
Fig. 6. Integration of human and mouse data for rare disease genetics.
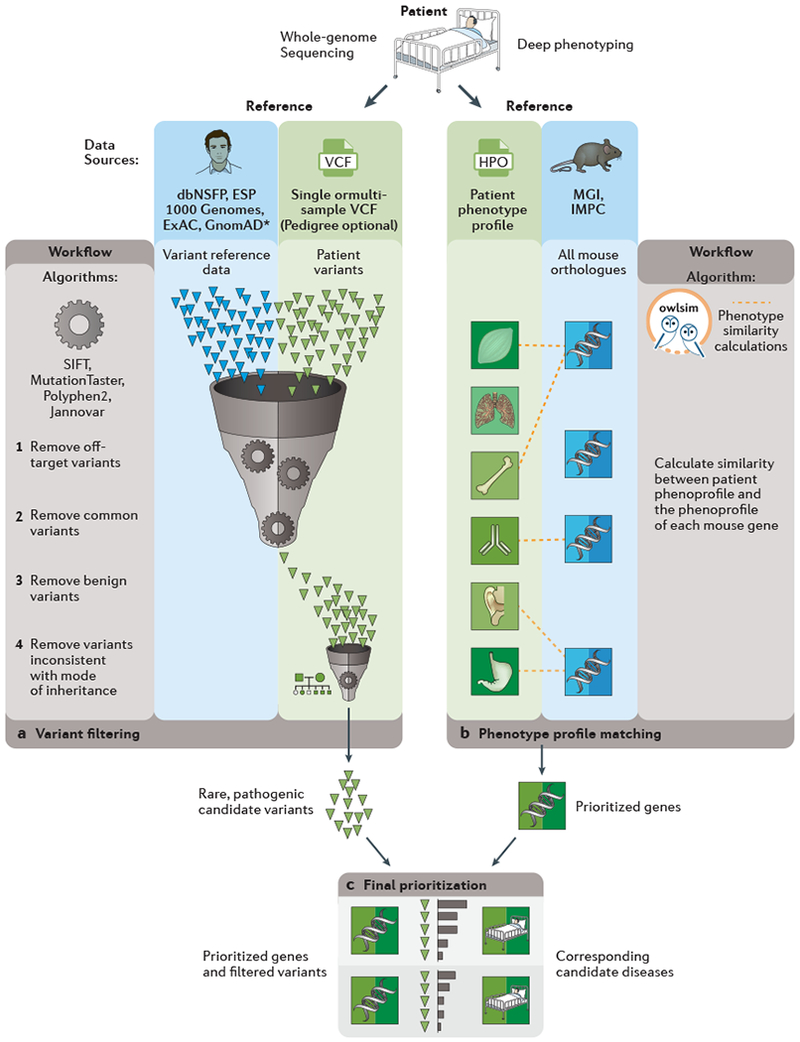
The figure exemplifies the data sources and algorithms available from the Monarch Initiative portal and variant prioritization software suite (Exomiser). Candidate, rare, pathogenic variants from patient genomes are identified by comparison against reference variant sources such as the Exome Aggregation Consortium (ExAC) to determine the population frequency of variants, and the use of algorithms such as Jannovar and Polyphen2 for predicting which variants are likely to have deleterious, potentially pathogenic, effects. Candidate genes are identified by semantic comparisons of the patient’s phenotypic profile against reference genotype-to-phenotype datasets for human disease as well as model organisms as produced by phenomics programmes, such as the International Mouse Phenotyping Consortium (IMPC). The final set of prioritized, rare pathogenic variants in genes with functional evidence from the phenotype comparisons are presented back to the clinician for a final diagnostic decision
dbNSFP, database of nonsynonymous SNPs and their functional predictions
ESP, Exome Sequencing Project;
GnomAD, Genome Aggregation Database;
1000g, 1000 Genomes Project;
HPO, Human Phenotype Ontology;
MGI, Mouse Genome Informatics;
SIFT, Sorts Intolerant From Tolerant database;
VCF, variant call format.