


Abstract
Recent studies on CRISPR adaptation revealed that priming is a major pathway of spacer acquisition, at least for the most prevalent type I systems. Priming is guided by a CRISPR RNA which fully/partially matches the invader DNA, but the plasticity of this RNA guide has not yet been characterized. In this study, we extensively modified the two conserved handles of a priming crRNA in Haloarcula hispanica, and altered the size of its central spacer part. Interestingly, priming is insusceptible to the full deletion of 3′ handle, which seriously impaired crRNA stability and interference effects. With 3′ handle deletion, further truncation of 5′ handle revealed that its spacer-proximal 6 nucleotides could provide the least conserved sequence required for priming. Subsequent scanning mutation further identified critical nucleotides within 5′ handle. Besides, priming was also shown to tolerate a wider size variation of the spacer part, compared to interference. These data collectively illustrate the high tolerance of priming to extensive structural/size variations of the crRNA guide, which highlights the structural flexibility of the crRNA-effector ribonucleoprotein complex. The observed high priming effectiveness suggests that primed adaptation promotes clearance of the fast-replicating and ever-evolving viral DNA, by rapidly and persistently multiplexing the interference pathway.
INTRODUCTION
CRISPR-Cas provides adaptive immunity against diverse foreign genetic elements in prokaryotes (1–4). CRISPR is an array of direct DNA repeats interspaced by variable foreign DNA sequences termed spacers. Cas-encoding genes are usually organized as an operon, and physically associated with CRISPR arrays. To date, two classes (class 1 and class 2), six types (types I–VI), and dozens of subtypes have been defined for this highly diversified system (5,6). Despite this diversity, the principle of CRISPR immunity generally involves three functional stages (4,7). The first stage, ‘adaptation’, refers to the process by which CRISPR acquires new spacers from the invader (8). During the second stage, CRISPR transcripts are processed into unit-sized mature CRISPR RNAs (crRNAs) (9–13), which, during the third stage, guide the effector protein (class 2) or the effector complex (class 1) to interfere the cognate foreign DNA/RNA (14–16). For the most prevalent type I systems, adaptation is special in that it has a priming pathway, which allows rapid spacer acquisition from the invaders that are fully or partially matched by the crRNA of a preexisting spacer (17–19). Like interference (20), priming also authenticates the protospacer adjacent motif (PAM), albeit with a relaxed stringency (21). Hence, each crRNA has the potential to guide both interference and priming, and the outcome depends on the spacer-protospacer matching (full or partial) and the PAM identity.
Transcripts of type I/III CRISPRs are usually processed by a Cas6-family endoribonuclease (9–11,22). Cas6 recognizes the repeat RNA in a sequence- and structure-specific manner (11) or via a wrap-around mechanism (23), and makes a single cleavage within the repeat. This cleavage generates two conserved handles (5′ or 3′) flanking the spacer guide. Interestingly, mature crRNAs purified from different effector complexes, including the Escherichia coli Cascade (9), the Pyrococcus furiosus CMR (15), the Staphylococcus epidermidis CSM (24), the Sulfolobus solfataricus CSM (25) and CMR (26), and the Thermus thermophilus CMR (27), usually partially or fully lack their 3′ handle. A recent in vitro study further showed that the crRNA-free Cascade purified from E. coli was able to incorporate a synthetic crRNA when only the single nucleotide immediately following its spacer part was retained (28). Besides, studies on crRNAs containing a shortened or extended spacer portion demonstrated an altered stoichiometry for the type I-E (29,30) or I-Fv (31) effector complex. These documents illustrated that the CRISPR machinery tolerates structural variations in the middle or 3′ part of the crRNA molecule during its assembly and/or functioning. However, the crRNA plasticity tolerated during priming adaptation has been rarely investigated.
Our previous studies on type I-B CRISPR in Haloarcula hispanica (18,32,33) demonstrated that efficient adaptation to a halovirus could be primed by a preexisting spacer. In this study, we showed that the crRNA of this priming spacer actually does not have a well-defined 3′ handle, which makes it highly unstable in vivo and impotent in interference. Using the archaeal transcription elements, we generated noncanonical crRNAs with both handles independent of Cas6 processing. By a series of mutagenesis analyses, we demonstrated that, compared to interference, primed adaptation is much more tolerant to extensive structural variations throughout the crRNA molecule, including 5′ handle truncation, spacer truncation/extension, and/or 3′ handle deletion/extension, which highlights the extraordinary robustness of the priming pathway and the structural flexibility of the crRNA-effector complex.
MATERIALS AND METHODS
Strains and culture conditions
The H. hispanica strains used in this study are listed in Supplementary Table S1. The uracil auxotrophic strain DF60 (H. hispanica ATCC 33960, ΔpyrF) and its derivative strains were cultured at 37°C in AS-168 medium (per liter, 200 g of NaCl, 20 g of MgSO4·7H2O, 2 g of KCl, 3 g of trisodium citrate, 1 g of sodium glutamate, 50 mg of FeSO4·7H2O, 0.36 mg of MnCl2·4H2O, 5 g of Bacto casamino acids, 5 g of yeast extract, pH 7.2) that contained uracil at a final concentration of 50 mg/l. Strains transformed by pyrF-expressing plasmids were cultured in yeast extract-subtracted AS-168 medium (unless otherwise specified).
The E. coli JM109 strain was used for cloning and cultured in Luria–Bertani medium. Ampicillin was added to a final concentration of 100 mg/l when necessary.
Plasmid and mutant constructions
The plasmids used in this study are listed in Supplementary Table S1. To replace the chromosomal CRISPR with a single-spacer mini-CRISPR (Sp1d or Sp13c), its upstream and downstream fragments (each ∼600 bp) were separately amplified, and then linked by overlap extension PCR (the mini-CRISPR was designed on the overlapping primers, e.g. Sp13c_UR and Sp13c_DF; see Supplementary Table S2). The final products containing the mini-CRISPR flanked by two homology arms were inserted into the suicide plasmid pHAR (predigested with BamHI and KpnI), and introduced into DF60 cells to replace the WT (wild-type) CRISPR via the pop-in-pop-out strategy (34). The expression plasmid pWL502 (35) was used to express mini-CRISPRs that target the virus (or the plasmid). The short-version (constitutive) promoter of the PHA synthesis-related gene phaR (36) was employed. Two DNA fragments respectively containing the promoter and the mini-CRISPR sequence were linked via overlap extension PCR. The products were cloned into pWL502 (predigested by BamHI and KpnI) and sequenced. Plasmid transformation was performed according to the online protocol (https://haloarchaea.com/wp-content/uploads/2018/10/Halohandbook_2009_v7.3mds.pdf).
CR-RT-PCR, primer extension and Northern blotting assay
Total RNA was extracted from stationary H. hispanica cultures using TRIzol reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer's instructions. For CR-RT-PCR (circularized RNA-reverse transcription-PCR) assay (37), 5–10 μg of total RNA was circularized using T4 RNA ligase (New England Lab, Massachusetts, USA) and reverse-transcribed using 200 U of M-MLV reverse transcriptase (Promega, Wisconsin, USA) with the CR-RT_R primer (Supplementary Table S2). The cDNA was amplified using the primer pair CR-RT_F/CR-RT_R with KOD-Plus DNA polymerase (TOYOBO, Osaka, Japan), and the PCR products were cloned into the pMD18-T for determination of the 5′ and 3′ extremities by DNA sequencing. For the primer extension assay, 5 μg of total RNA and 2.5 μg of 5′ FAM (6-carboxyfluorescein)-labeled primer (FAM-v10 in Supplementary Table S2; ordered from Thermo Fisher Scientific, MA, USA) were mixed and subjected to reverse transcription using 200 U of M-MLV reverse transcriptase (Promega, Wisconsin, USA) according to the manufacturer's instructions. The products were analyzed using an ABI3730xl DNA analyzer (Thermo Fisher Scientific, MA, USA) and the results were visualized using Genemapper (Version 4.1).
For Northern blotting analysis, 8–15 μg of total RNA, 20–40 fmol of 5′ biotin-labeled ssDNA (64 nt), and 2–4 μg of 100 to 1000 nt RNA Century-Plus marker (Thermo Fisher Scientific, MA, USA) were co-electrophoresed on an 8% polyacrylamide gel. The lane containing the RNA marker was excised, stained with ethidium bromide, and imaged using a ChemiDoc MP System (Bio-Rad, CA, USA). The lanes containing the RNA samples and ssDNA were transferred to a Biodyne B nylon membrane (Pall, NY, USA) using a Mini-Protean Tetra system (Bio-Rad, CA, USA). The membrane was then UV cross-linked. For hybridization, 25 pmol of 5′ biotin-labeled oligonucleotide probe (Supplementary Table S2; ordered from Thermo Fisher Scientific, MA, USA) was used. The pre-hybridization, hybridization, and washing procedures were performed as previously described (12). After hybridization, the samples were washed twice at room temperature and detection was performed using the Chemiluminescent nucleic acid detection module kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer's instructions. The membrane was imaged using a Tanon 5200 Multi chemiluminescent imaging system (Tanon Science & Technology, Shanghai, China). For each CRISPR variant, at least three independent replicates were performed.
Plasmid and virus interference assays
For the plasmid challenge assay, DF60 or its derivative strain was transformed with the target plasmid (pTTC1 or pTTC13), and the transformants were screened on yeast extract-subtracted AS-168 agar plates. For each target plasmid, at least three independent replicates were performed.
Virus interference was determined using a plaque test according to the online protocol (https://haloarchaea.com/wp-content/uploads/2018/10/Halohandbook_2009_v7.3mds.pdf). For each crRNA-expressing plasmid, at least three individual transformant colonies were picked and separately cultured in yeast extract-subtracted AS-168 medium. 200 μl of mid-log cell culture was mixed with 100 μl of 10-fold serial dilutions of the HHPV-2 virus and incubated for 15 min at room temperature. The mixture was added to 3 ml of molten yeast extract-subtracted AS-168 medium (maintained at 60°C; 0.7% agar), mixed and poured onto previously prepared overlay medium (1.2% agar). The plates were incubated for 7 days at 30°C for plaque formation. The plaque forming units (PFUs) on the lawns of H. hispanica cells containing either an empty plasmid (pWL502) or a crRNA-expressing plasmid were counted separately, and the ratio (empty: crRNA-expressing) was used to represent the relative virus immunity (RVI) that was conferred by the CRISPR construct.
Spacer acquisition assay
To monitor spacer acquisition from the plasmid DNA, the transformant of each target plasmid was inoculated into 3 ml of fresh AS-168 medium. Sub-inoculation was performed after 5-day culturing in a ratio of 1:15 (i.e., inoculating 200 μl of cell culture into 3 ml of fresh AS-168 medium). For each inoculation, colony PCR was performed after 5-day culturing. Briefly, 200 μl of cell culture was centrifuged at 12 000 rpm for 1 min, and then the sediment was lysed by 400 μl of distilled water, and 0.3 μl was used as the template for PCR analysis (using the primer pair test_F/test_R; Supplementary Table S2). The PCR program consists of the following steps: (i) 95°C for 5 min; (ii) 30 cycles of 95°C for 30 s, 54°C for 30 s, and 72°C for 30 s; (iii) 72°C for 10 min. The PCR products were separated on a 1.5% agarose gel. For each target plasmid, at least three transformant colonies were individually subjected to this analysis.
To monitor spacer acquisition from the viral DNA, 10-fold serial dilutions of HHPV-2 and 200 μl of mid-log cell culture were mixed at a multiplicity of infection (MOI) ranging from 1:1 to 1:10 000, and the mixture was inoculated into 3 ml of fresh yeast extract-subtracted AS-168 medium. After 5 days of culturing, colony PCR was performed as described above, and sub-inoculation was then performed at a ratio of 1:15 with addition of fresh virus dilutions at the same MOI. For each crRNA-expressing plasmid, at least three independent replicates were subjected to this analysis.
Quantitative analysis of CRISPR expansion was performed using the Quantity One software (Bio-Rad, CA, USA). For each gel lane, the parental and expanded bands were detected after background subtraction, and subjected to quantification using the Gaussian modeling method. For each lane, the intensity of expanded band(s) was divided by that of all bands to get the percentage of expanded products. At least three independent replicates were analyzed for each assay, and the average percentage was obtained with a standard deviation (SD).
Spacer loss assay
Two individual colonies of DF60 were separately inoculated into the AS-168 medium (containing uracil) and cultured for 4 days. Serial sub-inoculation was performed by repeatedly inoculating (10 μl of the cell culture into 10 ml of fresh medium) and culturing (for 4 days). After sub-inoculation was performed for 20 times, the cell culture was challenged by 1 μg of pTTC11 (Supplementary Table S1) via plasmid transformation. The CRISPR contents of the survivors were analyzed by DNA sequencing.
RESULTS
crRNA production and interference were hardly detected for the terminal spacer that primes efficient adaptation
The repeats from a single CRISPR array usually hold stringent sequence conservation, but the most leader-distal one is exceptional and known as the ‘degenerate repeat’ (see Figure 1A for an example). Due to this degeneration, the terminal spacer is usually expected to be non-functional, e.g. such a spacer from a type II CRISPR was shown incapable of guiding interference in Enterococcus faecalis (38). However, our previous studies (18,21,32,33) showed that the terminal spacer (spacer13 in Figure 1A) of the H. hispanica CRISPR functions well in priming efficient adaptation to the halovirus HHPV-2. To test whether it also functions well during interference, we constructed two target plasmids, each carrying a PAM sequence (5′-TTC-3′) and the protospacer of spacer1 (pTTC1) or that of spacer13 (pTTC13). Notably, transformation of the H. hispanica DF60 cells (which carry the WT CRISPR) with pTTC1 resulted in very few colonies, while pTTC13 showed a transformation rate comparable to that of the empty plasmid pWL502 (Figure 1C), suggesting that the interference activity of spacer13 is completely lost. Unexpectedly, we failed to detect any crRNA signals using a spacer13-specific probe during repeated Northern blotting assays (even after very long exposure), while the mature crRNA of spacer1 (s1-crRNA) was readily detected in abundance (Figure 1B). To rule out potential polar effects (CRISPR transcription may attenuate or terminate prematurely), we constructed two single-spacer mini-CRISPRs, Sp1d (previously designated as Δsp1-14, in which spacer1 is followed by a degenerate repeat) (18) and Sp13c (in which spacer13 is followed by a conserved repeat) (Figure 1A). The mini-CRISPR was introduced into DF60 cells using the suicide plasmid pHAR, and the WT CRISPR was replaced via the pop-in-pop-out strategy (34). The production of s13-crRNA and its interference against pTTC13 in the Sp13c cells were readily detected by Northern blotting and plasmid transformation assays, respectively (Figure 1B and C). In contrast, the blotting signal of s1-crRNA and its interference against pTTC1 both disappeared in Sp1d cells. Notably, when pTTC1 was introduced into the Sp1d cells, the mini-CRISPR (Sp1d) rapidly acquired new spacers (Figure 1D), suggesting that the undetected s1-crRNA molecules primed efficient adaptation to the target plasmid. Therefore, the crRNA stability and interference function of the terminal spacer are seriously impaired, while its priming activity stubbornly persists.
Figure 1.

The crRNA production and interference effects were not detected for the terminal spacer. (A) Schematic representation of the WT CRISPR on the H. hispanica genome and two single-spacer mutants. In the WT CRISPR, the most leader-distal spacer (spacer13) is followed by a degenerate repeat (yellow), which differs from the consensus repeat sequence (orange) by 9 nucleotides (in bold and italic). In the mutants Sp1d and Sp13c, spacer1 and spacer13 are followed by the degenerate and the consensus repeat, respectively. Primers employed in panel D are indicated. (B) Northern analysis of the crRNA molecules of spacer1 (s1) and spacer13 (s13) in different strains. 7S RNA was probed as the internal control. M, a biotin-labeled 64-nt ssDNA. -, an empty control without RNA loading. (C) A plot showing the transformation rate of different target plasmids in DF60, Sp1d or Sp13c cells. The target plasmids pTTC1 and pTTC13 are pWL502 derivatives carrying the protospacer of spacer1 and that of spacer13, respectively. The protospacer sequence was designed immediately downstream of a canonical PAM, i.e. 5′-TTC-3′. CFU, colony-forming units. (D) Sp1d cells transformed by pTTC1 were inoculated into the liquid medium, and after 5-day culturing, cells were lysed and subjected to PCR analysis. M, dsDNA size marker. The ∼180 bp PCR products correspond to the parental Sp1d structure (Pa), while the larger-sized products represent those expanded by one or more new spacers (Ex). The plot below the gel shows the percentage of the intensity of ‘expanded’ bands (Ex/(Ex+Pa)). Error bars indicate the standard deviation (SD) calculated from three independent replicates.
The conserved 3′ handle is critical for crRNA stability in vivo
To introduce mutations into the conserved 3′ handle without abolishing crRNA production, we sought to generate a 3′ handle independent of Cas6-processing. A poly-T stretch (5-8 thymidines) has been reported to be an effective transcription terminator in different archaeal species (39–42). Hence, a run of eight thymidine residues (designated as T8) was employed to terminate transcription immediately downstream of the 22-nt 3′ handle (Figure 2A and B). We selected a 34-nt spacer denoted ‘v10’, which was acquired from the HHPV-2 rep gene (ORF1) during a previous adaptation assay (18) and has a canonical PAM (5′-TTC-3′) (Supplementary Figure S1). We first constructed two mini-CRISPRs, which were designated R-v10-R and R-v10-h22 (‘R’ denotes a repeat sequence and ‘h22’ indicates the 22 nucleotides of 3′ handle) (Figure 2A). Their transcription was driven by the strong promoter PphaR (36) and stopped by the T8 terminator. It should be noted that, as shown in Figure 2B, the 5′ untranslated sequence (5 bp) and the start codon (3 bp) of the phaR gene were both retained to ensure promoter activity (unless otherwise specified). The theoretical size of the mature WT v10-crRNA is 64 nt, which coincides with the blotting results of R-v10-R (Figure 2D). In contrast, R-v10-h22 is expected to produce noncanonical crRNAs of ∼72 nt due to the extra uridines tailing 3′ handle (Figure 2C), and a main RNA band corresponding to this size was observed, with an abundance similar to that of the WT v10-crRNA (Figure 2D). Apparently, these extra uridines did not affect crRNA stability. We also noticed that leaky termination of transcription frequently occurred to produce a longer precursor (Figure 2D; Supplementary Figure S2), but the corresponding mature crRNA, which should have a longer 3′ extension (∼20 nt), could not be observed. Presumably, a large number of extra nucleotides at the crRNA 3′ end would cause instability, which consists with the undetectable crRNA products of the terminal spacer (Figure 1B).
Figure 2.

The first 12 nt of 3′ handle decides crRNA stability and interference capability. (A) The experimental design to generate a processing-independent 3′ handle and to test its impacts on crRNA function. The Sp1d host cells carries a chromosomal CRISPR that cannot interfere or prime adaptation to HHPV-2. The v10 spacer targets the HHPV-2 genome, and may elicit virus interference and/or primed adaptation. Spacer incorporation into the chromosomal CRISPR Sp1d can be monitored by PCR analysis with the test_F/R primer pair. Transcription of the plasmid CRISPR is driven by the phaR promoter (PphaR) and stopped by the T8 terminator. Repeat sequences are in orange (the degenerate one is in yellow). The size (nt) of each part is given for each crRNA molecule. (B) The nucleotide sequences of R-v10-R (with the nucleotides in brackets) and R-v10-h22 (without the nucleotides in brackets). The promoter, 5′-UTR, and start codon of phaR are shaded blue, and the repeat sequences are shaded orange. The ‘TTTTCTT’ in red may act as another terminator (see Supplementary Figure S2). (C) CRISPR constructs producing crRNAs with different 3′ handles. The predicted size of the major crRNA product is given for each construct. (D) The RNA products were analyzed by Northern blotting using the v10-specific probe. 7S RNA was probed as the internal control. The question mark indicates an unexpected product. Two precursor RNA bands were detected for each sample due to leaky termination of CRISPR transcription (see Supplementary Figure S2). M1, the 100-nt ssRNA ladder. M2, a biotin-labeled 64-nt ssDNA primer. -, the empty pWL502 control. (E) The relative virus immunity (RVI) conferred by different CRISPR constructs in Sp1d cells. Error bars indicate the SD calculated from three independent replicates.
By modifying R-v10-h22, we further constructed R-v10-h16, R-v10-h12, R-v10-h10, R-v10-h8 and R-v10-h0, which produce the first 16, 12, 10, 8 or 0 nucleotides of 3′ handle, respectively (Figure 2C). It should be noted that these truncated 3′ handles would have lost the potential to form a hairpin (Supplementary Figure S3). Nevertheless, transcription termination in archaea appears to be independent of RNA secondary structures (39,41,42), and consistently, R-v10-h16 and R-v10-h12 produced large amounts of crRNAs of expected sizes (about 66 nt and 62 nt, respectively) (Figure 2D). In contrast, crRNA amounts from R-v10-h10, R-v10-h8 and R-v10-h0 dramatically decreased, with the latter two showing the least crRNA abundance. It seems that the first 12 conserved nucleotides of 3′ handle are important for crRNA stability in vivo. To exclude putative effects of crRNA size, we mutated R-v10-h12 by replacing its 3′ handle nucleotides with the 12-nt viral sequence that immediately follows the v10 protospacer (Supplementary Figure S1), giving rise to the structure R-v10-h12m (Figure 2C). In cells expressing this variant, the size of mature crRNA (∼62 nt) was not affected, but its amounts dramatically declined (Figure 2D), corroborating the significance of the first 12 nucleotides.
3′ Handle deletion strongly impairs interference but enhances the priming effects
To test the effects of these modified 3′ handles on the two guiding functions (in interference and primed adaptation), plasmids carrying these CRISPR structures were introduced into the Sp1d cells, which were then subjected to HHPV-2 infection (Figure 2A). The chromosomal CRISPR Sp1d contains only one spacer that shows no homology to HHPV-2 genome, thus it can neither interfere nor prime adaptation against this virus. Nevertheless, this structure contains an intact CRISPR leader preceding a repeat that can incorporate new spacers (32) when the plasmid-expressed crRNAs elicit a priming process (as illustrated in Figure 2A). The plaque forming units (PFUs) on the lawns of H. hispanica cells containing the empty plasmid or a CRISPR-bearing plasmid were separately counted, and their ratio (empty: CRISPR-bearing) was used to indicate the relative virus immunity (RVI) conferred by this CRISPR construct. Cells expressing R-v10-R, R-v10-h22, R-v10-h16 or R-v10-h12 showed a high-level immunity (RVI > 105), whereas this immunity was decreased by three orders of magnitude in cells expressing R-v10-h10 (103 > RVI > 102), and by nearly four orders of magnitude in cells expressing R-v10-h8, R-v10-h0 or R-v10-h12m (102 > RVI > 10) (Figure 2E). Hence, the interference effects showed a clear dependence on the crRNA in vivo stability/amounts observed in Figure 2D, suggesting that an abundance of crRNA molecules were required for substantial interference against this virus. When we employed another virus-targeting spacer, 3′ handle deletion resulted in complete loss of virus immunity (Supplementary Figure S1), indicating that the interfering effect of different spacers showed different tolerance to 3′ handle deletion. Overall, the first 12 nucleotides of 3′ handle seem to be very important, if not absolutely necessary, for crRNA to accumulate to a sufficient concentration that is required for virus interference.
To test the priming efficiency, we subjected liquid cultures of Sp1d cells expressing different v10-crRNAs to HHPV-2 infection, and then examined the expansion of the Sp1d structure (resulted from new spacer acquisition) via PCR analysis 5 days later (when required, sub-inoculation and reinfection were performed) (Figure 3A). Remarkably, PCR products corresponding to expanded Sp1d structures (hereinafter referred to as expanded products) were observed for cells expressing any of these v10-crRNA molecules, but not in those containing the empty plasmid (Supplementary Figure S4). This data supports that 3′ handle is dispensable for priming adaptation, as it was completely mutated or deleted in R-v10-h12m and R-v10-h0. Interestingly, when cells expressing R-v10-R or R-v10-h0 were infected at an MOI ranging from 1:1 to 1:10000, the latter structure appeared to prime more rapid and more sensitive adaptation to HHPV-2. During the 1st PCR analysis (Figure 3B), expanded PCR products were observed for R-v10-h0 cells, but not for R-v10-R cells. Only when infected twice at a higher MOI, expanded products could be observed for R-v10-R cells (Figure 3C). Moreover, when MOI was 1:1, 1:10 or 1:100, we observed a higher percentage of expanded products for R-v10-h0 than for R-v10-R cells (34.2% versus 26.7%, 31.8% versus 14.5%, and 21.5% versus 11.7%, respectively) during the second PCR analysis. Hence, 3′ handle deletion appears to enhance the priming effects while impairing the interfering effects during virus immunity. This observation fits the kinetic model (43) that attenuated interference may allow the fast-replicating viral DNA to provide more substrates for adaptation within a longer time period.
Figure 3.
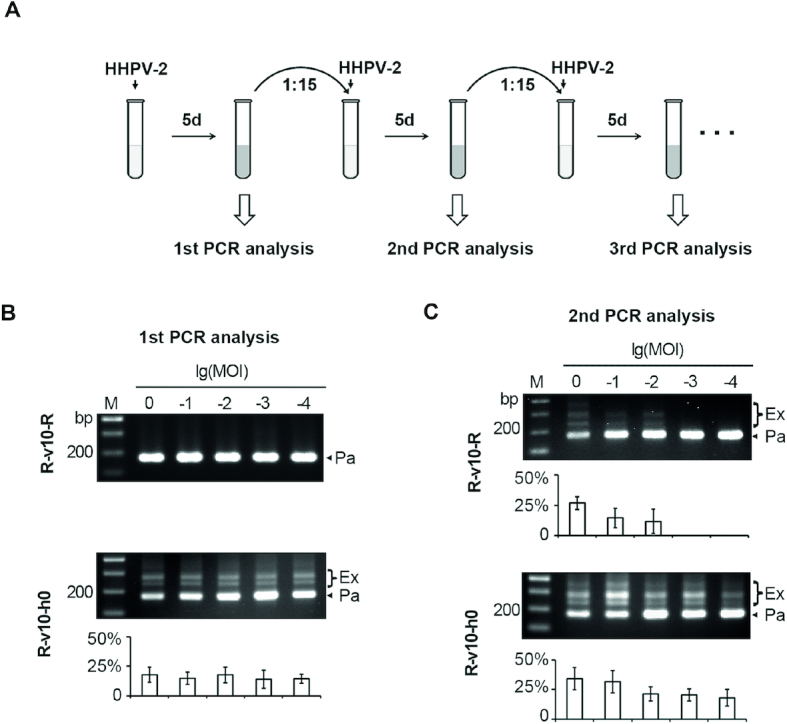
Primed adaptation became more evident when 3′ handle was deleted. (A) Schematic depiction of the procedure to monitor the v10-primed adaptation to HHPV-2. The mid-log culture of Sp1d cells expressing v10-crRNA was inoculated into fresh medium with HHPV-2 addition (MOI ranging from 1:1 to 1:10000). After 5-day culturing, expansion of the Sp1d CRISPR was monitored by PCR analysis (see Figure 2A for primer information). Sub-inoculation and reinfection were performed until Sp1d expansion was detected. The first (B) and the second (C) PCR analysis of spacer acquisition in Sp1d cells expressing R-v10-R or R-v10-h0. The log values of MOI are indicated. M, dsDNA size marker. The plots show the percentage of the intensity of ‘expanded’ bands (Ex/(Ex+Pa)) for each gel lane. Error bars indicate the SD calculated from three independent replicates.
The last 6 nt of 5′ handle is required for priming
During interference or priming, the effector complex must incorporate a crRNA by recognizing its conserved handle sequences. Given the dispensability of 3′ handle during priming adaptation, the 8-nt 5′ handle should contain necessary signals for Cascade recognition. To introduce mutations into this conserved element without abolishing crRNA production, we attempted to generate a processing-independent 5′ handle by controlling transcription initiation. We put the 5′ handle-transcribing 8 bp (5′-GTTGAAGC-3′) directly downstream of the phaR promoter, so that transcription starts precisely at its first nucleotide G (guanine), which was verified by primer extension and CR-RT-PCR analysis (Supplementary Figure S5). In this way, H8-v10-h22 was designed to produce a noncanonical crRNA that is independent of Cas6-processing (Figure 4A). Compared to R-v10-h22, a very low level of virus immunity (102 > RVI > 10) was conferred by H8-v10-h22 (Figure 4B). Consistently, Northern blotting revealed that the crRNA of H8-v10-h22 showed a significantly decreased accumulation compared to that of the mature crRNA from R-v10-h22, although these two RNA species are of the same size (72 nt) (Figure 4C). Theoretically, their only structural difference is the 5′ end group: the crRNA of H8-v10-h22 carries a 5′ triphosphate, which is a feature of a nascent RNA transcript, while the mature crRNA from R-v10-h22 carries a 5′ hydroxyl, which is a product of Cas6-processing. Given the in vitro observation that Cascade or Cmr assembly occurs only when the synthetic crRNA bears a 5′ hydroxyl (44,45), we predicted that the RNA transcripts of H8-v10-h22 should be subjected to cellular end-processing enzymes, such as phosphatases, to become a functional crRNA. In contrast to the low-level interference effects, adaptation to HHPV-2 was efficiently primed by H8-v10-h22: when Sp1d cells expressing this structure were subjected to HHPV-2 infection at an MOI ranging from 1:1 to 1:10 000 and cultured for 5 days, 10.7–16.1% of the PCR products from the Sp1d structure were observed to be ‘expanded’ (Figure 4D).
Figure 4.

The last 6 nt of 5′ handle is required for priming adaptation to HHPV-2. (A) The H8-v10-h22 and H8-v10-h0 constructs producing Cas6-independent crRNAs (predicted sizes are given). As indicated by the curved arrows, transcription was designed to initiate exactly from the beginning nucleotide of the DNA sequence for 5′ handle. (B) The relative virus immunity (RVI) provided by H8-v10-h22 and H8-v10-h0 in Sp1d cells. Data of R-v10-h22 (against a grey background) is shown again to facilitate comparison. (C) The v10-crRNA from H8-v10-h22 showed a largely decreased stability compared to that from R-v10-h22. 7S RNA was probed as the internal control. Adaptation to HHPV-2 was primed by H8-v10-h22 (readily detected in the first PCR analysis) (D) or H8-v10-h0 (hardly detected until the 4th PCR analysis) (E). (F) Constructs producing crRNAs with a truncated 5′ handle (and no 3′ handle). Note that RNA transcripts from these constructs carry a natural 5′ triphosphate, which may need to be removed prior to Cascade assembly. (G) HHPV-2 adaptation primed by constructs listed in panel F. –, the empty pWL502 control. Adaptation was monitored as illustrated in Figure 3A, and infection was performed at the specified MOIs. The plots show the percentage of the intensity of ‘expanded’ bands (Ex/(Ex+Pa)) for each gel lane. Error bars indicate the SD calculated from three independent replicates.
Subsequently, we modified H8-v10-h22 by deleting the DNA sequence of 3′ handle to generate the structure H8-v10-h0 (Figure 4A). This structure was unable to provide virus immunity (Figure 4B), suggesting that 5′ hydroxyl and the 3′ handle sequence additively contribute to the interfering effects. When cells expressing H8-v10-h0 were subjected to adaptation analysis, only after four rounds of (sub-)inoculation and (re)infection at a higher MOI (1:1, 1:10 and 1:100), expanded PCR products could be observed (with a percentage of 22.2%, 15.3% and 10.2%, respectively) (Figure 4E). Therefore, the RNA products from H8-v10-h0 were capable of priming, but the efficiency appeared to be dramatically decreased compared to those from H8-v10-h22. Then we gradually trimmed the DNA sequence of 5′ handle to generate a series of crRNAs with different-length (4-8 nt) 5′ handles (Figure 4F). It should be noted that these noncanonical crRNAs should consistently carry a 5′ triphosphate and no 3′ handle sequence. Sp1d cells expressing these crRNAs were subjected to rounds of virus infection (at an MOI of 1:1) and PCR analysis. When 5′ handle was 8nt, 7nt or 6nt, the expanded products accounted for 19.2%, 30.3% or 18.6%, respectively, during the fourth PCR analysis (Figure 4G). Notably, these ‘expanded’ products were not detected for a shorter 5′ handle, even during the sixth PCR analysis (data not shown). By sequencing the PCR products acquired from the H6-v10-h0 assay, we confirmed that the new spacers were derived from the virus genome (Supplementary Table S3). These data indicate that the minimal handle sequence required for priming is the last 6 nt of 5′ handle.
To confirm this requirement, we substituted the v10-spacer with a spacer (p1) targeting the pyrF gene which locates on the pWL502 backbone (Figure 5A), and expressed these 5′ handle-truncated p1-crRNAs in Δcas6 cells (so that the new spacers acquired from the pWL502 derivatives would not lead to plasmid interference). After inoculation into liquid mediums and culturing for 5 days, expansion of the WT CRISPR structure in these cells was monitored (Figure 5B) using the test_F/test_R primer pair (Figure 2A). When 5′ handle was 8 nt, 7 nt or 6 nt, products corresponding to the expanded CRISPR account for 20.5%, 32.3% or 10.3%, respectively (Figure 5C). In contrast, the expanded products were not detected for a shorter 5′ handle, even after sub-inoculation and another 5-day culturing (data not shown). These results are generally in line with the virus-targeting assay, confirming that the last 6 nt of 5′ handle is the minimal handle sequence required for priming.
Figure 5.

The last 6 nt of 5′ handle contain critical nucleotides for priming. (A) Design of a plasmid-targeting CRISPR that primes spacer acquisition from the plasmid backbone. The target sequence of the p1 spacer (p1 protospacer) locates within the pyrF gene. (B) The experimental procedure to monitor the adaptation primed by p1-crRNA in Δcas6 cells. Primers indicated in Figure 2A were used to amplify the WT CRISPR in the Δcas6 cells. (C) Adaptation primed by p1-crRNAs with a truncated 5′ handle and no 3′ handle. (D) Adaptation primed by p1-crRNAs carrying no 3′ handle and various single nucleotide substitutions in a 6-nt 5′ handle. M, dsDNA size marker. –, the empty pWL502 control. The plots show the percentage of the intensity of ‘expanded’ bands (Ex/(Ex+Pa)) for each gel lane, with error bars indicate the SD calculated from three independent replicates.
Subsequently, we performed single base substitutions throughout the 6-nt 5′ handle of H6-p1-h0. We found that primed adaptation was nearly insensitive to the C-1A mutation: in the first PCR analysis, its ‘expanded’ products account for 12.7%, which is a little higher than the WT (10.3%) (Figure 5D). This percentage was much lower in the case of G-2U (3.5%), A-4C (4.3%) and G-5U (3.1%) mutations, and became zero in the case of A-3C and U-6G mutations. During the second PCR analysis (after sub-inoculation and another 5-day culturing), the percentage of expanded products increased to 18.4%, 5.0%, 2.5%, 6.4%, and 3.6% in the C-1A, G-2U, A-3C, A-4C, G-5U samples, respectively; but expansion was still not detected for U-6G. In addition, when G-2U and A-3U substitutions were simultaneously introduced, expansion was not detected in the second PCR analysis. Therefore, conservation of at least G-2, A-3 and U-6 should be important for the priming function of a crRNA.
The majority size variation of newly acquired spacers is tolerated during priming
Though the spacers in a single CRISPR array are kept largely constant in size, spacer size variation can be substantially observed for CRISPR arrays of different subtypes, or for those of the same subtype but from different organisms (30,46). By high-throughput analysis of the spacer acquisition process in H. hispanica, our previous study revealed a considerable heterogeneity in the size of newly acquired spacers, which mainly varied from 32 to 39 bp (33). When re-analyzing these data, we noticed the rare occurrence (∼0.15%) of much shorter or longer spacers (Figure 6A), and wondered whether a spacer of such a rare size could lead to interfering or priming effects. Therefore, we modified the R-v10-h22 structure to produce a spacer ranging from 16 to 72 bp. By Northern blotting, we found that the mature crRNA products were similar in abundance when the spacer size varied between 16 and 44 bp and only mildly decreased when the spacer was 60 or 72 bp (Figure 6C). Surprisingly, when Sp1d cells expressing these CRISPRs were subjected to HHPV-2 infection, substantial immunity was observed as long as the spacer was longer than 30 bp (Figure 6B). This immunity declined by 3–4 orders of magnitude when the v10-spacer was shortened to 30 bp and almost disappeared when shortened to 28, 24 or 16 bp. The liquid cultures were further subjected to rounds of HHPV-2 infection (MOI = 1:1) and PCR analysis. When the v10-spacer varied from 28 to 72 bp, adaptation could be readily detected, with the percentage of expanded products ranging between 45.1% and 70.5% during the third PCR analysis (Figure 6D). This percentage was significantly compromised by further truncation of the spacer to 24 bp (10.5%). In contrast, adaptation primed by the 21-bp was not detected, even during the fifth PCR analysis (data not shown). Therefore, these data suggest that substantial interference can occur with a spacer longer than 30 bp, and priming can occur with a spacer no shorter than 24 bp. According to the size distribution shown in Figure 6A, we predict that 99.965% and 99.997% of new spacers should be proficient in interfering and priming, respectively, when only the factor of spacer size is considered.
Figure 6.

Priming tolerates the majority of the spacer size variation observed during acquisition. (A) The wide size variation observed for 37,638 new spacers acquired in a previous adaptation assay (33). (B) The relative virus immunity (RVI) conferred by R-v10-h22 derivatives, of which the spacer part varies between 16 and 72 bp. (C) The impact of spacer size on crRNA stability. The lowest band of each sample lane corresponded to their mature crRNA products, and the upper band(s) corresponded to precursors. 7S RNA was probed as the internal control. M1, the 100-nt ssRNA ladder. M2, a biotin-labeled 64-nt ssDNA primer. (D) HHPV-2 adaptation primed by the R-v10-h22 derivatives with a varying spacer size. M, dsDNA size marker. –, the empty pWL502 control. The plot below the gel shows the percentage of the intensity of ‘expanded’ bands (Ex/(Ex+Pa)) for each lane. Error bars indicate the SD calculated from three independent replicates.
DISCUSSION
Biochemical studies on Cas6 revealed that it remains tightly bound to 3′ handle after cleavage (10,47,48), thus led to the speculation that the Cas6-3′ handle binding serves as a nucleation point for Cascade assembly (49) which can protect nascent crRNA from cellular RNases. Our observation that 3′ handle (especially its first 12 nucleotides) is critical for crRNA stability provides in vivo evidence (Figure 2D). Note that the critical 12 nt coincide with the in vitro observation that the first 12 nt of the repeat RNA is required for Cas6 binding (10). Since 3′ handle is usually missing from the crRNA molecules purified from the effector complex (9,15,24–27), the Cascade function seemingly doesn’t require this element (28). Hence, the impacts of 3′ handle truncation/deletion on virus immunity that were observed in this study (Figure 2E) may be mainly contributed to its impacts on Cascade assembly and crRNA abundance. Similarly, the declined immunity observed when 5′ end group was altered from hydroxyl to triphosphate (Figure 4B) may also be derived from its impacts on Cascade assembly, because 5′ hydroxyl was reported to be essentially required for the in vitro assembly of this complex (45). Interestingly, truncation of the spacer part appears to compromise target recognition, instead of the assembly of the Cascade complex, because a short spacer of 28 or 24 bp provided no virus immunity while the abundance of its crRNA was almost uninfluenced (Figure 6). In contrast, priming showed a high tolerance to these structural variations, as well as truncation of 5′ handle. Significantly, an RNA molecule that begins with the last 6 nt of 5′ handle was able to prime adaptation to a cognate viral/plasmid DNA. Furthermore, except the uridine at position -6, mutation of any of the other 5 nucleotides was differently tolerated during priming adaptation to a plasmid target. Given that crRNA serves as the backbone of the Cascade complex, we infer that: (i) priming tolerates extensive structural variations of the Cascade complex, and some variations (e.g., truncation of spacer to 28 bp) may render Cascade inactive in interference but hyperactive in priming; (ii) a very low concentration of functional Cascade is required for efficient priming, which allows more prominent spacer acquisition when interference gets compromised by crRNA insufficiency (the viral DNA could replicate and persist for a longer time period to provide more spacer substrates), as observed in Figure 3.
The fact that the MOI used for plaque assays was usually below 1:1 (see Materials and Methods) suggests that each cell was generally infected by no more than one virus. Hence, the significant dependence of immunity on crRNA stability/Cascade assembly implies that an abundance of CRISPR effector complexes are required to guarantee the complete elimination of the fast-replicating viral DNA, even when only one copy enters the cell. This requirement gives an escape chance to the virus, especially to those encoding an anti-CRISPR protein that can lower down the concentration of functional Cascade (50,51). However, the much lower concentration of Cascade required for priming allows the persistent acquisition of new spacers from the infecting virus and the enrichment of Cascade effectors that specifically target this virus. Moreover, the host cell usually encodes additional defense systems, such as toxin-antitoxin systems which can elicit temporary cell dormancy upon infection (52), and BREX systems which can inhibit viral DNA replication (53,54). Such additional defense mechanisms will buy time for the robust and effective primed adaptation process and for the recovery of the concentration of effective Cascade complex. From this point of view, priming should substantially enhance CRISPR immunity not only by combating with the virus escape mutants, but also by assisting the complete elimination of the fast replicating wild-type viral DNA. This may help to explain the widest distribution of type I CRISPRs (primed adaptation has been reported only for type I systems).
Specifically, we investigated the noncanonical crRNA of the terminal spacer. This crRNA probably carries a largely extended 3′ handle due to the mutations within its downstream repeat (degenerated) that can abolish Cas6-processing. Though this crRNA is too low in abundance (below the detection limit of Northern blotting analysis) to support evident interference, primed adaptation could stubbornly and efficiently occur (Figure 1). These results again highlight the robustness of the priming process. Additionally, the base substitutions within the degenerate repeat might lead to the long-term preservation of the terminal spacer, as spacer loss via the recombination between this repeat and any of the other repeats within the CRISPR array may be largely avoided. In fact, we did observe the preservation of spacer13 during investigation of the spacer loss process in H. hispanica (Supplementary Figure S6). Preservation of the terminal spacer should be evolutionarily beneficial when it targets a conserved region, for example, spacer13 targets the critical rep gene that is required for viral genome replication. Conceivably, the preservation of spacer13 in H. hispanica CRISPR may allow it to adapt to a collection of related viruses that carry the conserved rep gene, via the robust priming pathway.
In summary, our study highlights the robustness of primed adaptation, which tolerates extensive structural variations within the crRNA molecule, including 5′ handle truncation, spacer truncation/extension, and 3′ handle deletion/extension, and also tolerates a very low concentration of crRNA-Cascade complex. This robustness reflects the extraordinary flexibility in the architecture of the crRNA-effector complex, which may be exploited to create simplified/modified tools for DNA/RNA manipulation.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31571283, 31771381]; Young Elite Scientists Sponsorship Program by CAST [2017QNRC001]; National Transgenic Science and Technology Program [2019ZX08010-001]. Funding for open access charge: National Natural Science Foundation of China [31571283].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Terns M.P., Terns R.M.. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011; 14:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wiedenheft B., Sternberg S.H., Doudna J.A.. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012; 482:331–338. [DOI] [PubMed] [Google Scholar]
- 4. Hille F., Richter H., Wong S.P., Bratovič M., Ressel S., Charpentier E.. The biology of CRISPR-Cas: Backward and forward. Cell. 2018; 172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 5. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al.. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wright A.V., Nuñez J.K., Doudna J.A.. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell. 2016; 164:29–44. [DOI] [PubMed] [Google Scholar]
- 7. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.V., van der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016; 353:aad5147. [DOI] [PubMed] [Google Scholar]
- 8. Sternberg S.H., Richter H., Charpentier E., Qimron U.. Adaptation in CRISPR-Cas systems. Mol. Cell. 2016; 61:797–808. [DOI] [PubMed] [Google Scholar]
- 9. Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carte J., Pfister N.T., Compton M.M., Terns R.M., Terns M.P.. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010; 16:2181–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A.. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010; 329:1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li M., Liu H.L., Han J., Liu J.F., Wang R., Zhao D.H., Zhou J., Xiang H.. Characterization of CRISPR RNA biogenesis and Cas6 cleavage-mediated inhibition of a provirus in the haloarchaeon Haloferax mediterranei. J. Bacteriol. 2013; 195:867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Charpentier E., Richter H., van der Oost J., White M.F.. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 2015; 39:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J.J., Severinov K.. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datsenko K.A., Pougach K., Tikhonov A., Wanner B.L., Severinov K., Semenova E.. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012; 3:945. [DOI] [PubMed] [Google Scholar]
- 18. Li M., Wang R., Zhao D.H., Xiang H.. Adaptation of the Haloarcula hispanica CRISPR-Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res. 2014; 42:2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richter C., Dy R.L., McKenzie R.E., Watson B.N.J., Taylor C., Chang J.T., McNeil M.B., Staals R.H.J., Fineran P.C.. Priming in the Type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014; 42:8516–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westra E.R., Semenova E., Datsenko K.A., Jackson R.N., Wiedenheft B., Severinov K., Brouns S.J.. Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013; 9:e1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li M., Wang R., Xiang H.. Haloarcula hispanica CRISPR authenticates PAM of a target sequence to prime discriminative adaptation. Nucleic Acids Res. 2014; 42:7226–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carte J., Wang R.Y., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang R., Preamplume G., Terns M.P., Terns R.M., Li H.. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011; 19:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatoum-Aslan A., Maniv I., Marraffini L.A.. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21218–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rouillon C., Zhou M., Zhang J., Politis A., Beilsten-Edmands V., Cannone G., Graham S., Robinson C.V., Spagnolo L., White M.F.. Structure of the CRISPR interference complex CSM reveals key similarities with cascade. Mol. Cell. 2013; 52:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J., Rouillon C., Kerou M., Reeks J., Brugger K., Graham S., Reimann J., Cannone G., Liu H.T., Albers S.V. et al.. Structure and mechanism of the CMR complex for CRISPR-Mediated antiviral immunity. Mol. Cell. 2012; 45:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Staals R.H.J., Agari Y., Maki-Yonekura S., Zhu Y.F., Taylor D.W., van Duijn E., Barendregt A., Vlot M., Koehorst J.J., Sakamoto K. et al.. Structure and activity of the RNA-Targeting type III-B CRISPR-Cas complex of Thermus thermophilus. Mol. Cell. 2013; 52:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beloglazova N., Kuznedelov K., Flick R., Datsenko K.A., Brown G., Popovic A., Lemak S., Semenova E., Severinov K., Yakunin A.F.. CRISPR RNA binding and DNA target recognition by purified Cascade complexes from Escherichia coli. Nucleic Acids Res. 2015; 43:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo M.L., Jackson R.N., Denny S.R., Tokmina-Lukaszewska M., Maksimchuk K.R., Lin W., Bothner B., Wiedenheft B., Beisel C.L.. The CRISPR RNA-guided surveillance complex in Escherichia coli accommodates extended RNA spacers. Nucleic Acids Res. 2016; 44:7385–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuznedelov K., Mekler V., Lemak S., Tokmina-Lukaszewska M., Datsenko K.A., Jain I., Savitskaya E., Mallon J., Shmakov S., Bothner B. et al.. Altered stoichiometry Escherichia coli Cascade complexes with shortened CRISPR RNA spacers are capable of interference and primed adaptation. Nucleic Acids Res. 2016; 44:10849–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gleditzsch D., Müller-Esparza H., Pausch P., Sharma K., Dwarakanath S., Urlaub H., Bange G., Randau L.. Modulating the Cascade architecture of a minimal Type I-F CRISPR-Cas system. Nucleic Acids Res. 2016; 44:5872–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang R., Li M., Gong L.Y., Hu S.N., Xiang H.. DNA motifs determining the accuracy of repeat duplication during CRISPR adaptation in Haloarcula hispanica. Nucleic Acids Res. 2016; 44:4266–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li M., Gong L., Zhao D., Zhou J., Xiang H.. The spacer size of I-B CRISPR is modulated by the terminal sequence of the protospacer. Nucleic Acids Res. 2017; 45:4642–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu H.L., Han J., Liu X.Q., Zhou J., Xiang H.. Development of pyrF-based gene knockout systems for genome-wide manipulation of the archaea Haloferax mediterranei and Haloarcula hispanica. J. Genet. Genomics. 2011; 38:261–269. [DOI] [PubMed] [Google Scholar]
- 35. Cai S.F., Cai L., Liu H.L., Liu X.Q., Han J., Zhou J., Xiang H.. Identification of the haloarchaeal phasin (PhaP) that functions in polyhydroxyalkanoate accumulation and granule formation in Haloferax mediterranei. Appl. Environ. Microbiol. 2012; 78:1946–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cai S., Cai L., Zhao D., Liu G., Han J., Zhou J., Xiang H.. A novel DNA-binding protein, PhaR, plays a central role in the regulation of polyhydroxyalkanoate accumulation and granule formation in the haloarchaeon Haloferax mediterranei. Appl. Environ. Microbiol. 2015; 81:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhn J., Binder S.. RT-PCR analysis of 5′ to 3′-end-ligated mRNAs identifies the extremities of cox2 transcripts in pea mitochondria. Nucleic Acids Res. 2002; 30:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hullahalli K., Rodrigues M., Palmer K.L.. Exploiting CRISPR-Cas to manipulate Enterococcus faecalis populations. Elife. 2017; 6:e26664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Santangelo T.J., Reeve J.N.. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J. Mol. Biol. 2006; 355:196–210. [DOI] [PubMed] [Google Scholar]
- 40. Brenneis M., Hering O., Lange C., Soppa J.. Experimental characterization of Cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet. 2007; 3:2450–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Santangelo T.J., Cubonová L., Skinner K.M., Reeve J.N.. Archaeal intrinsic transcription termination in vivo. J. Bacteriol. 2009; 191:7102–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dar D., Prasse D., Schmitz R.A., Sorek R.. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat. Microbiol. 2016; 1:16143. [DOI] [PubMed] [Google Scholar]
- 43. Severinov K., Ispolatov I., Semenova E.. The influence of copy-number of targeted extrachromosomal genetic elements on the outcome of CRISPR-Cas defense. Front Mol. Biosci. 2016; 3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hale C.R., Cocozaki A., Li H., Terns R.M., Terns M.P.. Target RNA capture and cleavage by the Cmr type III-B CRISPR-Cas effector complex. Genes Dev. 2014; 28:2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Majumdar S., Ligon M., Skinner W.C., Terns R.M., Terns M.P.. Target DNA recognition and cleavage by a reconstituted Type I-G CRISPR-Cas immune effector complex. Extremophiles. 2017; 21:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grissa I., Vergnaud G., Pourcel C.. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007; 35:W52–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sashital D.G., Jinek M., Doudna J.A.. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat. Struct. Mol. Biol. 2011; 18:680–687. [DOI] [PubMed] [Google Scholar]
- 48. Sternberg S.H., Haurwitz R.E., Doudna J.A.. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA. 2012; 18:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wiedenheft B., Lander G.C., Zhou K., Jore M.M., Brouns S.J.J., van der Oost J., Doudna J.A., Nogales E.. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011; 477:486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bondy-Denomy J., Garcia B., Strum S., Du M., Rollins M.F., Hidalgo-Reyes Y., Wiedenheft B., Maxwell K.L., Davidson A.R.. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015; 526:136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fineran P.C., Blower T.R., Foulds I.J., Humphreys D.P., Lilley K.S., Salmond G.P.. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goldfarb T., Sberro H., Weinstock E., Cohen O., Doron S., Charpak-Amikam Y., Afik S., Ofir G., Sorek R.. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015; 34:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gordeeva J., Morozova N., Sierro N., Isaev A., Sinkunas T., Tsvetkova K., Matlashov M., Truncaite L., Morgan R.D., Ivanov N.V. et al.. BREX system of Escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 2018; 47:253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.