

Abstract
Of all divalent metals, mercury (HgII) has the highest affinity for metallothioneins. HgII is considered to be enclosed in the α and β domains as tetrahedral α‐type Hg4Cys11‐12 and β‐type Hg3Cys9 clusters similar to CdII and ZnII. However, neither the four‐fold coordination of Hg nor the existence of Hg–Hg atomic pairs have ever been demonstrated, and the HgII partitioning among the two protein domains is unknown. Using high energy‐resolution XANES spectroscopy, MP2 geometry optimization, and biochemical analysis, evidence for the coexistence of two‐coordinate Hg‐thiolate complex and four‐coordinate Hg‐thiolate cluster with a metacinnabar‐type (β‐HgS) structure in the α domain of separate metallothionein molecules from blue mussel under in vivo exposure is provided. The findings suggest that the CXXC claw setting of thiolate donors, which only exists in the α domain, acts as a nucleation center for the polynuclear complex and that the five CXC motifs from this domain serve as the cluster‐forming motifs. Oligomerization is driven by metallophilic Hg⋅⋅⋅Hg interactions. Our results provide clues as to why Hg has higher affinity for the α than the β domain. More generally, this work provides a foundation for understanding how metallothioneins mediate mercury detoxification in the cell under in vivo conditions.
Keywords: cluster compounds, homeostasis, mercury, metallothionein, oligomerization
Introduction
How is 5d10 HgII bonded to the canonical αβ‐domains of metallothioneins (MTs) in living cells? This question is important to address for the understanding of the intracellular fate of this potent toxin. If HgII is tetrahedrally coordinated to four cysteine residues (Hg(Cys)4), by analogy with 4d10 CdII and 3d10 ZnII (Figure 1),1 how can one explain that extended X‐ray absorption fine structure (EXAFS) spectroscopy only detects a linear two‐coordination (Hg(Cys)2) with a prototypical Hg‐S distance of 2.33 Å?2 Also, if the metal tetrahedra are incorporated as tetranuclear Hg4(Cys)11–12 (abbreviated as Hg4S11‐12) clusters in the α domain and trinuclear Hg3(Cys)9 (abbreviated as Hg3S9) cluster in the β domain for a total of seven Hg per molecule (Hg7‐MT), similar to ZnII in Zn7‐MT and CdII in Cd7‐MT,1a,1b, 3 why does not EXAFS detect a higher Hg shell beyond the thiolate ligands?2 Have answers to these questions failed because of structural disorder,4 mixture of the mononuclear and multinuclear bonding environments, or did HgII alter the tertiary structure of MTs to the point of suppressing the α and β domains to form a supercoiled peptide chain5 and Hg(Cys)2 complexes only?6 Also, do the molecular structures determined in vitro at saturation (i.e., Hg7‐MT) or in excess (e.g., Hg18‐MT) of the seven tetrahedral sites by X‐ray absorption and nuclear magnetic resonance (NMR) spectroscopy and by X‐ray crystallography represent how Hg is incorporated in vivo?1d, 2, 7
Figure 1.
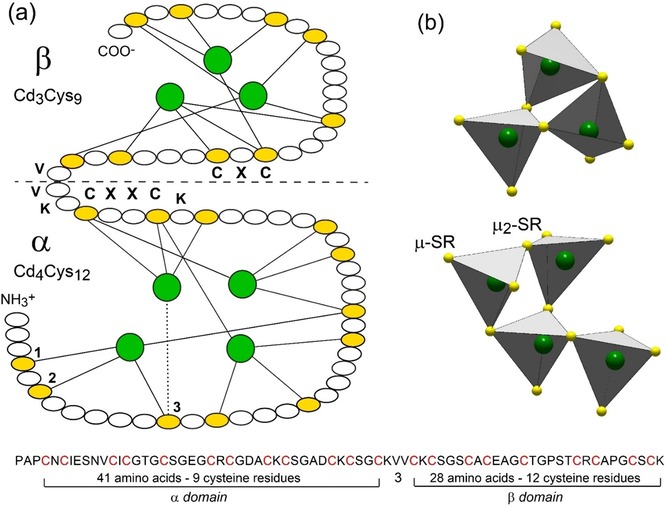
Primary structure of Cd7‐MT from mussel MT‐10 and Cd‐thiolate connectivity in the two clusters. (a) Amino acid sequence of the α‐ and β‐domains,14a showing the 21 Cys residues (in yellow) and Cd3Cys9 and Cd4Cys12 clusters3b (in green). Cysteine represents 21/72=29 % of the total amino acids. The Cys residues are arranged in nine CXC, one CXXC, and five CXXXC motifs, where X can be any amino acid. The solid lines denote the CdII‐Cys bonds, and the dotted line indicates an ambiguous assignment between the Cys residues 1, 2, and 3 (only the last possibility is represented for clarity). The horizontal dashed line denotes the boundary between the two domains connected by a flexible linker segment of three amino acids (KVV). (b) Polyhedral representation of the connectivity of the Cd(Cys)4 tetrahedra in each domain. The Cd3Cys9‐β cluster has three bridging (μ2‐SR) and six terminal (μ‐SR) cysteinyl sulfur atoms, and the Cd4Cys12‐α cluster has four μ2‐SR and eight μ‐SR.
Here, we show that direct insight into HgII binding to MTs in living cells is provided by the new application of high energy‐resolution X‐ray absorption near‐edge structure (HR‐XANES) spectroscopy at extreme dilution.8 Compared to EXAFS, XANES spectroscopy provides geometric information,4, 9 is less sensitive to structural disorder around the photoabsorbing atom,4, 10 and has superior elemental sensitivity.11 Therefore, high spectral resolution allows more precise identification of the plurality of the bonding environments in the α and β domains, either within the same MT molecule, if all molecules are structurally equivalent, or among the MT molecules, if various coordination environments coexist in a mixture.
HR‐XANES, applied earlier for the determination of the binding site of inorganic and organic mercury in human hair,12 is employed here to characterize the chemical form of mercury in whole blue mussel Mytilus edulis (ME) and its MT extract. This abundant and widely distributed filter‐feeder mollusk is often used as a sentinel organism for marine pollutants.13 Also, the strong inducibility of its MTs14 and proximity of the cysteine amino acid sequence to those in mammals3b, 7b make this particular mussel a model organism of choice for studying Hg toxicity. The level of MT in ME was increased previously by exposure to cadmium or mercury, and two isoforms of apparent molecular weight 12 kDa (MT‐10) and 20 kDa (MT‐20) were isolated by polyacrylamide gel electrophoresis.15 Here, MTs were induced at Hg(NO3)2 concentration (100 μg HgII L−1 or 0.1 ppm Hg) and exposure time (9 days) broadly similar to those in previous HgCl2 (0.01 to 0.04 ppm for up to 21 days16) and CdCl2 experiments (0.1 ppm for 11 days,14b 0.1 ppm for 3–4 months,17 0.2 ppm for 14 days,14d 0.2 ppm for 3–4 months,14a and 0.4 ppm for 22 days18) and extracted by thermal denaturation (see the Experimental Section).
Results
Molecular weight and metal content
The metallothionein extracts from the Hg exposed (MT) and unexposed (control) mussels have similar sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) patterns (Figure 2, Experimental Section). Four main bands are observed, one faint at 10 kDa from monomers (MT‐10), one smeared at 18 kDa from dimers (MT‐20) followed by a faint and narrower band at 21 kDa, another faint at 30 kDa from trimers (MT‐30), and an extremely intense last one at 37 kDa from tetramers (MT‐40). The MT‐20/30/40 oligomers were also detected in the initial mussel homogenate before the MT purification step by thermal denaturation of the cell proteins. The MT oligomer with a molecular weight of 21 kDa is unidentified.
Figure 2.
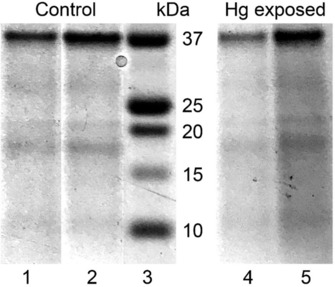
SDS‐PAGE electropherograms of the MT extracts after thermal denaturation under argon. Lane 1=5 μL and lane 2=10 μL from the mussel control, lane 3=mass markers in kDa, lane 4=5 μL and lane 5=10 μL from the Hg exposed mussels (MT).
Considering that MTs are metal‐inducible proteins, Hg‐exposed mussels should contain more MTs than the reference mussels, which we verified with a Bradford protein assay performed on the supernatant after thermal denaturation. The weight concentration of MTs in exposed mussels is 76 nmol MT g−1 wet weight (w.w.) mussel, compared to 57 nmol g−1 w.w. mussel under basal conditions. The two values are on the same order as those reported previously in the gills of M. edulis (35–56 nmol MT g−1 w.w19), and whole Pacific oyster Crassostrea gigas (38–91 nmol g−1 w.w20). The enhancement of MT induction in response to Hg exposure reached (76−57)/57=33 %. The Hg:Cu:Zn molar stoichiometry, as determined from elemental analysis of the MT extracts using inductively coupled plasma mass spectrometry (ICP‐MS), is 0:1:1 in the control and 2:2:5 in the Hg mussels. The constitutive levels of Cu and Zn in naturally occurring MTs are consistent with previous observations14d, 21 and with the role of MTs in the intracellular homeostasis of the two essential metals.22 The total number of metals bound per molecule of MT was calculated by dividing the sum of the metals concentration in the MT extracts by the weight concentration of MTs in the same extracts and taking a MT‐10 molecular weight of 7.24 kDa.18 The metal to MT molecular ratios thus obtained are 4 % for the control and 7 % for the mussels exposed to Hg. Unmetallated apo‐MT, or thionein,23 is therefore the major form in solution after thermodenaturation. This form has probably two origins, the thioneins existing in vivo in the mussels and the thioneins generated by the release of metals (Cu, Zn, Hg) during the heating step. Some of the in vivo thioneins were certainly denatured, but their yields have no bearing on XANES results. The proportion of the metallated MTs is even lower if the metals are not randomly distributed among all MT molecules but clustered in the α‐ and β‐domains of a few molecules. HR‐XANES results show that the Hg atoms are indeed partly clustered in vivo, despite their vanishingly low amount (3.4 10−6 g Hg mL−1 MT extract, or ppm), supporting the finding that thionein largely prevails.
Evidence for dithiolate and tetrathiolate Hg complexes
The HR‐XANES spectrum of the MT aqueous extract containing 3.4 10−6 g Hg mL−1 (ppm) is distinctly different from that of the tissues from whole Hg mussels containing 317 10‐6 g Hg g‐1 tissue or ppm (ME; Figure 3). Two observations stand out. First, the two spectra exhibit a near‐edge peak at about 12279.3 eV. This “indicator” region, denoted A in Figure 3, is characteristic of HgII linearly coordinated to two thiolate groups [Hg(Cys)2 complex].24 Its intensity is lower in MT, which indicates that this coordination is less abundant in this sample and coexists in a mixture with either three‐ or four‐coordinate Hg (Figure 4). Observation of two‐coordinate Hg was expected in ME because it is the most common geometry in mercury chemistry and it occurs in biological systems in complexes with cysteine, peptides, and proteins.7a, 12, 25 It was somewhat expected in the MT extract because the α domain has a conserved CXXC motif (single‐letter amino acid code, where X can be any amino acid) known to bind mercury linearly in metalloproteins (Figure 1).1d, 7a, 25a,25b Molecular mechanics (MM2) simulations for a mammalian MT model metallated with seven Hg atoms (Hg7‐MT consisting of Hg4S11‐α and Hg3S9‐β clusters) by isomorphous substitution of Cd and Zn in the X‐ray diffraction structure showed that the α cluster is too big to fit inside the protein and that some thiolate groups are exposed to the solvent (Figure S1, Supporting Information). These sulfur atoms could be engaged in trithiolate or tetrathiolate coordination inwardly and dithiolate linear coordination outwardly. The RS‐Hg‐SR coordination could behave as anchors to intermolecular bridging between MT‐10 monomers forming MT‐20/30/40.14a We show below from the quantitative analysis of the HR‐XANES spectrum for MT that the amount of two‐coordinate Hg is too high to arise significantly from intermolecular RS‐Hg‐SR crosslinks. In addition, compelling evidence for RS‐SR crosslinks (in place of RS‐Hg‐SR) between monomeric subunits exists.14a
Figure 3.
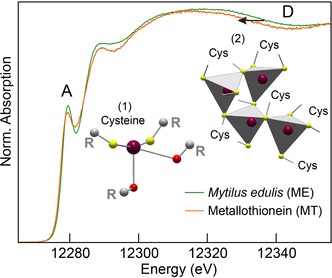
Mercury L3‐edge HR‐XANES spectra of metallothionein (MT) and Mytilus edulis tissues (ME). Two Hg species (represented in inset) are identified by HR‐XANES and structure modeling, (1) a two‐coordinate thiolate complex with secondary Hg⋅⋅⋅ O interactions from carbonyl oxygen donors, and (2) a tetrahedrally coordinated Hg‐thiolate cluster [Hgx(Cys)y].
Figure 4.
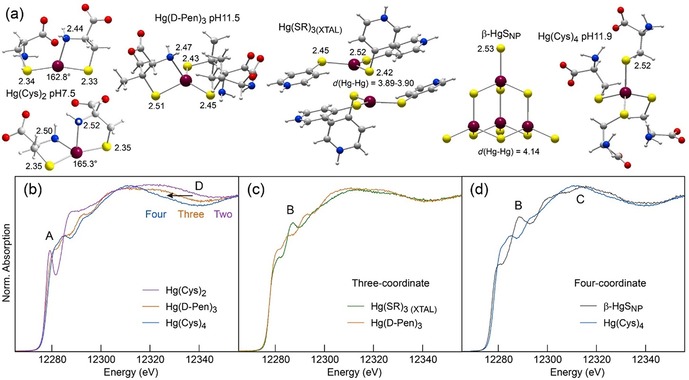
Sensitivity of L3‐edge HR‐XANES to the bonding environment of Hg. (a) Structure of the Hg complexes. (b–d) Spectra of two‐, three, and four‐coordinate Hg‐thiolate and Hg‐sulfide references. The Hg(Cys)2 complex was prepared at a Cys:Hg molar ratio of 2 and pH 7.5, the Hg(d‐Pen)3 complex at d‐Pen:Hg=10.0 and pH 11.5, and Hg(Cys)4 at Cys:Hg=10.0 and pH 11.9. Their structure was optimized geometrically (MP2‐RI/def2‐TZVP‐ecp67), and the Hg(SR)3 complex and β‐HgSNP model are X‐ray crystal structures. Peak B is diagnostic of Hg–Hg pairs. Bond lengths are in angstroms. Dark red, Hg; yellow, S; blue, N; red, O; gray, C; light gray, H.
Second, ME has a broader absorption edge maximum and the trailing spectral edge of the MT extract is shifted to lower energy (“indicator” region D in Figure 3). According to the Natoli rule,26 the energy shift of the post‐edge maximum results from an increase of the interatomic distance (i.e., coordination number) between the photoabsorbing ion (Hg) and the neighboring atoms (S). Similar observations regarding the sensitivity of XANES to the nearest ligand distance were reported for the coordination geometry of cadmium in MTs9 and for Hg coordinated to two [d(Hg−S)≈2.33 Å] and four [d(Hg−S)≈2.52 Å] sulfur atoms (Figures 4 and S2 in the Supporting Information).24a Thus, region D confirms that MT has less dithiolate Hg complex than ME, but still is not conclusive on the coordination of the second Hg species, which can be three‐ or four‐fold. With an average Hg−S distance of 2.44 Å in thiolate complexes,27 three‐coordinate Hg has indeed a trailing edge intermediate between those from the two and four coordination modes (Figure 4 b). This question can be resolved with a two‐component fit of ME and MT. The best fits were obtained with 91 % Hg(Cys)2 + 9 % Hg(Cys)4 for ME and 82 % Hg(Cys)2 + 18 % Hg(Cys)4 for MT (Figure 5 a). The model fits reproduce well the data in regions A, C, and D, but fail to reconstruct peak B. We conclude that Hg is both two‐ and four‐coordinate, and that MT has a higher proportion of the second species.
Figure 5.
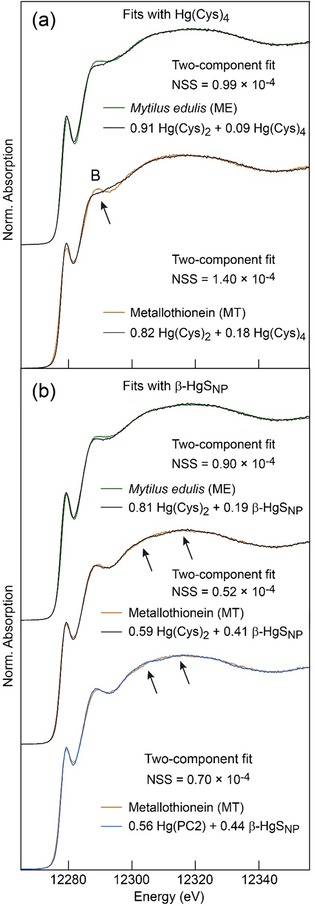
Least‐squares fits of the two mussel spectra (ME and MT). (a) Fits with the two‐ and four‐coordinate Hg‐thiolate references from Figure 4. MT, which has more tetrathiolate Hg coordination (18 mol %), also has a more intense peak B, which is not reproduced by the fit model. (b) Fits with the tetrathiolate Hg(Cys)4 reference replaced by β‐HgSNP in which Hg is also four‐fold coordinated but which contains in addition Hg–Hg pairs. Peak B is now well reproduced, but the crystalline reference introduces small modulations in the top edge region C (Figure 4) not observed on data. The little bumps are n‐order Hg↔Hg multiple scattering paths that occur in medium‐range ordered β‐HgSNP and are not present in trinuclear and tetranuclear HgxSy MT clusters. The bell‐shape of the MT spectrum puts a constrain on the size of the Hg clusters and is an indication that the incorporation of the HgII atoms did not alter the domain structure of metallothionein. Fits were optimized by minimizing the normalized sum‐square difference between data and fit, NSS=Σ(datai−fiti)2/Σ(datai)2. The precision on the fractional amount of each reference spectrum is estimated to be 6 % of total Hg.12.
Evidence for Hg–Hg pairs in the tetrathiolate complex
Peak B is associated with the four‐coordinate species because its intensity is higher in MT (Figure 5 a). It is however absent in Hg(Cys)4 (Figure 4 d). The best shape among the tetrahedral compounds from our extended spectral database (ref. 11 and references therein) that best matched peak B was obtained with nanoparticulate metacinnabar (β‐HgSNP;24b Figure 4 d). In β‐HgS, HgII is coordinated to four sulfide atoms at 2.53 Å and surrounded by higher shells of Hg atoms.28 The β‐HgSNP reference was obtained by aging a Hg‐(l‐Cys‐OEt)2 complex.24b The β‐HgSNP HR‐XANES spectrum is much different from the unaged Hg‐(l‐Cys‐OEt)2 spectrum and cannot be reconstructed with a mixture of Hg‐(l‐Cys‐OEt)2 and well crystallized β‐HgS or any combination of other references (Figure S3 a, Supporting Information). This material has broad β‐HgS X‐ray diffraction peaks24b and damped HR‐XANES structures (Figures S2 a and S3 b, Supporting Information) characteristic of highly defective material with a wide range of both angles and bond lengths, as commonly observed for metal thiolate clusters in proteins.7a, 9, 29 β‐HgSNP has been identified previously in natural organic matter and soils,24b in plants,11 and in pyrite (FeS2).30 The β‐HgSNP crystallites imaged by HRTEM are about 5 nm in diameter (Figure S3 b, Supporting Information).24b, 30a
β‐HgSNP has little bumps in region C (Figure 4 d). These features, which result from multiple scattering events of the photoelectron on higher Hg shells (CN=12), are more intense in well crystallized β‐HgS (Figure S3 b, Supporting Information), and should vanish when the β‐HgSNP nanocrystals are smaller. We verified this hypothesis with a trithiolate complex [Hg(SR)3(XTAL)], in which the Hg atoms have two nearest Hg neighbors at 3.89–3.90 Å (Figure 4 c). The top edge region C is now loosely structured. Peak B is still present, although it is absent when the trithiolate complex is mononuclear [Hg(d‐Pen)3]31 (Figure 4 c). We conclude from this that peak B provides a signature for Hg–Hg pairs in three‐ and four‐coordinate Hg.
Best‐fit results of the two HR‐XANES spectra were obtained with 81 % Hg(Cys)2 + 19 % β‐HgSNP for ME and 59 % Hg(Cys)2 + 41 % β‐HgSNP for MT (Figure 5 b). The accuracy of estimation of the fit components is 6 mol % of total Hg.12, 24b We show below that the inorganic mercury sulfide reference β‐HgSNP is a computationally tractable structural analogue to the mercury thiolate “mineral core” in MT, thus providing the first spectroscopic proof for the existence of Hg–Hg pairs. Metallothioneins are produced in all tissues in bivalves,16 which explains their detection (19±6 mol %) in whole mussel. Furthermore, the fit of the ME and MT spectra with only two and the same Hg references serves as internal consistency check for the reliability of our analysis and negates the possibility that a new Hg species formed during the purification procedure.
The diminution of the proportion of Hg(Cys)2 from 81 % in ME to 59 % in MT, and the corresponding augmentation of β‐HgSNP from 19% to 41 % may have two origins. One is the elimination during MT purification of thermolabile dithiolate forms from Hg‐metalloproteins, such as metallochaperones.7a, 25b SDS‐PAGE shows that heat treatment effectively removed undesired high molecular weight proteins. A second is the transformation of some native Hg(Cys)2 forms to Hgx(Cys)y clusters at the elevated purification temperature of 95 °C used here. This hypothesis was tested by thermal denaturation of an air‐equilibrated mussel homogenate. In contact with oxygen, non‐metallated cysteine residues from MT are prone to oxidation to cystine and are unlikely to form new Hgx(Cys)y clusters. In addition, metallochaperones, which in the cell would deliver metals to MTs, are unlikely to do it ex vivo in the homogenate, and even less so in the presence of oxygen. The HR‐XANES spectra from the argon and air isolates are essentially the same, differing only in the intensity of the near‐edge peak (Figure S4 a, Supporting Information). The best two‐component fit of the air isolate yielded 63% Hg(Cys)2 + 37% β‐HgSNP, values which coincide closely with those for the argon isolate (Figures 5 and S4 b, Supporting Information). The repeatability of the results under two distinct experimental conditions is a strong indication that the enrichment of β‐HgSNP in MT (41 %) relative to ME (19 %) results dominantly, if not only, from the denaturation at 95 °C of thermolabile dithiolate forms.
The dithiolate MT complex, represented here by Hg(Cys)2 at pH 7.5, features a digonal coordination to two nearest thiolate ligands at about 2.33 Å completed by one to two secondary amine ligands (Hg[(SR)2+N1‐2] coordination, Figures 4 and S2, Supporting Information).12 Intra‐ and intermolecular Hg−N bonding complementary to cysteine‐derived Hg−S bonds has been observed in keratin12 and for Zn in metallothioneins, typically with histidine side chains.24b, 32 The dithiolate species could be also an oxothiolate complex, modeled here with the phytochelatine reference Hg([γGlu−Cys]2Gly, abbreviated Hg(PC2) (Figure 5 b). The Hg(PC2) reference is a bis‐six‐membered ring chelate, through two cysteinyl S atoms and two carbonyl oxygen atoms (C=O) at 2.6–3.0 Å from the peptide backbone (Figure 6 a).
Figure 6.
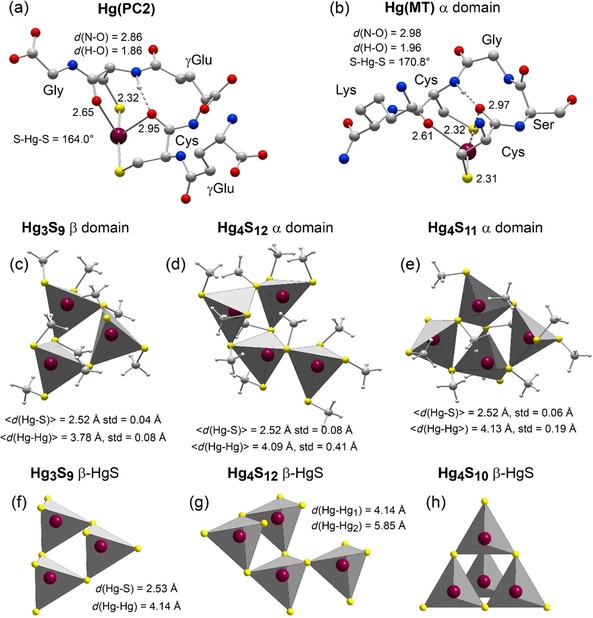
Geometry‐optimized structure models. (a, b) Hg oxothiolate complex in phytochelatine PC2 and the MT‐α domain. The amino acid sequence of the binding site is CysγGluCys (CγGC) for PC2 and CysSerGlyCys (CSGC) for MT‐α. The two dithiolate sites have approximately the same dimension and therefore bind Hg similarly. Hg is bonded to the two cysteinyl S and O atoms forming a double six‐membered bis‐oxothiolate ring chelate (Hg(SR+O)2 coordination). The bis‐chelate is further stabilized by an hydrogen bond between a carbonyl oxygen and an amide proton from the cysteine residues. The amine groups for Hg(MT‐α) were not protonated and an amide group has been added to the carboxyl terminus to make the complex neutral. Protons, other than the hydrogen‐bonded amide proton, are not represented for clarity. Cartesian coordinates, and atomic charges calculated by natural population analysis (NPA40) are given in the Supporting Information. (c–e) Connectivity of the HgS4 tetrahedra from the HgxSy clusters in the α and β domains of metallothionein optimized geometrically (MP2/TZVP‐ecp). Cys residues from the metallothionein clusters were modeled with methanethiolate (CH3S−) (Figure S7, Supporting Information). (f–h) Connectivity of the HgS4 tetrahedra in metacinnabar (β‐HgS).28 Dark red, Hg; yellow, S; blue, N; red, O; gray, C.
Discussion
Nature and function of the MT‐20/30/40 oligomers
In SDS‐PAGE, non‐covalent interactions among individual polypeptide chains are disrupted, whereas subunits held together covalently by disulfide bonds between cysteine residues from two different chains remain attached. Mussels MT‐10 have 21 Cys residues and the monomeric subunits of MT‐20, known to be inducible by Cd‐exposure, have 23 Cys residues.14a, 18, 33 In mussel Cd7‐MT‐10, twelve out of the 21 Cys residues form tetranuclear Cd4S12‐α clusters, and the other nine form trinuclear Cd3S9‐β clusters (Figure 1).3b In the related Cd‐experiment, the two additional cysteinyl sulfurs from MT‐20 were considered to bridge the subunits through RS−Cd−SR or disulfide RS−SR links, thus leaving the 21 other Cys residues for the formation of the polynuclear Cd4S12‐α and Cd3S9‐β clusters.14a Here, stabilization of the quaternary structure of MT‐20/30/40 by RS‐Hg‐SR crosslinks formed during the Hg exposure can be dismissed because the MT oligomers are observed also in the mussel control. Therefore, our results suggest the presence of intermolecular RS−SR links within the mussel cytosol. Care was taken (Ar saturation of all solutions) during the MT purification to avoid the oxidation of thiolates to cystine. If it occurred, oxidation of sulfur atoms exposed to the solvent in clefts on each domain should have been limited, and therefore cannot explain the vast amount of tetrameric MT‐40 in the two isolates (Figure 2). In addition, our results agree with the previous observation of Zn‐ and Cd‐rich MT‐20, and a Zn‐rich MT‐40 fraction, in the gel‐permeation chromatograms of the cytosol extracts from mussels exposed to Cd.14a
The MT‐20 and MT‐40 oligomers have been observed previously in the Mytilidae following exposure to HgII and CdII.14a,14b,14d, 17, 18, 34 Results on the inducibility of MT expression suggest that MT‐10 is specialized in the metabolism of essential ZnII and Cu(I/II) ions, whereas MT‐20 participates in the detoxification of heavy metal cations and hydroxyl radicals.35 This suggestion is supported, in particular, by the low concentration of MT‐20 in mussels exposed to Cu, and its considerable enhancement in mussels exposed to Cd, and to a lesser extent to Hg.14b, 35, 36 However, the finding that MT monomers would be involved in physiological functions and MT oligomers in biological effects of metal exposure is controversial,37 as shown for example by the high Zn content and low Cd content of MT‐40 in M. edulis exposed to Cd.14a This contradictory view is confirmed here by the detection of MT‐20/30/40 in control mussels.
Structure of the dithiolate complex
Divalent mercury is most commonly linearly coordinated to two proximal cysteines (C) separated by two amino acids (X) in living cells.38 The recurring CXXC motif is found in a wide variety of metalloproteins, including metallochaperones and metal‐transporting ATPases. The three‐dimensional structures of several Hg‐bound proteins have been solved by NMR and X‐ray crystallography, including MerP, MerA, and Atx1.25a,25b,25e In these proteins, the dicysteinate metal complex (HgII and CuI) is stabilized by secondary bonding interactions, typically with oxygen atoms from side chains (e.g., from Thr and Ser) and water molecules, and with an extended H‐bond network. In M. edulis, the CXXC motif is found at the end of the α domain, just before the short LysValVal linker connecting the two domains (Figure 1). Its sequence from the N‐terminus is LysCysSerGlyCysLys (KCSGCK motif).
The geometry of the HgII binding site was modeled computationally at the MP239 level of the molecular orbital theory (Figure 6 b). The two cysteine residues coordinate the metal ion with their thiolate ligand at 2.31–2.32 Å and their carbonyl oxygen atoms at 2.61 and 2.97 Å. The predicted Hg−O distances are between the sums of the Hg and O covalent radii (1.98 Å) and the Hg and O van der Waals radii (3.07 Å). The closest oxygen atom bends the S‐Hg‐S angle to 170.8°, a value significantly higher than the 163–165° angular values for the Hg(Cys)2 and Hg(PC2) references (Figures 4 and 6 a). The O ligands are almost perpendicular to the S‐Hg‐S bond direction, as reported previously for the NH2 and RSR sulfide/thioether secondary ligands.24a Bond analysis in terms of natural population analysis (NPA40) shows that a carbonyl oxygen is a little more nucleophilic (partial atomic charge −0.6 to −0.7 e) than an amide nitrogen from the ‐CO−NH‐ peptide bond (−0.6 e), and these two atoms are much less than an amine nitrogen (−0.9 e) (Figures S2 and S5, see Supporting Information).12 Therefore, an amine group is more likely to bond Hg than a peptide bond. However, other factors besides the electronic charge of the donor are involved in the stability of a macromolecular complex, such as its conformation and hydrogen bonds. Regarding the chelate structure, the Hg(SR+O)2 coordination forms a double six‐membered bis‐oxothiolate ring. The two rings are linked by the two intervening residues Ser and Gly and the cage stabilized by one ‐NH⋅⋅⋅O=C‐ hydrogen bond between the amide group of one cysteine and the carbonyl oxygen of the other cysteine (Figure 6 b). In the absence of Hg, the two SH groups protrude from the plane containing the CXXC motif by 1.8 Å like crab‐claws (Figure S6, see Supporting Information). This claw setting of thiolate donors has the potential to adapt both geometrically and electronically to the linear coordination requirement of a mercuric ion, which it can capture with extremely high affinity.
The Hg complex in phytochelatine PC2 is also a Hg[6‐S/O‐ring]2 chelate (Figure 6 a). In PC2, the binding motif is CXC and the intervening amino acid is γGlu. Its length is close to that of the SerCys dipeptide, therefore the loops of the PC2 and MT‐α complexes have nearly the same size. Phytochelatins mediate metal detoxification in plants.41 Their synthesis is also induced by Hg exposure,42 but they are enzymatically synthesized from glutathione (GSH) by PC synthase, not transcriptionally induced from DNA like MTs. Still, plants can also express MTs genetically, and plant MTs also possess CXXC motifs like animal MTs.43 Thus, the same Hg[6‐S/O‐ring]2 detoxification chelate appears to exist in the two kingdoms. However, plants have adopted it through two synthesis pathways, one catalyzed by enzymes (PC2) and the other from gene expression (MTs).
A metacinnabar‐type (β‐HgS) mineral core
The coordination geometry of the tetranuclear Cd4S12‐α and trinuclear Cd3S9‐β clusters is known for Cd7‐MT‐10 of Mediterranean mussel Mytilus galloprovincialis.3b They consist of CdS4 tetrahedra connected through their apices by bridging cysteinyl sulfurs for a total of 21 Cys residues (Figure 1 and Figures 6 c–e for the equivalent Hg species). In the Cd3S9‐β cluster, each tetrahedron comprises two bridging (μ2‐SR) and two terminal (μ‐SR) thiolates, as commonly observed in animals and plants MTs (Figure 6 c).29a, 43, 44 In contrast, in the Cd4S12‐α cluster, one of the four tetrahedra has only one bridging thiolate, thus being attached to a single tetrahedron (Figure 6 d). This configuration differs from the so‐called boat‐like conformation29a of the Cd4S11‐α cluster in vertebrates which, unlike mussels, have 20 MT Cys residues in total (S11‐α + S9‐β, Figure 6 e). In Cd4S11‐α, two tetrahedra are each linked to the three other tetrahedra from the four‐metal cluster through three bridging thiolates, and the two other tetrahedra are linked to only two tetrahedra through two bridging thiolates. With its singly‐bonded CdS4 tetrahedron, the Cd4S12‐α mussel cluster is therefore less compact than the Cd4S11‐α vertebrate cluster.
The Cd4S12‐α and Cd3S9‐β clusters have the same tetrahedral association as β‐HgS which features a sphalerite‐type (ZnS) lattice (Figures 6 f and g). Thus, poorly crystalline β‐HgSNP is a good mineral proxy for the Hg7‐MT mussel structure on the local scale. Similarities between metal thiolate clusters and inorganic structures and complexes are also observed for CuI, AgI, CdII, and ZnII.29a, 45 As an aside, we note that the connectivity in MeII 4S11‐α has no direct equivalent in ZnS/β‐HgS, in contrast to MeII 4S12‐α and MeII 3S9‐β. In a tetranuclear β‐HgS‐type cluster, each tetrahedron shares three bridging sulfurs yielding an adamantane‐type cage (Figure 6 h). In MeII 4S11‐α, only two tetrahedra have three bridging sulfurs and the two others have two bridging sulfurs. The four tetrahedra and five bridging sulfurs of MeII 4S11‐α form two fused six‐membered rings (2×[S‐Hg‐S‐Hg‐S‐Hg]) with a distorted boat conformation (Figure 6 e). This topology cannot be represented as a portion of the ZnS/β‐HgS lattice that has only chair conformation. Therefore, the recurrent description in the literature of MeII 4S11‐α in terms of an adamantane‐type structure is incorrect, as pointed out previously.29a
Structure of the mineral core
The geometry of the Hg4S12‐α and Hg3S9‐β mussel clusters and the Hg4S11‐α vertebrate cluster were optimized at the MP2 level using the connectivity of the Cd4S12‐α and Cd3S9‐β clusters in M. galloprovincialis 3b as structural templates (Figure 1). The model thiolate was methanethiolate CH3S−, as in our previous computational studies.11, 46 This substitution marginally changes the geometry of a complex or the effective atomic charge on the sulfur and mercury atoms (Figure S7, Supporting Information). The three clusters have a predicted Hg−S bond length of 2.52±0.04–0.08 Å, close to crystallographic values for inorganic Hg(SR)4 complexes (2.566±0.047 Å27) and well‐crystallized β‐HgS (2.53 Å28) (Figure 6 f). The predicted Hg−Hg distances across the shared corners are 3.78±0.08 Å for Hg3S9‐β and 4.09±0.41 Å for Hg4S12‐α, compared to 4.14 Å in β‐HgS. The Hg−Hg distances in Hg4S11‐α (4.13±0.19 Å) approach on average the Hg−Hg distance in β‐HgS, but are extremely unequal. Although the Hg4S11‐α and Hg4S12‐α clusters do not have the same polyhedral connectivity (Figures 6 d and e), and thus the same degree of angular flexibility, this computational study shows that a tetranuclear cluster is more disordered than a trinuclear cluster. The EXAFS photoelectron wave of the Hg–Hg pairs for the Hg4S11‐α cluster was calculated to determine if disorder (σ=0.19 Å) could have prevented their detection with this technique by Jiang et al.2 The large dispersion of the Hg−Hg distances leads to a wave beating at k=π/(2 ΔR)=π/(2×0.16 Å)=10 Å−1 and, as a result, to the extinction of the Hg‐Hg peak on the radial distribution function (Figure S8, Supporting Information). The disordered β‐HgSNP reference used in HR‐XANES analysis adequately accounts for the distribution of the Hg−Hg distances in MTs. However, being a solid phase β‐HgSNP features ideally twelve Hg–Hg pairs at 4.14 and six at 5.85 Å,28 whereas these numbers are extremely small in nanosized MT clusters, even zero for the second Hg atomic shell at about 5.8 Å in Hg3S9‐β. Approximating a MT cluster by the β‐HgSNP model manifests in the reconstruction of the MT spectra as a significant misfit near the edge maximum at 12 300–12 320 eV (arrows in Figure 5 b), which is the region where β‐HgSNP exhibits a modulation of the absorption signal from distant Hg–Hg pairs (Figure 4 d). These modulations are absent in the mussel spectra which in contrast have a bell‐shape top edge, thus negating the presence of Hg neighbors at a medium distance. Based on XANES calculation,47 the nuclearity of the MT Hg clusters is less than seven. A source of uncertainty is the effect of the peptide environment on the Hg coordination geometry because the enfolding of the two clusters by the polypeptide chain was not taken into account in our modeling as it would be too costly at the MP2 level of the molecular orbital theory.
Partitioning of the mineral core among the α‐ and β‐domains
Strong compelling evidence from X‐ray diffraction, NMR spectroscopy, UV/pH titration, and circular dichroism (CD) spectroscopy, indicate that CdII and ZnII are partitioned among the α and β domains of mammals MTs, with CdII preferentially bound in the MeII 4S11‐α cluster and ZnII in the MeII 3S9‐β cluster.1b, 3a, 7b, 21, 48 Copper, which forms CuI 6S11‐α and CuI 6S9‐β clusters in mammals,49 also has a marked preference for the β domain when it is mixed with CdII.50 With reference to their dynamic behavior, metal positions exchange much faster in the β than the α domain.21, 51 The metal‐binding selectivity and distinct chemical reactivity of the two domains leads to the hypothesis for a physiological specialization, with the β domain having a role in metal homeostasis and the α domain having a role in metal detoxification.21
In keeping with the view that metal ions are bound with different affinities in each domain according to their chemical properties and biological functions, we speculate that the polynuclear HgxSy species identified by HR‐XANES is Hg4(Cys)12‐α. This species would be located in the same domain, but clearly not the same MT molecules, as the Hg(Cys)2 species. Mercury atoms have a marked propensity to aggregate through relativistic effects.52 Mercuration, like auriphilic and argentophilic interactions, occurs in solution at room temperature because of the similarity in magnitude of metallophilic interactions with hydrogen bonding.53 A mercury atom bound to the two cysteines of the CXXC motif is therefore a nucleation center for the formation of HgxSy oligomers through mercuration, similar to the formation of β‐HgS from Hg(SR)2 complex in natural organic matter.46b Furthermore, the five CXC motifs from the α domain offer the correct spacing for the formation of the Hg4S12‐α cluster, similarly to Cd4S12‐α3b (Figure 1). The suggested nucleation mechanism has a parallel in the case of the CuI 4S6 adamantane‐type cluster formation in the human copper chaperone for the superoxide dismutase (CCS)54 and the yeast copper transporter Ctr1,55 which both involve CXC motifs. With its 3d10 configuration, CuI also forms a linear complex with thiolate ligands. One could assert that CuI should also be bound preferentially to the α domain rather than the β domain. This assertion is undermined by two observations. First, the stability constant of the linear Cu‐CXXC complex is two orders of magnitude lower than that of the Hg‐CXXC complex.56 Second, the consensus CXC motif has the highest metal selectivity for CuI because it is a nucleation center for highly stable Cu clusters.54, 55
Thermodynamic calculation adds further support to the α domain occupation for the HgxSy species identified by HR‐XANES. The Gibbs free energy (ΔG) of formation of multinuclear n[Hg(SR)2] complexes from the condensation of bis‐Hg‐thiolate complexes Hg(SR)2 increases with the nuclearity (n) of the cluster.24a Thus, a tetranuclear Hg‐α cluster is more stable than a trinuclear Hg‐β cluster. The energetic penalty associated with an alteration of nuclearity from four to three is probably more elevated for relativistic Hg atoms than non‐relativistic Cd and Zn atoms, as corroborated by their co‐occurrence in [Cd4]α[CdZn2]β‐MT.1d, 3a One could also advocate that the highly defective β‐HgSNP reference, which displays broad X‐ray diffraction peaks,24b better describes the Hg4S12‐α cluster because it has more incoherent Hg−Hg distances than the Hg3S9‐β cluster (Figure 6 c). However, this interpretation is not unique because it is not possible by HR‐XANES to differentiate a disorder effect from a particle size effect due to both effects producing a damping of the fine structure in the edge spectrum. In summary, we postulate that Hg is incorporated preferentially in the α domain, whereas the constitutive CuI and ZnII metal ions are in the β domain in agreement with previous studies on (Cd,Zn,Cu)‐MTs.48d, 50, 57 The β domain is unlikely to be heteronuclear because the CuI 6S9‐β and ZnII 3S9‐β clusters do not have the same structure, CuI being trihedrally coordinated49 and ZnII tetrahedrally3a, 48b coordinated. According to this scheme, CuI and ZnII are bound to different MT molecules. The Cu‐MT and Zn‐MT molecules may hold a Hg4S12 cluster in their α domain, albeit with limited likelihood, given the low proportion of metallated MT (7 %) relative to thionein (93 %).
Conclusion
Although these findings illuminate significant aspects of HgII detoxification in cells, they also give rise to challenging questions and suggest future research directions. Additional studies will be required to investigate the stability of the MeII 4S12‐α cluster in bivalves (21 Cys) relative to the far more abundant MeII 4S11‐α cluster (20 Cys) present in some invertebrates and all vertebrates, and to know if these differences endow functional differences among the two clusters with regard to metal detoxification.58 Does the MeII 4S12‐α core derive from the MeII 3S9‐β core by attachment of a trigonal MeS3 complex to one terminal cysteinyl sulfur, as suggested by the conformational similarity of the two cores (Figures 6 c and d)? Similarly, does the MeII 4S11‐α core derive from the MeII 3S9‐β core by attachment of a digonal MeS2 complex to two terminal cysteinyl sulfur (Figures 6 c and e)? Bridging thiolates are an important factor for the high thermodynamic stability of metal thiolate clusters, and a correlation has been suggested between the ratio of bridging (μ2‐SR) to terminal (μ‐SR) thiolates and the stability of a cluster.59 With a μ2:μ ratio of 5:6, the MeII 4S11‐α cluster would be more stable than the MeII 4S12‐α cluster, which has a ratio of 4:8. Under these premises, did evolution select the more closely packed and thermodynamically more stable MeII 4S11‐α structure relative to MeII 4S12‐α? Structure likely controls stability over the number of cysteine residues. If the MeII 4S11‐α cluster structure confers superior resistance to metal toxicity, does it mean that bivalves are more sensitive to HgII intoxication than other animals?
How does MT‐40 form? Although circumstantial evidence exists for its presence in vivo, an aggregation of the MT‐10 units during purification cannot be totally dismissed. What is the amino acid sequence of the MT‐10 units (primary structure) and how are they covalently bonded in MT‐40 (quaternary structure)? Do the monomeric units from MT‐40 contain 21 cysteines, as in canonical MT‐10,3b or 23 cysteines, as in the monomeric MT‐10 units of MT‐20?14a Lastly, are the biogenic MT clusters more stable than the metallothionein‐like β‐HgS nanoparticles formed naturally in natural organic matter24b, 46b, 60 and plant leaves?11 All these questions could be asked similarly to 4d10 AgI and 5d10 AuI, which, like 5d10 HgII, form distorted thiolate clusters in metalloproteins and linear coordination with thiolate ligands when they are embedded in sterically complex and demanding macromolecules. All these important biometals and intriguing questions are now leading to investigations in living matter by HR‐XANES down to, and below, the ppm level.8
Experimental Section
Materials: Wild blue mussels (Mytilus edulis) were collected from the Charente–Maritime (France) coastline in March 2016. They were cleaned from fouling and acclimatized to laboratory conditions for two weeks in a 150 L aquarium filled with filtered seawater. Conditions were adjusted to be as close as possible to those in the marine environment: salinity=35 p.s.u., temperature=15±0.3 °C, pH 8.1±0.1, light/dark=12 h/12 h. During the acclimatization and experimental periods, mussels were fed every two or three days with a mix of six marine microalgae (Shellfish Diet 1800®). Considering that body size (age) affects metal bioaccumulation in marine organisms, only individuals with homogeneous size were used in the experiment. After the acclimatization period, 60 mussels were equally distributed in two 50 L aquaria, one as control and one for exposure to 100 μg HgII L−1 for 9 days. A stock solution of mercury nitrate at 1 g Hg L−1 was used as the source of mercury (ASTASOL®). Addition of Hg(NO3)2 did not change pH value nor salinity. Concentrations of dissolved Hg in the two aquaria were monitored daily by Hg analyses with an Altec AMA 254 spectrophotometer. Thus, the Hg concentration in the experiment tank was readjusted as needed to 100 μg HgII L−1 to compensate losses resulting from bioaccumulation by the mussels and adsorption on the aquaria walls. At the end of the exposure period, mussels were rapidly dissected, frozen with liquid nitrogen and maintained at −20 °C until further analyses. No mortality was observed during our experiment.
Metallothionein purification: Mussels were defrosted and dried on filter paper. MTs being contained in gills, digestive gland, kidney and mantle, the whole mussel tissues were crudely homogenized in a mixer at 4 °C and pH 8.1 in 100 mmol L−1 Tris buffer, then finely mixed in a glass‐Teflon® homogenizer. The homogenate was centrifuged at 20 000 g for 10 min. The supernatant was deaerated with an argon flow, not under gas bubbling to avoid protein denaturation. The Ar saturated solution was gently stirred with a magnetic bar and heated at 95 °C for 15 min under a continuous flow of Ar after previously described procedure.15, 61 Protein denaturation at 95 °C for 15 min was considered a suitable trade‐off between the extent of MT purification,61 and the amount of remaining MT material needed for spectroscopy (3.4 ppm Hg in 300 μL extract). Thermolabile proteins and lipids precipitated whereas heat stable MTs remained soluble. A control experiment was performed in which the MT was purified in aerated conditions on an air‐equilibrated homogenate. After thermal denaturation, the glass vessel was rapidly cooled in ice and the soluble fraction was separated by centrifugation at 30 000 g for 20 min. 5–10 mL of the MT solution was dialyzed against 5 L of pure water in Spectra/Por® dialysis tubing of 3.5 kDa molecular weight cut‐off. Dialysis was repeated four times every three hours to eliminate salts (mussels were in seawater and Tris buffer was used for the homogenization step), and metals (Cu, Zn, and Hg) which were either free or complexed to small organic molecules, such as cysteine and glutathione, and which may not have precipitated during the heating step. After the dialysis step, all the Hg atoms were bound to metallothioneins. Protein concentration was determined by the Bradford's method using bovine serum albumin as reference. The Bradford reagent reacts mainly with NH3 + groups, and their content is different in BSA and MT. No correction was applied, however, because the reactivity of the NH3 + groups in the two proteins is unknown and is another source of uncertainty in quantification.62 The quality control of the protein extract was assessed by dynamic light scattering (DLS). Less than 6 % of the protein was in aggregate form. Purity and oligomerization state of the MTs were examined by SDS‐PAGE electrophoresis.15 To this end, the MT solution was concentrated about ten times using Spin‐X UF ultracentrifugation concentrators of 5 kDa molecular weight cut‐off (Corning). One aliquot of 5 μL and another of 10 μL were mixed with denaturing buffer (BIO‐RAD) and heated at 90 °C for 5 minutes. Electrophoresis migration was performed at 180 V for 30 min on Criterion XT (4–12 %) precast gel (BIO‐RAD) with 50 mm MES running buffer (pH 6.5). The protein bands were visualized with Coomassie Brilliant Blue.
Metal analysis: Hg, Cu, and Zn concentrations in the control and MT extract were measured by quadrupole ICP‐MS (ThermoFisher iCAPQ) at masses 200 and 202, 63, and 64, respectively, after calibration with SPEXCertiPrep standards. Analyses were carried out at low concentration to avoid a memory effect and in triplicate with appropriate blank subtraction.
Complex Hg(SR)3(XTAL): The formulation of the new crystal is [Hg2(C5H5NS)6](SO4)2⋅4 H2O and its structure is represented in Figure S9 (see Supporting Information). Mercury nitrate (Hg(NO3)2⋅H2O, 238 mg, 0.695 mmol) was dissolved in methanol (6 mL) and added dropwise to 4‐mercaptopyridine (CAS 4556‐23‐4, 95 % purity, 300 mg, 2.56 mmol) in a solution of methanol (15 mL), distilled water (15 mL), and ammonium tetrafluoroborate (CAS 13826‐83‐0, 99 % purity, 210 mg, 1.98 mmol). Then 0.1 m sulfuric acid was added dropwise to reach a pH of 1.0. After 24 h of slow evaporation of the light yellow homogenous solution at room temperature in the dark, a fine yellowish precipitate formed with yellow and colorless crystals. The crystals were isolated by filtration and washed with distilled water. The crystalline quality of the yellow crystals was too poor for structural determination. A colorless single crystal (0.22×0.11×0.06 mm) was mounted on a Bruker Kappa CCD diffractometer using monochromatic AgKα radiation (λ=0.56087 Å). Crystal data at 293 K: C30H38Hg2N6O12S8, M w=1332.32, space group P‐1 (no. 2), a=10.418(2) Å, b=14.320(1) Å, c=15.556(1) Å, α=77.41(1)°, β=74.13(1)°, γ=79.04(1)°, V=2157.4(4) Å3, Z=2, D x=2.05 g cm−3, μ=41 cm−1, 48326 measured reflections, 9784 unique reflections (R int =0.10), R1=0.06 (with 6115 I>2σ(I) and wR2=0.14, 548 parameters refined, GOF=1.07, max. min−1 residual peaks 1.58/−1.77 e Å3. Data were corrected for Lorentz and polarization effects and empirical absorption (SADABS from Bruker/Siemens). The crystal structure was solved by direct methods with the SIR9263 program and refined by full matrix least‐squares, based on F 2, using the SHELXL64 software through the WinGX65 program suite. The refinement was performed with anisotropic thermal parameters for all non‐hydrogen atoms. Only (N−H) hydrogen atoms of mercaptopyridium were localized on difference Fourier map and refined with isotropic thermal parameters. The other hydrogen atoms were generated at idealized positions, riding on the carbon carrier atoms, with isotropic thermal parameters. Hydrogen atoms of water molecules were not localized. CCDC 1838782 contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
HR‐XANES spectroscopy: All Hg L3‐edge HR‐XANES spectra were measured in high energy‐resolution fluorescence yield detection mode with high‐luminosity analyzer crystals8 on beamline ID26 at the European Synchrotron Radiation Facility (ESRF). Three hundred microliters of the concentrated MT extract (MT sample, [Hg]=3.4 ppm) were placed in a polyether ether ketone (PEEK) holder designed for solutions, and the holder was frozen immediately in liquid nitrogen (LN2) and stored in a LN2 Dewar until its transfer into the liquid helium cryostat of the ID26. Tissues of whole mussel (ME sample) were freeze‐dried to concentrate the sample, placed in a PEEK holder designed for pellets, sealed with poly (4,4′‐oxydiphenylene‐pyromellitimide) (Kapton®) tape, and stored in a desiccator until transfer into the liquid helium cryostat of the of beamline. We checked that the process of freeze‐drying a frozen tissue and preparing a pellet did not alter the HgII speciation itself by comparing the HR‐XANES spectrum of an as‐prepared fish sample to the spectrum of a snap‐frozen hydrated sample from the same fish. The two data are statistically identical (Figure S10, Supporting Information), in agreement with previous report.66 Spectra were collected at a temperature of 10–15 K and a scan time of 15 s to reduce exposure, and repeated at different pristine positions on the sample to increase the signal‐to‐noise ratio. Scans were monitored carefully for any evidence of radiation damage. The incident energy was scanned from 12260 to 12360 eV in 0.2 eV steps and the spectra were normalized to unity at E=12360 eV. More information on the beamline optics and data acquisition is given in the Supporting Information.
Geometry optimization: Conformations were energy‐optimized at the MP2 level with the resolution of identity (RI) approximation using the def2‐TZVP basis set and the def2‐TZVP/C auxiliary basis set for S, C, and H, and the def2‐TZVP‐ecp basis set for Hg. ORCA 3.0.367 was used for all calculations. Geometries were optimized in the aqueous phase with the implicit solvation method COSMO.68 Thermal energy corrections to the free energy were also calculated at the RI‐MP2 level for the Hg3(SMe)9 and Hg4(SMe)11 clusters (Figure 6) to verify that the conformations corresponded to minima (i.e., no negative/imaginary frequencies). The Hg4(SMe)12 frequency calculation was too costly in terms of computation time to be performed. This computational Scheme has been tested previously on the modeling of the structure and stability of mononuclear and multinuclear Hg‐thiolate complexes.24a, 46
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support was provided to A.M., S.P., J.P.B., I.G.L., C.L., M.G.R., and A.H. by the French National Research Agency (ANR) under grant ANR‐12‐BS06‐0008‐01, to A.M., M.R., and P.G. by the ANR under Grant ANR‐10‐EQPX‐27‐01 (EcoX Equipex), and to P.B. by the Institut Universitaire de France (IUF). The Froggy platform of the CIMENT infrastructure (ANR Grant ANR‐10‐EQPX‐ 29‐01) provided computing resources. Pierre Girard provided his expertise in parallel scientific processing, Emmanuel Dubillot collected mussels and maintained the aquaria, and Alice Simionovici created the Frontispiece
A. Manceau, P. Bustamante, A. Haouz, J. P. Bourdineaud, M. Gonzalez-Rey, C. Lemouchi, I. Gautier-Luneau, V. Geertsen, E. Barruet, M. Rovezzi, P. Glatzel, S. Pin, Chem. Eur. J. 2019, 25, 997.
References
- 1.
- 1a. Arseniev A., Schultze P., Worgotter E., Braun W., Wagner G., Vasak M., Kagi J. H. R., Wuthrich K., J. Mol. Biol. 1988, 201, 637–657; [DOI] [PubMed] [Google Scholar]
- 1b. Schultze P., Worgotter E., Braun W., Wagner G., Vasak M., Kagi J. H. R., Wuthrich K., J. Mol. Biol. 1988, 203, 251–268; [DOI] [PubMed] [Google Scholar]
- 1c. An Y., Li G. P., Ru B. G., Acta Crystallogr. Sect. D 1999, 55, 1242–1243; [DOI] [PubMed] [Google Scholar]
- 1d. Romero-Isart N., Vasak M., J. Inorg. Biochem. 2002, 88, 388–396. [DOI] [PubMed] [Google Scholar]
- 2. Jiang D. T., Heald S. M., Sham T. K., Stillman M. J., J. Am. Chem. Soc. 1994, 116, 11004–11013. [Google Scholar]
- 3.
- 3a. Robbins A. H., McRee D. E., Williamson M. E., Collett S. A., Xuong N. H., Furey W. F., Wang B. C., Stout C. D., J. Mol. Biol. 1991, 221, 1269–1293; [PubMed] [Google Scholar]
- 3b. Digilio G., Bracco C., Vergani L., Botta M., Osella D., Viarengo A., J. Biol. Inorg. Chem. 2009, 14, 167–178. [DOI] [PubMed] [Google Scholar]
- 4. Penner-Hahn J. E., Coord. Chem. Rev. 2005, 249, 161–177. [Google Scholar]
- 5.
- 5a. Cai W. H., Stillman M. J., J. Am. Chem. Soc. 1988, 110, 7872–7873; [Google Scholar]
- 5b. Lu W. H., Kasrai M., Bancroft G. M., Stillman M. J., Inorg. Chem. 1990, 29, 2561–2563. [Google Scholar]
- 6. Stillman M. J., Thomas D., Trevithick C., Guo X. J., Siu M., J. Inorg. Biochem. 2000, 79, 11–19. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Wernimont A. K., Huffman D. L., Lamb A. L., O'Halloran T. V., Rosenzweig A. C., Nat. Struct. Mol. Biol. 2000, 7, 766–771; [DOI] [PubMed] [Google Scholar]
- 7b. Meloni G., Zovo K., Kazantseva J., Palumaa P., Vasak M., J. Biol. Chem. 2006, 281, 14588–14595. [DOI] [PubMed] [Google Scholar]
- 8. Rovezzi M., Lapras C., Manceau A., Glatzel P., Verbeni R., Rev. Sci. Instrum. 2017, 88, 013108. [DOI] [PubMed] [Google Scholar]
- 9. Chan J., Merrifield M. E., Soldatov A. V., Stillman M. J., Inorg. Chem. 2005, 44, 4923–4933. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Marcus M. A., Chen H. S., Espinosa G. P., Tsai C. L., Solid State Commun. 1986, 58, 227–230; [Google Scholar]
- 10b. Crozier E. D., Nucl. Instrum. Methods Phys. Res. Sect. B 1997, 133, 134–144. [Google Scholar]
- 11. Manceau A., Wang J., Rovezzi M., Glatzel P., Feng X., Environ. Sci. Technol. 2018, 52, 3935–3948. [DOI] [PubMed] [Google Scholar]
- 12. Manceau A., Enescu M., Simionovici A., Lanson M., Gonzalez-Rey M., Rovezzi M., Tucoulou R., Glatzel P., Nagy K. L., Bourdineaud J.-P., Environ. Sci. Technol. 2016, 50, 10721–10729. [DOI] [PubMed] [Google Scholar]
- 13. Goldberg E. D., Environ. Monit. Assess. 1986, 7, 91–103. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Mackay E. A., Overnell J., Dunbar B., Davidson I., Hunziker P. E., Kagi J. H. R., Fothergill J. E., Eur. J. Biochem. 1993, 218, 183–194; [DOI] [PubMed] [Google Scholar]
- 14b. Lemoine S., Bigot Y., Sellos D., Cosson R. P., Laulier M., Mar. Biotechnol. 2000, 2, 195–203; [DOI] [PubMed] [Google Scholar]
- 14c. Isani G., Andreani G., Kindt M., Carpene E., Cell. Mol. Biol. 2000, 46, 311–330; [PubMed] [Google Scholar]
- 14d. Ivankovic D., Pavicic J., Kozar S., Raspor B., Helgol. Mar. Res. 2002, 56, 95–101; [Google Scholar]
- 14e. Amiard J. C., Amiard-Triquet C., Barka S., Pellerin J., Rainbow P. S., Aquat. Toxicol. 2006, 76, 160–202. [DOI] [PubMed] [Google Scholar]
- 15. Geret F., Cosson R. P., Arch. Environ. Contam. Toxicol. 2002, 42, 36–42. [DOI] [PubMed] [Google Scholar]
- 16. Fang Y., Yang H. S., Liu B. Z., Ecotoxicology 2012, 21, 1593–1602. [DOI] [PubMed] [Google Scholar]
- 17. Frazier J. M., Environ. Health Perspect. 1986, 65, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barsyte D., White K. N., Lovejoy D. A., Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 1999, 122, 287–296. [DOI] [PubMed] [Google Scholar]
- 19. Pellerin J., Amiard J. C., Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 2009, 150, 186–195. [DOI] [PubMed] [Google Scholar]
- 20. Boutet I., Tanguy A., Auffret M., Riso R., Environ. Toxicol. Chem. 2002, 21, 1009–1014. [PubMed] [Google Scholar]
- 21. Nettesheim D. G., Engeseth H. R., Otvos J. D., Biochemistry 1985, 24, 6744–6751. [DOI] [PubMed] [Google Scholar]
- 22. Brady F. O., Trends Biochem. Sci. 1982, 7, 143–145. [Google Scholar]
- 23. Yang Y., Maret W., Vallee B. L., Proc. Natl. Acad. Sci. USA 2001, 98, 5556–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Manceau A., Lemouchi C., Rovezzi M., Lanson M., Glatzel P., Nagy K. L., Gautier-Luneau I., Joly Y., Enescu M., Inorg. Chem. 2015, 54, 11776–11791; [DOI] [PubMed] [Google Scholar]
- 24b. Manceau A., Lemouchi C., Enescu M., Gaillot A.-C., Lanson M., Magnin V., Glatzel P., Poulin B. A., Ryan J. N., Aiken G. R., Gautier-Luneau I., Nagy K. L., Environ. Sci. Technol. 2015, 49, 9787–9796. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Steele R. A., Opella S. J., Biochemistry 1997, 36, 6885–6895; [DOI] [PubMed] [Google Scholar]
- 25b. Rosenzweig A. C., Huffman D. L., Hou M. Y., Wernimont A. K., Pufahl R. A., O'Halloran T. V., Structure 1999, 7, 605–617; [DOI] [PubMed] [Google Scholar]
- 25c. Mah V., Jalilehvand F., Chem. Res. Toxicol. 2010, 23, 1815–1823; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25d. Luczkowski M., Zeider B. A., Hinz A. V. H., Stachura M., Chakraborty S., Hemmingsen L., Huffman D. L., Pecoraro V. L., Chem. Eur. J. 2013, 19, 9042–9049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25e. Lian P., Guo H. B., Riccardi D., Dong A. P., Parks J. M., Xu Q., Pai E. F., Miller S. M., Wei D. Q., Smith J. C., Guo H., Biochemistry 2014, 53, 7211–7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bianconi A., Dell′Ariccia M., Gargano A., Natoli C. R. in Bond length determination using XANES, Vol. 27 (Eds.: A. Bianconi, A. Incoccia, S. Stipcich), Springer, Berlin, 1983, pp. 57–61. [Google Scholar]
- 27. Manceau A., Nagy K. L., Dalton Trans. 2008, 11, 1421–1425. [DOI] [PubMed] [Google Scholar]
- 28. Rodic D., Spasojevic V., Bajorek A., Onnerud P., J. Magn. Magn. Mater. 1996, 152, 159–164. [Google Scholar]
- 29.
- 29a. Henkel G., Krebs B., Chem. Rev. 2004, 104, 801–824; [DOI] [PubMed] [Google Scholar]
- 29b. Voronova A., Meyer-Klaucke W., Meyer T., Rompel A., Krebs B., Kazantseva J., Sillard R., Palumaa P., Biochem. J. 2007, 408, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Deditius A. P., Utsunomiya S., Reich M., Kesler S. E., Ewing R. C., Hough R., Walshe J., Ore Geol. Rev. 2011, 42, 32–46; [Google Scholar]
- 30b. Manceau A., Merkulova M., Murdzek M., Batanova V., Baran R., Glatzel P., Saikia B. K., Paktunc D., Lefticariu L., Environ. Sci. Technol. 2018, 52, 10286–10296. [DOI] [PubMed] [Google Scholar]
- 31. Leung B. O., Jalilehvand F., Mah V., Dalton Trans. 2007, 4666–4674. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Torreggiani A., Domenech J., Atrian S., Capdevila M., Tinti A., Biopolymers 2008, 89, 1114–1124; [DOI] [PubMed] [Google Scholar]
- 32b. Freisinger E., Chimia 2010, 64, 217–224; [DOI] [PubMed] [Google Scholar]
- 32c. Tarasava K., Freisinger E., J. Inorg. Biochem. 2015, 153, 197–203. [DOI] [PubMed] [Google Scholar]
- 33. Leignel V., Laulier M., Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 2006, 142, 12–18. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Frazier J. M., George S. S., Overnell J., Coombs T. L., Kagi J., Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 1985, 80, 257–262; [DOI] [PubMed] [Google Scholar]
- 34b. Erk M., Ivankovic D., Raspor B., Pavicic J., Talanta 2002, 57, 1211–1218. [DOI] [PubMed] [Google Scholar]
- 35. Dondero F., Piacentini L., Banni M., Rebelo M., Burlando B., Viarengo A., Gene 2005, 345, 259–270. [DOI] [PubMed] [Google Scholar]
- 36. Zorita I., Bilbao E., Schad A., Cancio I., Soto M., Cajaraville M. P., Toxicol. Appl. Pharmacol. 2007, 220, 186–196. [DOI] [PubMed] [Google Scholar]
- 37. Le T. T. Y., Zimmermann S., Sures B., Environ. Pollut. 2016, 212, 257–268. [DOI] [PubMed] [Google Scholar]
- 38. Opella S. J., DeSilva T. M., Veglia G., Curr. Opin. Chem. Biol. 2002, 6, 217–223. [DOI] [PubMed] [Google Scholar]
- 39. Møller C., Plesset M. S., Phys. Rev. 1934, 46, 618–622. [Google Scholar]
- 40. Glendening E. D., Landis C. R., Weinhold F., J. Comput. Chem. 2013, 34, 1429–1437. [DOI] [PubMed] [Google Scholar]
- 41. Cobbett C., Goldsbrough P., Annu. Rev. Plant Biol. 2002, 53, 159–182. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Iglesia-Turino S., Febrero A., Jauregui O., Caldelas C., Araus J. L., Bort J., Plant Physiol. 2006, 142, 742–749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42b. Krupp E. M., Mestrot A., Wielgus J., Meharg A. A., Feldmann J., Chem. Commun. 2009, 28, 4257–4259; [DOI] [PubMed] [Google Scholar]
- 42c. Chen L. Q., Yang L. M., Wang Q. Q., Metallomics 2009, 1, 101–106. [Google Scholar]
- 43. Freisinger E., J. Biol. Inorg. Chem. 2011, 16, 1035–1045. [DOI] [PubMed] [Google Scholar]
- 44. Peroza E. A., Schmucki R., Guntert P., Freisinger E., Zerbe O., J. Mol. Biol. 2009, 387, 207–218. [DOI] [PubMed] [Google Scholar]
- 45. Stillman M. J., Coord. Chem. Rev. 1995, 144, 461–511. [Google Scholar]
- 46.
- 46a. Enescu M., Manceau A., Theor. Chem. Acc. 2014, 133, 1457; [Google Scholar]
- 46b. Enescu M., Nagy K. L., Manceau A., Sci. Rep. 2016, 6, 39359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.
- 47a. Ankudinov A. L., Rehr J. J., Low J. J., Bare S. R., J. Chem. Phys. 2002, 116, 1911–1919; [Google Scholar]
- 47b. Demchenko I. N., Denlinger J. D., Chernyshova M., Yu K. M., Speaks D. T., Olalde-Velasco P., Hemmers O., Walukiewicz W., Derkachova A., Lawniczak-Jablonska K., Phys. Rev. B 2010, 82, 075107. [Google Scholar]
- 48.
- 48a. Otvos J. D., Engeseth H. R., Wehrli S., Biochemistry 1985, 24, 6735–6740; [DOI] [PubMed] [Google Scholar]
- 48b. Vasak M., Worgotter E., Wagner G., Kagi J. H. R., Wuthrich K., J. Mol. Biol. 1987, 196, 711–719; [DOI] [PubMed] [Google Scholar]
- 48c. Braun W., Vasak M., Robbins A. H., Stout C. D., Wagner G., Kagi J. H. R., Wuthrich K., Proc. Natl. Acad. Sci. USA 1992, 89, 10124–10128; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48d. Zhou Y. J., Li L. Y., Ru B. G., BBA-Gen. Sub. 2000, 1524, 87–93. [DOI] [PubMed] [Google Scholar]
- 49. Presta A., Fowle D. A., Stillman M. J., J. Chem. Soc. Dalton Trans. 1997, 6, 977–984. [Google Scholar]
- 50. Duncan K. E. R., Ngu T. T., Chan J., Salgado M. T., Merrifield M. E., Stillman M. J., Exp. Biol. Med. 2006, 231, 1488–1499. [DOI] [PubMed] [Google Scholar]
- 51. Otvos J. D., Liu X., Li H., Shen G., Basti M. in Dynamic aspects of metallothionein structure (Eds.: K. T. Suzuki, N. Imura, M. Kimura), Birkhäuser, Basel, 1993, pp. 57–74. [Google Scholar]
- 52.
- 52a. Schwerdtfeger P., Li J., Pyykko P., Theor. Chim. Acta 1994, 87, 313–320; [Google Scholar]
- 52b. Pyykkö P., Straka M., Phys. Chem. Chem. Phys. 2000, 2, 2489–2493. [Google Scholar]
- 53.
- 53a. Lin J. C. Y., Tang S. S., Vasam C. S., You W. C., Ho T. W., Huang C. H., Sun B. J., Huang C. Y., Lee C. S., Hwang W. S., Chang A. H. H., Lin I. J. B., Inorg. Chem. 2008, 47, 2543–2551; [DOI] [PubMed] [Google Scholar]
- 53b. Schmidbaur H., Schier A., Chem. Soc. Rev. 2008, 37, 1931–1951; [DOI] [PubMed] [Google Scholar]
- 53c. Doerrer L. H., Dalton Trans. 2010, 39, 3543–3553. [DOI] [PubMed] [Google Scholar]
- 54. Stasser J. P., Siluvai G. S., Barry A. N., Blackburn N. J., Biochemistry 2007, 46, 11845–11856. [DOI] [PubMed] [Google Scholar]
- 55. Xiao Z. G., Loughlin F., George G. N., Howlett G. J., Wedd A. G., J. Am. Chem. Soc. 2004, 126, 3081–3090. [DOI] [PubMed] [Google Scholar]
- 56. Rousselot-Pailley P., Seneque O., Lebrun C., Crouzy S., Boturyn D., Dumy P., Ferrand M., Delangle P., Inorg. Chem. 2006, 45, 5510–5520. [DOI] [PubMed] [Google Scholar]
- 57. Vasak M., Biodegradation 1998, 9, 501–512. [DOI] [PubMed] [Google Scholar]
- 58. Vergani L., Grattarola M., Borghi C., Dondero F., Viarengo A., FEBS J. 2005, 272, 6014–6023. [DOI] [PubMed] [Google Scholar]
- 59. Jiang L. J., Vasak M., Vallee B. L., Maret W., Proc. Natl. Acad. Sci. USA 2000, 97, 2503–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nagy K. L., Manceau A., Gasper J. D., Ryan J. N., Aiken G. R., Environ. Sci. Technol. 2011, 45, 7298–7306. [DOI] [PubMed] [Google Scholar]
- 61. Bataille C., Baldacchino G., Cosson R. P., Coppo M., Trehen C., Vigneron G., Renault J. P., Pin S., Biochim. Biophys. Acta Gen. Subj. 2005, 1724, 432–439. [DOI] [PubMed] [Google Scholar]
- 62. Sedmak J. J., Grossberg S. E., Anal. Biochem. 1977, 79, 544–552. [DOI] [PubMed] [Google Scholar]
- 63. Altomare A., Cascarano G., Giacovazzo C., Guagliardi A., Burla M. C., Polidori G., Camalli M., J. Appl. Crystallogr. 1994, 27, 343–350. [Google Scholar]
- 64. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Farrugia L. J., J. Appl. Crystallogr. 1999, 32, 837. [Google Scholar]
- 66. George G. N., Pickering I. J., Pushie M. J., Nienaber K., Hackett M. J., Ascone I., Hedman B., Hodgson K. O., Aitken J. B., Levina A., Glover C., Lay P. A., J. Synchrotron Radiat. 2012, 19, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Neese F., WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- 68. Klamt A., Schuurmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary