

Abstract
We are entering a new era of mouse phenomics, driven by large-scale and economical generation of mouse mutants coupled with increasingly sophisticated and comprehensive phenotyping. These studies are generating large, multi-dimensional gene–phenotype datasets, which are shedding new light on the mammalian genome landscape and revealing many hitherto unknown features of mammalian gene function. Moreover, these phenome resources provide a wealth of disease models and can be integrated with human genomics data as a powerful approach for the interpretation of human genetic variation and it relationship to disease. For the future, the development of novel phenotyping platforms allied to improved computational approaches, including machine learning, for the analysis of phenotype data will continue to enhance our ability to develop a comprehensive and powerful model of mammalian gene–phenotype space.
Table of contents blurb:
Although the field of functional genomics is increasingly adopting genome-scale approaches, a comprehensive understanding of gene functions requires the parallel development of deep phenotyping platforms. This Review discusses strategies for broad-based mouse phenomics, applied both to gene-knockout collections and to diverse strains harbouring natural genetic variation. The authors discuss technical challenges, analysis pipelines and insights into human disease genetics.
Introduction
Deciphering the genetic networks underlying disease and biology will require a comprehensive multi-species approach that combines an increasingly sophisticated genetic analysis with deep and robust phenotyping approaches and advanced analytics. This paradigm is well exemplified by the use of the mouse as a key model organism for gene function studies and the elucidation of disease mechanisms.
The mouse provides a highly advanced tool box for genome manipulation that enables the generation of a wide variety of genetic variants and alleles at each and every locus across the mouse genome 1,2,3. The advent of CRISPR–Cas9 technology is a further step forward in the rapid and economical generation of complex and targeted mouse mutations4,5,6. Coupling large-scale mutagenesis programmes with broad-based phenotyping pipelines offers the possibility of generating multi-dimensional datasets that relate mutant alleles to biochemical, physiological and developmental changes across the phenotype landscape7–11,12. This provides a fundamental knowledge base on which we can dissect and analyse genetic variation in the context of functional mechanisms using new statistical approaches and machine-learning algorithms. As such the generation of a comprehensive picture of gene function in the mouse has a natural synergy with the development of comparable multi-dimensional datasets and biobanks in humans, which involves the study of the relationship between genetic variation and phenotype across diverse cohorts and populations.
However, the history of mouse genetics and the application of mouse genomic tools to date highlights the many challenges that we face in delivering comprehensive and robust datasets that inform on gene function. These include, firstly, the relatively poor state of legacy data on gene function in mouse genetics that often arises from a failure to record detailed experimental procedures or metadata, and at the same time underlines future requirements for standardization. Second, investigations of mouse mutants often reflect the interests and expertise of the investigator, thus failing to identify pleiotropy (the multiple functions of a gene) to its fullest extent.
Uncovering pleiotropy is central to elaborating gene function and the pathological basis of disease (Fig. 1). There is an increasing recognition that pleiotropy is ubiquitous 13. Pleiotropy is visible in numerous genetic phenomena, from variable expressivity14, phenotypic expansion15, to the genetic networks revealed through genome-wide association studies (GWAS)16 and phenome-wide association studies (PheWAS)17 (Fig. 1). It is possible that pleiotropy emerges from the highly interconnected gene networks in cells and tissues that extend beyond core loci, which have direct roles in traits and disease in particular tissues, to the modest regulatory effects of many peripheral genes. This might be termed network pleiotropy 16. The implication is that there are few genes expressed in a particular tissue that do not have some effect on a disease phenotype associated with that tissue. Whatever the mechanism, pleiotropy is a fundamental property of genetic networks and disease, and the manifestation of pleiotropy will vary with the genetic context. Extending phenotyping approaches to capture the full panoply of pleiotropic effects will be key to revealing a profound understanding of gene function and the genetic bases for disease. Moreover, the acquisition of more extensive phenotyping datasets in any species will improve our ability to compare phenotypes across species, to identify causal associations and to dig deeper into pathobiological mechanisms using a comparative approach.
Fig. 1. Pleiotropy is central to our understanding of mammalian gene function.
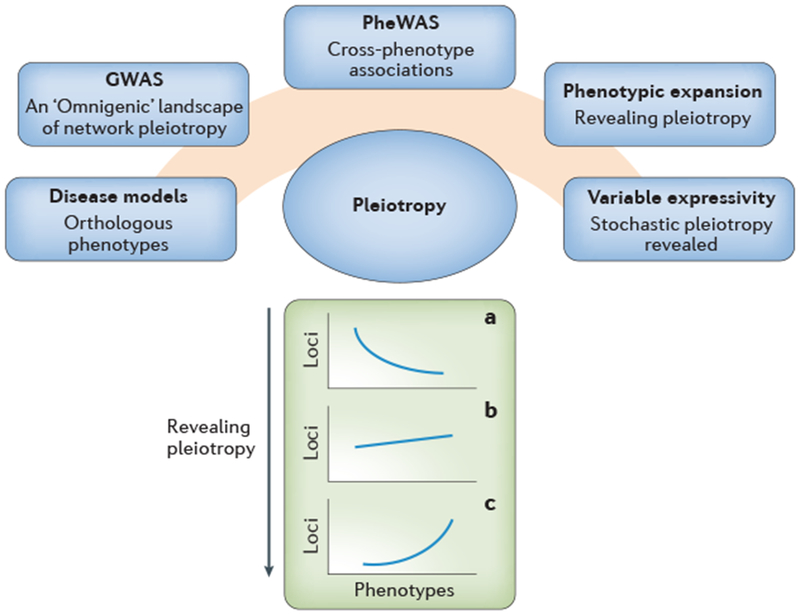
Pleiotropy, the multiple functions of a gene, is manifest through the exploration of disease models and a variety of other phenomena. These include genome-wide association studies (GWAS) where for complex traits the association signals are widely spread across numerous genes and not simply in core disease pathways. The implication is that network pleiotropy is rife and that all genes expressed in a particular tissue are likely to affect phenotype outcome – the “omingenic” hypothesis16. Pleiotropy is also revealed in phenome-wide association studies (PheWAS) where the associations of individual genetic variants with multiple phenotypes, known as cross-phenotype associations, are uncovered17. The well-known phenomenon of phenotypic expansion in human genetics also exemplifies the pervasiveness of human pleiotropy15. Finally, the well-known phenomenon of variable expressivity by which the expression of different aspects of phenotype varies across individuals with identical genotype is also revealing of pleiotropy. Uncovering pleiotropy to its fullest extent is a critical ambition for high-throughput mouse phenomics with the aim of improving our knowledge of pleiotropy and developing datasets where multiple functions are documented. Currently, for most loci we have limited knowledge of pleiotropy and for most genes our knowledge of phenotypes is limited (a). The challenge for genetics is to extend our knowledge of the multiple functions of genes to an increasing number of loci (b) and ultimately to most of the genes in the genome (c).
Ultimately, the development of comprehensive catalogues of mouse gene function and the integration of those phenotype datasets with other species requires a step change in phenotype approaches, as well as novel informatics tools and approaches, in order to realise the value of the extensive genome-wide analyses that are being undertaken across human and other model organisms. For both these aspects there is enormous current activity and interest in fundamental and novel developments that build upon international consortia and collaborations, and these alliances are laying the foundation for a new phenomics in the mouse.
This article describes the ongoing and future developments in genetic and phenotyping analysis in the mouse, including the development of new phenotyping technologies, which will enhance a comprehensive, systems-wide view of gene function. We discuss the key challenge of developing new analytical tools to enable data integration across species, particularly for integration of phenotype data. We also highlight how a comprehensive catalogue of gene function in the mouse will underpin the dissection of the parallel multi-dimensional datasets generated in human.
Phenotypic screens of laboratory mice
Over the past century, the characterization of phenotypes and the identification of the underlying genetic bases of spontaneously occurring variants in colonies of laboratory mice has contributed greatly to our current knowledge of gene function. Early detection of spontaneous variants depends upon careful observations of breeding colonies for the identification of phenodeviants demonstrating visible phenotypes such as eye anomalies, changes in coat colour or texture, and behavioural changes (such as hyperactivity or circling)18. Meticulous inspection of mouse stocks still remains an important tool for identifying new mutants in laboratory colonies 19 and an essential universal test for high-throughput phenotyping screens 20,21. However, assessing gene function through the manifestation of physical or behavioural anomalies in mouse colonies will only dissect a very limited segment of the genome landscape. The recent history of mouse genetics over the past few decades is characterized by the implementation of a range of mutagenesis tools — radiation 22, chemical 23,8 and genetic 2 — which enable efficient induction and recovery of mutants. High-throughput mouse mutagenesis in combination with increasingly sensitive phenotype screens is delivering a plethora of mouse mutant strains and adding substantially to the gene function catalogue. Increasingly, the focus has been on ever more sophisticated phenotyping platforms covering all body systems in order to deliver insights into a diverse range of biological mechanisms.
Hypothesis-free phenotyping screens
Mouse genetics was transformed by the implementation of high-throughput mutagenesis programmes utilizing two contrasting approaches: random N-ethyl-N-nitrosourea (ENU) chemical mutagenesis7,8 and gene-trap and transposon technologies9. For both modalities, it is feasible to generate large numbers of mutant animals that can be screened by a range of phenotype tests. Even for gene-based technologies the screens are applied in a hypothesis-free and unbiased manner without making any a priori assumptions about the function of the underlying gene that was mutated. The expectation was that novel aspects of gene function would be efficiently revealed. Indeed, this promise was fulfilled and the first hints of extensive pleiotropy in the mammalian genome uncovered7,8. Importantly, the identification of phenotypes for genes of unknown function gathered pace, and henceforth it was recognized that an important limitation to discovery lay with the availability, sensitivity and logistics of phenotyping screens.
The majority of all mouse screens involve elements of cage-side observations that reveal visible phenotypes including dysmorphologies and size differences24,25. For example, many neurological disorders can be identified by experienced mouse handlers noting differences in movement and activity26, and the appearance of unusual behaviours such as fitting27, jerky movements28 and circling24. Building on these simple observational protocols, a whole plethora of phenotyping tests have emerged over the past few decades in parallel to the emergence of the mouse mutagenesis toolkit. There is not the space here to exhaustively cover the full range of phenotyping platforms (many of which are illustrated in Fig. 2), but several areas serve to illustrate the pace of change. Neurological and behavioural systems have been an areas of focus, with a variety of test environments employed for the identification of novel mutants with defects in characteristics such as motor function29,30, balance 31 and various behavioural measures such as anxiety 32,33. More complex test environments such as wheel running activity to measure circadian profiles34, pre-pulse inhibition (PPI) profiling35 and neurohistopathology36 have enabled the discovery of complex behavioural phenotypes modelling many human neurological28 and neuropsychiatric disorders37. Sensory systems have been investigated by extending the test environment to include the optokinetic drum 38 and auditory brainstem response 39. Furthermore, imaging systems have been applied to allow the capture of diverse parameters on bone structure and body composition40. Clinical chemistry and haematological analyses have developed to allow the assessment of a wide range of parameters relating to multiple organs and systems including blood chemistry and urinalysis7,8,25,41,42,43, haematology44 and components of the murine immune system45,46,47. Importantly, each of these phenotyping platforms has a relatively low impact on the mouse tested and can therefore be used in combination with other platforms and also repeated during the animal’s lifetime. Ultimately, phenotypic screens end with terminal assays, which include histopathology 48 or terminal imaging, and the assessment of morphology by a variety of modalities at embryonic stages for lethal mutations49–51,52. In addition, tissues can be banked and utilized for a variety of omics studies; however, partly due to cost, these omics analyses have not featured extensively in the past or current phenotyping pipelines. Finally, broad-based phenotyping would be most efficiently and economically delivered at mouse genetics centres with a diverse range of skills and equipment, the so-called mouse clinics53. The mouse clinics would take advantage of the inherent economies of scale and operate a phenotyping pipeline that delivers a comprehensive assessment of gene–phenotype relationships at reasonable cost. The economies of scale also speak to the extensibility of the pipeline; incorporating new tests and paradigms (as discussed below) can be delivered at modest expense.
Fig. 2. The IMPC phenotyping pipeline.
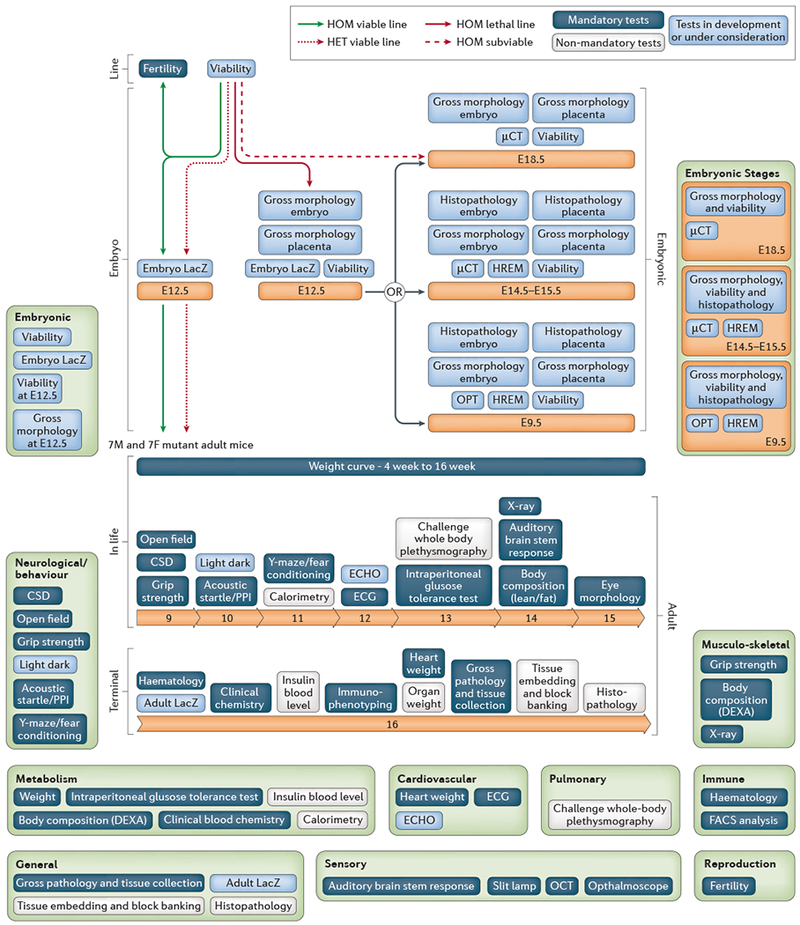
The International Mouse Phenotyping Consortium (IMPC) pipeline provides an exemplar of the potential of high-throughput pipelines for the acquisition of broad-based phenotype data at both embryonic and adult time-points. The range of phenotyping platforms ensures the recovery of phenotype data across multiple systems and disease states. The key systems areas that are analysed are indicated along with the relevant phenotype tests that impact upon that area. Each phenotyping test is underpinned by a standard operating procedure (SOP) in the International Mouse Phenotyping Resource of Standardised Screens (IMPReSS) database (www.mousephenotype.org/impress) that defines the phenotyping procedure and the associated metadata that is required.
μCT, micro-computed tomography;
CSD, combined SHIRPA (SmithKline Beecham, Harwell, Imperial College, Royal London Hospital, phenotype assessment) and dysmorphology;
DEXA, dual-energy X-ray absorptiometry;
E, embryonic day,
ECG, electrocardiography;
ECHO, echocardiography;
FACS, fluorescence-activated cell sorting;
HREM, high-resolution episcopic microscopy;
OCT, optical coherence tomography;
OPT, optical projection tomography;
PPI, pre-pulse inhibition.
Launch of multi-centre broad-based phenotyping
The increasingly diverse phenotyping platforms that emerged for use in large-scale mutagenesis screens required a critical appraisal of both the utility of the phenotyping procedures and how they might be employed. First, it is desirable to assess and, if necessary, improve procedures to ensure reproducibility across laboratories and across time. The European Union Mouse Genetics Research for Public Health And Industrial Applications (EUMORPHIA) programme tested and developed a robust set of phenotyping standard operating procedures (SOPs) that is available through the European Mouse Phenotyping Resource for Standardized Screens (EMPReSS) database54 (Table 1). Importantly, the standardised SOPs incorporate the capture of metadata (including feed, environment, person performing the test etc.) that takes into account possible confounders and gene–environment interactions55, and is critical for reproducibility as well as improving the statistical power to detect significant associations. Second, it is recognized that the application of a full range of tests to each mouse line is essential to reveal pleiotropy and provide a more profound understanding of gene function in the context of diverse biological systems.
Table 1.
Major high-throughput knockout mutagenesis and phenotyping programmes and consortia.
| Programme | Date | Aims | Mutant allele | Refs |
|---|---|---|---|---|
| International Knockout Mouse Consortium (IKMC)a | 2003–2015 | Generate mutations in embryonic stem cells for every mouse gene | 1. Gene trap alleles 2. Knock-out first, conditional ready tm1ab alleles 3. KOMP Regeneron allelesc |
56,57 |
| European Union Mouse Genetics Research for Public Health and Industrial Applications (EUMORPHIA) | 2002–2006 | Develop and test a robust set of phenotyping SOPs, reproducible across multiple centres (EMPReSS) | Employed a panel of standard inbred strains to demonstrate between-centre reproducibility | 54 |
| European Mouse Disease Clinic (EUMODIC) | 2007–2012 | Multi-centre programme generating and analysing hundreds of mouse mutant lines employing standardized EUMORPHIA procedures | Used tm1ab alleles available from IKMC | 11 |
| Mouse Genetics Programme, Sanger Institute (MGP) | 2007–present | Analysed hundreds of mutant lines employing standardized EUMORPHIA procedures and other SOPs | Used both tm1ab, tm1bb alleles and KOMP Regeneronc alleles | 10 |
| International Mouse Phenotyping Consortium (IMPC) | 2011–present | Analysed thousands of mutant lines employing standardized IMPReSS procedures | Used tm1bb, KOMP Regeneronc alleles as well as CRISPR–Cas9 critical exon deletion alleles | 12 |
The IKMC incorporated a number of programmes including European Conditional Mouse Mutagenesis Program (EUCOMM), the Knockout Mouse Project (KOMP), the North American Conditional Mouse Mutagenesis Project (NorCOMM) and the Texas A&M Institute for Genomic Medicine (TIGM).
The IKMC most commonly produced targeted mutation is the tm1a knock-out first, conditional ready allele used by several programmes. The tm1a allele can be converted by Cre excision into a tm1b null mutant allele. Both the tm1a and tm1b alleles incorporate a lacZ reporter that enables determination of expression patterns of the disrupted gene. However, for the current CRISPR–Cas9 critical exon deletion alleles generated by IMPC a lacZ reporter is not incorporated.
As part of the IKMC programme a number of targeted alleles were produced using large bacterial artificial chromosome (BAC) targeting vectors leading to the ablation of the entire gene, so-called KOMP Regeneron alleles. These were used in various programmes, but were usually a small minority of alleles. EMPReSS, European Mouse Phenotyping Resource of Standardised Screens54; IMPReSS, International Mouse Phenotyping Resource of Standardised Screens (http://www.mousephenotype.org/impress). SOPs, standard operating procedures.
It was appreciated that mouse clinics also provide the infrastructure for large-scale mutagenesis, the ability to generate and breed many mouse lines simultaneously, that when allied to broad-based phenotyping pipelines could analyse hundreds, perhaps thousands, of genes over reasonable timescales. The activity of the clinics need not be focused entirely on single gene mutations, but could also assess any genetic resource from inbred lines to outbred populations. The aim would be to gather multi-dimensional genetic and phenotype data that would reveal the genetic networks at play across diverse systems.
Phenomics for every mouse gene
Critical to the emergence of a comprehensive catalogue of mammalian gene function is the generation of a mutant resource for every gene in the mouse genome. The International Knockout Mouse Consortium (IKMC) set out to generate mutations in embryonic stem cells for every gene in the mouse genome56 (Table 1). Using both high-throughput gene trapping and gene targeting approaches57, they developed mutant embryonic stem (ES) cell lines for more than 18,500 genes representing over 90% of mouse protein-coding loci. Most of the mutations are conditional, where mutations can be induced in a temporal or spatial manner.
The focus of mouse mutagenesis and phenotyping pipelines on null and severe loss-of-function mutations raises the question of the relevance of these alleles and their phenotypes to our understanding of the contributions of human genetic variants (which are probably alleles of modest effect) at these loci to complex disease. The primary rationale for the use of null mutations is to reveal important functional contributions to phenotypes across diverse biological and disease systems and to provide a fundamental baseline for gene function. Indeed, it is clear from studies in the human population that there are thousands of associations between Mendelian and complex diseases that are manifest in a phenotype code linking Mendelian loci to complex disorders58. As such, human variants associated with complex disease are enriched in genes that encapsulate this Mendelian code. This underlines the rationale and utility of analysing and cataloguing mouse null mutations. In a similar vein, there are numerous examples where for human complex disorders the generation of null mutants for strong candidate disease genes has proven valuable in validating genes and elucidating constituent endophenotypes that may contribute to the pathology of a complex disease. To take one example, null mouse mutations for autism candidate genes Shank159,60, Shank2 61 and Chd8 62,63 result in autistic-like behaviour, which exemplifies the utility of mouse knockouts in assessing fundamental and relevant disease phenotypes.
The IKMC mutant ES cell resource was the basis for two major pilot programmes to assess broad-based phenotyping across hundreds of mouse lines (Table 1). At the Sanger Institute, UK, the Mouse Genetics Programme (MGP) undertook the generation of hundreds of mutant lines and applied a diverse range of phenotype screens10. At the same time, the European Mouse Disease Clinic (EUMODIC) programme undertook a multi-centre programme (involving MRC Harwell, Helmholtz München, ICS Strasbourg and the Sanger Institute) generating and analysing hundreds of lines through a common phenotyping pipeline employing standardized EUMORPHIA procedures11. The use of common reference lines in the EUMODIC programme allowed an assessment of the reproducibility of phenotype outcome across centres, and demonstrated the robustness of the phenotype platforms employed. Importantly, both the MGP and the EUMODIC programmes revealed extensive pleiotropy across hundreds of genes and uncovered phenotypes for many genes with hitherto unknown function. These programmes underlined the critical role for broad-based screens in any future initiative to scale to a genome-wide effort.
Current systematic phenotyping efforts
International Mouse Phenotyping Consortium.
In 2011, the International Mouse Phenotyping Consortium (IMPC) was formed with the goal of generating a comprehensive catalogue of mouse gene function by generating and characterizing null mutations for every mouse gene12. Until recently, the IMPC has generated mouse mutants using the tm1b null mutant allele from IKMC 57 (Table 1). IMPC now employs CRISPR–Cas9 to generate null mutations by the deletion of an early critical exon. Importantly, all mutants are generated on a coisogenic C57BL/6N background. Homozygous mutants enter a broad-based, standardized adult phenotyping pipeline64 (Fig. 2). Cohorts of male and female mutant mice undergo a wide range of phenotype tests from 9–16 weeks, followed by a variety of terminal tests. The tests cover a wide range of system areas including neurological, behavioural, metabolic, cardiovascular, pulmonary, fertility, sensory, and musculo-skeletal function. Homozygotes that are embryonic lethal undergo characterization through an embryonic pipeline14 that assesses the stage of lethality, as well as applying various high-resolution imaging modalities (such as optical projection tomography (OPT), micro-computed tomography (μCT) and high-resolution episcopic microscopy (HREM)) to elaborate morphological defects49–51,52. In the case of embryonic lethal mutants, heterozygotes enter the adult phenotyping pipeline. The tm1b allele carries a lacZ reporter that enables the determination of tissue expression patterns of each disrupted gene. In IMPC, both embryonic and adult expression profiles have been acquired for many lines. To date, IMPC has generated over 7,000 mutant lines and phenotype data has been collected on over 5,000 lines.
The value of a broad-based phenotype approach is strongly supported by the global analysis of the multi-dimensional datasets that are emerging. A number of key findings are transforming our view of the mammalian genome landscape. First, an extraordinary number of novel phenotypes and models are revealed 64. 90% of the gene–phenotype annotations described by IMPC have not previously been reported, emphasizing the noteworthy and widespread pleiotropy. For the first 3,328 genes analysed, 889 known human disease genes in Online Mendelian Inheritance in Man (OMIM) and Orphanet have an orthologous IMPC mouse strain with at least one phenotype. Given that the IMPC pipeline is broad but shallow, not focussed on particular disease areas, and incomplete for some of the lines, it was remarkable that 360 IMPC lines (40%) have phenotypic overlap with the 889 human disease genes, and the majority (279, 78%) are the first reported mouse model for these diseases. A major analysis of embryonic lethal lines from the first 1,751 knockouts provided several profound, novel insights into essential (lethal) genes 14. The analysis confirmed a strong enrichment for human disease genes within the set of embryonic lethal, essential genes. But perhaps the most intriguing finding was the high degree of variable expressivity observed in embryonic lethals, despite the uniform, co-isogenic background of the mutant lines. These observations were also reflected in the significant number of subviable lines that demonstrate variable lethality. Mutant genes resulting in subviability, as with standard non-essential genes, were significantly more likely to have a paralogue compared to essential genes. The implication is that subviability reflects stochastic variation that is induced in normally buffered pathways65. This is another example of pleiotropy where differing components (and phenotypes) of the disrupted genetic network are variably manifest in the mutant. It might be termed ‘stochastic pleiotropy’.
The phenotyping of both male and female cohorts (mutant and wild-type) through the IMPC pipeline has enabled for the first time an in-depth analysis of the extent of sexual dimorphism across the entire mammalian genome 66. Analysis of 2,186 mutant lines for up to 234 traits found that nearly 18% of mutant phenotypes are influenced by sex, demonstrating that sexual dimorphism is extraordinarily pervasive. This is an important finding as, in the past, for many individual mutants and traits phenotype analysis was confined to one sex.
Finally, in two disease areas, deafness and metabolism, analysis of relevant disease-specific phenotypes for a large number of IMPC mutant lines has uncovered numerous novel genetic loci. These findings substantially expand our understanding of the genetic landscape associated with these disease states 67,68.
Inbred, recombinant inbred and outbred lines.
In addition to the analysis of single-locus, typically null mutations, mouse geneticists have employed the considerable genetic variation between inbred strains to study genetic systems, and in particular to dissect the genes involved in complex traits. These complex trait resources have been analysed through many phenotyping platforms, and it is very pertinent to consider their application to high-throughput phenomics. There are essentially two classes of mouse resources for the analysis and mapping of complex traits. The first class includes the huge panel of extant inbred strains and the Recombinant Inbred (RI) lines69, which includes the Collaborative Cross (CC) lines70. Inbred and RI lines have been phenotyped incrementally for multiple traits over many years, and together these resources have been an important tool for quantitative trait locus (QTL) mapping71,72. The Hybrid Mouse Diversity Panel (HDMP) combines inbred strains and RI lines for fine mapping of diverse phenotypes 72,73. The CC lines have also been extensively phenotyped at a variety of centres for a number of traits 74,75. However, there is considerable potential and economies to be gained by a comprehensive analysis of all the CC lines through a high-throughput, broad-based pipeline similar to that operated by IMPC.
The second class of mouse resource for QTL mapping includes various outbred populations of mice generated by pseudo-random breeding from inbred strains. This class covers the Heterogeneous Stock (HS) and Diversity Outbred (DO) populations. A high-throughput phenotyping pipeline for HS populations has already been developed76 and applied to the genetic analysis of diverse traits including asthma, type 2 diabetes, obesity, anxiety, immunological, biochemical and haematological phenotypes77. DO mice are derived from outbreeding of different CC lines and are a potent tool for high-resolution trait mapping78,79 and a resource that would be amenable to analysis in the high-throughput pipelines currently being applied to single-gene mutations. Lastly, commercial outbred mice have been used for fine mapping of specific traits80. Recently, the same outbred population (CFW) has been employed coupled to a high-throughput phenotyping pipeline for large-scale trait discovery and mapping81. From the analysis of more than 1,800 animals from the outbred population, 156 unique QTLs for 92 phenotypes were discovered, of which around a fifth were mapped at gene-level resolution. Thus broad-based phenotyping and the development of standardized, high-throughput pipelines is an important tool not only in the analysis of specific gene variants, but also for the discovery and dissection of complex traits.
Current challenges in mouse phenotyping
Two of the most demanding challenges with respect to mouse phenotyping are reproducibility and inter-species comparability between mice and humans. In order to address these issues it is necessary to overcome both genetic and species differences, thereby ensuring the data collected from mouse models is increasingly biologically relevant to human clinical studies. By delivering robust and comparable phenotype data in both mice and humans we will increasingly build upon our understanding of genetic and disease systems. At the same time, it is imperative to develop new modalities for phenotyping mice that address avenues for revealing hitherto unexplored physiological and behavioural areas.
Standardized genetic systems for reproducibility.
Inconsistencies in mouse phenotyping data between laboratories can often be attributed to uncontrolled genetic backgrounds, arising from differences between the background strain used to generate a mutant and the strain used in subsequent breeds 82. In recent years this has led to the demand to generate co-isogenic strains of genetically altered (GA) mice, delivering experimental cohorts where the genetic background is standardized and phenotyping data are reproducible. Phenotyping co-isogenic mutants provides robust data in the context of a single defined genome and is the essential foundation for the investigation of gene function 11,10. However, progress in gene editing and the development of extensive series of recombinant inbred lines 83,70 will enable a wider range of standardized genetic backgrounds in which a mutation can be examined. The analysis of individual CRISPR mutations on a diversity of inbred backgrounds is likely to uncover further pleiotropic diversity (in a similar manner to phenotypic expansion in humans), elaborating the biology of gene function and complexity of gene interactions 84.
Whereas for inbred strains of mice the issue of genetic variation has been all but eliminated, this does not remove other unwanted sources of variation, which need to be assessed (as discussed above). Increasingly, we need to pay attention to the mouse microbiota — the commensal, symbiotic and pathogenic microorganisms found living in or on laboratory mice — and understand how they contribute to phenotypic variation. Undoubtedly the microbiota is influential in many85 but all not phenotyping tests. The logistics of characterizing and stabilizing the microbiomes of an experimental cohort of mice and developing experimental mouse models of the human microbiome represent considerable challenges for the future 86, including new statistical methods that can adjust for the microbiome variation.
Humanizing the test environment.
One of the most exciting prospects is the development of new phenotyping technologies that ‘humanizes’ the testing environment. For example, metabolic phenotyping has begun to address the comparative differences in surface area to volume ratio of mice and humans by establishing testing paradigms that run at thermoneutrality, thereby circumventing differences in the regulation of heat production in the mouse 87. In addition, the use of home-cage monitoring systems to detect changes in feeding patterns 88 is likely to replace more conventional metabolic cages as they offer improvements in the complexity of data acquisition including duration, number of feeding bouts and timing of feeds in combination with activity data, replacing the rather crude single measurement of food intake.
Arguably the most controversial field in terms of reproducibility in mouse phenotyping data has been behavioural assays 89. A future goal is not just to reduce variability caused by subjectivity or non-standardized testing (largely addressed by the widespread use of automation such as tracking systems) but also to increase the sensitivity and diversity of parameters measured. Extrapolating mouse behaviour to humans is problematic, with many behavioural tests assessing simple paradigms that fail to exemplify the complexity of human behaviour. More-complex protocols often require weeks of training and acclimatization. However, new home-cage monitoring technologies bring extraordinary opportunities in behavioural phenotyping that will enable researchers to explore new but different complex paradigms and improve the assessment and development of behavioural models (Fig. 3). In addition, they are adaptable to high-throughput phenotyping pipelines. By recording data including video from the home cage, it is possible to avoid issues of anxiety in the test environment and explore sequences of voluntary activity longitudinally90 over many days including behaviours associated with well-being91. In contrast to some conventional apparatus, it is now possible to monitor activity continuously and during the entirety of the dark/active phase of the mouse92 and to reveal previously unrecognized behaviours93, including with cage-mates present94. The specialist equipment required for such analysis is costly and requires both substantial infrastructure and skills to handle the large multidimensional datasets generated. However, the rapid expansion of both supervised and unsupervised machine-learning technologies in animal behavioural studies 95 promises to deliver objective, longitudinal and sensitive data and a rich stream of novel behavioural phenotypes (see below and Fig. 4).
Fig. 3. Home-cage monitoring and machine learning.
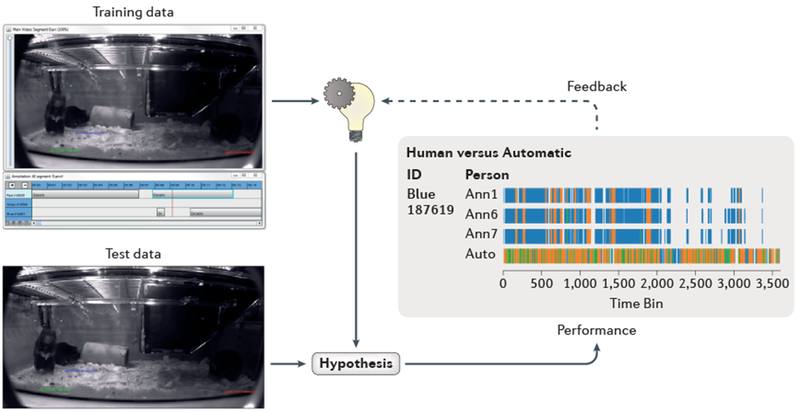
The figure illustrates a supervised learning feedback loop. This type of automation is essential in order to analyse longitudinal changes in the patterns of behaviour in genetically altered (GA) mice, potentially extending into months and years. Experienced animal researchers and technical staff watch many hours of video recording during which time they record the specific behaviours of individual mice (such as climbing, feeding and drinking). Subsequent machine learning from the manual annotation data generates algorithms that are validated by using test data and performance analysis. This is represented by the data plot (bottom right), where the pattern of behaviours detected by human annotation is compared to those of the machine-learning algorithms. Where the data is non-comparable, further refinement of the algorithms ensues.
Fig. 4. Ageing as a new dimension of high-throughput mouse phenotyping.
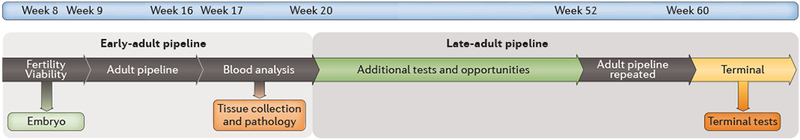
There is increasing interest in the use of ageing pipelines to reveal novel phenotypes, particularly those that might model age-related disease. We illustrate a typical plan for an ageing phenotyping pipeline (mouse age: week 8 to week 60). Cohorts of mutant mice would as usual enter a phase of early-adult phenotyping, including where appropriate embryo analyses. At the end of early-adult phenotyping, some mice from the cohort may be removed from the pipeline for terminal assays. The remaining cohort proceeds to ageing, and subsequently, at around one year or older, adult phenotyping is repeated (late-adult phenotyping) followed by terminal tests. The intervening period between early and late adult phenotyping provides a window for additional phenotyping tests that might not be part of the standard adult phenotyping pipeline.
Environmental perturbations.
There is considerable interest in the application of environmental perturbations or challenges to large-scale phenotyping screens, thus enriching the phenotypes that might be revealed. Challenges might include diet (such as high fat 96), noise 97, infection 98 or other immunological perturbations 99, although inevitably careful attention is required to consider potential indirect and confounding effects of a challenge across multiple phenotypic domains. As such it will often be prudent to utilize separate cohorts of animals for some challenges.
Age-related phenotypes.
The assessment of phenotype throughout the animal’s lifetime is the focus of an increasing number of mouse genetics studies. There have been systematic efforts to determine the phenotypes of aged inbred lines and GWAS to reveal loci involved with age-related disease 100,101. In addition, a recent report describes a large-scale phenotype-driven ENU mutagenesis screen for age-related disease loci involving the recurrent screening of mutagenized pedigrees as the mice aged 38. A considerable number of novel age-related phenotypes and genes were uncovered that would not have been revealed from screens at earlier time-points. Fig. 4 illustrates a typical pipeline that might be used for the discovery of age-related phenotypes from large-scale screens, involving early-adult phenotype screens of mouse cohorts followed by an intervening period while mice are aged, and a subsequent round of late-adult phenotyping at one year of age or more.
Analysis of multi-dimensional phenotype data
High-throughput, broad-based phenotyping pipelines present a number of profound challenges in terms of phenotype analysis. First, the diverse nature of the data, from categorical to continuous measures, requires differing presentational and statistical approaches that are tuned to the data and analysis requirements. Second, metadata parameters need to be incorporated within the defined SOPs for each phenotyping test to ensure standardization and reproducibility. Metadata should also include the production and entry of control cohorts into the phenotyping pipeline. Moreover, it is important to document randomization and blinding schemes. Overall, it is critical that statistical models are developed that capture the experimental design and any significant sources of variation. It is also important that the reporting of experiments meets the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines for animal experimentation, providing transparency that underpins reproducibility102. Finally, the use of phenotyping pipelines in a multi-centre programme benefits from the incorporation of common reference lines, which enables measures of phenotype concordance to be calculated and provides confidence in standardization and reproducibility11.
The high-throughput, multi-centre nature of the IMPC exemplifies the challenges that are faced and the extraneous factors that must be incorporated into the data analysis. Initial approaches with t-tests and Mann–Whitney tests have given way to more sophisticated linear regression and Bayesian approaches that model the impact of other sources of variation beyond genotype and phenotype11,103. As might be expected, evidence has emerged of significant batch effects, litter effects and interday variability that have the potential for confounding effects on phenotype readouts. For example, female Expitm1a(KOMP)Wtsi mutant mice were initially observed to have significantly decreased circulating chloride levels. Further investigation showed that all the knockout females were phenotyped on a single day without concurrent controls, and all data collected on that day were low compared to other test days. It is difficult and costly to test littermate controls in a high-throughput pipeline, and also not always feasible to test controls concurrently with mutants. Thus centres tend to use a multi-batch workflow. As a consequence IMPC employs a linear mixed model, which has been shown to have the appropriate power and sensitivity for multi-batch workflows, minimizing the risk of genotype effects being confounded by temporal variation 103.
The interpretation of data from the wide array of image data generated from adult and embryonic pipelines remains a substantial roadblock, but progress is being made. For example, in the current IMPC pipelines, 2D and 3D images are generated ranging from lacZ expression patterns to μCT data. The manual annotation of these data by specialists is time consuming and prone to bias or error so it is therefore crucial to develop automated image analysis approaches that can robustly identify phenotypes. The development of methodologies to date has largely focused on the 3D data, which is nearly impossible to manually annotate. However, the data are amenable to image registration methods. Automated pipelines have been developed 49,50,51 which allow anatomical differences in embryonic day (E) 15.5 micro-CT data to be detected. Computer-automated image registration algorithms consist of three analyses (intensity, deformation and atlas-based) which detect missing anatomical structures and differences in volume of whole organs. These initial efforts provide a platform on which to develop tools for other embryonic stages and to manage the large volumes of data that will emerge from comprehensive embryonic phenotyping at multiple developmental time-points. Nevertheless, progress in the methods for the analysis of imaging data from other phenotyping modalities such as X-ray and high-resolution scans of histopathology slides is a priority. Novel techniques such as deep convolutional neural networks, a supervised machine learning method, are demonstrating applications in mouse phenotyping and have to date robustly segmented a number of mammalian cells. Methods such as this can be applied to automatically annotating slides in high-throughput phenotyping 104
These multi-dimensional data sets and associated analysis from high-throughput mouse phenotyping are disseminated by the FAIR principles of data management — making data Findable, Accessible, Interoperable and Reusable 105. The application of these principles builds off the experience from other large, collaborative biological projects that include biobanks as well as the Roadmap Epigenomics106 and Functional Annotation of the Mammalian Genome (FANTOM)107 consortia that have had their data reapplied in thousands of published studies108 (Fig 5). A key process in adhering to FAIR data principles is annotation of data with community developed ontologies to facilitate data discovery. The key ontology used by the high-throughput mouse phenotyping groups is the mammalian phenotype (MP) ontology109, with other integrated ontologies such as the Edinburgh Mouse Atlas Project (EMAP)110 and the Phenotype And Trait Ontology (PATO)111. These ontology annotations allow integration with the diverse array of functional datasets in the public domain and opens up the potential for assigning function to poorly characterized genes in the mouse genome. By taking the annotated gene–phenotype data from knockouts and leveraging datasets such as the Genotype–Tissue Expression (GTEx) project 112, Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 113, and Orphanet114, we can look to build new pathways based on network analysis of our phenotype data, informing us on potential disease mechanisms and therapies based on orphan drugs
Fig. 5. Overview of data flow for large-scale, broad-based mouse phenotyping programmes.
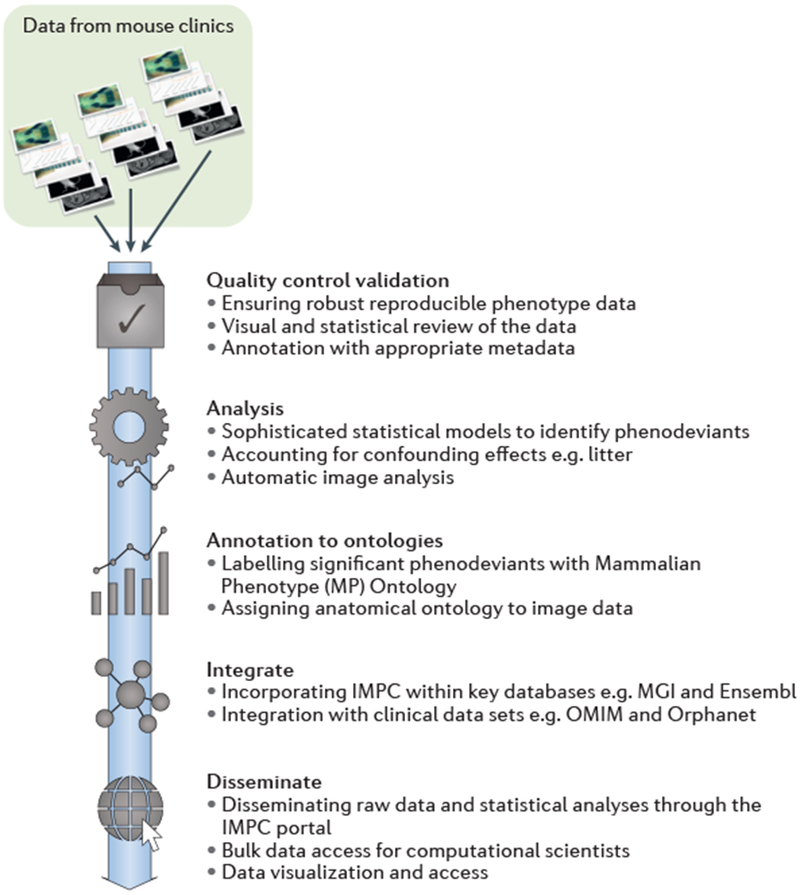
Mouse clinics acquire diverse multi-dimensional datasets, including categorical and continuous data alongside a variety of image data. Data is uploaded routinely to a data coordination centre where it undergoes processing through a standardized pipeline including data validation, robust quality control, statistical analysis, annotation and data integration followed by dissemination to the scientific community.
MGI, Mouse Genome Informatics
OMIM, Online Mendelian Inheritance in Man
New statistical machine learning methods will be developed that can integrate the multiple phenotype data-modalities into a single joint model. In human genetics, these multiple-phenotype models will increase statistical power to detect genetic variants by averaging out confounding variation found in any one phenotype, similar to ‘enrichment analysis’ used in GWAS115, thus enhancing comparisons with mice. Latent-factor models and related methods will help to identify low-dimensional structure within the high-dimensional phenotype data116, providing novel statistical targets for association testing. Moreover, the use of molecular phenotypes from omics data including RNA expression and proteomics will facilitate the identification of causal genetic variants of unknown function. Real-time cage monitoring will demand scalable computational methods for processing and compressing hours of video capture of home-cage environment117. New bioinformatics techniques will collate video recordings of group-housed mice in their home-cage environment and use advanced visualization tools and machine-learning analytics to extract comprehensive datasets and automatically classify behaviours from a spectrum of mouse strains and mutants (Fig 3). Classification and mapping of phenotypes into networks and hierarchies — e.g. of behavioural types, and sub-phenotypes — has the potential to increase the power to detect variants affecting biologically related traits 118.
Integration with human clinical genetics
The extensive multi-dimensional datasets emerging from large-scale mouse phenotyping programmes are an important foundation for the analysis of mammalian gene function. As we elaborate above, these datasets are already resetting our view of the mammalian genomics landscape. In addition, they are a powerful tool for cross-species analysis and integration that will shed further light on gene–phenotype relationships in diverse species. This is particularly true for human and clinical genetics and the community’s ability to assess gene–phenotype relationships in humans, and to relate DNA variation in humans to disease outcome. Advances in DNA sequencing technologies have revolutionized diagnosis and discovery of new gene associations in human disease since the first successes in 2010, particularly for rare conditions involving Mendelian inheritance119,120. Based on these successes, numerous national programmes have been or are being initialized to perform large-scale whole-exome or whole-genome sequencing of patients alongside comprehensive collections of clinical data to address the long diagnostic odysseys that many patients with rare diseases undergo, as well as personalized treatments of cancer patients. For example, in the UK the 100,000 Genomes Project [www.genomicsengland.co.uk] is embedding genomics into the mainstream of a national healthcare system for the first time. In the US, the NIH Undiagnosed Disease Network 121, Centers for Mendelian Genomics15 and Precision Medicine Initiative 122 are leading the way, and major national initiatives are underway in Japan, China, Singapore, Canada, Australia, Denmark, Norway, the Netherlands and France.
Despite all these advances, most large-scale projects can provide a genetic diagnosis for only around 25% of cases123,124,125 with many patients presumed to carry as yet uncharacterized variants within, or regulating, known or novel disease genes. Here, the potential of mouse phenomics programmes to shed light on genes carrying rare, potentially pathogenic variants but with little or no previous functional knowledge can be critical. Clinical geneticists routinely interrogate such data through searching the literature as well as model organism databases and integrated portals provided by the IMPC126, model organism aggregated resources for rare variant exploration (MARRVEL) 127 and the Monarch Initiative 128. However, manual searches are not scalable for routine healthcare pipelines such as that of the 100,000 Genomes Project, hence it is valuable to have programmatic access as provided by the Monarch Initiative through their portal or automated variant prioritization tools that include comparison of a patient’s clinical phenotypes to those observed in model organisms including the IMPC (Figure 6) 129. Examples of such approaches used in the 100,000 Genomes Project, and other disease sequencing efforts, include Exomiser 130, Genomiser 131 and Phevor 132. Routine inclusion of mouse and fish model organism data in Exomiser and application to the NIH Undiagnosed Disease Programme has been demonstrated to increase diagnostic yield by 10–20% in the NIH Undiagnosed Disease Programme133. Collections of deep patient phenotype data and reproducible mouse phenotypes in a standardized structure using ontologies such as the Human Phenotype134 and Mammalian Phenotype Ontology 109 are critical to these approaches, as well as the semantic similarity algorithms to compare and rank phenotypic profiles between human and mouse developed by the Monarch Initiative 135. Similarly, standardized collections of other multi-dimensional datasets, such as transcriptomics, is emerging as a critical step as researchers seek to further improve variant analysis.
Fig. 6. Integration of human and mouse data for rare disease genetics.
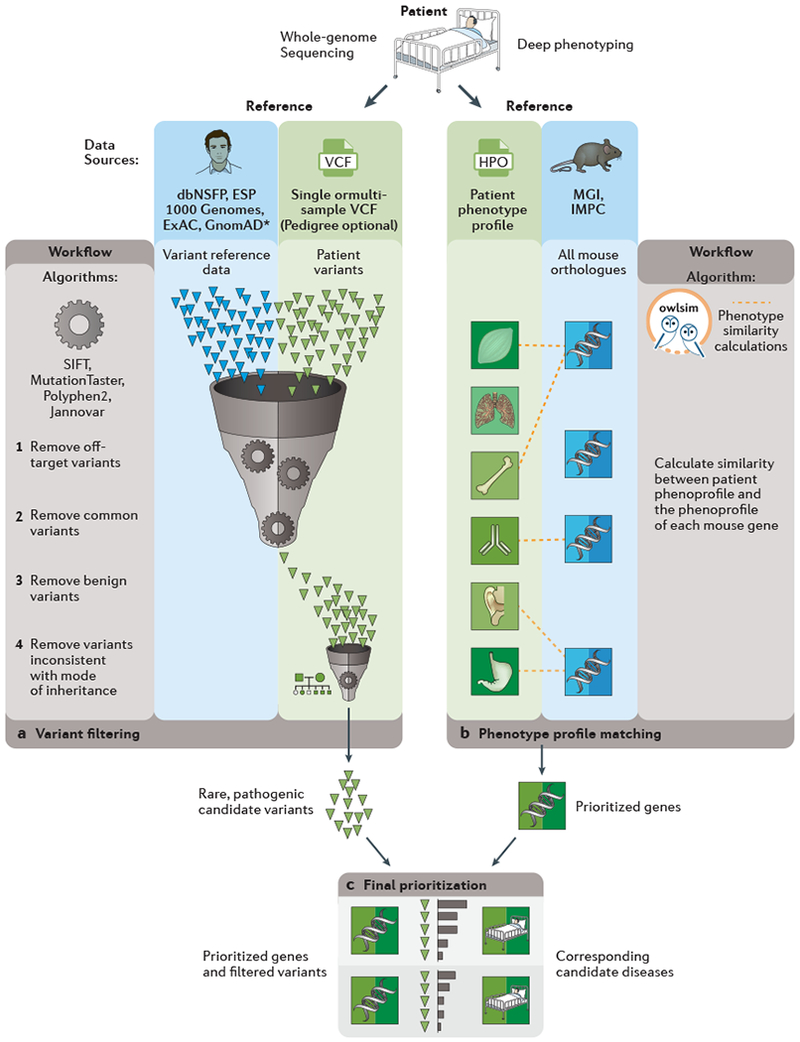
The figure exemplifies the data sources and algorithms available from the Monarch Initiative portal and variant prioritization software suite (Exomiser). Candidate, rare, pathogenic variants from patient genomes are identified by comparison against reference variant sources such as the Exome Aggregation Consortium (ExAC) to determine the population frequency of variants, and the use of algorithms such as Jannovar and Polyphen2 for predicting which variants are likely to have deleterious, potentially pathogenic, effects. Candidate genes are identified by semantic comparisons of the patient’s phenotypic profile against reference genotype-to-phenotype datasets for human disease as well as model organisms as produced by phenomics programmes, such as the International Mouse Phenotyping Consortium (IMPC). The final set of prioritized, rare pathogenic variants in genes with functional evidence from the phenotype comparisons are presented back to the clinician for a final diagnostic decision
dbNSFP, database of nonsynonymous SNPs and their functional predictions
ESP, Exome Sequencing Project;
GnomAD, Genome Aggregation Database;
1000g, 1000 Genomes Project;
HPO, Human Phenotype Ontology;
MGI, Mouse Genome Informatics;
SIFT, Sorts Intolerant From Tolerant database;
VCF, variant call format.
As well as variants in unknown disease genes, variants of uncertain significance (VUS) in known disease genes often overwhelm disease sequencing studies. Here, the tremendous advances in gene engineering through CRISPR–Cas9 5,6 are lowering the barrier to rapid, efficient and cost-effective validation of such variants. Moreover, despite the numerous successes of genomics in identifying causative disease variants, an understanding of the pathobiological mechanisms and development of therapeutic options for rare disease patients remains as slow as ever. The ready availability of well-characterized and reproducible mouse models of human disease from large-scale mouse phenomics programmes is key to addressing this challenge at the basic research and translation level. Personalized mouse lines containing the same functional variants as human patients will accelerate the investigation of mechanisms and pre-clinical screening of therapeutics. The single gene, mouse KO resources such as IMPC are obviously most applicable to the study of Mendelian disorders, especially where the mechanism is loss of function and pathogenic variations in the human gene have not been previously described. Overarching the single-gene mutant resources are mouse inbred and outbred resources and their power to identify the loci and networks involved in complex traits, which can be explored in more depth by the generation and phenotyping of mutations at individual loci. Importantly, however, it will be insufficient to simply generate a comparable allele in the mouse genome. High-throughput and broad-based phenotyping pipelines will need to be utilized to reveal comprehensive phenotype data, uncover pleiotropy across diverse disease systems, and enable clinical geneticists to use the available tools to assess phenotype matches and compare pathobiology.
Outlook
High-throughput mouse phenomics has been a powerful engine for biology and biomedical sciences, with a number of transformative impacts on genetics and genomics. Increasingly, however, mouse mutagenesis and phenotyping will be integrated with the demands and strategic directions of human biology and disease studies, including Mendelian disorders, precision medicine and complex disease. Already the power of linking rich, multi-dimensional mouse phenomics data with that of humans is persuasive. Moreover, the ability through CRISPR to alter the mouse genome at will highlights the opportunities to utilize the vast expertise of the mouse genetics community and its facilities and pipelines in the functional analysis of human variation, including point mutations both in coding and regulatory sequences. The functional analysis of non-coding sequences remains a formidable challenge, but one that will need tackling by both mouse and human geneticists in the drive to understand the totality of human variation in complex disease. The determination of the function of non-coding sequences, including regulatory and non-coding transcripts must be a critical ambition for mouse phenomics. The scale of the endeavour is enormous, but initially over the next few years we expect mouse genetics to turn its attention in this direction, undertaking comprehensive studies into the phenotypes of a substantial number of non-coding variants and transcripts.
Acknowledgements
We are grateful to Julie McMurry for help with Figure 6 which is a composition of several images previously licensed under CC0 in Pixabay and some CC-BY 4.0 in https://github.com/jmcmurry/open-illustrations. We also thank all of our colleagues in the International Mouse Phenotyping Consortium, who have contributed in no small measure to the thinking and future of mouse phenomics. We would also like to thank many colleagues who have participated with us in other consortia involving mouse phenomics, including EUMORPHIA, EUMODIC and EUCOMM. The views expressed in our article are built on the many discussions and insights that have emerged within these consortia over many years. Lastly, we are grateful to the Medical Research Council, UK (SDMB, CCH, A-MM, SW), the NIH (A-MM, TF, DS), and the Engineering and Physical Sciences Research Council, UK (CCH) for funding support.
Glossary terms
- Variable expressivity
Differing phenotypic features among individuals with the same genotype
- Phenotypic expansion
The expanding array of phenotypes that may be associated with mutations in a specific gene
- Genome-wide association studies
(GWAS). Genome-wide analyses of single nucleotide polymorphisms (SNPs) in human cohorts to test for association between SNPs and traits
- Phenome-wide association studies
(PheWAS). Testing genetic variants for an association with multiple phenotypes or traits (the phenome) in human cohorts
- Pre-pulse inhibition
(PPI). Used to assess sensorimotor gating. In the PPI test, sensorimotor gating is assessed by measuring the innate reduction of the startle reflex induced by a weak prestimulus (prepulse) prior to a subsequent strong startle stimulus (pulse). Deficits in PPI responses are noted in patients suffering from a range of illnesses including schizophrenia
- Optokinetic drum
Assesses the threshold of visual acuity by placing a mouse in the centre of a rotating drum and measuring reflexive head turning in response to the rotation of stripes which subsequently decrease in width and distance of separation
- Auditory brain stem response
(ABR). Measures the electrical response in the auditory nerve and brain stem to either a defined frequency, or a longer, complex auditory stimulus. This allows frequency-specific auditory thresholds to be determined
- Gene trapping
A random insertional mutation into an intron or exon of a gene that disrupts expression of the trapped gene
- Gene targeting
Targeting by homologous recombination into embryonic stem (ES) cells to introduce mutations ranging from single base-pair substitutions to large deletions
- Endophenotype
A heritable and measurable component of a phenotype, intermediate between gene and disease.
- Coisogenic
Isogenic strains differing only at a single locus are coisogenic strains. Thus, all International Mouse Phenotyping Consortium (IMPC) lines are coisogenic on the C57BL/6N background
- Optical projection tomography
(OPT). An optical computed tomography technique that is used to acquire 3D images of early embryo morphology in the mouse
- Micro-computed tomography
(μCT). High-resolution X-ray computed tomography to acquire 3D images of embryo morphology in the mouse, usually during later stages of development
- High-resolution episcopic microscopy
(HREM). A method for the determination of the 3D structure of embryos using recurrent block surface (episcopic) imaging of sections from histological samples
- Subviable lines
Mouse mutant lines for which some individual mice show embryonic lethality, whereas others of identical genotype survive
- Paralogue
Paralogues are pairs of genes that derive from a common ancestral gene, and may undertake similar functions
- Recombinant inbred
Recombinant inbred (RI) mouse lines are derived by the intercrossing and subsequent inbreeding of two distinct inbred lines. Each line carries a differing patchwork of chromosome segments from the two parental lines, allowing us to relate phenotypic differences between the parental inbred strains to the underlying genetic loci involved
- Collaborative Cross
Collaborative Cross (CC) lines are a multi-parental recombinant inbred panel derived from crosses between 8 inbred lines (including 3 wild-derived inbred strains), capturing a greater genetic diversity more evenly spread across the genome
- Quantitative trait locus
(QTL). A locus that contributes some proportion of the total phenotypic variance of the quantitative trait. Many quantitative traits are determined by multiple genes (or QTLs), each of which may have small or large effects on the phenotype
- Heterogeneous Stock
(HS). HS populations enable fine-resolution mapping of traits, and are created by the intercrossing of inbred or recombinant inbred lines followed by mating schemes that minimize inbreeding
- Diversity Outbred
(DO). The DO population is a Heterogeneous Stock that was derived by random mating of 144 partially inbred Collaborative Cross lines, providing single-gene mapping resolution
- Ontology
Phenotype ontologies encompass the naming, description and interrelationship of phenotypes
- Orphan drugs
Drugs that are developed to treat a rare medical condition, an orphan disease
Footnotes
Further information
International Mouse Phenotyping Consortium (IMPC): http://www.mousephenotype.org
International Mouse Phenotyping Resource of Standardised Screens (IMPReSS): http://www.mousephenotype.org
Online Mendelian Inheritance in Man (OMIM): http://www.omim.org
Orphanet: http://www.orpha.net
Hybrid Mouse Diversity Panel (HDMP): https://systems.genetics.ucla.edu/
Edinburgh Mouse Atlas Project (EMAP): http://www.emouseatlas.org/
Phenotype And Trait Ontology (PATO): https://bioportal.bioontology.org/ontologies/PATO
Genotype–Tissue Expression (GTEx) Project: https://www.gtexportal.org/home/
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING): https://string-db.org/
Monarch Initiative: https://monarchinitiative.org/
100K Genomes Project: https://www.genomicsengland.co.uk/
NIH Undiagnosed Diseases Network (UDN): https://undiagnosed.hms.harvard.edu/
Mouse Genome Informatics: http://www.informatics.jax.org
Competing interests statement
The authors declare no competing interests.
Subject categories
Biological sciences / Genetics / Genetic association study [URI /631/208/205]
Biological sciences / Genetics / Functional genomics [URI /631/208/191]
Biological sciences / Genetics / Genome / Genetic variation [URI /631/208/726/649]
Biological sciences / Biological techniques / Experimental organisms / Model vertebrates / Mouse [URI /631/1647/334/1874/345]
Biological sciences / Biological techniques / Genetic techniques / Gene targeting [URI /631/1647/1513/1967]
Biological sciences / Genetics / Mutation [URI /631/208/737]
Biological sciences / Genetics / Clinical genetics / Disease genetics [URI /631/208/2489/144]
Biological sciences / Biological techniques / Genetic engineering [URI /631/1647/1511]
Biological sciences / Biological techniques / High-throughput screening [URI /631/1647/2163]
References
- 1.Brown SD, Wurst W, Kuhn R & Hancock JM The functional annotation of mammalian genomes: the challenge of phenotyping. Annu Rev Genet 43, 305–333, doi: 10.1146/annurev-genet-102108-134143 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Doyle A, McGarry MP, Lee NA & Lee JJ The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res 21, 327–349, doi: 10.1007/s11248-011-9537-3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouabe H & Okkenhaug K Gene targeting in mice: a review. Methods Mol Biol 1064, 315–336, doi: 10.1007/978-1-62703-601-6_23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918, doi: 10.1016/j.cell.2013.04.025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez A, Josa S & Montoliu L A history of genome editing in mammals. Mamm Genome, doi: 10.1007/s00335-017-9699-2 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Birling MC, Herault Y & Pavlovic G Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm Genome, doi: 10.1007/s00335-017-9703-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrabe de Angelis MH et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet 25, 444–447, doi: 10.1038/78146 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Nolan PM et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet 25, 440–443, doi: 10.1038/78140 (2000). [DOI] [PubMed] [Google Scholar]; References 7 and 8 were instrumental in demonstrating the power of comprehensive phenotyping pipelines in large-scale mutagenesis screens.
- 9.Takeda J, Keng VW & Horie K Germline mutagenesis mediated by Sleeping Beauty transposon system in mice. Genome Biol 8 Suppl 1, S14, doi: 10.1186/gb-2007-8-s1-s14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JK et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154, 452–464, doi: 10.1016/j.cell.2013.06.022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Angelis MH et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat Genet 47, 969–978, doi: 10.1038/ng.3360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 10 and 11 describe the generation and phenotyping of hundreds of knockout mouse lines, revealing extensive pleiotropy and laying the groundwork for the IMPC.
- 12.Brown SD & Moore MW The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome 23, 632–640, doi: 10.1007/s00335-012-9427-x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visscher PM & Yang J A plethora of pleiotropy across complex traits. Nat Genet 48, 707–708, doi: 10.1038/ng.3604 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Dickinson ME et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514, doi: 10.1038/nature19356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uncovers mouse embryonic lethal (essential) genes and their relationship to human disease loci from large-scale phenotyping and analysis of hundreds of knockout mutations.
- 15.Chong JX et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet 97, 199–215, doi: 10.1016/j.ajhg.2015.06.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186, doi: 10.1016/j.cell.2017.05.038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bush WS, Oetjens MT & Crawford DC Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 17, 129–145, doi: 10.1038/nrg.2015.36 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Schlager G & Dickie MM Natural mutation rates in the house mouse. Estimates for five specific loci and dominant mutations. Mutat Res 11, 89–96 (1971). [DOI] [PubMed] [Google Scholar]
- 19.Davisson MT, Bergstrom DE, Reinholdt LG & Donahue LR Discovery Genetics - The History and Future of Spontaneous Mutation Research. Curr Protoc Mouse Biol 2, 103–118, doi: 10.1002/9780470942390.mo110200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers DC et al. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8, 711–713 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Rogers DC et al. SHIRPA, a protocol for behavioral assessment: validation for longitudinal study of neurological dysfunction in mice. Neurosci Lett 306, 89–92 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Russell LB, Russell WL, Popp RA, Vaughan C & Jacobson KB Radiation-induced mutations at mouse hemoglobin loci. Proc Natl Acad Sci U S A 73, 2843–2846 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell WL et al. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc Natl Acad Sci U S A 76, 5818–5819 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a key publication describing the use of ENU to efficiently induce point mutations in mice and facilitate broad-based forward genetic screens.
- 24.Nolan PM et al. Implementation of a large-scale ENU mutagenesis program: towards increasing the mouse mutant resource. Mamm Genome 11, 500–506 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Arnold CN et al. ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res Notes 5, 577, doi: 10.1186/1756-0500-5-577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliver PL & Davies KE New insights into behaviour using mouse ENU mutagenesis. Hum Mol Genet 21, R72–81, doi: 10.1093/hmg/dds318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boles MK et al. A mouse chromosome 4 balancer ENU-mutagenesis screen isolates eleven lethal lines. BMC Genet 10, 12, doi: 10.1186/1471-2156-10-12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X et al. ENU mutagenesis screen to establish motor phenotypes in wild-type mice and modifiers of a pre-existing motor phenotype in tau mutant mice. J Biomed Biotechnol 2011, 130947, doi: 10.1155/2011/130947 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tucci V et al. Reaching and grasping phenotypes in the mouse (Mus musculus): a characterization of inbred strains and mutant lines. Neuroscience 147, 573–582, doi: 10.1016/j.neuroscience.2007.04.034 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Zimprich A et al. Analysis of locomotor behavior in the German Mouse Clinic. J Neurosci Methods, doi: 10.1016/j.jneumeth.2017.05.005 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Wilson L et al. Random mutagenesis of proximal mouse chromosome 5 uncovers predominantly embryonic lethal mutations. Genome Res 15, 1095–1105, doi: 10.1101/gr.3826505 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flint J et al. A simple genetic basis for a complex psychological trait in laboratory mice. Science 269, 1432–1435 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Wada Y et al. ENU mutagenesis screening for dominant behavioral mutations based on normal control data obtained in home-cage activity, open-field, and passive avoidance tests. Exp Anim 59, 495–510 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Vitaterna MH et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264, 719–725 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandillo S et al. Reliability, robustness, and reproducibility in mouse behavioral phenotyping: a cross-laboratory study. Physiol Genomics 34, 243–255, doi: 10.1152/physiolgenomics.90207.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the importance of cross-centre standardization and validation of phenotyping platforms.
- 36.Isaacs AM et al. A mutation in Af4 is predicted to cause cerebellar ataxia and cataracts in the robotic mouse. J Neurosci 23, 1631–1637 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clapcote SJ et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402, doi: 10.1016/j.neuron.2007.04.015 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Potter PK et al. Novel gene function revealed by mouse mutagenesis screens for models of age-related disease. Nat Commun 7, 12444, doi: 10.1038/ncomms12444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Ageing and recurrent broad-based screening of mutants from a large-scale mutagenesis programmes reveals novel gene functions underlying age-related disease.
- 39.Hardisty-Hughes RE, Parker A & Brown SD A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat Protoc 5, 177–190, doi: 10.1038/nprot.2009.204 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Esapa CT et al. N-ethyl-N-Nitrosourea (ENU) induced mutations within the klotho gene lead to ectopic calcification and reduced lifespan in mouse models. PLoS One 10, e0122650, doi: 10.1371/journal.pone.0122650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpinelli MR et al. Suppressor screen in Mpl−/− mice: c-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc Natl Acad Sci U S A 101, 6553–6558, doi: 10.1073/pnas.0401496101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aigner B et al. Diabetes models by screen for hyperglycemia in phenotype-driven ENU mouse mutagenesis projects. Am J Physiol Endocrinol Metab 294, E232–240, doi: 10.1152/ajpendo.00592.2007 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Hough TA et al. Novel phenotypes identified by plasma biochemical screening in the mouse. Mamm Genome 13, 595–602, doi: 10.1007/s00335-002-2188-1 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Aigner B et al. Generation of N-ethyl-N-nitrosourea-induced mouse mutants with deviations in hematological parameters. Mamm Genome 22, 495–505, doi: 10.1007/s00335-011-9328-4 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Hoebe K & Beutler B Forward genetic analysis of TLR-signaling pathways: an evaluation. Adv Drug Deliv Rev 60, 824–829, doi: 10.1016/j.addr.2008.02.002 (2008). [DOI] [PubMed] [Google Scholar]; This paper reviews how forward genetic mouse screens revealed the pathways that activate the innate immune system.
- 46.Miosge LA, Blasioli J, Blery M & Goodnow CC Analysis of an ethylnitrosourea-generated mouse mutation defines a cell intrinsic role of nuclear factor kappaB2 in regulating circulating B cell numbers. J Exp Med 196, 1113–1119 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelms KA & Goodnow CC Genome-wide ENU mutagenesis to reveal immune regulators. Immunity 15, 409–418 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Adissu HA et al. Histopathology reveals correlative and unique phenotypes in a high-throughput mouse phenotyping screen. Dis Model Mech 7, 515–524, doi: 10.1242/dmm.015263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong MD, Dorr AE, Walls JR, Lerch JP & Henkelman RM A novel 3D mouse embryo atlas based on micro-CT. Development 139, 3248–3256, doi: 10.1242/dev.082016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong MD, Maezawa Y, Lerch JP & Henkelman RM Automated pipeline for anatomical phenotyping of mouse embryos using micro-CT. Development 141, 2533–2541, doi: 10.1242/dev.107722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong MD et al. 4D atlas of the mouse embryo for precise morphological staging. Development 142, 3583–3591, doi: 10.1242/dev.125872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weninger WJ et al. Phenotyping structural abnormalities in mouse embryos using high-resolution episcopic microscopy. Dis Model Mech 7, 1143–1152, doi: 10.1242/dmm.016337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gailus-Durner V et al. Introducing the German Mouse Clinic: open access platform for standardized phenotyping. Nat Methods 2, 403–404, doi: 10.1038/nmeth0605-403 (2005). [DOI] [PubMed] [Google Scholar]; This paper illuminates the important concept of the mouse clinic as a centre for mutant generation and broad-based phenotyping.
- 54.Brown SD, Chambon P, de Angelis MH & Eumorphia C EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet 37, 1155, doi: 10.1038/ng1105-1155 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Tucci V et al. Gene-environment interactions differentially affect mouse strain behavioral parameters. Mamm Genome 17, 1113–1120, doi: 10.1007/s00335-006-0075-x (2006). [DOI] [PubMed] [Google Scholar]
- 56.Bradley A et al. The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23, 580–586, doi: 10.1007/s00335-012-9422-2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skarnes WC et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342, doi: 10.1038/nature10163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 56 and 57 describe the IKMC mouse mutant resource, which has been key to the development of high-throughput mouse phenomics.
- 58.Blair DR et al. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell 155, 70–80, doi: 10.1016/j.cell.2013.08.030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; By data mining medical records of over 100 million patients, the authors revealed a correlation between rare genetic diseases and complex disease, demonstrating that highly penetrant phenotypic alleles can help understand the genetic aetiology of common disorders.
- 59.Sungur AO, Schwarting RK & Wohr M Early communication deficits in the Shank1 knockout mouse model for autism spectrum disorder: Developmental aspects and effects of social context. Autism Res 9, 696–709, doi: 10.1002/aur.1564 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Sungur AO, Schwarting RKW & Wohr M Behavioral phenotypes and neurobiological mechanisms in the Shank1 mouse model for autism spectrum disorder: A translational perspective. Behav Brain Res, doi: 10.1016/j.bbr.2017.09.038 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Schmeisser MJ et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260, doi: 10.1038/nature11015 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Katayama Y et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679, doi: 10.1038/nature19357 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Platt RJ et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep 19, 335–350, doi: 10.1016/j.celrep.2017.03.052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meehan TF et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat Genet 49, 1231–1238, doi: 10.1038/ng.3901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided extensive novel insights into gene function along with numerous new disease models from the work of the International Mouse Phenotyping Consortium.
- 65.Raj A, Rifkin SA, Andersen E & van Oudenaarden A Variability in gene expression underlies incomplete penetrance. Nature 463, 913–918, doi: 10.1038/nature08781 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karp NA et al. Prevalence of sexual dimorphism in mammalian phenotypic traits. Nat Commun 8, 15475, doi: 10.1038/ncomms15475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; In a large study involving over 50,000 mice, the authors demonstrate that differences exist between male and female mice in the majority of phenotypic screens employed by the IMPC.
- 67.Bowl MR et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat Commun 8, 886, doi: 10.1038/s41467-017-00595-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rozman J et al. Identification of genetic elements in metabolism by high-throughput mouse phenotyping. Nat Commun 9, 288, doi: 10.1038/s41467-017-01995-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peirce JL, Lu L, Gu J, Silver LM & Williams RW A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 5, 7, doi: 10.1186/1471-2156-5-7 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Churchill GA et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36, 1133–1137, doi: 10.1038/ng1104-1133 (2004). [DOI] [PubMed] [Google Scholar]; This paper describes plans to create hundreds of independently bred, recombinant inbred mouse lines from eight inbred parental strains to study polygenic networks and interactions among genes that complement knockout mouse studies.
- 71.Paigen K & Eppig JT A mouse phenome project. Mamm Genome 11, 715–717 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Bennett BJ et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res 20, 281–290, doi: 10.1101/gr.099234.109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patterson M et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet 49, 1346–1353, doi: 10.1038/ng.3929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shusterman A et al. Genotype is an important determinant factor of host susceptibility to periodontitis in the Collaborative Cross and inbred mouse populations. BMC Genet 14, 68, doi: 10.1186/1471-2156-14-68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mao JH et al. Identification of genetic factors that modify motor performance and body weight using Collaborative Cross mice. Sci Rep 5, 16247, doi: 10.1038/srep16247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Solberg LC et al. A protocol for high-throughput phenotyping, suitable for quantitative trait analysis in mice. Mamm Genome 17, 129–146, doi: 10.1007/s00335-005-0112-1 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Valdar W et al. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet 38, 879–887, doi: 10.1038/ng1840 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Svenson KL et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics 190, 437–447, doi: 10.1534/genetics.111.132597 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Churchill GA, Gatti DM, Munger SC & Svenson KL The Diversity Outbred mouse population. Mamm Genome 23, 713–718, doi: 10.1007/s00335-012-9414-2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; A variation of the Collaborative Cross that allows population genetic studies using a heterogeneous stock where each mouse is genetically unique, and the extent of genetic variability is similar to that observed in humans
- 80.Pallares LF et al. Mapping of Craniofacial Traits in Outbred Mice Identifies Major Developmental Genes Involved in Shape Determination. PLoS Genet 11, e1005607, doi: 10.1371/journal.pgen.1005607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nicod J et al. Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing. Nat Genet 48, 912–918, doi: 10.1038/ng.3595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports comprehensive phenotyping pipelines applied to the genetic analysis of an outbred mouse population revealing numerous complex traits mapped at gene-level resolution.
- 82.Doetschman T Influence of genetic background on genetically engineered mouse phenotypes. Methods Mol Biol 530, 423–433, doi: 10.1007/978-1-59745-471-1_23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Broman KW The genomes of recombinant inbred lines. Genetics 169, 1133–1146, doi: 10.1534/genetics.104.035212 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sittig LJ et al. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 91, 1253–1259, doi: 10.1016/j.neuron.2016.08.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moon C et al. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93, doi: 10.1038/nature14139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nguyen TL, Vieira-Silva S, Liston A & Raes J How informative is the mouse for human gut microbiota research? Dis Model Mech 8, 1–16, doi: 10.1242/dmm.017400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Overton JM Phenotyping small animals as models for the human metabolic syndrome: thermoneutrality matters. Int J Obes (Lond) 34 Suppl 2, S53–58, doi: 10.1038/ijo.2010.240 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Robinson L & Riedel G Comparison of automated home-cage monitoring systems: emphasis on feeding behaviour, activity and spatial learning following pharmacological interventions. J Neurosci Methods 234, 13–25, doi: 10.1016/j.jneumeth.2014.06.013 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Spruijt BM, Peters SM, de Heer RC, Pothuizen HH & van der Harst JE Reproducibility and relevance of future behavioral sciences should benefit from a cross fertilization of past recommendations and today’s technology: “Back to the future”. J Neurosci Methods 234, 2–12, doi: 10.1016/j.jneumeth.2014.03.001 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Bains RS et al. Assessing mouse behaviour throughout the light/dark cycle using automated in-cage analysis tools. J Neurosci Methods, doi: 10.1016/j.jneumeth.2017.04.014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hong W et al. Automated measurement of mouse social behaviors using depth sensing, video tracking, and machine learning. Proc Natl Acad Sci U S A 112, E5351–5360, doi: 10.1073/pnas.1515982112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zarringhalam K et al. An open system for automatic home-cage behavioral analysis and its application to male and female mouse models of Huntington’s disease. Behav Brain Res 229, 216–225, doi: 10.1016/j.bbr.2012.01.015 (2012). [DOI] [PubMed] [Google Scholar]
- 93.Steele AD, Jackson WS, King OD & Lindquist S The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington’s and prion diseases. Proc Natl Acad Sci U S A 104, 1983–1988, doi: 10.1073/pnas.0610779104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bains RS et al. Analysis of Individual Mouse Activity in Group Housed Animals of Different Inbred Strains using a Novel Automated Home Cage Analysis System. Front Behav Neurosci 10, 106, doi: 10.3389/fnbeh.2016.00106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Valletta JJ, Torney C, Kings M, Thornton A & Madden J Applications of machine learning in animal behaviour studies. Animal Behaviour 124, 203–220, doi: 10.1016/j.anbehav.2016.12.005 (2017). [DOI] [Google Scholar]
- 96.Atamni HJ, Mott R, Soller M & Iraqi FA High-fat-diet induced development of increased fasting glucose levels and impaired response to intraperitoneal glucose challenge in the collaborative cross mouse genetic reference population. BMC Genet 17, 10, doi: 10.1186/s12863-015-0321-x (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Myint A et al. Large-scale phenotyping of noise-induced hearing loss in 100 strains of mice. Hear Res 332, 113–120, doi: 10.1016/j.heares.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferris MT et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog 9, e1003196, doi: 10.1371/journal.ppat.1003196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horsch M et al. Cox4i2, Ifit2, and Prdm11 Mutant Mice: Effective Selection of Genes Predisposing to an Altered Airway Inflammatory Response from a Large Compendium of Mutant Mouse Lines. PLoS One 10, e0134503, doi: 10.1371/journal.pone.0134503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sundberg JP et al. The mouse as a model for understanding chronic diseases of aging: the histopathologic basis of aging in inbred mice. Pathobiol Aging Age Relat Dis 1, doi: 10.3402/pba.v1i0.7179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sundberg JP et al. Approaches to Investigating Complex Genetic Traits in a Large-Scale Inbred Mouse Aging Study. Vet Pathol 53, 456–467, doi: 10.1177/0300985815612556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karp NA et al. Applying the ARRIVE Guidelines to an In Vivo Database. PLoS Biol 13, e1002151, doi: 10.1371/journal.pbio.1002151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karp NA et al. Impact of temporal variation on design and analysis of mouse knockout phenotyping studies. PLoS One 9, e111239, doi: 10.1371/journal.pone.0111239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Valen DA et al. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput Biol 12, e1005177, doi: 10.1371/journal.pcbi.1005177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilkinson MD et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 3, 160018, doi: 10.1038/sdata.2016.18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roadmap Epigenomics C et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This resource describes the collection, archiving and analysis of epigenomic data generated as part of The NIH Roadmap Epigenomics Consortium.
- 107.Kawaji H, Kasukawa T, Forrest A, Carninci P & Hayashizaki Y The FANTOM5 collection, a data series underpinning mammalian transcriptome atlases in diverse cell types. Sci Data 4, 170113, doi: 10.1038/sdata.2017.113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews the wide impact the 5th iteration of the RIKEN-led FANTOM consortium has had on understanding cell function by the generation of a comprehensive cellular transcription atlas.
- 108.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779, doi: 10.1371/journal.pmed.1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; A large scale resource that integrates extensive phenotypic and genotypic data from >500,000 participants to support investigations into the genetic and non-genetic determinants of the diseases of middle and old age.
- 109.Smith CL & Eppig JT The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm Genome 23, 653–668, doi: 10.1007/s00335-012-9421-3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hayamizu TF et al. EMAP/EMAPA ontology of mouse developmental anatomy: 2013 update. J Biomed Semantics 4, 15, doi: 10.1186/2041-1480-4-15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gkoutos GV, Schofield PN & Hoehndorf R The anatomy of phenotype ontologies: principles, properties and applications. Brief Bioinform, doi: 10.1093/bib/bbx035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Consortium GT et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213, doi: 10.1038/nature24277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szklarczyk D et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368, doi: 10.1093/nar/gkw937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rath A et al. Representation of rare diseases in health information systems: the Orphanet approach to serve a wide range of end users. Hum Mutat 33, 803–808, doi: 10.1002/humu.22078 (2012). [DOI] [PubMed] [Google Scholar]
- 115.Zhu X, Stephens M A large-scale genome-wide enrichment analysis identifies new trait-associated genes, pathways and tissues across 31 human phenotypes. bioRxiv, doi: 10.1101/160770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rukat T, Holmes CC, Titsias MK, Yau C Bayesian Boolean Matrix Factorisation. Proceedings of Machine Learning Research 70, 2969–2978 (2017). [Google Scholar]
- 117.Wiltschko AB et al. Mapping Sub-Second Structure in Mouse Behavior. Neuron 88, 1121–1135, doi: 10.1016/j.neuron.2015.11.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cortes A et al. Bayesian analysis of genetic association across tree-structured routine healthcare data in the UK Biobank. Nat Genet 49, 1311–1318, doi: 10.1038/ng.3926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ng SB et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42, 30–35, doi: 10.1038/ng.499 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H & Inoue I Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet 57, 621–632, doi: 10.1038/jhg.2012.91 (2012). [DOI] [PubMed] [Google Scholar]
- 121.Ramoni RB et al. The Undiagnosed Diseases Network: Accelerating Discovery about Health and Disease. Am J Hum Genet 100, 185–192, doi: 10.1016/j.ajhg.2017.01.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Collins FS & Varmus H A new initiative on precision medicine. N Engl J Med 372, 793–795, doi: 10.1056/NEJMp1500523 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Announcement of how the NIH will refocus efforts in the precision medicine era, including the need for more reliable models for preclinical testing.
- 123.Yang Y et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369, 1502–1511, doi: 10.1056/NEJMoa1306555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang Y et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879, doi: 10.1001/jama.2014.14601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee H et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887, doi: 10.1001/jama.2014.14604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koscielny G et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res 42, D802–809, doi: 10.1093/nar/gkt977 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang J et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am J Hum Genet 100, 843–853, doi: 10.1016/j.ajhg.2017.04.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mungall CJ et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res 45, D712–D722, doi: 10.1093/nar/gkw1128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smedley D & Robinson PN Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med 7, 81, doi: 10.1186/s13073-015-0199-2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Robinson PN et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res 24, 340–348, doi: 10.1101/gr.160325.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; An important algorithmic approach that uses genotype-to-phenotype data from model organism studies to assess the potential impact of exome variants identified in rare-disease patient sequencing.
- 131.Smedley D et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet 99, 595–606, doi: 10.1016/j.ajhg.2016.07.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Singleton MV et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet 94, 599–610, doi: 10.1016/j.ajhg.2014.03.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gall T et al. Defining Disease, Diagnosis, and Translational Medicine within a Homeostatic Perturbation Paradigm: The National Institutes of Health Undiagnosed Diseases Program Experience. Front Med (Lausanne) 4, 62, doi: 10.3389/fmed.2017.00062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kohler S et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 45, D865–D876, doi: 10.1093/nar/gkw1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mungall CJ et al. Integrating phenotype ontologies across multiple species. Genome Biol 11, R2, doi: 10.1186/gb-2010-11-1-r2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]