


Abstract
DNA polymerase ϵ (Pol ϵ), the major leading-strand DNA polymerase in eukaryotes, has a catalytic subunit (Pol2) and three non-catalytic subunits. The N-terminal half of Pol2 (Pol2CORE) exhibits both polymerase and exonuclease activity. It has been suggested that both the non-catalytic C-terminal domain of Pol2 (with the two cysteine motifs CysA and CysB) and Pol2CORE (with the CysX cysteine motif) are likely to coordinate an Fe–S cluster. Here, we present two new crystal structures of Pol2CORE with an Fe–S cluster bound to the CysX motif, supported by an anomalous signal at that position. Furthermore we show that purified four-subunit Pol ϵ, Pol ϵ CysAMUT (C2111S/C2133S), and Pol ϵ CysBMUT (C2167S/C2181S) all have an Fe–S cluster that is not present in Pol ϵ CysXMUT (C665S/C668S). Pol ϵ CysAMUT and Pol ϵ CysBMUT behave similarly to wild-type Pol ϵ in in vitro assays, but Pol ϵ CysXMUT has severely compromised DNA polymerase activity that is not the result of an excessive exonuclease activity. Tetrad analyses show that haploid yeast strains carrying CysXMUT are inviable. In conclusion, Pol ϵ has a single Fe–S cluster bound at the base of the P-domain, and this Fe–S cluster is essential for cell viability and polymerase activity.
INTRODUCTION
The duplication of DNA depends on numerous proteins that together replicate the genome. At the core of DNA replication are the DNA polymerases that with the guidance of a single-stranded template build a complementary DNA strand. In eukaryotes there are three replicative DNA polymerases – DNA polymerase α (Pol α), DNA polymerase δ (Pol δ), and DNA polymerase ϵ (Pol ϵ), each having different functions within the cell (1). Pol ϵ is the major contributor to leading-strand synthesis in Saccharomyces cerevisiae (1) and consists of a catalytic subunit (Pol2) and three non-catalytic subunits (Dpb2, Dpb3 and Dpb4) (2,3).
An increasing number of DNA replication and DNA repair enzymes have been shown to contain Fe–S clusters (4), and iron incorporation into the catalytic subunits of Pol α (Pol1), Pol δ (Pol3), Pol ϵ (Pol2), and the translesion polymerase Pol ζ (Rev3) of S. cerevisiae is dependent on the Fe–S assembly machinery in mitochondria (5). These eukaryotic family B DNA polymerases contain two conserved cysteine motifs, called CysA and CysB, in the C-terminal domain of the catalytic subunit (5) (Figure 1). CysB was found to bind an Fe–S cluster in the catalytic subunit, Pol3, of Pol δ. Mutations in CysB of Pol δ resulted in the loss of the Fe–S cluster along with the loss of accessory subunits, and as a result PCNA-dependent DNA synthesis was compromised. In contrast, the corresponding CysA mutant did not affect the iron content and the Pol δ complex remained intact, but PCNA-dependent DNA replication was sensitized to low levels of PCNA (5).
Figure 1.
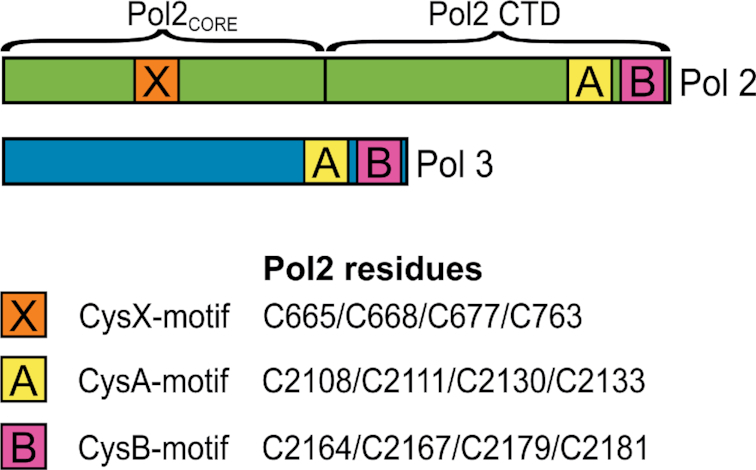
Schematic overview of Pol2 (Pol ϵ) and Pol3 (Pol δ), with the location of the cysteine motifs: CysA, CysB (5) and the CysX motif (8).
A sequence alignment of family B polymerases revealed that the non-catalytic C-terminal domains of Pol α and Pol ϵ are significantly larger in size and were predicted to have a very different structure compared to the C-terminal domains of Pol δ and Pol ζ (6). Complexes of a C-terminal fragment of the catalytic subunits and the B-subunits of the human family B DNA polymerases were over-expressed, purified, and characterized, and the results suggested the presence of an Fe–S cluster in the C-terminal domains of Pol3 and Rev3, but not in the C-terminal domains of Pol1 or Pol2 (6). In agreement with these observations, a crystal structure of human p261C-p59 (equivalent to the Pol2 C-terminal domain and Dpb2 in yeast) showed two Zn2+ ions bound to the CysA and the CysB motifs (7). However, the conclusion that human Pol2 CysA and CysB would not bind an Fe–S cluster was in conflict with the earlier report suggesting that the C-terminal domain of yeast Pol2 bound an Fe–S cluster (5).
Interestingly, the N-terminal catalytic part of yeast Pol2 (Pol2CORE) contains another cysteine motif conserved in its orthologues (8), and this is referred to by us as ‘CysX’ (C665/C668/C677/C763, Figure 1). Pol2CORE (lacking CysA and CysB) was shown to bind an Fe–S cluster, and it was suggested that the cysteines in CysX bind the Fe–S cluster (8). By replacing three of the four cysteines in CysX with serine, it was shown that these cysteines were essential for the polymerase activity of Pol2CORE, but not for its exonuclease activity (8). However, high-resolution crystal structures of Pol2CORE did not show electron density for an Fe–S cluster bound to the CysX motif (9,10), and a Zn2+ ion was modeled in that position (9).
Here we present two new crystal structures of Pol2CORE with an Fe–S cluster coordinated by the four cysteines in the CysX motif. To determine whether Fe–S clusters are specifically bound to CysX or also bound to CysA and/or CysB in yeast Pol ϵ, we systematically made cysteine to serine substitutions in the CysX, CysA and CysB motifs of yeast Pol2 and purified the variants in the form of four-subunit Pol ϵ and measured their iron contents. Our results suggest that there is only one Fe–S cluster associated with yeast Pol ϵ and that this cluster is not present in Pol ϵ CysXMUT (C665S/C668S). In contrast to Pol δ, the subunit composition was not affected when the CysA, CysB or CysX motif was mutated by replacing two of the cysteines in each motif with serines. Haploid yeast cells expressing Pol ϵ CysAMUT or Pol ϵ CysBMUT were viable, in contrast to Pol ϵ CysXMUT that does not support cell proliferation in haploid cells. Furthermore, the four-subunit Pol ϵ CysXMUT exhibited a strong decrease in polymerase activity but only a minor decrease in processive exonuclease activity.
MATERIALS AND METHODS
Purification, crystallization, and data collection of Pol2CORE
The catalytic domain of Pol2 (Pol2CORE, residues 1–1228) was expressed in yeast and purified as previously described (9). To obtain crystals, the ternary complex of Pol2CORE was formed with 11ddC/16 primer-template with dT as the templating base and dATP as the incoming nucleotide under the same crystallization conditions as described previously (9). The crystal was equilibrated in reservoir solution containing 15% glycerol and then flash frozen in liquid nitrogen. A complete dataset was collected at 100 K using the ID23 beamline at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and processed using Mosflm (11). An anomalous dataset was also collected at the Fe-edge (wavelength 1.7360 Å), which was processed using XDS (12) keeping the Friedel pairs separate.
A slightly shorter version of Pol2CORE (residues 1–1187) was expressed in Escherichia coli from a pET28a vector (a kind gift from Dr. Aneel K. Aggarwal (8)). The 6 × His-tagged Pol2CORE was bound to Ni2+-NTA beads in 25 mM Tris-Ac pH 7.4, 10% glycerol and 300 mM NaAc, called buffer T300 (300 denotes the NaAc concentration), and the eluted fractions were incubated with PreScission protease to remove the His-tag. To obtain the untagged purified protein, the protein sample was passed a second time over the Ni2+-NTA beads. The collected protein was loaded onto a Mono Q column and eluted with a linear salt gradient (T200–T1000). The peak fractions were collected, and the purified protein was adjusted to 25 mM HEPES pH 7.4, 800 mM NaAc, 10% glycerol, and 2 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) using a PD10 column. The ternary complex of Pol2CORE with the 11ddC/16 primer-template and dATP was formed as previously described (9). The crystals were obtained in crystallization conditions containing 10 mM Tris–HCl pH 8, 10 mM calcium chloride and 15% PEG8000, and were flash frozen in liquid nitrogen after equilibration with reservoir solution containing 15% glycerol. A complete dataset at 2.8 Å was collected using beamline ID30A-3 at the ESRF, and the data were processed with Mosflm (11). Data collection statistics are found in Supplementary Table S1.
Structure determination and refinement
Phaser (13) was used to solve the two structures of Pol2CORE by molecular replacement methods using PDB ID: 4m8o (9) as the model with a single ternary complex in the asymmetric unit. Coot (14) was used for model building, and the structures were refined using REFMAC (15) and Phenix refine (16). The final refined structures contained more than 95% of the residues in the most favored regions of the Ramachandran plot (Supplementary Table S1), and the model was validated using Coot (14) and MolProbity (17). PyMol (18) was used to create figures and to superimpose structures. The anomalous difference map was calculated using phases from the refined model (Figure 2A). The crystal structure of Pol2CORE (1–1228) was superimposed onto 4m8o (9) with a root mean square deviation (r.m.s.d.) of 0.4 Å for 975 Cα atoms.
Figure 2.

Crystal structures of the catalytic core of Pol ϵ showing an Fe–S cluster bound to the CysX motif. (A) Overall structure of Pol2CORE (1–1228) with an [4Fe–4S] cluster modelled at the base of the P-domain (PDB ID: 6h1v). An anomalous map over the Fe–S cluster, shown in blue mesh at 3σ supports the presence of a cubic Fe–S cluster. Pol2CORE (1–1228) was over-expressed in yeast. The P-domain is shown in slate blue cartoon. The N-terminal, palm, and thumb domains are in yellow, pink, and light blue surface representations, respectively. (B) Close-up view of the Fe–S cluster bound to the CysX motif at the base of the P-domain (PDB ID: 6h1v). An omit map at 3σ is shown in green mesh. (C) The crystal structure of Pol2CORE (1–1187, PDB ID: 6qib) shows the complete loop region folded at the base of the P-domain revealing the positions of all four cysteines in the CysX motif. Pol2CORE (1–1187) was over-expressed in E. coli.
Expression and purification of four-subunit Pol ϵ
Saccharomyces cerevisiae Pol ϵ wild-type, CysXMUT (Pol2C665S/C668S), CysAMUT (Pol2C2111S/C2133S), CysBMUT (Pol2C2167S/C2181S), Pol ϵ exo− (Pol2D290A,E292A) and Pol ϵ exo− CysXMUT were over-expressed in the S. cerevisiae strain PY116 from the pJL1 (containing POL2) and pJL6 (containing DPB2, DPB3, and DPB4) plasmids under the control of the GAL1-10 promoter (2). A 1xFLAG-tag was inserted into the pJL1 plasmid by introducing the sequence 5′-ATGGACTACAAAGACGATGACGACAAGGGCGCCAATGGAGGT-3′, which encodes a start codon, the FLAG-tag (DYKDDDDK), and a short flexible linker (GANGG) in front of the POL2 gene. Protein expression was performed essentially as described in (2). The cells were harvested by centrifugation for 10 min at 5000 × g at 4°C. Cell pellets were flash frozen in liquid nitrogen.
All purification steps were performed on ice or at 4°C, and all buffers were flushed with argon before use. The following buffers were used. 2 × Lysis buffer: 600 mM NaAc, 20% glycerol, 2 μM leupeptin, 2 μM pepstatin A, and 300 mM Tris-Ac, pH 7.8. Wash buffer: 300 mM NaAc, 10% glycerol, 1 μM leupeptin, 1 μM pepstatin A, and 50 mM Tris-Ac, pH 7.8. B700 buffer: 25 mM HEPES pH 7.6, 700 mM NaAc, 10% glycerol, 0.01% NP-40, 1 mM dithiothreitol, 2 μM leupeptin, 2 μM pepstatin A, and 10 mM NaHSO3. B1200 buffer: 25 mM HEPES pH 7.6, 1200 mM NaAc, 10% glycerol, 0.01% NP-40, 1 mM dithiothreitol, 2 μM leupeptin, 2 μM pepstatin A and 10 mM NaHSO3. Gel filtration buffer: 300 mM NaAc, 10% glycerol, 1 mM TCEP and 50 mM Tris-Ac pH 7.8.
Cells were disrupted in a SPEX SamplePrep freezer mill 6850 under liquid nitrogen in seven cycles with 2 min grinding at an impact frequency rate of 12. The cell powder was dissolved in an equal volume of 2× lysis buffer. (NH4)SO4 was added to a final concentration of 175 mM, followed by centrifugation at 125 000 x g for 1 h. The supernatant was retrieved and incubated for 1 h with M2 resin. The column was washed with ∼25 column volumes of wash buffer, and FLAG-Pol ϵ was eluted in wash buffer with 100 μg/ml 1 × FLAG-peptide (ApexBio). Next, 1 mM TCEP (final concentration) was added to the eluate before loading onto a 1 ml Mono Q column equilibrated in B700 buffer. The MonoQ column was washed with B700 buffer and eluted with a linear gradient (B700 – B1200). Peak fractions were concentrated on a 100 kDa cutoff filter (Amicon Ultra) and loaded onto a Superose 6 PC 3.2/30 column (GE Healthcare) equilibrated with gel filtration buffer.
Metal determination
The metal content was determined with a ferrozine assay (19,20). A total of 31 μl of purified Pol ϵ (1.1–4.8 μM in gel filtration buffer) was mixed with 3.1 μl 12 M HCl and boiled for 10 min at 100°C. Precipitated protein was removed by centrifuging at 21 000 × g for 10 min. A total of 33 μl of the supernatant was transferred to a 96-well plate. For the standard curve, 30 μl fractions of FeSO4 dissolved in gel filtration buffer were mixed with 3 μl 12 M HCl in the 96-well plate. To each sample 50 μl 3 M NaAc, 5 μl 1 M ascorbic acid, and 5 μl 10 mM ferrozine (3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-4′,4″-disulfonic acid sodium salt, Sigma-Aldrich) were added. The flat-bottom 96-well microtiter plate was incubated 2 h at 37°C, and absorbance was measured at 562 and 900 nm to correct for background (5).
In vitro activity assays
The primer extension studies in Figure 5, Supplementary Figures S4 and S5 were performed essentially as described in (9). Briefly, 10 μl reaction mix A (20 nM Pol ϵ and 20 nM primer or primer/template, 20 mM Tris–HCl pH 7.8, 40 mM NaAc, 0.1 mg/ml bovine serum albumine, and 0.5 mM dithiothreitol) was pre-incubated on ice then mixed with 10 μl reaction mix B (16 mM MgAc, 20 mM Tris–HCl pH 7.8, 0.1 mg/ml bovine serum albumine, 0.5 mM dithiothreitol, and with or without 0.2 mM dNTPs) and incubated for the indicated time at 30°C. The reactions were stopped by the addition of 20 μl stop solution (96% formamide, 20 mM EDTA, 0.1% bromophenol blue). The reactions were heated to 85°C for 15 min before the products were separated on a 10% denaturing polyacrylamide gel. The 50-mer primer strand used for all primer extension and exonuclease assays was labeled at the 5′ end by tetrachlorofluorescein (TET) to allow visualization of the DNA.
Figure 5.

Impact of the substitutions in the CysX motif (C665S/C668S) on Pol ϵ polymerase and exonuclease activity. Primer-extension assays in the presence of dNTPs and a DNA substrate with a fluorescently labeled 50-mer primer annealed to a perfectly matched 80-mer template. Exonuclease activity was assayed in the absence of dNTPs using the same double-stranded DNA substrate or only the single-stranded 50-mer.
The primer extension in Figure 6 was performed similarly with the modification that reaction mix B contained 0.2 or 2.0 mM dNTPs. For the primer extension assay in Supplementary Figure S2, 10 μl reaction mix A (10 nM Pol ϵ and 10 nM primer/template, 20 mM Tris–HCl pH 7.8, 40 mM NaAc, 0.1 mg/ml bovine serum albumine, and 1 mM dithiothreitol) was pre-incubated on ice and then mixed with 10 μl reaction mix B (16 mM MgAc, 20 mM Tris–HCl pH 7.8, 0.1 mg/ml bovine serum albumine, 1 mM dithiothreitol, and with physiologically balanced dNTPs (21) at 2× the indicated concentration) and incubated for 10 min. at 30°C, as also done in (22).
Figure 6.

Impact of the substitutions in the CysX motif (C665S/C668S) on Pol ϵ polymerase in the absence of a functional exonuclease activity. (A) Primer-extension assays in the presence of 0.1 or 1.0 mM dNTPs and a DNA substrate with a 50-mer primer annealed to a perfectly matched 80-mer template. (B) Quantification of the amount of DNA (as % of total) for the unextended primer (50 bp), and of the extended products in the presence of 1 mM dNTP. The fully extended product is 80–81 base pairs.
Holoenzyme assays were performed in a 15 μl reaction that contained 40 mM Tris–HCl pH 8, 1 mM dithiothreitol, 0.2 mg/ml bovine serum albumin, 8 mM MgAc2, 125 mM NaAc, 100 μM dGTP, 100 μM dATP and 100 μM dTTP, 50 μM dCTP, 2.9 μCi [α-32P] dCTP (Perkin Elmer), 70 fmol single-primed pBluescript II SK(+), 237 fmol DNA polymerase, 10.5 pmol RPA, 80 fmol RFC and 0–1.15 pmol PCNA. The reactions were incubated at 30°C for 8 min and terminated by adding 1 μl of 10% SDS. The DNA was purified on illustra MicroSpin G-25 columns (GE Healthcare). Samples were loaded on a 0.8% alkaline agarose gel that contained 30 mM NaOH and 2 mM EDTA. The gel was run at 40 V for 16 h and fixed in 5% TCA for 1 h. Afterward, it was dried at 55°C, incubated with a phosphoimager screen (Fuji) and scanned with a Typhoon 9400 phosphoimager (GE Healthcare).
In vivo analysis
A linearized integration plasmid (23) carrying mutations resulting in a CysX (C665S, C668S), CysA (C2111S, C2133S) or CysB (C2167S, C2181S) variant, was integrated into a diploid E134 yeast strain (MATα/MATa ade5-1/ade5-1 lys2::InsEA14/lys2::InsEA14 trp1-289/trp1-289 his7-2/his7-2 leu2-3,112/leu2-3,112 ura3-52/ura3-52). URA3 was used for selection of integration. Four integrants from each transformation were isolated and then patched on YPD over night to allow for the looping out of the URA3 marker, leaving the specific POL2 mutation on the chromosome. Patched clones were then replica-plated on 5-FOA plates to select for clones that had lost URA3. Three 5-FOAr clones from each patch were picked and streaked for single colonies on YPD. PCR was used to screen colonies for the desired mutation and positive diploid clones were sequenced across the POL2 gene to confirm that the selected mutation was correctly integrated in the heterozygote strain and to verify that there were no additional mutations. At least 8 tetrads from each strain were dissected as previously described (24).
The isogenic haploid cells with pol2 CysAMUT or Pol2 from one tetrad, or with pol2 CysBMUT or Pol2 from another tetrad, were grown to saturation overnight in liquid YPD media to a cell density of A600 nm = 10–12. Differences in cell density were adjusted and then the cells were serially diluted in MilliQ water by two 10-fold dilutions followed by five 3-fold dilutions. 5 μl from the serial dilutions except the first, were spotted on YPD plates with or without 100 mM hydroxyurea. The plates were incubated at 30 or 37°C.
RESULTS
Crystal structures of Pol2CORE with an Fe–S cluster
It was earlier suggested that C665, C668, C677 and C763 (the CysX motif) in the catalytic core domain of Pol2 might bind a [4Fe–4S] cluster (8), even though the available crystal structures did not show any electron density for it (9,10). To clarify whether the reported Fe–S cluster in Pol ϵ was bound to the CysX motif in yeast Pol2CORE, we over-expressed wild-type Pol2CORE in yeast and crystallized the protein as previously described (9). Fe–S clusters are known to be sensitive to radiation damage (4), and to minimize such damage the crystals were not pre-screened on our home source. Instead, crystals were screened directly at the ESRF, and a complete dataset at 2.7 Å resolution was collected with the shortest possible exposure time to minimize radiation damage. After solving the new structure (PDB ID 6h1v, Supplementary Table S1) using PDB ID: 4m8o (9) as a molecular replacement model, a region of positive density was observed at the CysX site. To confirm that the positive density corresponded to an Fe–S cluster, an anomalous dataset was collected at the Fe edge (Supplementary Table S1). The anomalous difference map clearly suggested that Fe was bound to the CysX motif (Figure 2A). Based on the shape of the omit map, a [4Fe–4S] cluster was modeled into the CysX site and refined with an occupancy of 70% (Figure 2B). The electron density allowed us to correctly position three cysteines (C665, C677 and C763) that coordinate three of the iron atoms in the Fe–S cluster, while the fourth cysteine, C668, could not be modeled because it was located in a disordered loop region 666–675 (Figure 2B).
In addition, we expressed Pol2CORE in Escherichia coli, and we crystallized a ternary complex under new crystallization conditions. A diffraction dataset was collected and processed to 2.8 Å resolution in the same C2 space group, but with slightly different unit cell dimensions (PDB ID 6qib, see Supplementary Table S1). This crystal structure also showed a region of positive electron density at the CysX site (Figure 2C), where a [4Fe–4S] cluster was modeled and refined with an occupancy of 80%. For this structure, electron density was observed for the entire loop region (amino acids 666–675) at the base of the P-domain. Residues 664–668, containing C665 and C668, were found to be folded into a short α-helix. In this structure, all four cysteines in CysX could be seen to coordinate the four iron atoms of the possible [4Fe–4S] cluster (Figure 2C). The observed electron density for the entire region (residues 665–675) at the base of the P-domain might be the consequence of a slightly different packing arrangement of macromolecules in this crystal when compared to previously reported structures.
Comparison of Pol2CORE structures
The two new crystal structures of Pol2CORE, purified from yeast and E. coli, superimposed with an r.m.s.d. of 0.4 Å (983 Cα atoms) suggesting almost identical structures. Superimposition of the two new structures (PDB IDs: 6h1v and 6qib) containing a [4Fe–4S] cluster onto previous Pol2CORE structures without an Fe–S cluster (PDB IDs: 4m8o (9) and 4ptf (10)) did not reveal any large structural changes (r.m.s.d. < 0.5 Å). Zinc can be an opportunistic binder, and the zinc observed in 4m8o might have replaced the Fe–S cluster in 4m8o due to radiation damage to the protein. The superimposition of only the P-domains of the new structures onto the previously published 4m8o structure (9) suggested that the P-domain was also largely unchanged (r.m.s.d. = 1.39 Å for 84 Cα-atoms), except for differences at the base containing the CysX motif (Supplementary Figure S1).
Determining the number of Fe–S clusters in Pol ϵ
The reports on Fe–S clusters in the N-terminal (8) and C-terminal (5) domains of Pol2 suggested that there might be two Fe–S centers in Pol ϵ. However, the CysA and CysB motifs that are located in the C-terminal domain of Pol2 are not present in the Pol2CORE structures discussed above, because Pol2CORE only includes the N-terminal half of Pol2. To clarify the number of Fe–S clusters bound to Cys motifs in four-subunit Pol ϵ, each of the three conserved Cys motifs (CysX, CysA and CysB) in Pol2 were altered by site-directed mutagenesis to block any binding of a potential Fe–S cluster to each specific Cys motif. Two of the four cysteines in CysX (C665 and C668), CysA (C2111 and C2133) and CysB (C2167 and C2181) were substituted with serine (resulting in Pol ϵ CysXMUT, Pol ϵ CysAMUT and Pol ϵ CysBMUT, respectively). The Pol ϵ variants were over-expressed separately in yeast and purified. To minimize the loss of the Fe–S cluster due to oxidation, a new purification protocol was designed where FLAG-tagged Pol ϵ was purified in buffers that were flushed with argon. All three Pol ϵ variants, Pol ϵ CysXMUT, Pol ϵ CysAMUT and Pol ϵ CysBMUT gave a stable four-subunit complex (Figure 3).
Figure 3.
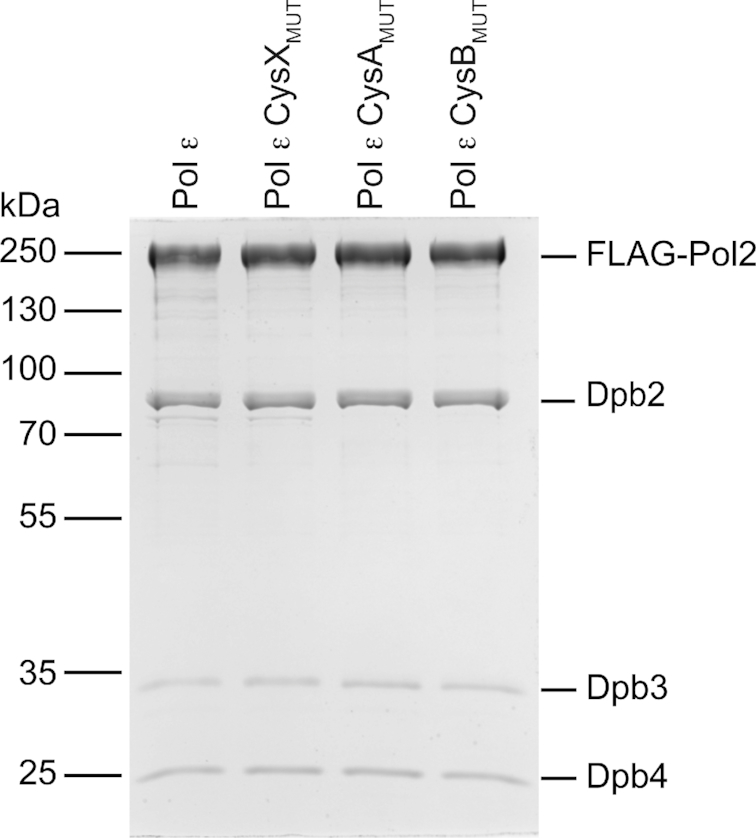
Coommassie-stained SDS-PAGE showing FLAG-tag purified Pol ϵ complexes. The complexes, Pol ϵ, Pol ϵ CysXMUT (C665S/C668S), Pol ϵ CysAMUT (C2111S/C2133S) and Pol ϵ CysBMUT (C2167S/C2181S) were overexpressed in yeast with FLAG-tagged Pol2. Each lane was loaded with ∼1 μg of purified protein.
We observed that the purified protein of Pol ϵ CysXMUT, was colorless and did not show any absorbance ∼400 nm during the purification, while wild-type Pol2CORE and Pol ϵ fractions were yellow to brown at high concentrations. Furthermore, UV–Vis spectra of wild-type Pol ϵ, Pol ϵ CysAMUT and Pol ϵ CysBMUT showed a single broad peak around 400 nm with an extinction coefficient of ∼14 mM−1 cm−1 (Figure 4A) indicative of a single [4Fe–4S] or [3Fe–4S] cluster (5,25,26). In contrast, Pol ϵ CysXMUT showed only background absorption at wavelengths of 320–600 nm (even at 34 μM) (Figure 4A). It was therefore important to quantify the amount of iron in wild-type Pol ϵ and the Pol ϵ CysXMUT by an independent method. Iron quantification with the help of ferrozine gave 3.7 ± 1.1 Fe2+/Pol ϵ (mol/mol protein, n = 9, error is the standard deviation) for wild-type Pol ϵ, which correlated well with a single [4Fe–4S] or [3Fe–4S] cluster in Pol ϵ (Figure 4B). The measured iron level in Pol ϵ CysXMUT was 0.1 ± 0.3 Fe2+/Pol ϵ (mol/mol protein, n = 7, error is the standard deviation) (Figure 4B), indicating that an Fe–S cluster was indeed absent in this variant. The UV–Vis measurements and ferrozine assay thus both suggested that there is a single [4Fe–4S] or [3Fe–4S] cluster in four-subunit Pol ϵ and this cluster is bound by the CysX motif.
Figure 4.
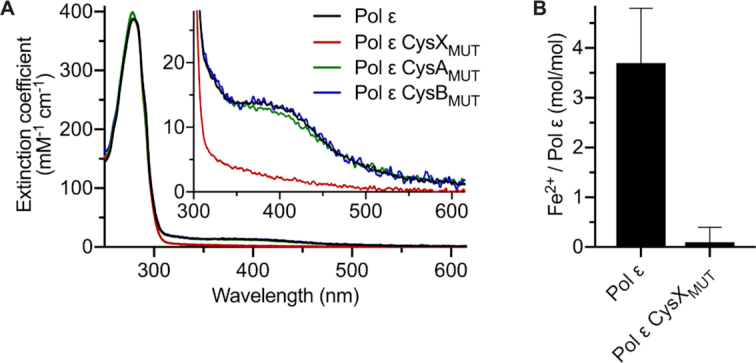
Quantification of the Fe–S cluster in Pol ϵ and Pol ϵ variants. (A) UV–Vis absorption spectra of wild-type Pol ϵ (black), Pol ϵ CysAMUT (C2111S/C2133S, green) or Pol ϵ CysBMUT (C2167S/C2181S, blue) show a broad absorbance peak around 400 nm (with an extinction coefficient of ∼14 mM−1 cm−1), which is absent in the Pol ϵ CysXMUT (C665S/C668S, red). (B) The amount of Fe per four-subunit Pol ϵ as determined by the ferrozine assay. The error bars indicate the standard deviation, and n = 7 and n = 9 for the wild type and the variant, respectively.
In vitro characterization of Pol ϵ cysteine motif variants
To determine the impact of the cysteine motifs on the function of Pol ϵ, we first measured the ability of Pol ϵ CysXMUT, Pol ϵ CysAMUT and Pol ϵ CysBMUT to extend or degrade a primer (Figure 5, Supplementary Figures S4 and S5). The polymerase activity of Pol ϵ CysXMUT (C665S/C668S) was found to be severely compromised, with less than 1% of the primer extended after 8 minutes (Figure 5). Instead, the balance was shifted toward the exonuclease site resulting in degradation products of the complementary primer with no mismatches (Figure 5). In fact, >80% of the primer was degraded after 8 min even in the presence of 100 μM dNTPs. The Pol ϵ CysXMUT was equally efficient in degrading the primer in the absence or presence of 100 μM dNTPs. This could suggest that Pol ϵ CysXMUT either has an excessive exonuclease activity per se or has a lower affinity for dNTPs in the polymerase active site.
We therefore investigated whether a very high concentration of dNTPs could stabilize the 3′-end of the nascent strand in the polymerase site and as a result suppress the exonuclease activity. A series of primer extension reactions with increasing levels of dNTPs showed that Pol ϵ CysXMUT mainly degrades the DNA at dNTP concentrations up to 27x higher than the estimated physiological dNTP concentration (Supplementary Figure S2). Only at the highest tested dNTP concentration (with 891 μM dGTP, 3159 μM dCTP, 5346 μM dATP and 16038 μM dTTP), was the exonuclease activity suppressed in Pol ϵ CysXMUT. However, the amount of fully extended primer remained very low (<1%, compared to ∼56% for wild-type Pol ϵ at the highest dNTP concentration).
To determine whether the lack of polymerase activity was due to an excessive exonuclease activity we purified exonuclease deficient Pol ϵ and exonuclease deficient Pol ϵ CysXMUT to compare their polymerase activity (Figure 6). In contrast to exonuclease deficient Pol ϵ, Pol ϵ exo– CysXMUT extended the primer with a very low efficiency at 100 μM dNTP (Figure 6A). In the presence of 1 mM dNTP up to 30% of the primer was extended by Pol ϵ exo− CysXMUT compared to 78% for Pol ϵ exo– (Figure 6B). However, most elongation products were short, not extended beyond 10 nucleotides, while for Pol ϵ exo– most products were full length (Figure 6B). In conclusion, DNA has access to the polymerase active site in Pol ϵ CysXMUT and the polymerase activity is stimulated at high concentrations of dNTP suggesting that the affinity for dNTP is decreased in Pol ϵ CysXMUT.
Both wild-type Pol ϵ and the Pol ϵ CysXMUT variant were efficiently degrading the primer when it was annealed to a matched DNA template (Figure 5). The amount of full-length primer that was degraded was comparable for Pol ϵ and Pol ϵ CysXMUT, but Pol ϵ gave shorter products suggesting either a decreased rate or decreased processivity in Pol ϵ CysXMUT (Figure 5 and Supplementary Figure S4). The exonuclease activities of Pol ϵ CysXMUT and wild-type Pol ϵ on single-stranded DNA suggested that the catalytic exonuclease efficiency was unaltered in Pol ϵ CysXMUT and that the reduction in processive exonuclease activity on a primed template might be related to the capacity of the enzyme to present single-stranded DNA in the exonuclease active site without dissociating from the DNA.
The primer extension experiments were replicated with Pol ϵ CysAMUT and Pol ϵ CysBMUT (Supplementary Figures S4 and S5). Both Pol ϵ CysAMUT and Pol ϵ CysBMUT showed polymerase and exonuclease activity comparable to wild-type Pol ϵ. It was earlier shown that Pol δ depends on the CysA and CysB motifs for a fully functional interaction with the PCNA clamp (5). To determine whether the functional interaction between Pol ϵ and PCNA depends on the CysA or CysB motifs, holoenzyme assays were carried out with a single-primed circular single-stranded DNA (Bluescript). Both Pol ϵ CysAMUT and Pol ϵ CysBMUT could still fully extend the primer at PCNA concentrations ≥19 nM (Figure 7). The impact of substitutions in the CysB motif in Pol ϵ differed from the corresponding substitutions in Pol δ (5), as they neither affected protein complex formation (unaltered subunit composition in Pol ϵ CysBMUT (Figure 3) compared to a complete loss of subunits in Pol δ (5)), nor affected the enzyme activity in the holoenzyme assay (wild-type activity for Pol ϵ CysBMUT (Figure 7) compared to no observed activity of Pol δ (5)). The addition of PCNA could not rescue the low polymerase activity of Pol ϵ CysXMUT (Figure 7).
Figure 7.

PCNA-dependent DNA synthesis by wild-type Pol ϵ, Pol ϵ CysXMUT, Pol ϵ CysAMUT, and Pol ϵ CysBMUT in holoenzyme assays. The products of each reaction with 16 nM Pol ϵ, 4.7 nM single-primed pBluescript template, 700 nM RPA, 5.3 nM RFC, and varying concentrations of PCNA (as indicated) were separated on an alkaline agarose gel.
Importance of CysA, CysB and CysX in vivo
To determine the impact of each of the three cysteine motifs on cell fitness, heterozygote diploid strains of S. cerevisiae were established with mutations that result in CysAMUT, CysBMUT, or CysXMUT Pol ϵ variants. Tetrad analysis showed that spores expressing only Pol ϵ CysXMUT were unable to produce colonies (see Figure 8A). In contrast, spores that expressed Pol ϵ CysAMUT or Pol ϵ CysBMUT were viable and gave at 30°C colonies of similar size as those expressing wild-type Pol ϵ. Cells that only expressed Pol ϵ CysAMUT were, in contrast to cells expressing only Pol ϵ CysBMUT, temperature sensitive and showed slower growth at 37°C (Figure 8B). This growth defect at 37°C was enhanced when 100 mM hydroxyurea was added, but neither Pol ϵ CysAMUT nor Pol ϵ CysBMUT expressing strains showed hydroxyurea sensitivity when grown at 30°C (Figure 8B). In conclusion, a functional CysX motif is essential for the polymerase activity of Pol ϵ and for cell viability, whereas cysteine to serine substitutions in the CysA and CysB motifs do not affect cell growth at 30°C.
Figure 8.

Impact of each of the three cystein motifs on cell fitness. (A) Tetrad analysis of diploid yeast strains heterozygous for POL2/pol2 CysXMUT,POL2/pol2 CysAMUT, or POL2/pol2 CysBMUT alleles. The results from two independent tetrads are presented from each heterozygous strain (≥8 were analyzed see material and methods). (B) Haploid cells with pol2 CysAMUT (CysA) or POL2 (WT) from one tetrad, and with pol2 CysBMUT (CysB) or POL2 (WT) from another tetrad were analyzed for temperature and hydroxyurea sensitivity by comparing their growth at 30°C and 37°C on YPD plates with or without 100 mM hydroxyurea. Images of the plates without hydroxyurea are taken after 24 h, while the shown image from the 30°C 100 mM HU plate is from 40 h and the 37°C 100 mM HU plate is from 72 h after plating.
DISCUSSION
Here, we have shown that there is a single Fe–S cluster in four-subunit Pol ϵ and that this cluster is bound to the Cys-motif (CysX) at the base of the P-domain in the catalytic subunit, Pol2. Fe–S clusters have been reported to stabilize protein structures and subunit interactions, but also to regulate substrate binding and/or catalysis in a redox state-dependent manner (27–33). In the case of the two replicative eukaryotic polymerases, Pol δ and Pol ϵ, there are clear differences when comparing the function of their Fe–S clusters.
The Fe–S cluster in Pol δ is bound to the CysB motif in Pol3 and was shown to be important for stabilizing the interaction with the B-subunit, Pol31 (5). Our analyses of Pol ϵ CysAMUT and Pol ϵ CysBMUT suggest that Pol ϵ is not dependent on an Fe–S cluster in the CysA or CysB motifs for subunit interactions. In support of that, our biochemical data suggests that Pol ϵ CysA and CysB do not coordinate an Fe–S cluster (Figure 4A). However, it should be noted that both cysteine motifs in a recent structure of the human B-subunit (Dpb2 in yeast) in complex with a C-terminal fragment of the catalytic subunit (amino acids 2142–2286) were coordinating zinc ions and this structure suggested that they are located near the dimer interface (7). There is neither a growth defect nor hydroxyurea sensitivity observed when cells expressing Pol ϵ CysAMUT or Pol ϵ CysBMUT are grown at 30°C (Figure 8). However, haploid cells expressing only Pol ϵ CysAMUT are temperature sensitive, showing a growth defect at 37°C (Figure 8B). A previous study using a plasmid shuffling approach did report hydroxyurea sensitivity when altering amino acids in the CysA-motif to alanines, but all experiments were done at 37°C (34). We also see a weak growth inhibition when adding 100 mM hydroxyurea at 37°C, but this might not be surprising since the cells already have a proliferation defect at this temperature, caused by the altered leading strand polymerase. A plausible explanation for the observed sensitivity to elevated temperature could be that the essential interaction between the Pol2 C-terminus and Dpb2 is weakened in Pol ϵ CysAMUT. In summary, the cysteine to serine substitutions in the CysA or CysB motif in Pol ϵ did not affect protein complex formation (Figure 3), enzyme activity (Figure 7, Supplementary Figures S4 and S5), or cell proliferation at 30°C (Figure 8).
The only Fe–S cluster found in four-subunit Pol ϵ is located at the CysX motif in the N-terminal catalytic domain of Pol2 (Figures 4 and 2). Anomalous data (Figure 2A) collected from the Pol2CORE (1–1228) crystal showed that iron is bound by the CysX motif, consisting of residues C665, C668, C677, and C763, positioned at the base of the P-domain. The Fe–S omit maps calculated in two independent structures are shaped as a cube (Figure 2B and C) and are consistent with a [3Fe–4S] or [4Fe–4S] cluster, but not with a [2Fe-2S] cluster, which would result in a planar density. Previously reported experimental data from FT-EXAFS and EPR on the Fe–S cluster in Pol2CORE, was consistent with a [4Fe–4S] cluster or a [2Fe-2S], but ruled out a [3Fe–4S] cluster (8). The UV-Vis absorbance spectra and the measured molar ratio of Fe2+ over Pol ϵ (Figure 4) support the presence of a single [4Fe–4S] cluster in Pol ϵ. All combined, our structural and biochemical data and the previously reported data based on FT-EXAFS and EPR (8), suggest that the identified cluster in Pol ϵ is a [4Fe–4S] cluster.
The activity assays with the CysX variant in Pol2CORE (8) and in four-subunit Pol ϵ (Figures 5 and 6) show that Pol ϵ depends on the Fe–S cluster to synthesize DNA, but not for its exonuclease activity. Haploid yeast cells carrying the Pol ϵ CysXMUT allele were not viable, as expected based on the model that Pol ϵ is responsible for the bulk synthesis on the leading strand during DNA replication. This observation that loss of DNA polymerase activity of Pol ϵ causes cell death is in agreement with an earlier study where it was shown that the growth of yeast was reduced as the balance was shifted from the polymerase to the exonuclease site in Pol ϵ (22). Thus, the previously presented model (35) where the primary role for Pol ϵ at the replication fork is to proofread errors made by Pol δ is not supported by these results. If the role of Pol ϵ was primarily as an exonuclease, strains carrying the Pol ϵ CysXMUT (Figure 8A), the pol2-K967A allele (residue stabilizing the 3′-end of the new strand in the polymerase site) (22), and the pol2-D875A/D677A allele (polymerase catalytic residues) (34) would all be viable and show at the most a minor growth defect because these mutations only have a negative impact on the polymerase activity. This is, however, not what we observe. Instead, the results are in agreement with a model where Pol ϵ replicates most of the leading strand during DNA replication (36–41).
The CysX motif in Pol ϵ is conserved in Pol ϵ orthologues but is absent in all other known DNA polymerases. Interestingly, it is located at the base of the P-domain, which is also unique to Pol ϵ orthologues. The Fe–S cluster thus might function as an ‘iron-staple’ (42) because one of the cysteines in the motif is present at the C-terminal end of the P-domain while the other three are at the N-terminal end of the P-domain. However, only small differences were observed between the structures with or without the Fe–S cluster (P-domain r.m.s.d. < 1.39 Å for 84 Cα atoms). It is possible that the P-domain remains stable in the crystal after the Fe–S cluster was lost during data collection for PDB ID 4m8o, or the cluster might only play a role in the ab initio folding of the P-domain. Alternatively the small differences found in the loop that contains the CysX motif (Supplementary Figure S1) might be sufficient to inactivate the polymerase function. This loop is far away from the polymerase site (30 Å), but near the hinge region of the finger domain. Therefore it might be involved in controlling the finger movement as proposed earlier (43). The finger domain, consisting of two long α-helices, cycles between an ‘open’ state, which allows the nucleotide to enter the polymerase site, and a ‘closed’ state, in which the nucleotide is stabilized and incorporated into the growing DNA strand. A protein variant where the finger domain is more often in the ‘open’ state, would be expected to have reduced affinity for dNTPs. The observed polymerase activity of Pol ϵ CysXMUT at very high dNTP concentrations (Figure 6 and Supplementary Figure S2) may therefore suggest a functional link between the Fe–S cluster and the movement of the finger domain. It can be speculated that the geometry or oxidation state of the bound metal might modulate the predisposition of Pol ϵ for exonuclease proofreading by destabilizing the closed conformation of the finger domain (43). It should, however, be considered that amino acid substitutions leading to a loss of the Fe–S cluster may have a greater effect than an altered redox-state of the Fe–S cluster.
The Fe–S cluster in Pol ϵ could play a direct role in DNA replication as more and more DNA repair and polymerase enzymes have been shown to contain such a redox-active cluster. It has been proposed that the redox states of these clusters can be regulated by electron transport through the DNA (30,44). This could provide a way to block DNA replication under oxidative stress and/or when too many breaks or mismatches are present in the DNA. The progression of the cell cycle in yeast can indeed be directly reduced or accelerated by the addition of oxidizing or reducing agents, respectively (45). Furthermore, under nutrient-limiting conditions, which mimic yeast growth in the wild, cell growth and DNA replication are confined to the reductive phase of the yeast metabolic cycle. This is regulated by a gene that also regulates the cell cycle and DNA damage response in mammalian cells (45). It was indeed observed that replication forks are slowed in cancer cells with elevated levels of reactive oxygen species (ROS) and in human cells in which ROS are artificially increased (46).
We conclude that the cysteine motif CysX, positioned at the base of the P-domain in Pol ϵ is important for cell function and for polymerase activity in vitro. Furthermore, Pol ϵ binds a single Fe–S cluster, most likely a [4Fe–4S] cluster, and this cluster is coordinated by the CysX motif. More experiments are currently under way to determine if the Fe–S cluster in Pol ϵ has a redox-active role.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank as PDB ID: 6h1v and PDB ID: 6qib.
Supplementary Material
ACKNOWLEDGEMENTS
Data were collected at beamline ID 23 and ID30A-3 of the European Synchrotron Radiation Facility.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Sven and Lilly Lawski Foundation (to J.t.B.); Insamlingsstiftelsen för medicinsk forskning (to E.J.); Carl Tryggers stiftelse (to E.J.); Swedish Cancer Foundation (to E.J.); Swedish Research Council (to E.S.E and E.J.). Funding for open access charge: Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Burgers P.M.J., Kunkel T.A.. Eukaryotic DNA replication fork. Annu. Rev. Biochem. 2017; 86:417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chilkova O., Jonsson B.-H., Johansson E.. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J. Biol. Chem. 2003; 278:14082–14086. [DOI] [PubMed] [Google Scholar]
- 3. Hamatake R.K., Hasegawa H., Clark A.B., Bebenek K., Kunkel T.A., Sugino A.. Purification and characterization of DNA polymerase II from the yeast Saccharomyces cerevisiae. Identification of the catalytic core and a possible holoenzyme form of the enzyme. J. Biol. Chem. 1990; 265:4072–4083. [PubMed] [Google Scholar]
- 4. Fuss J.O., Tsai C.-L., Ishida J.P., Tainer J.A.. Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim. Biophys. Acta. 2015; 1853:1253–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Netz D.J.A., Stith C.M., Stümpfig M., Köpf G., Vogel D., Genau H.M., Stodola J.L., Lill R., Burgers P.M.J., Pierik A.J.. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011; 8:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baranovskiy A.G., Lada A.G., Siebler H.M., Zhang Y., Pavlov Y.I., Tahirov T.H.. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012; 287:17281–17287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baranovskiy A.G., Gu J., Babayeva N.D., Kurinov I., Pavlov Y.I., Tahirov T.H.. Crystal structure of the human Polε B-subunit in complex with the C-terminal domain of the catalytic subunit. J. Biol. Chem. 2017; 292:15717–15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jain R., Vanamee E.S., Dzikovski B.G., Buku A., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. An Iron–Sulfur cluster in the polymerase domain of yeast DNA polymerase ϵ. J. Mol. Biol. 2014; 426:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hogg M., Osterman P., Bylund G.O., Ganai R.A., Lundström E.-B., Sauer-Eriksson A.E., Johansson E.. Structural basis for processive DNA synthesis by yeast DNA polymerase ϵ. Nat. Struct. Mol. Biol. 2014; 21:49–55. [DOI] [PubMed] [Google Scholar]
- 10. Jain R., Rajashankar K.R., Buku A., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. Crystal structure of yeast DNA polymerase ϵ catalytic domain. PLoS One. 2014; 9:e94835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Battye T.G.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G.W.. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 16. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schrödinger LLC. The PyMOL molecular graphics system. 2015; Version 2.0.
- 19. Stookey L.L. Ferrozine-A new spectrophotometric reagent for iron. Anal. Chem. 1970; 42:779–781. [Google Scholar]
- 20. Kahle M., ter Beek J., Hosler J.P., Ädelroth P.. The insertion of the non-heme FeB cofactor into nitric oxide reductase from P. denitrificans depends on NorQ and NorD accessory proteins. Biochim. Biophys. Acta (BBA) - Bioenergetics. 2018; 1859:1051–1058. [DOI] [PubMed] [Google Scholar]
- 21. Sabouri N., Viberg J., Goyal D.K., Johansson E., Chabes A.. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 2008; 36:5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ganai R.A., Bylund G.O., Johansson E.. Switching between polymerase and exonuclease sites in DNA polymerase ϵ. Nucleic Acids Res. 2015; 43:932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kirchner J.M., Tran H., Resnick M.A.. A DNA polymerase epsilon mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast. Genetics. 2000; 155:1623–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Isoz I., Persson U., Volkov K., Johansson E.. The C-terminus of Dpb2 is required for interaction with Pol2 and for cell viability. Nucleic Acids Res. 2012; 40:11545–11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ollagnier de Choudens S., Barras F.. Genetic, biochemical, and biophysical methods for studying Fe-S proteins and their assembly. MethodsEnzymol. 2017; 595:1–32. [DOI] [PubMed] [Google Scholar]
- 26. Freibert S.-A., Weiler B.D., Bill E., Pierik A.J., Mühlenhoff U., Lill R.. Biochemical reconstitution and spectroscopic analysis of iron-sulfur proteins. Methods Enzymol. 2018; 599:197–226. [DOI] [PubMed] [Google Scholar]
- 27. Tsai C.-L., Tainer J.A.. Robust production, crystallization, structure determination, and analysis of [Fe–S] proteins: uncovering control of electron shuttling and gating in the respiratory metabolism of molybdopterin guanine dinucleotide enzymes. Methods Enzymol. 2018; 599:157–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson D.C., Dean D.R., Smith A.D., Johnson M.K.. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005; 74:247–281. [DOI] [PubMed] [Google Scholar]
- 29. Beinert H., Holm R.H., Münck E.. Iron-sulfur clusters: nature's modular, multipurpose structures. Science. 1997; 277:653–659. [DOI] [PubMed] [Google Scholar]
- 30. Tse E.C.M., Zwang T.J., Barton J.K.. The oxidation state of [4Fe4S] clusters modulates the DNA-binding affinity of DNA repair proteins. J. Am. Chem. Soc. 2017; 139:12784–12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009; 460:831–838. [DOI] [PubMed] [Google Scholar]
- 32. Lill R., Broderick J.B., Dean D.R.. Special issue on iron-sulfur proteins: Structure, function, biogenesis and diseases. Biochim. Biophys. Acta. 2015; 1853:1251–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fan L., Fuss J.O., Cheng Q.J., Arvai A.S., Hammel M., Roberts V.A., Cooper P.K., Tainer J.A.. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008; 133:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dua R., Levy D.L., Campbell J.L.. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol epsilon and its unexpected ability to support growth in the absence of the DNA polymerase domain. J. Biol. Chem. 1999; 274:22283–22288. [DOI] [PubMed] [Google Scholar]
- 35. Johnson R.E., Klassen R., Prakash L., Prakash S.. A major role of DNA polymerase δ in replication of both the leading and lagging DNA Strands. Mol. Cell. 2015; 59:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pursell Z.F., Isoz I., Lundström E.-B., Johansson E., Kunkel T.A.. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007; 317:127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clausen A.R., Lujan S.A., Burkholder A.B., Orebaugh C.D., Williams J.S., Clausen M.F., Malc E.P., Mieczkowski P.A., Fargo D.C., Smith D.J. et al.. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol. 2015; 22:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding J., Taylor M.S., Jackson A.P., Reijns M.A.M.. Genome-wide mapping of embedded ribonucleotides and other noncanonical nucleotides using emRiboSeq and EndoSeq. Nat. Protoc. 2015; 10:1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daigaku Y., Keszthelyi A., Müller C.A., Miyabe I., Brooks T., Retkute R., Hubank M., Nieduszynski C.A., Carr A.M.. A global profile of replicative polymerase usage. Nat. Struct. Mol. Biol. 2015; 22:192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garbacz M.A., Lujan S.A., Burkholder A.B., Cox P.B., Wu Q., Zhou Z.-X., Haber J.E., Kunkel T.A.. Evidence that DNA polymerase δ contributes to initiating leading strand DNA replication in Saccharomyces cerevisiae. Nat. Commun. 2018; 9:858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeeles J.T.P., Janska A., Early A., Diffley J.F.X.. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell. 2017; 65:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeeles J.T.P., Cammack R., Dillingham M.S.. An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. J. Biol. Chem. 2009; 284:7746–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zahn K.E., Doublié S.. Look Ma, no PCNA: how DNA polymerase e synthesizes long stretches of DNA without a processivity factor. Nat. Struct. Mol. Biol. 2014; 21:12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bartels P.L., Stodola J.L., Burgers P.M.J., Barton J.K.. A redox role for the [4Fe4S] cluster of yeast DNA polymerase δ. J. Am. Chem. Soc. 2017; 139:18339–18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Z., Odstrcil E.A., Tu B.P., McKnight S.L.. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science. 2007; 316:1916–1919. [DOI] [PubMed] [Google Scholar]
- 46. Somyajit K., Gupta R., Sedlackova H., Neelsen K.J., Ochs F., Rask M.-B., Choudhary C., Lukas J.. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science. 2017; 358:797–802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank as PDB ID: 6h1v and PDB ID: 6qib.