymerase. Three different markers, LEU2, URA3 and ADE2 from pTYE378, pTYE379 and pTYE380, respectively, were available (Supplementary Table S1B) (23).
Iterative introduction of multiple intronless mutations into the yeast genome
Intronless strains were mainly constructed from the parental strain BY418 [MATαade2Δ::hisG his3Δ200 leu2Δ1 lys2Δ202 met15Δ0 trp1Δ63 ura3-52] (24). On the other hand, we used W303-1A for intron removal from the genes encoding tRNALeuUAG because BY418 lacks tL(UAG)L2, one of the three synonymous tRNALeuUAG genes, due to accidental deletion during introduction of met15Δ mutation. Of note, BY418 was also found to lack tL(CAA)C, one of the 10 genes encoding tRNALeuCAA, near the LEU2 locus. To integrate an intronless tRNA gene into the yeast chromosome, a plasmid with the intronless gene and a dual marker cassette was first digested with appropriate restriction enzymes to release the entire insert of the plasmid from its vector part. The resulting linear DNA fragments were introduced into yeast cells according to Gietz et al. (25), and then transformants were selected on an appropriate selection medium corresponding to the introduced auxotrophic marker. Replacement of the corresponding chromosomal tRNA gene with the intronless tRNA allele was confirmed by genomic PCR with an appropriate set of primers shown in Supplementary Table S3 followed by AGE with a 2.5%(w/v) agarose gel.
The dual marker cassette has a toxic GIN11M86 gene placed under the control of the GAL1 promoter (26). Because this gene and the auxotrophic marker are sandwiched by two hisG sequences, yeast clones that lost the marker cassette via homologous recombination between the hisG sequences were selected as galactose resistant clones on the galactose medium. Subsequently, another locus of the same tRNA species was converted into the intronless allele again as described as above. If necessary, the corresponding wild-type tRNA gene on a multicopy vector with a URA3 marker was introduced into the intermediate constructs, and then iterative replacement of chromosomal copies of the tRNA genes in question was continued until all the chromosomal copies became intronless. Finally, the URA3 plasmid was cured by plating the strain on the 5′-fluoroorotic acid (5′-FOA) medium.
To reintroduce mitochondrial DNA into TYSC1974, the original tRNALeuCAA intronless isolate, we performed cytoduction between TYSC1974 and TYSC774 (27). The resulting strains are summarized in Supplementary Table S5.
RNA preparation and northern blotting
Small RNAs were prepared either by the guanidine thiocyanate/phenol method or by the Na-acetate/phenol method. For the guanidine thiocyanate/phenol method (28), crude RNAs were extracted with a 1:1 mixture of GTE Buffer [0.10 M Tris–HCl, pH 7.6, 4.0 M guanidine thiocyanate and 10 mM ethylenediaminetetraacetic acid (EDTA)] and water-saturated phenol from mid log-phase yeast cells at 65°C, and the water phase was separated by the addition of chloroform and subsequent centrifugation. After phenol chloroform extraction of the aqueous phase followed by chloroform extraction, RNAs were precipitated with 2-propanol, and the final pellets were dissolved in TE [10 mM Tris–HCl, pH 7.5 and 1.0 mM EDTA]. The crude small RNA fractions were separated on an 8% (w/v) or a 10% (w/v) TBE-urea polyacrylamide gel. For the analysis of tRNA aminoacylation, crude RNAs were prepared by the Na-acetate/phenol method, and the resulting RNA fractions were analyzed with an acid-urea gel as described previously (29). As a deacylation control, a portion of the RNA sample was treated with 50 mM Tris–HCl, pH 9.0 at 37°C for 1 h. The crude RNA fractions were separated on an 8% (w/v) or a 10% (w/v) acid-urea gel at 4°C. RNAs in the TBE-urea and acid-urea gels were transferred onto the Hybond-N+ charged nylon membrane (GE Healthcare, Chicago, Illinois, USA) using 50 mM Na-acetate, pH 5.5 as a transfer buffer. For analysis of rRNA intermediates, total RNAs were prepared with the Na-acetate/sodium dodecyl sulphate (SDS) buffer [50 mM Na-acetate, pH 5.2, 10 mM EDTA and 1.0% (w/v) SDS] and acidic phenol chloroform [phenol:chloroform = 5:1, pH 4.5] instead of GTE to recover longer RNA molecules. The RNAs were separated on a 1.2% (w/v) agarose gel with 2.2 M formaldehyde in the MOPS buffer, and then transferred onto Hybond-N+ membranes by capillary transfer in 20 × SSC. Hybridization with digoxigenin (DIG)-labeled probes was carried out in Hybridization Solution [0.50 M Na2HPO4, 0.34% (v/v) H3PO4, 7.0% (w/v) SDS, 1.0 mM EDTA, pH 7.0] or DIG Easy Hyb (Roche Diagnostics, Basel, Switzerland) at 42°C or at 37°C. The probes whose sequences are summarized in Supplementary Table S4 were labeled with DIG Oligonucleotide Tailing Kit (Roche).
Immunofluorescence microscopy
Logarithmically growing cells were fixed in 3.7% (w/v) formaldehyde for 1 h at 30°C. The cells were pelleted by centrifugation at 1600 × g for 3 min, washed in SHA Buffer [0.90 M sorbitol, 0.10 M K-Pi, pH 6.5 and 5.0 mM NaN3] and then recovered by the same centrifugation step. The cells were resuspended in SHA Buffer to a final concentration of 2.5 OD600, and their cell walls were digested with Zymolyase 100T (Nacalai Tesque, Kyoto, Japan). The resulting spheroplasts were gently washed with SHA Buffer, and placed onto poly-L-lysine-coated multi-well slides. The spheroplasts were permeabilized with 0.10% (w/v) Triton X-100, and blocked for 10 min in phosphate-buffered saline (PBS) with 1.0% (w/v) bovine serum albumin (BSA) in a humid chamber, and then incubated with an anti-Nop1p mouse monoclonal antibody 28F2 (GeneTex, Inc., Irvine, California, USA) for 1 h at room temperature. After a number of washes with PBS containing 0.10% (w/v) BSA, the spheroplasts were incubated with anti-mouse IgG antibodies labeled with Alexa 594 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 1 h in the dark. The spheroplasts were washed with PBS containing 0.10% (w/v) BSA, and mounted in Mounting Solution [90% (v/v) glycerol and 0.10% (w/v) p-phenylenediamine] after staining with 4′,6-diamidino-2-phenylindole (DAPI). Images were obtained using an Olympus BX60 fluorescence microscope (Olympus Corp., Tokyo, Japan) with a CSU-10 confocal unit (Yokogawa Electric, Musashino, Japan). Three-dimensional images were constructed from z-stacks of confocal images by MetaMorph software (Molecular Devices, San Jose, California, USA).
Purification and mass spectrometric analysis of tRNAs
tRNAIleUAU was purified from the wild-type, intronless and pus1Δ strains by the chaplet column chromatography method (30). Briefly, a small RNA fraction was prepared from the above yeast cells cultured in YPD until the mid-log phase by the guanidine thiocyanate/phenol method described previously. The small RNA fraction was heat-denatured at 65°C, and hybridized with a biotinylated anti-tRNAIleUAU oligonucleotide (biotin-5′-CCACGACGGTCGCGTTATAAGCACGAAGCT-3′) that was immobilized on a HiTrap Streptavidin HP pre-packed column (1 ml bed volume, GE Healthcare) in Binding Buffer [30 mM HEPES-KOH, pH 7.5, 15 mM EDTA and 1.2 M NaCl] through gradual cooling from 65°C to the room temperature under continuous circulation of the applied RNA sample. After washing the column with Washing Buffer [2.5 mM HEPES-KOH, pH 7.5, 1.25 mM EDTA and 0.10 M NaCl], the tRNA was eluted with TE at 65°C. The eluate was ethanol-precipitated, further purified by preparative TBE-urea PAGE and finally dissolved in RNase-free water.
Details of tRNA analysis by mass spectrometry (MS) were described in (31,32). To analyze pseudouridylation, an aliquot of the purified tRNAIleUAU was subjected to cyanoethylation of Ψ residues with acrylonitrile, as described by Megen-Jørgensen and Kirpekar (33). Subsequently, 250 femtomoles of the cyanoethylated and control tRNAIleUAU were digested with RNase T1 (Epicentre, Madison, Wisconsin, USA), and analyzed with a system consisting of a capillary liquid chromatography (LC) (DiNa, KYA Technologies, Tokyo, Japan) and a nano-electrospray ionization (ESI)–mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific). The digested tRNA fragments were desalted on a trapping column (0.5 × 1 mm, KYA Technologies) and separated with a C18 capillary column (HiQ Sil C18; 3 μm, 100 Å pore size; 0.15 × 50 mm, KYA Technologies, Tokyo, Japan), and the eluate was sprayed from an energized nanosprayer tip into the mass spectrometer. Ions were scanned with a negative polarity mode over an m/z range of 600 to 2000 throughout the separation. To identify ncm5U in tRNAIleUAUΔint, enzymatically digested nucleosides were analyzed by a micro-flow LC/MS system consisting of a Q Exactive (Thermo Fisher Scientific) with an ESI source and an Ultimate 3000 LC system (Dionex/Thermo Fisher Scientific) through a hydrophilic interaction LC (HILIC) column (SeQuant ZIC-cHILIC, 3 μm, 2.1 × 150 mm, Merck Millipore, Burlington, Massachusetts, USA) with a guard column (SeQuant ZIC-cHILIC guard kit, 3 μm, 2.1 × 20 mm, Merck Millipore) as described previously (34). Deprotonated nucleoside ions were monitored with a negative polarity mode over an m/z range of 110–700 throughout the separation.
In vivo mis-decoding assay
To evaluate misdecoding of an AUG codon to Ile, a c-myc tag sequence with an AUA codon as its critical 5th Ile residue and its derivative containing AUG instead of AUA were introduced at the terminus of the EGFP ORF (see Figure 5F). A set of two 38 nt oligoDNAs (see Supplementary Table S4) were annealed to form ds oligoDNA encoding the c-Myc tag. Another ds oligoDNA encoding the mutant (c-Myc Ile5Met; Supplementary Table S4) was also prepared similarly. These two oligoDNAs were inserted into the BglII/EcoRI site of the plasmid pTYSC425, which harbors an EGFP gene under the control of the ADH1 promoter, to yield pSM002 and pSM004, respectively (Supplementary Table S1B). The plasmids were introduced into appropriate strains, their yeast lysates for western blotting were prepared by alkaline lysis (35), and subjected to western blotting with the anti-c-Myc antibody, 9E10 (Wako Pure Chemicals, Osaka, Japan). For signal detection, Cy3-conjugated anti-mouse IgG antibodies and Cy5-conjugated anti-rabbit IgG antibodies (Molecular Probes, Eugene, Oregon, USA) were used as the secondary antibodies, and fluorescence signals were read with Typhoon FLA-7000 Fluorescence Scanner (GE Healthcare).
Figure 5.

tRNAIleUAU receives intron-dependent pseudouridylation in vivo, and the pseudouridylation prevents the tRNA from aberrant ncm5U modification. (A) Intron-dependency of Ψ34 and Ψ36 formation on tRNAIleUAU by Pus1p was investigated by MS analyses. As described in the ‘Materials and Methods’ section, tRNAIleUAU purified from the wild-type (WT), intronless (Δintron) and PUS1-deletion (pus1Δ) strains was subjected to LC-MS analysis with or without cyanoethylation. The panels in the top row represent the LC elution patterns by base peak chromatograms, and the panels in the other three rows represent the MS signals of extracted ion chromatograms (XICs) corresponding to the non-, mono- and di-cyanoethylated RNase T1 fragments (C32-G40) of tRNAIleUAU, respectively. Information about the corresponding MS peak analyzed in each row is indicated at the top of each panel on the left. Cyanoethylated derivatives were not detected in the MS samples without acrylonitrile treatment (data not shown). (B) A typical mass spectrum of the C32–G40 fragment prepared from the tRNAIleUAU intronless strain. The m/z values of the expected mass peaks are shown in gray, and those of the unknown peaks are in black. (C) XICs of divalent negative ions at m/z 1510.7 and m/z 1539.2 (middle and bottom row, respectively) of tRNAIleUAU fragments from the wild-type (WT), intronless (Δintron) and PUS1-deletion (pus1Δ) strains. (D) Collision-induced dissociation (CID) analyses of the RNase T1 fragments with molecular masses of 3023.4 Da (upper panel) and 3080.4 Da (lower panel) derived from the tRNAIleUAU in the intronless yeast. Product ions for c- and y-series are assigned in the CID spectra and fragment sequences. (E) Identification of aberrant U + 57 Da by nucleoside analysis. The top row represents a trace of UV absorbance at 254 nm, and the second row represents XIC at m/z 300.083 corresponding to a deprotonated anion of ncm5U from co-injection of tRNAIleUAUΔint and yeast total RNA. The third and fourth rows represent XICs at the same m/z from tRNAIleUAU purified from the intronless strain and yeast total RNA alone, respectively. (F) Investigation of misdecoding of an AUG codon to Ile using a c-Myc epitope. EGFP fused with the wild-type (Wt) or an AUA-to-AUG mutant form (mut) of the c-Myc tag was expressed in the wild-type (WT) or the tRNAIleUAU intronless (Δintron) cells. The mutant c-Myc contained an AUG codon instead of the AUA codon for the 5th residue of Ile in the c-Myc epitope (possible decoding patterns of the two reporters are shown at the top). The same blot was decorated with both the anti-c-Myc 9E10 mouse monoclonal antibody (anti-c-Myc) and the anti-GFP rabbit antibodies (anti-GFP). Srp1p on the same blot was also detected as a loading control (anti-Srp1p). ‘Vec’ is a negative control without expressing any EGFP fusion. Two independent clones (‘1’ and ‘2’) of each strain were subjected to western blotting.
RESULTS
All of the tRNA species encoded by intron-containing genes can be expressed from the intronless allele in S. cerevisiae
We previously demonstrated that the intron in tRNATrp genes is dispensable for yeast growth (23), following the report that the intron of tRNASerCGA is dispensable (21). Then, we extended our analysis to the other tRNA species on the yeast genome. As summarized in Table 1, 10 tRNA species are encoded by intron-containing genes with various gene numbers. Iterative gene replacement with cassettes consisting of positive and negative selection markers (23,26) was used to generate yeast strains in which all of the genes encoding a single tRNA species were converted into intronless alleles. Proper replacement of the chromosomal genes with intronless versions was confirmed by genomic PCR (Supplementary Figure S1A and B). During the iterative gene replacement, a wild-type gene of the same tRNA species was maintained on a URA3 plasmid to compensate possible deleterious effects of intron removal. As described in ‘Material and Methods’ section, most of intronless strains were constructed from BY418 as the wild-type strain with the exception that the intronless strain of tRNALeuUAG was constructed from W303-1A, because one of the three tRNALeuUAG genes, tL(UAG)L2, was accidentally deleted on introduction of an auxotrophic marker next to the tRNA gene during the construction of BY418. The genome of S. cerevisiae has two un-annotated possible intron-containing tRNA genes with high homology to tRNASerGCU genes, tX(XXX)D and tX(XXX)L, which have only one nucleotide deletion in the mature tRNASerGCU sequence. Thus, we also removed the introns from these two genes, and used the resulting quadruple mutant that lost all the introns from tS(GCU)F, tS(GCU)O, tX(XXX)D and tX(XXX)L loci as the tRNASerGCU intronless strain.
Table 1.
Summary of the growth phenotypes of all the intronless mutants on agar media
| C source | Glucose | Glycerol | Galactose | Ethanol | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Temperature (°C) | 17 | 23 | 30 | 33 | 37 | 23 | 30 | 23 | 30 | 23 | 30 |
| W303-1A | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| LeuUAGΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| BY418 | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| SerCGAΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| IleUAUΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++/++ | +++ | +++ | +++ | +++/++ |
| SerGCUΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| TrpCCAΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| LysUUUΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++/++ | +++ | +++ | +++ | +++/++ |
| TyrGUAΔint | + | ++ | ++ | ++/+++ | +++ | - | ++ | ++ | +++/++ | +/- | ++/+ |
| LeuCAAΔint | ++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| ProUGGΔint | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| PheGAAΔint | + | ++ | ++ | +++ | +++ | - | - | ++ | +++ | - | - |
+++ indicates growth similar to that of the corresponding wild-type strain (W303-1A or BY418). ++ and + indicate slight and mild growth defects, respectively. – indicates a severe growth retardation or lethality under these conditions. The tRNALeuCAAΔint strain analyzed here was the ρ+ strain (see the main text).
We could construct intronless strains of all the 10 tRNA species, and it was found that they could lose the URA3 plasmids with the corresponding wild-type genes. Figure 1 shows growth of the intronless strains without any additional copy of corresponding wild-type genes under various growth conditions. When growth of the intronless strains were compared with corresponding wild-type strains on YPD (rich glucose medium), the tRNASerGCU, tRNAIleUAU, tRNALeuUAG, tRNALysUUU and tRNAProUGG intronless strains grew similarly to the corresponding wild-type strains (Figure 1A), as well as the tRNASerCGA and tRNATrp intronless strains, whose normal growth phenotypes were reported previously (21,23). On the other hand, the intronless strains of tRNATyr and tRNAPhe showed marginal growth defects especially at low temperatures (Figure 1A). The tRNALeuCAA intronless strain also showed slight growth retardation at 17°C (Figure 1A). Next, we examined the growth of the intronless strains on different carbon sources. As shown in Figure 1B, the tRNASerGCU, tRNALeuUAG, tRNALeuCAA, tRNAProUGG, tRNASerCGA and tRNATrp intronless mutants did not show any growth defects on media with glycerol, ethanol or galactose, whereas the tRNAIleUAU and tRNALysUUU intronless strains grew slightly slowly on the glycerol and ethanol media at 30°C. The tRNATyr mutant, which exhibited a growth defect at the low temperatures on the glucose medium also displayed slow-growth phenotypes on the medium with glycerol, ethanol or galactose. Notably, the tRNAPhe intronless strain barely grew on the non-fermentable carbon sources, glycerol and ethanol. These growth phenotypes of the intronless strains are summarized in Table 1.
Figure 1.

Growth of the tRNA-intronless strains. (A) Growth of yeast strains in which all the tRNA genes encoding a certain tRNA species (shown on the right) were converted into an intronless allele was compared at various temperatures. Saturated cultures were serially diluted 10-fold as shown in the bottom, and 4 μl aliquots of each diluent were spotted onto YPD plates. The plates were incubated at the indicated temperatures. As described in the text, two wild-type strains were grown as controls: W303-1A as the parental strain of the tRNALeuUAG intronless strain and BY418 as the parental strain of the other intronless strains. Growth of TYSC2148 (ρ+ strain) is shown for the tRNALeuCAA intronless strain. (B) Growth of the tRNA intronless strains on different carbon sources. Serially diluted cultures of the two wild-type and 10 intronless strains were spotted onto YPGly (glycerol), YPEt (ethanol) and YPGal (galactose) plates as described in (A). The plates were incubated at 30°C.
We also analyzed growth of the tRNAPhe, tRNATyr, tRNAIleUAU and tRNALysUUU intronless strains more in detail in liquid media. The doubling times (tD) of these mutants grown in liquid YPD were not significantly different from those of the wild-type either at 30°C or at 23°C (Figure 2A). On the other hand, apparent slow growth and cold-sensitive phenotypes were seen for the tRNATyr intronless and tRNAIleUAU intronless mutants grown in YPGly (the tRNATyr intronless strain, tD = 9.3 ± 0.3 h at 30°C and 25.0 ± 3.9 h at 23°C; the tRNAIleUAU intronless strain, tD = 7.1 ± 0.1 h at 30°C and 20.2 ± 1.0 h at 23°C; BY418, tD = 6.3 ± 0.5 h at 30°C and 17.9 ± 1.4 h at 23°C) (Figure 2B). The tRNAPhe intronless and tRNALysUUU intronless mutants exhibited more interesting phenotypes, biphasic growth defects in which growth retardation was exaggerated at a certain timepoint of their growth. Therefore, we calculated two doubling times (early phase and late phase) for these strains. The doubling times of the tRNAPhe intronless strain during the early growth phase in YPGly were longer than those of the wild-type strain (Figure 2B; 12.3 ± 0.6 h versus 6.3 ± 0.5 h at 30°C, and 27.0 ± 3.9 h versus 17.9 ± 1.4 h at 23°C, respectively). This slow growth phenotype of the tRNAPhe intronless strain became exacerbated at the later timepoint (31.3 ± 2.4 h at 30°C and 43.3 ± 4.8 h at 23°C). In the case of the tRNALysUUU intronless mutant, it grew rather normally until a certain point (early phase tD = 6.9 ± 0.1 h at 30°C and 16.9 ± 0.2 h at 23°C), but its growth slowed down severely thereafter (late phase tD = 57.8 ± 0.0 h at 30°C and 53.0 ± 4.8 h at 23°C).
Figure 2.

Growth curve of the tRNAPhe, tRNATyr, tRNAIleUAU and tRNALysUUU intronless mutants in liquid YPD or YPGly. (A) Saturated yeast cultures of the wild-type strain (BY418) and the four intronless strains of tRNAPhe, tRNATyr, tRNAIleUAU and tRNALysUUU were inoculated into the liquid YPD medium and grown at 30°C (left) or 23°C (right). The OD660 traces of the wild-type, tRNAPhe intronless and tRNATyr intronless strains were shown in the upper panels. Those of the wild-type, tRNAIleUAU intronless and tRNALysUUU intronless strains were in the lower panels. A color code and the doubling time under the log phase growth of each strain are indicated in the graph. (B) Similar experiments as those described in (A) were performed using the non-fermentable liquid medium, YPGly, instead of YPD. When the tRNAPhe intronless and tRNALysUUU intronless strains were cultured in YPGly, they showed biphasic growth defects so that their doubling time in each growth phase is indicated (a, Early phase; b, Late phase).
Together with previous results, our results clearly reveal that none of the tRNA introns on the S. cerevisiae genome are essential for the yeast growth on fermentable carbon sources under laboratory conditions. However, the introns in specific tRNA genes, especially those of tRNAPhe, have crucial for survival and proper growth for yeast cells on non-fermentable carbon sources. Our original isolates of the tRNALeuCAA intronless strain grew poorly at all the temperatures we tested, but were found to be ρ−, where the yeast strain exhibits a growth defect in non-fermentable media but not lethal in fermentable media because of its loss of mitochondrial DNA (data not shown). Subsequently, we tested whether or not re-introduction of mitochondrial DNA into this strain via cytoduction could complement the growth defect. Indeed, the tRNALeuCAA intronless yeast supplied with mitochondrial DNA grew normally both in fermentable and non-fermentable media, while the strain showed the slight but reproducible growth defect at 17°C, as mentioned above. We used this ρ+ derivative, TYSC2148, in the following analyses.
Intron removal has minor effects on tRNA production
Next, we investigated expression of each tRNA species in the wild-type and intronless strains by northern blotting. Low-molecular weight RNA fractions were prepared from each strain using the guanidine thiocyanate/phenol extraction method to measure overall tRNA amounts. Figure 3 summarizes quantitative northern blotting after TBE-based urea-PAGE of RNA samples from the wild-type and intronless strains. The expression level of tRNAAspGUC was used as an internal control. As expected from the intron removal of all the tRNATrp genes, removal of the introns did not affect total amounts of tRNASerCGA, tRNAIleUAU, tRNASerGCU, tRNALeuUAG, tRNALysUUU or tRNAPhe. By contrast, tRNATyr, tRNALeuCAA and RNAProUGG in the corresponding intronless strains were expressed 67 ± 3%, 75 ± 3% and 73 ± 10% of those in the wild-type strain, respectively (Figure 3). Although the changes were not remarkable, the reductions in the levels of these tRNAs were reproducible (Student’s t-tests: P = 0.0011, 0.0001 and 0.0427, respectively). These results suggest that the introns of some yeast tRNA genes are required to maintain proper levels of the corresponding tRNAs in vivo. It should be noted that the tRNATyr intronless strain displayed growth defects (Figure 1) in addition to reduced tRNATyr levels, suggesting that the growth defects may come from this much of tRNATyr shortage.
Figure 3.

Intron removal from some isoacceptor tRNAs has weak effects on expression of the corresponding tRNAs. Northern blotting of tRNA levels in the wild-type (W) and intronless (Δ) strains. Small RNA fractions (2.0 μg each) were analyzed by TBE-based urea-PAGE/northern blotting for each tRNA isotype. The expression level of a major tRNA, tRNAAsp, was used as an internal control. The upper bar graph shows quantification of the northern blotting data. The amount of a tRNA species shown in the graph bottom was first normalized by that of tRNAAsp, and is expressed as relative abundance where the normalized amount of each tRNA in the wild-type strain is set to 1. Data represent the mean plus standard deviation (error bar) of at least three biological replicates. The lower images show a set of typical northern blotting images of the RNA in question (upper) and the control tRNAAsp (lower); *, P < 0.05 and **, P < 0.001 by Student's t-tests.
Then, we examined aminoacylation efficiency of tRNAs in the intronless mutants. With the exception of tRNASerGCU, aminoacylation statuses of tRNAs derived from the intronless tRNA genes were quite similar to those from the wild-type genes in the acidic urea-PAGE (Supplementary Figure S2), implying that aminoacylation is unaffected by the presence or absence of the tRNA introns in tRNA precursors. In the case of the tRNASerGCU intronless mutant, a new molecular species migrating faster than the wild-type tRNASerGCU was detected in the acidic urea-PAGE (Supplementary Figure S2, band indicated by ‘*’) while no such a fast-migrating molecular species was detected in TBE-based urea-PAGE (Figure 3). The position of the fast-migrating band was shifted upon alkaline treatment (Supplementary Figure S2, band indicated with ‘**’), suggesting that the fast-migrating tRNA species seems to be almost completely aminoacylated. This fast-migrating band was only detected in the quadruple intronless mutant [ts(gcu)fΔint ts(gcu)oΔint tx(xxx)dΔint tx(xxx)lΔint] but not in the triple intronless mutant [ts(gcu)fΔint ts(gcu)oΔint tx(xxx)lΔint tX(XXX)D] (data not shown).
Alteration in 5.8S rRNA production and nucleolar morphology in the tRNALeuCAA intronless mutant
During the analysis of tRNA amounts in the intronless strains, we noticed that the intronless mutants of tRNALeuCAA and tRNAPhe exhibited lower amounts of 5.8S rRNA than the wild-type strain. Indeed, quantitative northern blotting showed that the amounts of 5.8S rRNA in the tRNALeuCAA and tRNAPhe intronless mutants decrease to 70 ± 3% and 79 ± 10% of that of the wild-type strain, respectively (Figure 4A; Student’s t-tests, P = 0.0058 and 0.0193, respectively). Because rRNAs constitute a large portion of the total RNA fraction, a small reduction in the rRNA level would result in relative increases in the levels of the other RNA species. This effect explained the slight increase in the amount of the control tRNAAsp in the tRNALeuCAA and tRNAPhe intronless strains (Figure 4A). The 5.8S rRNA levels in the other mutants were not significantly different from those in the wild-type strains (Figure 4A).
Figure 4.
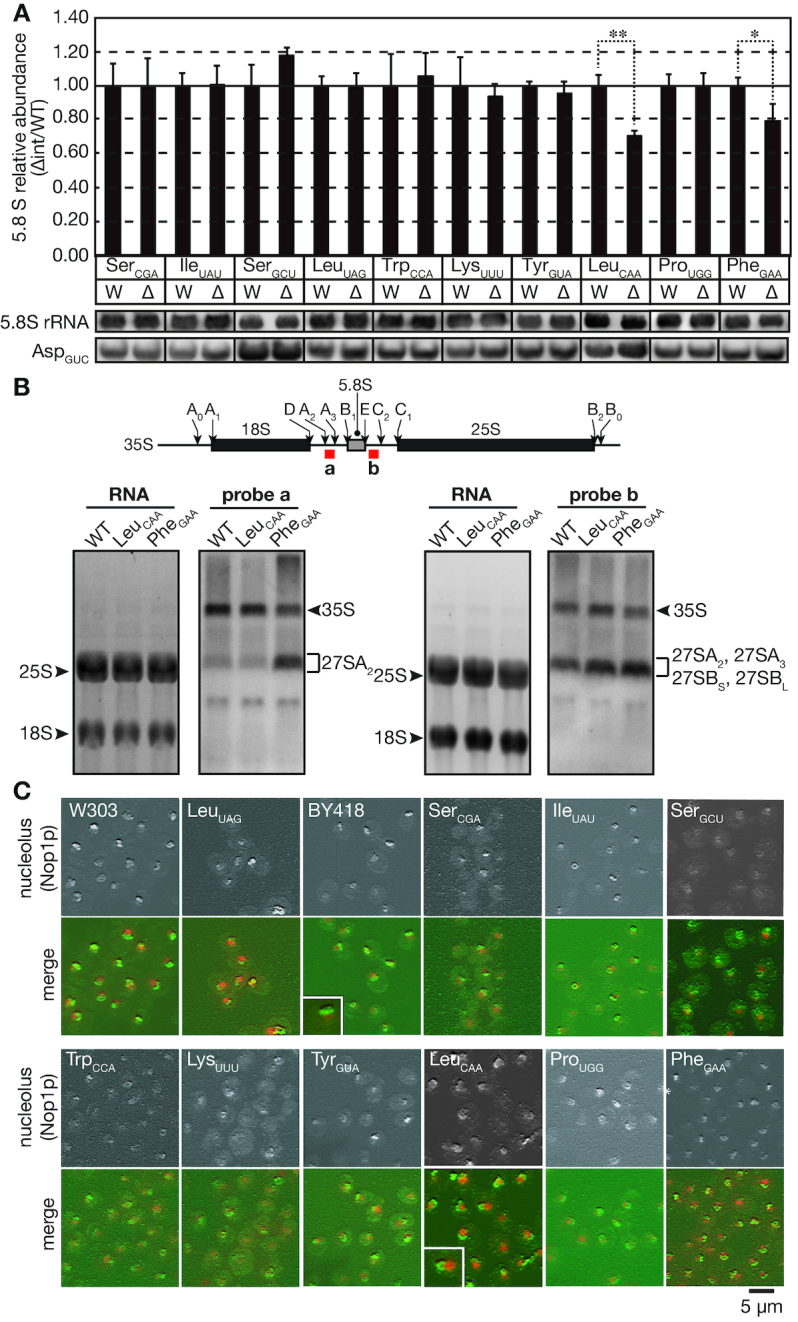
Intron removal from tRNALeuCAA genes reduces the 5.8S rRNA amount and alters nucleolar morphology. (A) Northern blotting analyses of 5.8S rRNA levels in the wild-type (W) and intronless (Δ) strains. Small RNA fractions were prepared, analyzed and quantified by northern blotting as described in Figure 3. Data represent the mean plus standard deviation (error bar) of three biological replicates. Because the RNA samples analyzed here were the same as those used in Figure 3, the same northern blotting data for tRNAAsp were used as an internal control. See the Figure 3 legend in detail. (B) Upper panel: Schematic illustration of the major processing sites of the 35S rRNA primary transcript. The hybridization regions of the probes are indicated by red boxes. Lower panel: RNAs from the wild-type (WT), tRNALeuCAA intronless and tRNAPhe intronless strains were analyzed by northern hybridization with probes against the A2-A3 region in ITS1 (probe a, second panel) and the E-C2 region in ITS2 (probe b, fourth panel) of the 35S rRNA precursor. Total RNA staining with SYBR Green II of the agarose gels is shown in the first and third panels. Assignment of intermediates was according to (37). (C) Immunofluorescence images of nucleolar morphology in the wild-type strains (W303-1A, BY418) and the indicated intronless strains. Yeast strains grown in YPD at 30°C were fixed and subjected to immunofluorescence with an anti-Nop1p (nucleolar marker) antibody, and DAPI was used to stain the nucleus. The upper panel in each set of images represents a 3D reconstruction image of the Nop1p stain, and the lower panel is the merged image of the Nop1p stain (green) and the DNA stain (red). The small boxed areas are enlarged images of the nucleolus in the wild-type and that of the typical nucleolus with an abnormal shape in the tRNALeuCAA intronless mutant.
Next, we examined the effects of removal of the introns from the tRNALeuCAA and tRNAPhe genes on other aspects of rRNA production. tRNA genes tend to gather around the nucleolus (36), and such nuclear/nucleolar organization may be affected by removal of the introns from tRNA genes. This may have some effects on rRNA synthesis and/or processing. First, we analyzed rRNA processing in the tRNALeuCAA intronless and tRNAPhe intronless mutants by northern blotting with intermediate-specific probes; ‘probe a’ hybridized with a region between the A2 and A3 processing sites in ITS1 of the 35S rRNA primary transcript, and ‘probe b’ hybridized with a region between the E and C2 sites in ITS2 (Figure 4B, upper panel). In northern hybridization with probe a, no accumulation of the 27SA2 rRNA intermediate was observed in the tRNALeuCAA intronless mutant, while significant signal accumulation of 27SA2 was observed in the tRNAPhe mutant (Figure 4B, second panel; assignment of intermediates was according to (37)). The total amount of ∼27S rRNA intermediates recognized by probe b increased slightly both in the mutants (Figure 4B, fourth panel). These results suggest that some nucleolar RNA processing events might be retarded in these mutants, and that the processing steps affected by the mutants are somehow different between the two. Next, we assessed the nucleolar morphology in all 10 tRNA intronless mutants by immunofluorescence using Nop1p as the nucleolar marker. Three-dimensional images were acquired via z-stack scanning of nucleoli (Figure 4C; upper panels), and were merged with a DAPI-staining image (Figure 4C; lower panels) to reveal the nucleolar structure and positioning. As seen in the wild-type strains (W303-1A and BY418), the cup-shaped nucleolus (green) usually closely attaches with the nucleoplasm containing chromosomes (red), and forms a part of the round or oval nuclear shape. Nine of the tRNA mutants, including tRNAPhe, displayed a normal nucleolus tightly adjoined their nucleoplasm. On the other hand, some of the tRNALeuCAA mutant cells had an atypical nucleolus of which part was detached from the nucleoplasm and formed a protrusion into the cytoplasm (Figure 4C and Supplementary Figure S4). Although only a small proportion of the mutant cells harbored this type of the atypical nucleolus, such aberrant nucleoli were seldom observed in the wild-type and the other intronless yeast cells. Indeed, 7.2 ± 0.7% of tRNALeuCAA intronless cells had the aberrant nucleolus while only 1.3 ± 1.1% of the wild-type cells have aberrant one (Student’s t-test, P = 0.0046). Hence, intron removal from the tRNA genes encoding tRNALeuCAA affects the nucleolar morphology to some degree, and this might be related to the reduction in the amount of 5.8S rRNA.
To examine the impact of tRNA intron removal from tRNA genes, especially those of tRNALeuCAA and tRNAPhe, on global translation in vivo, we analyzed polysome profiles in all 10 tRNA intronless mutants (Supplementary Figure S3). Many of the intronless strains, including tRNALeuCAA and tRNAPhe, had similar polysome/monosome ratios to those of the corresponding parent strains, indicating that intron removal from tRNA genes does not greatly affect global translation, even though some mutants show minor but obvious defects in ribosome biosynthesis.
The tRNA intron prevents aberrant modification in the anticodon
Post-transcriptional modifications in tRNAs are characteristic structural features of tRNAs. The anticodon of S. cerevisiae tRNAIleUAU contains two pseudouridines (Ψ) at positions 34 and 36 (15). These Ψs are introduced in the precursor form of tRNAIleUAU in an intron-dependent manner, because pseudouridylase Pus1p recognizes the anticodon in the double-stranded form containing the intron (16). Because a ΨAΨ anticodon in tRNAIleUAU is supposed to recognize an AUA codon specifically, the intron-dependent pseudouridylation plays a critical role in accurate decoding during protein synthesis (38).
To analyze the tRNA modification status, we isolated tRNAIleUAU from the wild-type, intronless and pus1Δ strains by the chaplet column chromatography method (30). Because Ψ is a mass-silent modification, the isolated tRNA was treated with acrylonitrile to derivatize Ψs by cyanoethylation (33), and then digested by RNase T1, and subjected to capillary LC-nano-ESI-mass spectrometry (RNA-MS) (31,32). In the wild-type strain, we clearly detected the anticodon-containing fragment (C32–G40) with the increased molecular mass by the addition of one or two cyanoethyl groups (Figure 5A, third and fourth rows), indicating the presence of Ψ34 and Ψ36 in this fragment as reported previously (16). In the tRNAIleUAU intronless strain, a non-cyanoethylated fragment was mainly detected, although a small peak representing the mono-cyanoethylated fragment was also observed (Figure 5A). Because the mono-cyanoethylated fragment was also detected slightly in the same tRNA isolated from the pus1Δ strain, it is likely that unmodified uridines were non-specifically cyanoethylated to some extent. Thus, as expected, these findings indicate that tRNAIleUAU isolated from the intronless strain contained no Ψs in the anticodon. Furthermore, we happened to detect the anticodon-containing fragment of tRNAIleUAU isolated from the intronless strain with the molecular mass 57 Da larger than that of the wild-type tRNA (Figure 5B and C, third row). This fragment was further probed by collision-induced dissociation (CID), confirming that the increased mass resided at position 34 in the anticodon (Figure 5D). According to its molecular mass, the modification was assumed to be 5-carbamoylmethyluridine (ncm5U). To confirm the chemical structure, we conducted an LC/MS nucleoside analysis of tRNAIleUAU isolated from the intronless strain (Figure 5E). As expected, the modified uridine formed in the tRNAIleUAU isolated from the intronless strain was coeluted with the ncm5U nucleoside in yeast total RNA (Figure 5E, third and second panels, respectively), confirming that ncm5U was introduced at position 34 of tRNAIleUAU isolated from the intronless strain. Curiously, ncm5U was also found in the same tRNA isolated from the pus1Δ strain, indicating that Pus1p-mediated ΨAΨ formation prevents ncm5U formation. Judging from the peak height ratio of the anticodon-containing fragments (Figure 5C), ∼40% of the tRNAIleUAU population contained the ncm5U modification.
Because ncm5U has an ability to read G-ending codons in vivo (39), we suspected that the tRNAIle with the ncm5UAU anticodon tends to misdecode AUG as Ile. To test this possibility, we expressed an EGFP reporter with a c-Myc tag (EQKLISEEDL). The c-Myc tag contains a critical Ile residue to be recognized by the mouse monoclonal antibody, 9E10; hence, we mutated this Ile codon to an AUG codon. If the AUG codon in the c-Myc tag was indeed mistranslated as Ile, the product would be detected by the 9E10 antibody. In the wild-type strain, the EGFP–c-Myc fusion protein was efficiently detected by western blotting with 9E10, whereas the mutant construct was hardly detected (Figure 5F). On the contrary to our expectation, similar results were obtained for the intronless strain. The results indicate that the tRNAIle with the aberrant ncm5UAU anticodon barely misdecodes the AUG codon in this reporter.
DISCUSSION
Most of the eukaryotes whose genomes have been sequenced have at least one isoacceptor/isodecoder tRNA whose genes contain a canonical intron. In an extreme case, all the tRNA species of an organism include at least one isodecoder encoded by intron-containing genes. Such canonical introns exist in all of the synonymous genes encoding a certain isoacceptor/isodecoder tRNA on a genome. These facts predicted that some tRNA introns might be essential for cell viability of the organism. However, we successfully deleted the introns from all the synonymous genes encoding any one of the 10 tRNA species in S. cerevisiae, which are the only tRNA species produced from intron-containing genes. These strains were all variable, and most of the mutants grew normally as the wild-type yeasts on the glucose media. Therefore, our results negate the above prediction, at least in S. cerevisiae, and are consistent with the recent report that the lethal effects of deleting an essential gene encoding tRNA ligase or 2′-phosphotransferase are bypassed by expressing a set of the ‘prespliced’ intronless versions of the 10 intron-containing tRNAs in S. cerevisiae (40).
On the other hand, the intron removal from some tRNA genes resulted in various phenotypes. Complete removal of the introns from the genes encoding tRNAIleUAU, tRNALysUUU, tRNATyr and tRNAPhe caused unexpected growth defects on respiratory media (Table 1; Figures 1 and 2). In addition, intron removal reduced the amounts of tRNALeuCAA, tRNATyr and tRNAProUGG in the mutant cells (Figure 3), but did not affect aminoacylation levels of all the 10 tRNAs. Surprisingly, the levels of 5.8S rRNA were reduced in the intronless mutants of tRNALeuCAA and tRNAPhe, and 27S rRNA intermediates were slightly accumulated in these mutants (Figure 4B). Especially, the tRNAPhe intronless but not tRNALeuCAA intronless mutant accumulated the 27SA2 rRNA intermediate. The intronless mutant of tRNALeuCAA showed a weak but apparent abnormality in nucleolar morphology (Figure 4C). As expected from in vitro studies (15,16), pseudouridylation of U34 and U36 on tRNAIleUAU was also intron-dependent in vivo, and tRNAIleUAU derived from the intronless genes underwent an unpredicted ncm5U34 modification.
Intron removal reduced the tRNA amount in three out of ten mutants by 23–33% (Figure 3). This may come from either transcriptional repression and/or enhancement of degradation. Although distance between the two internal promoter elements, box A and box B, in the tRNA genes has only a minor effect on recognition by TFIIIB and TFIIIC, because of the flexibility of these complexes (41), the absence or presence of the intron in a tRNA gene alters its ability of the tRNA gene to act as an insulator, a barrier for spreading of silenced chromatin, via stable recruitment of TFIIIC and condensin (42–44). Indeed, wild-type SUP53 (one of the tRNALeuCAA genes) itself has no insulator activity when positioned downstream of the HMR silencer, but its intronless allele can stop the spread of silencing from the silencer (44), meaning that the presence of the intron somehow compromises proper recruitment of TFIIIC and condensin on the tRNALeuCAA genes to build an insulator. These facts suggest that altering the distance between box A and box B by removing the intron may affect transcription factor recruitment and transcription efficiency, resulting in a reduction in tRNA levels observed in Figure 3. Since all the intronless mutants displayed steady state levels of tRNA aminoacylation similar to those of the wild-type strains (Supplementary Figure S2) and exhibited normal polysome/monosome ratios (Supplementary Figure S3), the aminoacyl-tRNAs derived from the intronless alleles seem to be produced and utilized properly. Thus, we consider that intron removal confers only minor impacts on the functions of tRNAs in translation.
To our surprise, the tRNALeuCAA intronless mutant had reduced levels of 5.8S rRNA and abnormality in nucleolar morphology despite its normal growth under laboratory conditions (Figures 1, 2 and 4). It is well known that tRNA genes prefer to locate near the nucleolus (36,45), and, as mentioned above, the presence of an intron in a tRNA gene can affect its function as an insulator (42). Therefore, removal of intron from a major group of tRNA genes, such as 9–10 synonymous genes encoding tRNALeuCAA, may affect structural organization of the nucleolus or the peri-nucleolar region via alteration of chromosome organization and/or chromatin status by introducing new insulators. In S. cerevisiae, a bulged nucleus, in which the nucleolus slightly stays away from the nucleoplasm, is thought to be an intermediated step between flared and round nuclei during mitosis, and is often observed in mitotically arrested cells (46,47). The abnormal nucleolar shape observed in the tRNALeuCAA mutant may be related to this phenomenon, and can be interpreted as retardation of a certain step of nucleolar dynamism during cell cycle progression. This kind of abnormality may cause a slight slowdown of some rRNA processing events in the nucleolus. We observed the major accumulation of the 27SA2 rRNA intermediate, which is specifically recognized by probe a, only in the tRNAPhe intronless mutant but not in the tRNALeuCAA intronless mutant. On the other hand, the total amount of several ∼27S rRNA intermediates including 27SA2, 27SA3, 27SBL and 27SBS slightly increased in both the tRNALeuCAA and tRNAPhe intronless mutants (Figure 4B). Thus, intron removal from tRNA genes of certain types may have tRNA-type specific effects on nuclear/nucleolar functions in rRNA maturation. In addition to our findings, several reports have demonstrated that defects in the pre-tRNA splicing machinery compromise rRNA processing; Sen34p depletion results in delayed pre-rRNA processing (48), and mitochondrial-located Sen2p is required for efficient pre-rRNA processing (49). Thus, the status of tRNA intermediates in the nucleus might also affect the efficient production of rRNA.
We confirmed intron-dependent pseudouridylation of tRNAIleUAUin vivo. Surprisingly, a considerable portion of tRNAIleUAU produced from the intronless allele underwent aberrant ncm5U modification at the wobble position (Figure 5). Thus, the tRNAIleUAU intron acts as a positive key to allow U34 pseudouridylation and a negative key to exclude U34 5-carbamoylmethylation. The same ncm5U modification was observed in the pus1Δ strain, in which intron-containing pre-tRNAIleUAU was transcribed as the wild-type. Eleven proteins including the Elp complex (Elp1–6p) required for 5-carbamoylmethylation of U34 in tRNAs are localized in the nucleus (50,51). Because the Elp complex does not seem to recognize intron-containing pre-tRNAIleUAU, its 5-carbamoylmethylation in the pus1Δ cells must take place after splicing, which occurs in the cytoplasm near mitochondria in S. cerevisiae (28). Consequently, ncm5U34 acquired in the pus1Δ cells should be the results of retrograde transport of spliced tRNAIleUAU from the cytoplasm (52,53), like the case of N1-methylation of G37 on tRNAPhe by Trm5p during wybutosine formation (54). Thus, for tRNAIleUAU, having an intron at the canonical position is not sufficient to avoid unwanted 5-carbamoylmethylation of U34, but efficient pseudouridylation of that position by Pus1p before recognition of the spliced anticodon by the Elp complex is also essential (Figure 6, upper panel; the wild-type cell). Loss of the introns from the tRNAIleUAU genes results in loss of these ‘double stoppers’ against the Elp complex to yield the aberrant modification (Figure 6, lower panel; the intronless mutant cell). In other words, the tRNA intron controls priority of modification enzymes acting on the corresponding tRNA. Although Ψ and ncm5U are predicted to have opposite effects on the wobble decoding (38,39), the tRNAIleUAU intronless cells normally distinguished the AUA codon from the AUG codon in our reporter system (Figure 5F). Because the minor tRNAIleUAU containing the ncm5UAU anticodon has to compete with the abundant tRNAMetelongation for recognition of AUG codons, it is possible that a low level of tRNAIlencm5UAU was not sufficient to compete for AUG-decoding in our experiments. We also examined translation of our EGFP-c-Myc reporters in tRNAIleUAU-overproducing strains, but saw no enhancement of AUG-to-Ile misdecoding, suggesting that ncm5U34 on tRNAIleUAU has a minimal impact on decoding specificity of this tRNA on our reporter mRNA (data not shown). Since we did not measure how much of total tRNAIleUAU was modified into the tRNAIlencm5UAU form in this intronless tRNAIleUAU overproducer, it is possible that only a very small portion of the tRNA receives ncm5U34 in this strain. Nevertheless, we think that our inability to detect misdecoding of AUG to Ile in our reporter system cannot be simply attributed to the molar ratio between tRNAIlencm5UAU and tRNAMetelongation. In some organisms, non-modified U34 in tRNAIleUAU can distinguish between AUA and AUG at the ribosomal A site (55), meaning that the overall structure of tRNAIleUAU has an additional role in exact discrimination between A and G at the wobble position. Such an effect might dominate codon recognition even in the case of yeast tRNAIlencm5UAU. To obtain the final answser of the effect of this altered modification, further in vivo analyses of the translatome/proteome from the whole mRNA transcriptome in the mutant yeast or in vitro analyses of decoding of AUA and AUG codons by tRNAIleUAU variants are required.
Figure 6.
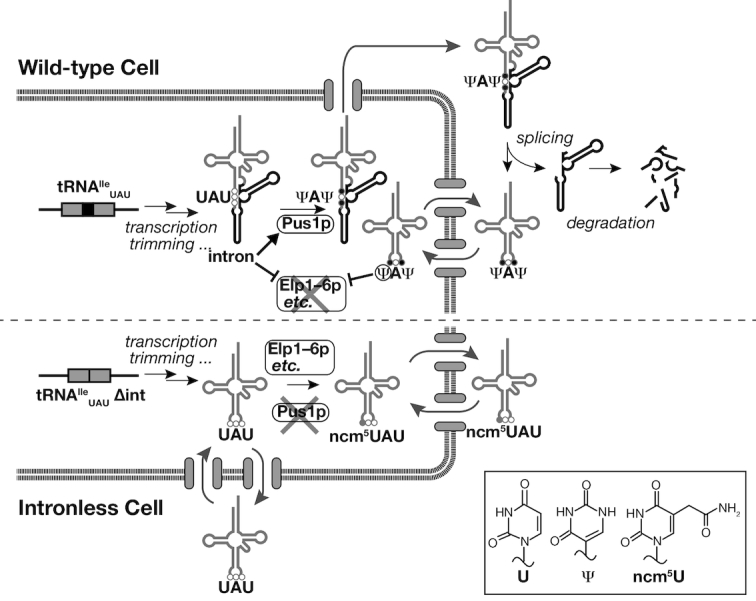
Physiological functions of introns in tRNA maturation. Schematic representation of the physiological functions of introns in tRNA maturation in Saccharomyces cerevisiae. Maturation of tRNAIleUAU in the wild-type yeast (upper) and the intronless (lower) cells is shown as an example. The gray and black parts of the tRNA represent two exons and an intron, respectively. The three circles represent the anticodon; open circle, unmodified nucleotide; black filled circle, Ψ introduced by Pus1p; gray filled circle, ncm5U introduced by the Elp complex and other factors. The chemical structures of U and its derivatives are shown in the inset. The thin black arrows represent reactions related to tRNA biosynthesis and maturation. The gray arrows represent intracellular dynamics of tRNA species during maturation. The intron of tRNAIleUAU promotes U34-to-Ψ34 modification but suppresses ncm5U34 formation during the early maturation step in the nucleus. Note that spliced tRNAIleUAU without Ψ modification, which is produced in pus1Δ cells, can be a substrate for ncm5U modification enzymes after its retrograde movement into the nucleus, but this feature is not shown in the figure for simplicity. Thus, Ψ34 formation is also essential for full protection against ncm5U34 mismodification.
Previous studies reported that tRNALeuCAA and tRNATyr undergo intron-dependent modifications (17,56). During our aminoacylation analysis of tRNAs, we found that removal of the intron from the tX(XXX)D locus, which may encode tRNASerGCU produced an unexpected tRNA species that migrated slightly faster than normal tRNASerGCU on the acid-urea gel. The unexpected tRNA was aminoacylated and migrated to a position similar to that of authentic tRNASerGCU in TBE-based gel electrophoresis, suggesting that some alkali-labile modification but still resistance to treatment at pH 9.0 and at 37°C might cause difference in electrophoretic mobility in the acid-urea PAGE. Alternatively, this new modification may introduce a functional group with pKa between 8.3 (the pH of TBE-based PAGE) and 5.0 (the pH of acidic PAGE), and the protonation state of the functional group affects electrophoretic mobility of the tRNA. Although the precise chemical natures of this new molecular species are still to be analyzed, it is possible that this modification on tRNASerGCU-like RNA is usually prevented by the canonical intron. The triple intron-deletion mutant of tRNASerGCU [ts(gcu)fΔint ts(gcu)oΔint tx(xxx)lΔint] did not produce the unknown fast-migrating species migrating faster in acid-urea-PAGE despite the fact that tX(XXX)L and tX(XXX)D have the same coding sequences. At present, we do not have enough pieces of evidence to explain this result precisely. One possible explanation is that the tX(XXX)L locus is somehow inactivated on the yeast chromosome and only tX(XXX)D is active. The set of our intronless mutants generated here will be valuable to analyze both intron-dependent modifications and modification avoidance in the nucleus by the introns.
In summary, this and previous reports clearly showed that the introns in genes encoding tRNAs on S. cerevisiae’s genome are dispensable for yeast viability, and their removal have minimal impacts on tRNA levels and functionality. However, some tRNA intronless mutants exhibit specific phenotypes related to various physiological aspects. Intron removal from the genes encoding certain tRNA species, such as tRNATyr and tRNAPhe, causes growth defects under respiratory conditions. Is there any relation between these growth phenotypes and mitochondrial localization of splicing machinery in the yeast? We are also wondering why the intronless mutant of tRNALeuCAA shows defects in 5.8S rRNA production and nucleolar morphology. The intron of tRNAIleUAU, and probably others, appears to control anticodon modification both positively and negatively. Still we do not have complete explanation of relationship between the growth defects and other individual phenotypes. However, these unexpected findings in the intronless mutants provide clues for novel interaction of the tRNA introns with respiration, the complexity of RNA modifications, and ribosomal properties, etc. The findings will also bring other unknown cellular roles of the introns to light. Further studies are required to understand the detailed functionalities of tRNA introns and their mechanisms of action, and our intronless strains constructed here will be powerful tools for this expedition.
Supplementary Material
ACKNOWLEDGEMENTS
We thank to our current and previous lab members for their support, especially, Prof. Toshiya Endo in Kyoto Sangyo University and Prof. Shuichi Nishikawa in Niigata University for fruitful discussion. We also thank to Ryota Ibusuki for preliminary analysis of possibility of AUG misdecoding by intronless tRNAIleUAU.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
KAKENHI for Scientific Research (C), Japan Society for the Promotion of Science [JP17K07289, JP17KT0113]; KAKENHI for Scientific Research on Innovative Areas, Ministry of Education, Culture, Sports, Science and Technology Japan [JP17H05672]; Hyogo Science and Technology Association, Japan [28144]. Funding for open access charge: JSPS.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kirchner S., Ignatova Z.. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015; 16:98–112. [DOI] [PubMed] [Google Scholar]
- 2. Hani J., Feldmann H.. tRNA genes and retroelements in the yeast genome. Nucleic Acids Res. 1998; 26:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heinemann I.U., Söll D., Randau L.. Transfer RNA processing in archaea: unusual pathways and enzymes. FEBS Lett. 2010; 584:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fujishima K., Kanai A.. tRNA gene diversity in the three domains of life. Front. Genet. 2014; 5:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshihisa T. Handling tRNA introns, archaeal way and eukaryotic way. Front. Genet. 2014; 5:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trotta C.R., Miao F., Arn E.A., Stevens S.W., Ho C.K., Rauhut R., Abelson J.N.. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell. 1997; 89:849–858. [DOI] [PubMed] [Google Scholar]
- 8. Phizicky E.M., Schwartz R.C., Abelson J.. Saccharomyces cerevisiae tRNA ligase. Purification of the protein and isolation of the structural gene. J. Biol. Chem. 1986; 261:2978–2986. [PubMed] [Google Scholar]
- 9. Culver G.M., McCraith S.M., Consaul S.A., Stanford D.R., Phizicky E.M.. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J. Biol. Chem. 1997; 272:13203–13210. [DOI] [PubMed] [Google Scholar]
- 10. Kosmaczewski S.G., Edwards T.J., Han S.M., Eckwahl M.J., Meyer B.I., Peach S., Hesselberth J.R., Wolin S.L., Hammarlund M.. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 2014; 15:1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang K.J., Guo L., Hou X.L., Gong H.-Q., Liu C.-M.. ZYGOTE-ARREST 3 that encodes the tRNA ligase is essential for zygote division in Arabidopsis. J. Integr. Plant Biol. 2017; 59:680–692. [DOI] [PubMed] [Google Scholar]
- 12. Wu J., Hopper A.K.. Healing for destruction: tRNA intron degradation in yeast is a two-step cytoplasmic process catalyzed by tRNA ligase Rlg1 and 5′-to-3′ exonuclease Xrn1. Genes Dev. 2014; 28:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujishima K., Sugahara J., Kikuta K., Hirano R., Sato A., Tomita M., Kanai A.. Tri-split tRNA is a transfer RNA made from 3 transcripts that provides insight into the evolution of fragmented tRNAs in archaea. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:2683–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clouet d’Orval B., Bortolin M.L., Gaspin C., Bachellerie J.P.. Box C/D RNA guides for the ribose methylation of archaeal tRNAs. The tRNATrp intron guides the formation of two ribose-methylated nucleosides in the mature tRNATrp. Nucleic Acids Res. 2001; 29:4518–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szweykowska-Kulinska Z., Senger B., Keith G., Fasiolo F., Grosjean H.. Intron-dependent formation of pseudouridines in the anticodon of Saccharomyces cerevisiae minor tRNAIle. EMBO J. 1994; 13:4636–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Motorin Y., Keith G., Simon C., Foiret D., Simos G., Hurt E., Grosjean H.. The yeast tRNA:pseudouridine synthase Pus1p displays a multisite substrate specificity. RNA. 1998; 4:856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Motorin Y., Grosjean H.. Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: identification of the gene and substrate specificity of the enzyme. RNA. 1999; 5:1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brzezicha B., Schmidt M., Makalowska I., Jarmolowski A., Pienkowska J., Szweykowska-Kulinska Z.. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNALeu(CAA). Nucleic Acids Res. 2006; 34:6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agris P.F., Eruysal E.R., Narendran A., Väre V.Y.P., Vangaveti S., Ranganathan S.V.. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2018; 15:537–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ranjan N., Rodnina M.V.. tRNA wobble modifications and protein homeostasis. Translation (Austin). 2016; 4:e1143076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho C.K., Abelson J.. Testing for intron function in the essential Saccharomyces cerevisiae tRNASer(UCG) gene. J. Mol. Biol. 1988; 202:667–672. [DOI] [PubMed] [Google Scholar]
- 22. Joardar A., Gurha P., Skariah G., Gupta R.. Box C/D RNA-guided 2′-O methylations and the intron of tRNATrp are not essential for the viability of Haloferax volcanii. J. Bacteriol. 2008; 190:7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mori S., Kajita T., Endo T., Yoshihisa T.. The intron of tRNA-TrpCCA is dispensable for growth and translation of Saccharomyces cerevisiae. RNA. 2011; 17:1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D.. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998; 14:115–132. [DOI] [PubMed] [Google Scholar]
- 25. Gietz R.D., Schiestl R.H., Willems A.R., Woods R.A.. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995; 11:355–360. [DOI] [PubMed] [Google Scholar]
- 26. Akada R., Hirosawa I., Kawahata M., Hoshida H., Nishizawa Y.. Sets of integrating plasmids and gene disruption cassettes containing improved counter-selection markers designed for repeated use in budding yeast. Yeast. 2002; 19:393–402. [DOI] [PubMed] [Google Scholar]
- 27. Adams A., Gottschling D., Kaiser C., Stearns T.. Methods in Yeast Genetics, A Cold Spring Harbor Laboratory Course Manual. 1997; 1997 edition. NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 28. Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T.. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol. Biol. Cell. 2003; 14:3266–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steiner-Mosonyi M., Leslie D.M., Dehghani H., Aitchison J.D., Mangroo D.. Utp8p is an essential intranuclear component of the nuclear tRNA export machinery of Saccharomyces cerevisiae. J. Biol. Chem. 2003; 278:32236–32245. [DOI] [PubMed] [Google Scholar]
- 30. Suzuki T., Suzuki T.. Chaplet column chromatography: isolation of a large set of individual RNAs in a single step. Methods Enzymol. 2007; 425:231–239. [DOI] [PubMed] [Google Scholar]
- 31. Ohira T., Suzuki T.. Precursors of tRNAs are stabilized by methylguanosine cap structures. Nat. Chem. Biol. 2016; 12:648–655. [DOI] [PubMed] [Google Scholar]
- 32. Suzuki T., Ikeuchi Y., Noma A., Suzuki T., Sakaguchi Y.. Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 2007; 425:211–229. [DOI] [PubMed] [Google Scholar]
- 33. Mengel-Jørgensen J., Kirpekar F.. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 2002; 30:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakaguchi Y., Miyauchi K., Kang B.-i, Suzuki T.. Nucleoside analysis by hydrophilic interaction liquid chromatography coupled with mass spectrometry. Methods Enzymol. 2015; 560:19–28. [DOI] [PubMed] [Google Scholar]
- 35. Yaffe M.P., Schatz G.. Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:4819–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson M., Haeusler R.A., Good P.D., Engelke D.R.. Nucleolar clustering of dispersed tRNA genes. Science. 2003; 302:1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kressler D., De La Cruz J., Rojo M., Linder P.. Fal1p is an essential DEAD-box protein involved in 40S-ribosomal-subunit biogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997; 17:7283–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Senger B., Auxilien S., Englisch U., Cramer F., Fasiolo F.. The modified wobble base inosine in yeast tRNAIle is a positive determinant for aminoacylation by isoleucyl-tRNA synthetase. Biochemistry. 1997; 36:8269–8275. [DOI] [PubMed] [Google Scholar]
- 39. Johansson M.J., Esberg A., Huang B., Björk G.R., Byström A.S.. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008; 28:3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cherry P.D., White L.K., York K., Hesselberth J.R.. Genetic bypass of essential RNA repair enzymes in budding yeast. RNA. 2018; 24:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagarajavel V., Iben J.R., Howard B.H., Maraia R.J., Clark D.J.. Global ‘bootprinting’ reveals the elastic architecture of the yeast TFIIIB-TFIIIC transcription complex in vivo. Nucleic Acids Res. 2013; 41:8135–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Donze D., Adams C.R., Rine J., Kamakaka R.T.. The boundaries of the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev. 1999; 13:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D’Ambrosio C., Schmidt C.K., Katou Y., Kelly G., Itoh T., Shirahige K., Uhlmann F.. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev. 2008; 22:2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donze D., Kamakaka T.R.. RNA polymerase III and RNA polymerase II promoter complexes are heterochromatin barriers in Saccharomyces cerevisiae. EMBO J. 2001; 20:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Belagal P., Normand C., Shukla A., Wang R., Léger-Silvestre I., Dez C., Bhargava P., Gadal O.. Decoding the principles underlying the frequency of association with nucleoli for RNA polymerase III-transcribed genes in budding yeast. Mol. Biol. Cell. 2016; 27:3164–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Witkin K.L., Chong Y., Shao S., Webster M.T., Lahiri S., Walters A.D., Lee B., Koh J.L., Prinz W.A., Andrews B.J. et al.. The budding yeast nuclear envelope adjacent to the nucleolus serves as a membrane sink during mitotic delay. Curr. Biol. 2012; 22:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walters A.D., May C.K., Dauster E.S., Cinquin B.P., Smith E.A., Robellet X., D’Amours D., Larabell C.A., Cohen-Fix O.. The yeast polo kinase Cdc5 regulates the shape of the mitotic nucleus. Curr. Biol. 2014; 24:2861–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Volta V., Ceci M., Emery B., Bachi A., Petfalski E., Tollervey D., Linder P., Marchisio P.C., Piatti S., Biffo S.. Sen34p depletion blocks tRNA splicing in vivo and delays rRNA processing. Biochem. Biophys. Res. Commun. 2005; 337:89–94. [DOI] [PubMed] [Google Scholar]
- 49. Dhungel N., Hopper A.K.. Beyond tRNA cleavage: novel essential function for yeast tRNA splicing endonuclease unrelated to tRNA processing. Genes Dev. 2012; 26:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang B., Johansson M.J.O., Byström A.S.. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005; 11:424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang B., Lu J., Byström A.S.. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA. 2008; 14:2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takano A., Endo T., Yoshihisa T.. tRNA actively shuttles between the nucleus and cytosol in yeast. Science. 2005; 309:140–142. [DOI] [PubMed] [Google Scholar]
- 53. Shaheen H.H., Hopper A.K.. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11290–11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohira T., Suzuki T.. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10502–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taniguchi T., Miyauchi K., Nakane D., Miyata M., Muto A., Nishimura S., Suzuki T.. Decoding system for the AUA codon by tRNAIle with the UAU anticodon in Mycoplasma mobile. Nucleic Acids Res. 2013; 41:2621–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson P.F., Abelson J.. The yeast tRNATyr gene intron is essential for correct modification of its tRNA product. Nature. 1983; 302:681–687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.