

Bullfrog saxiphilin:STX complex defines a toxin “molecular sponge” blueprint for STX recognition and toxin resistance.
Abstract
Dinoflagelates and cyanobacteria produce saxitoxin (STX), a lethal bis-guanidinium neurotoxin causing paralytic shellfish poisoning. A number of metazoans have soluble STX-binding proteins that may prevent STX intoxication. However, their STX molecular recognition mechanisms remain unknown. Here, we present structures of saxiphilin (Sxph), a bullfrog high-affinity STX-binding protein, alone and bound to STX. The structures reveal a novel high-affinity STX-binding site built from a “proto-pocket” on a transferrin scaffold that also bears thyroglobulin domain protease inhibitor repeats. Comparison of Sxph and voltage-gated sodium channel STX-binding sites reveals a convergent toxin recognition strategy comprising a largely rigid binding site where acidic side chains and a cation-π interaction engage STX. These studies reveal molecular rules for STX recognition, outline how a toxin-binding site can be built on a naïve scaffold, and open a path to developing protein sensors for environmental STX monitoring and new biologics for STX intoxication mitigation.
INTRODUCTION
Saxitoxin (STX), a bis-guanidinium small molecule produced by brackish and freshwater cyanobacteria and oceanic dinoflagelates associated with red tides, is one of the most lethal neurotoxins, causes paralytic shellfish poisoning (PSP), and is the only marine toxin that is declared a chemical weapon (1, 2). Its toxicity is thought to arise primarily from inhibition of select voltage-gated sodium channel (NaV) isoforms (3), although STX may affect other channels (4, 5) and enzymes (6). PSP caused by STX represents a notable public health and commercial hazard that is an increasing problem due to climate change (7). Consequently, shellfish for human consumption are monitored globally for PSP toxins. Current STX detection methods use a cumbersome mouse viability assay (8). Thus, there is great interest in developing better analytical techniques for measuring STX and related cogeners in food sources (7–9).
Nature uses diverse strategies to counter toxin exposure that include target protein resistance mutations, toxin sequestration, and toxin removal (10, 11). Although the best understood toxin resistance strategies involve target protein mutations (10, 12, 13), frogs display unusual resistance to STX poisoning (14, 15) that is not believed to involve altered responses of frog NaVs to STX (16). Saxiphilin (Sxph), a 91-kDa transferrin homolog, is an STX-binding protein from American bullfrog (Rana catesbeiana) heart and plasma (16–20). This soluble, dual-function protein has a single high-affinity STX-binding site [Kd (dissociation constant), ~0.2 nM] that recognizes certain STX derivatives (16, 21, 22) and has two ~60 residue thyroglobulin type I (Thy1) repeats (18) that act as potent [Ki (inhibition constant), ~1 nM] cysteine protease inhibitors (23). Other soluble STX-binding proteins have been identified in pufferfish (24, 25), cockles (26), and crabs (27), and STX-binding activity has been reported in the plasma, hemolymph, and tissues from arthropods (19), amphibians (28), fish (19), and reptiles (19). Hence, it is thought that Sxph and analogous toxin-binding proteins constitute a second, less well-characterized toxin resistance mechanism involving toxin sequestration (10, 16, 19).
Here, we present high-resolution x-ray crystal structures of apo-Sxph and STX-bound Sxph. These structures reveal key Sxph architectural features, how Sxph recognizes STX, and how the Thy1 domains may engage proteases. Remarkably, the two best-characterized high-affinity STX targets, Sxph and NaVs (29), share a core molecular recognition motif that defines a fingerprint for STX molecular recognition. This information should serve as a touchstone for understanding how STX interacts with channels and other targets (6) and provide guidance for the design of new molecular sensors for STX and related toxins.
RESULTS
Sxph crystal structure reveals a modified transferrin fold
The 2.5-Å resolution x-ray crystal structure of American bullfrog (R. catesbeiana) Sxph (Fig. 1A, fig. S1, A and B, and table S1) revealed a bilobal organization similar to transferrins (30, 31), consisting of an N-lobe (residues 1 to 88 and 232 to 465) and a C-lobe (residues 470 to 825) connected by a linker. Both lobes contain two subdomains, designated N1 (residues 1 to 88 and 389 to 465), N2 (residues 232 to 388), C1 (residues 470 to 557 and 726 to 825), and C2 (residues 558 to 725). The N1 and C1 subdomains comprise discontinuous polypeptides into which N2 and C2 are inserted, respectively (fig. S2). Unlike other transferrin family members, Sxph has a 143-residue (residues 89 to 231) insertion between N1 and N2 that encodes two Thy1 repeats (fig. S2) (18, 23). The two Sxph protomers in the asymmetric unit are essentially identical except for a displacement of the first thyroglobulin repeat by ~24° (fig. S1B). Hence, our description focuses on the more complete protomer B.
Fig. 1. Sxph structure.
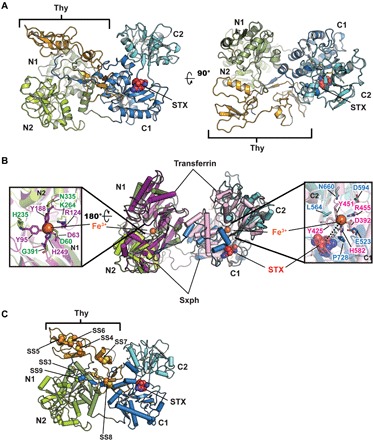
(A) R. catesbeiana Sxph:STX: complex ribbon diagram. Domains are indicated and are colored as follows: N1 (smudge), N2 (limon), thyroglobulin (Thy; bright orange), C1 (marine), and C2 (cyan). STX (red) is shown as space filling. (B) Superposition of Sxph and rabbit transferrin [Protein Data Bank (PDB): 1JNF] (32). Transferrin N-lobe and C-lobe are colored purple and pink, respectively. Sxph Thy1 repeats are not shown. Insets show transferrin Fe3+ ligands and Sxph equivalents as sticks. N domain: transferrin (purple) and Sxph (green); C domain: transferrin (pink) and Sxph (blue). STX (red) is shown as space filling. Right hand inset shows distance between the STX center and transferrin Fe3+. (C) Cartoon diagram showing unique Sxph disulfide bonds in space filling representation: SS3 (Cys27 to Cys417) SS4 (Cys91 to Cys111), SS5 (Cys122 to Cys129), SS6 (Cys131 to Cys153), SS7 (Cys161 to Cys183), SS8 (Cys203 to Cys225), and SS9 (Cys234 to Cys825). Colors and labels are the same as in (A).
The organization of the Sxph core architecture (N1, N2, C1, and C2) is conserved with transferrin (Fig. 1B). Sxph has 21 disulfides. Fourteen are conserved in the transferrin family. Seven are unique (Fig. 1C), of which five are in the Thy1 repeats (SS4 to SS8). The other two connect the α1N1 and α4N1 C-terminal ends (SS3) and the interdomain linker at the start of N2 to the C2 C-terminal tail (SS9) (Fig. 1C and fig. S2). The two Sxph lobes are related by a rigid body motion around the intersubdomain hinge that involves both a closure (~30°) and a twist (~60°) between the relatively closed (N-lobe) and open (C-lobe) conformations, respectively, (fig. S1C) that resembles the lobe conformations defined by apo- and Fe3+-bound transferrin (32). Similar to other transferrin family structures, there is a small (558 Å2) mainly hydrophobic interface between N-lobe and C-lobe.
Consistent with the inability of Sxph to bind Fe3+ (18, 31), almost all of the residues in each lobe required to coordinate Fe3+ and an associated carbonate (33) differ substantially from the conserved transferrin ligands (Fig. 1B and fig. S3). There are also no interlobe γ-turns, a feature of all avian and mammalian transferrins that is thought to aid interlobe cleft opening for Fe3+ binding (32, 34). Together, the structural data establish that Sxph has a transferrin-like fold having numerous modifications and demonstrate why Sxph lacks the classic transferrin Fe3+-binding function (35).
Sxph Thy1 repeats resemble Thy1 protease inhibitors
The ~60 residue thyroglobulin motif occurs in diverse proteins such as thyroglobulin, insulin-like growth factor–binding proteins, and the p41 invariant (Ii) chain involved in major histocompatibility complex class II maturation (36–39). The Sxph thyroglobulin domain forms an independent structure protruding from the N-lobe core (Fig. 1A). The two Thy1 repeats, Thy1-1 and Thy1-2 (Fig. 2A), adopt similar folds containing an α-helix and two antiparallel β-strands. Thy1-1 conforms to the Thy1 type 1A motif stabilized by three disulfides (40), whereas Thy1-2 lacks the inter–β-strand disulfide and conforms to the Thy1 type 1B motif (40). Both Sxph Thy1 repeats have a wedge shape presenting three loops from one end and bear strong structural similarity to each other and the p41 Ii Thy1 repeat (Fig. 2B) (38).
Fig. 2. Comparison of Sxph Thy1-1 and Thy1-2 with p41 Ii.
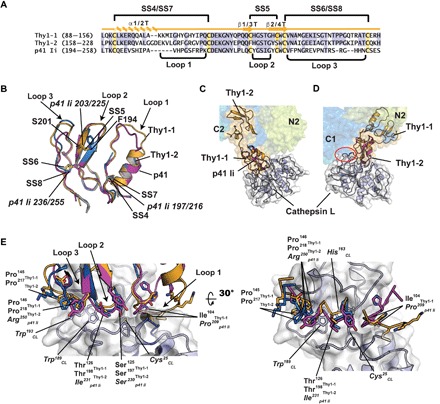
(A) Sequence comparison. Thy1-1 and Thy1-2 secondary structure elements and disulfide bonds are indicated. Cysteines and conserved residues are highlighted yellow and blue, respectively. (B) Cartoon diagram superposition of Thy1-1 (light orange), Thy1-2 (marine), and p41 Ii (magenta) (PDB: 1ICF) (38). Disulfide bonds (italics) and select residues are labeled. Thy1-1 and Thy1-2 have root mean square deviation of Cα position (RMSDCα) = 0.61 and 0.64 Å over 43 and 42 residues, respectively. p41 Ii has RMSDCα = 0.57 Å over 53 residues of Sxph Thy1-1 and Thy1-2. (C and D) Superposition of Sxph on the p41 Ii:cathespin L complex (PDB: 1ICF) (38) using the (C) Thy1-1 and (D) Thy1-2 domains. In (D), red oval indicates cathepsin L and Sxph C1 clash. Sxph colors are the same as in Fig. 1A. (E) Superposition of Sxph Thy1-1 (light orange), Thy1-2 (marine), and p41 Ii (magenta) in the context of the p41 Ii:cathepsin L interface.
Similar to p41 Ii, Sxph is a potent cysteine protease inhibitor, affecting cathepsin L, cathepsin B, and papain with nanomolar potency (23). The two Thy1 repeats appear to enable Sxph to engage these targets with different stoichiometries (1:1 for cathepsin L and cathepsin B and 1:2 for papain) (23). Superposition of the p41:cathepsin L complex on the Thy1-1 and Thy1-2 domains (Fig. 2, C and D) indicates that the Thy1-1 repeat can bind the protease unhindered, whereas binding to Thy1-2 incurs clashes (Fig. 2D and fig. S4A). Superposition of papain, an enzyme very similar to cathepsin L but that Sxph binds with a 1:2 stoichiometry (23), shows similar clashes (fig. S4, A and B). Hence, it seems likely that local rearrangements mitigate these clashes. Comparison of the Sxph Thy1 domains with the p41:cathepsin L complex (38) highlights the features that likely contribute to Sxph Thy1 domain:protease interaction specificity (Fig. 2E). The conserved central Loop 2 (Fig. 2A) can be well positioned over the cathepsin L active site cysteine (Fig. 2E). Loop 3 of both repeats is longer than in p41 Ii (Fig. 2, A and B). However, this difference would not interfere with enzyme binding, as the p41 Loop 3 interactions between Arg250 and the cathepsin L pocket lined by Trp189 and Trp193 are replaced by similar Sxph loop 3 tight turns in Thy1-1 and Thy1-2 (Fig. 2E). Loop 1 has the most varied conformations among Thy1 domains (fig. S4, C and D) and, in Thy1-1 and Thy1-2, has different mobilities, as Thy1-2 Loop 1 lacks visible electron density that indicates disorder. Comparison with the p41:cathepsin L complex shows that Thy1-1 Loop 1 occupies the protease S2 pocket in a manner in which any observed clashes could be relieved by modest reorganization. Loop 1, the point of highest sequence divergence between Thy1-1 and Thy1-2 (Fig. 2A), is likely responsible for binding specificity differences. Together, these analyses indicate how variations in Thy1 Loop 3 can recognize a common target, support the role of Loop 1 in protease recognition specificity (40), and suggest why Sxph binds similar proteases with different stoichiometries.
Structure of the Sxph:STX complex reveals a novel STX-binding site
Sxph binds STX with high affinity (Kd, 0.2 nM) and a 1:1 stoichiometry (20). Crystals made by soaking apo-Sxph crystals with STX or by cocrystallization diffracted x-rays to 2.50- and 2.12-Å resolutions, respectively (table S1). The asymmetric units in both contained two essentially identical Sxph:STX complexes (root mean square deviation of Cα position, 0.5 Å) that provide a high-resolution view of the Sxph:STX interaction (table S1). Because of its superior resolution and completeness, our description focuses on molecule B of the Sxph:STX cocrystallization complex.
Sxph and STX interact with a 1:1 stoichiometry matching biochemical studies (16, 21). However, contrary to the idea that the STX-binding site is made by a remodeled Fe3+-binding cavity in the C1-C2 Sxph interdomain cleft (20), the Sxph:STX structure shows that a novel, solvent-exposed surface C1 pocket of ~14 Å away from the C1-C2 cleft forms the STX-binding site (Fig. 1A and figs. S1A and S5A) comprising C1 domain elements α3C1, β4C1, α6C1, and the β6C1-α6C1 loop (Fig. 3A). Counter to previous proposals (20, 22), there are no large-scale conformational changes between apo-Sxph and Sxph:STX (fig. S5A). Hence, the Sxph:STX interaction occurs in an entirely unanticipated manner.
Fig. 3. Sxph STX-binding site.
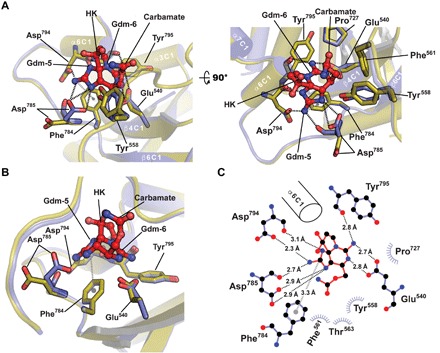
(A) Apo-Sxph (olive) and STX-bound Sxph (slate) superposition cartoon diagram. STX-interacting residues are shown as sticks. Key secondary structure elements are labeled. Black and gray dashed lines indicate hydrogen bond networks and the cation-π interaction, respectively. STX is shown as red sticks. Gdm-5, Gdm-6, and HK indicate the five- and six-membered guanidinium rings and hemiketal, respectively. (B) STX-binding site highlighting the cation-π interaction (gray) and Asp785 movement. (C) LIGPLOT diagram of the STX-binding site. α6C1 is shown for orientation.
STX binds the STX pocket in an orientation in which its five- and six-membered guanidinium rings engage the protein, while the C12 hydrated ketone and carbamoyl group point toward solvent (Fig. 3, A to C, and fig. S5B). The STX pocket uses a set of charged side chains that directly engage the toxin together with contributions from backbone hydrogen bonding groups, van der Waals interactions, and a cation-π interaction (Fig. 3, A to C). A set of side chain carboxyls coordinate the five- and six-membered guanidinium rings engaging all available STX guanidinium nitrogen atoms. Asp785 and Asp794 form a network that encompasses the three nitrogen atoms of the guanidinium group on the five-membered ring (Fig. 3, A to C), whereas the Glu540 side chain interacts with both available six-membered ring guanidinium nitrogens (Fig. 3, A to C). This intimate involvement of acidic side chains agrees with the observation that Sxph treatment with trimethyloxonium tetrafluorborate, a reagent that methylates aspartate and glutamate carboxylates, inhibits STX binding (20). Backbone carbonyl interactions from Asp785 with the five-membered ring N16 atom and Tyr795 with the six-membered ring N13 atom augment the side chain networks (Fig. 3C). Phe784 forms the STX-binding pocket base and makes a cation-π interaction with the STX five-membered ring guanidinium (Fig. 3, B and C). Thus, rather than hydrophobic interactions as proposed (22), multiple sets of complimentary charged-based interactions comprise the Sxph:STX complex.
Although there are no large-scale changes between apo-Sxph and Sxph:STX (fig. S5A), some local rearrangements in the STX-binding pocket are evident from the initial difference maps (fig. S1A) and structure refinement. Asp785 shows the largest change and acts like a latch that faces away from the apo-Sxph pocket and that closes to interact with the STX N7 atom (Fig. 3, A and B, fig. S1A, and movie S1). The remaining changes involve a small movement of the Glu540 away from the pocket and a ~15° rotation of the Tyr558 ring (Fig. 3B and movie S1). Hence, Sxph:STX high-affinity interaction results from capture of the rigid toxin by an essentially preformed binding site.
Radioligand competitive displacement studies of STX derivatives (fig. S5B) (19) match the observed binding pose and side chain interactions. In line with the observation that STX carbamoyl moiety does not interact with Sxph, removal of this group (decarbamoyl STX; fig. S5B) or its modification with a sulfate (gonyautoxin V; fig. S5B) had only a ~2-fold and no impact on affinity, respectively (19). By contrast, hydroxylation of the six-membered ring N1 atom (neosaxitoxin; fig. S5B) reduced affinity by ~550-fold, supporting the importance of the interaction between N1 and Glu540. Further, sulfation of the C11 atom in the STX C1 derivative (fig. S5B) reduced binding relative to the STX B1 parent by ~150-fold, consistent with a clash between the sulfate and α6C1. The excellent agreement of these biochemical studies validates the observed binding pose and outlines how Sxph recognizes diverse naturally occurring STX derivatives.
STX-binding site elements are absent from transferrins and Sxph N-lobe
Transferrins do not bind STX (35), and the STX-binding pocket resides in a region that has not been reported to bind small molecules in other transferrin family members. How then did nature sculpt the STX-binding pocket from a naïve transferrin scaffold? Comparison of Sxph with two exemplar transferrins, Fe3+-bound rabbit serum transferrin [Protein Data Bank (PDB): 1JNF] (41) (Fig. 4A) and apo-human serum transferrin (PDB: 2HAU) (32) (Fig. 4B), reveals a shallow “proto-pocket” on the transferrin C1 subdomain surface at the location of the STX-binding site. Notably, the structural homologs of the residues that form Sxph:STX interactions differ considerably (fig. S3). In the transferrins, positively charged and small hydrophobic residues are found at the positions of the Sxph acidic residues that coordinate the STX five-membered (Asp785 and Asp794) and six-membered (Glu540) rings, respectively. Further, residues corresponding to Phe784, the amino acid responsible for the cation-π interaction, are all branched, hydrophobic residues incapable of making this type of interaction.
Fig. 4. Sxph STX-binding site and transferrin proto-pocket.
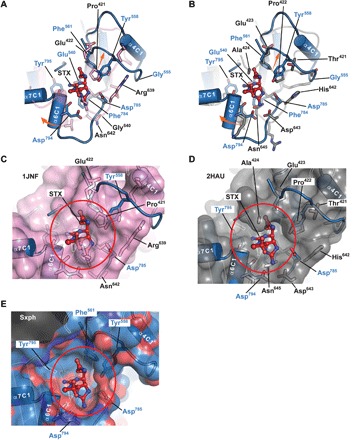
(A and B) Superposition of the Sxph (marine) STX-binding site with (A) Fe3+-bound rabbit serum transferrin (PDB: 1JNF) (41) (pink) and (B) apo-human serum transferrin (PDB: 2HAU) (32) (gray). STX (red) is shown as sticks. Select residues are shown. Blue labels indicate Sxph residues. Orange arrows indicate changes between transferrin and Sxph. (C to E) Transferrin proto-pocket and Sxph STX-binding pocket comparisons. (C) to (E) show apo-transferrin (pink), transferrin (gray), and Sxph (marine) surfaces, respectively. In (C) and (D), labels indicate Sxph residues that break through the transferrin surface. Red circle highlights the STX-binding site. STX is shown as space filling. Sxph surface is colored by atom type, where red and blue denote oxygen and nitrogen, respectively.
Besides lacking the residues to coordinate STX, the rather shallow transferrin C1 proto-pocket is too small to accommodate a molecule the size of STX. Structural comparisons highlight changes in Spxh elements on opposite sides of the pocket that expand its size (Fig. 4, C to E). Namely, the Sxph α6C1 helix position differs from the corresponding transferrin helices (residues 643 to 648 and 646 to 650 in rabbit and human transferrin, respectively; Fig. 4, A and B), and the loop adjacent to the STX carbamate (residues 555 to 561) differs relative to the corresponding transferrin structure (residues 418 to 423 and 419 to 424 in rabbit and human transferrin, respectively; Fig. 4, A and B). This Sxph loop is buttressed by the α4C1 helix, which is absent in transferrin (Fig. 4, A and B, and fig. S3). Collectively, these changes create a site ringed by a set of negatively charged atoms that complement the dicationic STX (Fig. 4E).
Sxph N1 and C1 are structurally similar (fig. S6A), and N1 has a proto-pocket that corresponds to the C1 STX-binding site. This proto-pocket is more open than in the transferrins and is framed on one side by two helices, α5N1 and α6N1, that match the C1 domain α6C1 and α7C1 (fig. S6B). However, α6N1 and Tyr82 on the opposite side of the narrow N1 proto-pocket would clash with the toxin near the C12 hemiketal and C13 carbamate, respectively (fig. S6A). Most importantly, Sxph N1 lacks residues that could coordinate STX. The C1 residues that engage the two guanidinium rings, Glu540, Asp794, Asp785, and Phe784, are replaced in N1 by Ala79, Leu448, Ser441, and Leu440, respectively, rendering the site incapable of making the necessary electrostatic and cation-π interactions (fig. S6A). Further, the N1 proto-pocket is occluded by α8C1, β7C1, and β5T (fig. S6C). Hence, a multitude of factors prevents Sxph from using the N1 subdomain proto-pocket as a second STX-binding site.
A frog Sxph homolog has the STX-binding motif
Although other STX-binding proteins have been reported (24–27), none are related to Sxph. We identified Sxph-like sequences in an invertebrate, springtail (Folsomia candida); two fish, Nile tilapia (Oreochromis niloticus) and Northern pike (Esox lucius); and the High Himalaya frog (Nanorana parkeri) (fig. S7). All share the transferrin fold core with Sxph and carry mutations that would prevent Fe3+ binding. The springtail and High Himalaya frog sequences also have two Thy1 repeat insertions between the N1 and N2, making them closer homologs to Sxph than the fish sequences. Notably, the High Himalaya frog sequence has the key features for STX recognition, namely, C1 equivalents of Glu540, Asp785, Asp794, and Phe784 that match the STX recognition fingerprint. Hence, bullfrog Sxph is not unique and has a counterpart in a frog from a distantly related family (Dicroglossidae) that we name SxphNP.
Sxph and NaVs recognize STX using a similar molecular logic
NaVs are the primary target for the paralytic effects of STX and bind STX with nanomolar affinity, similar to Sxph (1, 20). To ask whether NaVs and Sxph use a common STX recognition strategy, we compared structures of the STX-binding sites of Sxph and the cockroach NaV NaVPaS:STX complex determined at 3.2-Å resolution by single-particle cryo-electronmicroscopy (cryo-EM; Fig. 5, A and B) (29). Although the two proteins are unrelated and some recognition details appear to differ, namely, at the site of the hydrated C12 ketone (29, 42, 43), there are remarkably common STX molecular recognition themes. The channel and Sxph both engage the five- and six-membered guanidinium rings using side chain carboxylates. NaVPaS Glu378 recognizes the five-membered ring similar to Sxph Asp794, and NaVPaS Glu704 coordinates the two available nitrogens of the six-membered guanidinium similar to Sxph Glu540 (Fig. 5, C and D, and fig. S8A). Both glutamates are important for the NaV:STX interaction (44, 45). The two proteins also use an aromatic ring to engage the STX concave face through a cation-π interaction; however, the recognized STX ring differs. NaVPaS Tyr376, a residue important for STX binding (45, 46), engages the STX six-membered ring, whereas Sxph Phe784 interacts with the five-membered ring (fig. S8B). Similar interactions are found in the recent 3.2-Å cryo-EM structure of a human NaV1.7:STX complex (Fig. 5E and fig. S8, B to D) (47). Although the resolutions of the NaVPaS:STX and NaV1.7:STX structures and issues regarding acidic side chain definition by cryo-EM relative to x-ray studies (48–50) place limits on a very detailed comparison of STX binding, it is obvious that the two completely unrelated proteins, Sxph and NaVs, share general STX recognition rules (Fig. 5E).
Fig. 5. Sxph and NaVs share STX recognition strategies.
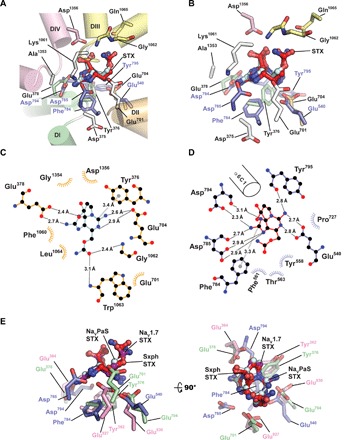
(A) NaVPaS:STX (PDB: 6a91) (29) and Sxph:STX STX-binding site superposition. NaVPaS is shown as a cartoon viewed from the central channel cavity. Pore domains are colored as follows: DI, green; DII, orange; DIII, yellow; and DIV, pink. STX coordinating and selectivity filter “DEKA” motif (white) residues are shown as sticks. Sxph STX-binding site side chains are blue. STX from Sxph:STX (red) and NaVPaS:STX (cyan) are superposed. (B) Closeup view of the STX-binding sites from (A). (C) Diagram of the NaVPaS:STX interactions. (D) Diagram of the Sxph:STX interactions. (E) Comparison of common STX interactions for Sxph (blue), NaVPaS (green), and NaV1.7 (magenta). STX from the Sxph:STX complex (red), NaVPaS:STX complex (cyan), and NaV1.7:STX complex (violet) are indicated. (C) and (D) were generated using LIGPLOT (67) and a 3.35-Å cutoff. Hydrogen bonding networks (black dashed lines) and cation-π interactions (gray dashed lines) are indicated. (D) is the same as Fig. 3C.
DISCUSSION
The Sxph structure defines a paradigm for molecular recognition of STX, one of nature’s most lethal poisons (1, 3, 6) at atomic resolution. The Sxph core is built from the transferrin fold (Fig. 1B), a family of soluble proteins best known for Fe3+ transport (30, 31), that has been modified to act as a “dual-function” protein that can make high-affinity interactions with STX and cysteine proteases. Contrary to expectations (20), the STX-binding site is not a remodeled version of the Fe3+-binding site but resides at a unique locale on the C1 domain. This high-affinity binding site undergoes minimal conformational changes upon binding the rigid STX scaffold. Remarkably, the general blueprint for STX recognition by Sxph using side chain carboxylates, a cation-π interaction, and a largely rigid binding site is shared with NaVs (1, 13, 29, 44–47). This commonality between a 91-kDa soluble protein and a ~200-kDa membrane protein ion channel unmasks an extraordinary convergence of STX molecular recognition strategies.
How proteins acquire new ligand-binding sites is an unresolved question for which surface pockets are thought to be a key factor (51, 52). Transferrins do not bind STX (35). Structural comparisons of Sxph and transferrins identify a transferrin proto-pocket at cognate position of the Sxph STX-binding site. This region is not known to bind small molecules in any transferrin family member. Nevertheless, the structural data suggest that a limited set of changes are required to reshape this naïve site into a high-affinity small-molecule binding site and provide an example of how a protein scaffold can acquire a new function.
Organisms are thought to cope with toxin exposure through diverse strategies that include target protein resistance mutations, toxin sequestration, and toxin removal (10, 11). Frogs are resistant to STX (14, 15), and Sxph may contribute to this property (16, 19). Although how toxin target mutations confer toxin resistance has been widely studied (12, 13, 53), understanding of alternative resistance mechanisms remains primitive. Our findings provide the first structural characterization of a toxin “molecular sponge,” identify an Sxph candidate in a distantly related frog family (Raindae versus Dicroglossidae), and provide a starting point for molecular dissection of toxin sequestration mechanisms. The observation that other organisms have soluble STX-binding proteins unrelated to Sxph (24–28) indicates that toxin sequestration is a general strategy (10, 16, 19) and that de novo STX-binding site creation has happened multiple times.
Defining how proteins recognize STX has implications for understanding its lethal effects, the mechanisms by which organisms evade intoxication, and the design of sensors that could monitor PSP toxins in the environment and food. The Sxph:STX complex structure defined here offers a new path to design protein-based assays for STX and related toxins (7–9, 54), provides a blueprint for STX target identification, and should aid development of STX intoxication countermeasures.
MATERIALS AND METHODS
Protein expression and purification
The gene for North American bullfrog, R. catesbeiana, Sxph (GenBank: U05246.1), including its N-terminal secretory sequence, was codon-optimized and synthesized by GenScript. The Sxph gene was cloned into the BamHI and HindIII multiple cloning sites of pFastBac1 (Invitrogen) in frame with a C-terminal 3C protease cleavage site, followed by green fluorescent protein (GFP) and a His10 tag. Bacmids and baculovirus were generated following the manufacturer’s protocol (Bac-to-Bac, Invitrogen). P2 baculovirus was used for transduction at dilution of 1:40 into Sf9 cells at cell density of 2 × 106 cells ml−1 in ESF921 media (Expression Systems). Cells were grown for 72 hours after transduction, and the expressed Sxph-GFP fusion protein was secreted into the growth media. Cells were removed by centrifugation, and the supernatant was adjusted to pH 8.0 with a final concentration of 50 mM Tris-HCl and treated with 1 mM NiCl2 and 5 mM CaCl2 to precipitate contaminants. Precipitants were removed by centrifugation, and the clarified supernatant was incubated with anti-GFP nanobody-conjugated sepharose beads (55, 56) for 5 hours at room temperature. Beads were washed with 20 column volumes of a buffer containing 300 mM NaCl and 30 mM Tris-HCl (pH 7.4). On-column cleavage of the GFP-His tag was achieved by incubating with 3C protease (0.1 mg ml−1) (57) overnight at 4°C.Cleaved sample was further purified by size exclusion chromatography using a Superdex 200 10/300 GL column in buffer containing 150 mM NaCl and 10 mM HEPES (pH 7.4).
Crystallization and structure determination
Purified Sxph was exchanged into a buffer of 10 mM NaCl and 10 mM HEPES (pH 7.4) and then concentrated to 65 mg ml−1 using a 50-kDa molecular weight cutoff Amicon Ultra centrifugal filter (Millipore) for crystallization screening by hanging drop vapor diffusion at 4°C using a 2:1 ratio of protein to screening solution. Apo-Sxph was crystallized from solution containing 0.1 M sodium cacodylate (pH 6.5), 5% PEG 8000, and 40% 2-methyl-2,4-pentanediol. Addition of 0.5% β-dodecyl maltoside or 10 mM sodium bromide to the crystallization solution further improved crystal quality of apo-Sxph. Crystals of the Sxph:STX complex were obtained by soaking STX (final concentration, 1 mM) into apo crystals for 5 hours before freezing. For cocrystallization, STX was added to Sxph in a molar ratio of 1.1:1 STX:Sxph. The sample was then incubated on ice for 1 hour before setting up crystallization. X-ray datasets for apo-Sxph and the Sxph:STX complex were collected at Advanced Light Source beamline 8.3.1 (Berkeley, CA) processed with XDS (58) and scaled and merged with Aimless (59). The apo-Sxph structure was determined by molecular replacement using Phaser from PHENIX (60) and the N-lobe of human serum transferrin (PDB: 1D4N) (61) as a search model. The resulting electron density map was further improved by rigid body refinement using phenix.refine and density modification using RESOLVE (62). The placed starting model was then subjected to model morphing in PHENIX using the prime-and-switch map generated from density modification (63). The morphed model thus allowed subsequent manual model building into the prime-and-switch map in COOT (64). Iterative model building, refinement, density modification, and model morphing allowed the apo-Sxph N-lobe and C1 domain to be built and refined (R-free of 31.7%) but left poor-quality electron density for the entire C2 domain. The feature-enhanced map (FEM) option (65) was applied in PHENIX to aid model building of the Sxph C2 domain. The C2 domain of rabbit serum transferrin (PDB: 1JNF) (41) was used as a starting model and placed into the FEM by rigid-body fitting. Subsequent model rebuilding and refinement were performed in COOT (64) and phenix.refine (60). The structure of the Sxph:STX complex was determined by molecular replacement using apo-Sxph as the search model in Phaser (60). Model building and refinement were carried out using COOT (64) and phenix.refine (60).
STX synthesis
STX was synthesized, purified, and validated as described in (66).
Supplementary Material
Acknowledgments
We thank C. Kimberlin and Y. Wu for help with molecular biology and protein expression, and F. Abderemane-Ali, K. Brejc, and J. Dumbacher for comments on the manuscript. Funding: This work was supported by grants NIH-NHLBI R01-HL080050 to D.L.M., NIH-NIGMS GM117263-01A1 to J.D.B., and an NSF Graduate Research Fellowship to R.T.-T. Author contributions: T.-J.Y. and D.L.M. conceived the study and designed the experiments. T.-J.Y. performed molecular biology experiments, expressed, purified, and crystallized the proteins, and collected diffraction data. T.-J.Y. and M.L. determined the structures. R.T.-T. synthesized STX. J.D.B. and D.L.M analyzed data and provided guidance and support. T.-J.Y., M.L., J.D.B., and D.L.M. wrote the paper. Competing interests: J.D.B. is a cofounder and holds equity shares in SiteOne Therapeutics Inc., a start-up company interested in developing subtype-selective modulators of sodium channels. The other authors declare that they no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Coordinates and structure factors are deposited in the RCSB under accession codes 6O0D, 6O0E, and 6O0F for apo-Sxph, Sxph:STX (soaked), and Sxph:STX (cocrystallized), respectively, and will be released immediately upon publication. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaax2650/DC1
Table S1. Crystallographic data collection and refinement statistics.
Fig. S1. Sxph structural analysis.
Fig. S2. Sxph sequence, secondary structure, and disulfide map.
Fig. S3. Comparison of Sxph and representative transferrin family member sequences.
Fig. S4. Sxph thyroglobulin domain structural analysis.
Fig. S5. STX-binding site and STX derivatives.
Fig. S6. Structural comparison of Sxph N1 and C1 domains.
Fig. S7. Sequence comparison of Sxph and putative Sxph homologs.
Fig. S8. NaVPaS:STX and NaV1.7:STX interactions.
Movie S1. Sxph conformational changes upon STX binding.
REFERENCES AND NOTES
- 1.Thottumkara A. P., Parsons W. H., Du Bois J., Saxitoxin. Angew. Chem. Int. Ed. Engl. 53, 5760–5784 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Duran-Riveroll L. M., Cembella A. D., Guanidinium toxins and their interactions with voltage-gated sodium ion channels. Mar. Drugs 15, 303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.B. Hille, Ion Channels of Excitable Membranes (Sinauer Associates Inc., ed. 3, 2001). [Google Scholar]
- 4.Su Z., Sheets M., Ishida H., Li F., Barry W. H., Saxitoxin blocks L-type ICa. J. Pharmacol. Exp. Ther. 308, 324–329 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Wang J., Salata J. J., Bennett P. B., Saxitoxin is a gating modifier of HERG K+ channels. J. Gen. Physiol. 121, 583–598 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llewellyn L. E., Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 23, 200–222 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Campbell K., Rawn D. F., Niedzwiadek B., Elliott C. T., Paralytic shellfish poisoning (PSP) toxin binders for optical biosensor technology: Problems and possibilities for the future: A review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 28, 711–725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.AOAC, in Official Methods of Analysis (Arlington, VA, 1999), vol. 1. [Google Scholar]
- 9.Ruberu S. R., Langlois G. W., Masuda M., Kittredge C., Perera S. K., Kudela R. M., Receptor binding assay for the detection of paralytic shellfish poisoning toxins: Comparison to the mouse bioassay and applicability under regulatory use. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 35, 144–158 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Almabruk K. H., Dinh L. K., Philmus B., Self-resistance of natural product producers: Past, present, and future focusing on self-resistant protein variants. ACS Chem. Biol. 13, 1426–1437 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Hunter P., Do not poison thyself: Mechanisms to avoid self-toxicity could inspire novel compounds and pathways for synthetic biology and applications for agriculture. EMBO Rep. 19, e46756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarvin R. D., Borghese C. M., Sachs W., Santos J. C., Lu Y., O’Connell L. A., Cannatella D. C., Harris R. A., Zakon H. H., Interacting amino acid replacements allow poison frogs to evolve epibatidine resistance. Science 357, 1261–1266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bricelj V. M., Connell L., Konoki K., MacQuarrie S. P., Scheuer T., Catterall W. A., Trainer V. L., Sodium channel mutation leading to saxitoxin resistance in clams increases risk of PSP. Nature 434, 763–767 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Prinzmetal M., Sommer H., Leake C. D., The pharmacological action of “mussel poison”. J. Pharmacol. Exp. Ther. 46, 63–73 (1932). [Google Scholar]
- 15.Kao C. Y., Fuhrman F. A., Differentiation of the actions of tetrodotoxin and saxitoxin. Toxicon 5, 25–34 (1967). [DOI] [PubMed] [Google Scholar]
- 16.Mahar J., Lukács G. L., Li Y., Hall S., Moczydlowski E., Pharmacological and biochemical properties of saxiphilin, a soluble saxitoxin-binding protein from the bullfrog (Rana catesbeiana). Toxicon 29, 53–71 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Doyle D. D., Wong M., Tanaka J., Barr L., Saxitoxin binding sites in frog-myocardial cytosol. Science 215, 1117–1119 (1982). [DOI] [PubMed] [Google Scholar]
- 18.Morabito M. A., Moczydlowski E., Molecular cloning of bullfrog saxiphilin: A unique relative of the transferrin family that binds saxitoxin. Proc. Natl. Acad. Sci. U.S.A. 91, 2478–2482 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llewellyn L. E., Bell P. M., Moczydlowski E. G., Phylogenetic survey of soluble saxitoxin-binding activity in pursuit of the function and molecular evolution of saxiphilin, a relative of transferrin. Proc. R. Soc. B 264, 891–902 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llewellyn L. E., Moczydlowski E. G., Characterization of saxitoxin binding to saxiphilin, a relative of the transferrin family that displays pH-dependent ligand binding. Biochemistry 33, 12312–12322 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Morabito M. A., Llewellyn L. E., Moczydlowski E. G., Expression of saxiphilin in insect cells and localization of the saxitoxin-binding site to the C-terminal domain homologous to the C-lobe of transferrins. Biochemistry 34, 13027–13033 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Lewis P., Fritsch I., Gawley R. E., Henry R., Kight A., Lay J. O. Jr., Liyanage R., McLachlin J., Dynamics of saxitoxin binding to saxiphilin c-lobe reveals conformational change. Toxicon 51, 208–217 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenarčič B., Krishnan G., Borukhovich R., Ruck B., Turk V., Moczydlowski E., Saxiphilin, a saxitoxin-binding protein with two thyroglobulin type 1 domains, is an inhibitor of papain-like cysteine proteinases. J. Biol. Chem. 275, 15572–15577 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Yotsu-Yamashita M., Sugimoto A., Terakawa T., Shoji Y., Miyazawa T., Yasumoto T., Purification, characterization, and cDNA cloning of a novel soluble saxitoxin and tetrodotoxin binding protein from plasma of the puffer fish, Fugu pardalis. Eur. J. Biochem. 268, 5937–5946 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Yotsu-Yamashita M., Yamaki H., Okoshi N., Araki N., Distribution of homologous proteins to puffer fish saxitoxin and tetrodotoxin binding protein in the plasma of puffer fish and among the tissues of Fugu pardalis examined by Western blot analysis. Toxicon 55, 1119–1124 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Takati N., Mountassif D., Taleb H., Lee K., Blaghen M., Purification and partial characterization of paralytic shellfish poison-binding protein from Acanthocardia tuberculatum. Toxicon 50, 311–321 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Lin H., Zhang C., Liao J., Yang F., Zhong S., Jiang P., Chen X., Nagashima Y., Neutralizing effect of hemolymph from the shore crab, Thalamita crenata, on paralytic shellfish toxins. Toxicon 99, 51–57 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Tanaka J. C., Doyle D. D., Barr L., Sodium channels in vertebrate hearts. Three types of saxitoxin binding sites in heart. Biochim. Biophys. Acta 775, 203–214 (1984). [DOI] [PubMed] [Google Scholar]
- 29.Shen H., Li Z., Jiang Y., Pan X., Wu J., Cristofori-Armstrong B., Smith J. J., Chin Y. K. Y., Lei J., Zhou Q., King G. F., Yan N., Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 362, eaau2596 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Lambert L. A., Molecular evolution of the transferrin family and associated receptors. Biochim. Biophys. Acta 1820, 244–255 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Mizutani K., Toyoda M., Mikami B., X-ray structures of transferrins and related proteins. Biochim. Biophys. Acta 1820, 203–211 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Wally J., Halbrooks P. J., Vonrhein C., Rould M. A., Everse S. J., Mason A. B., Buchanan S. K., The crystal structure of iron-free human serum transferrin provides insight into inter-lobe communication and receptor binding. J. Biol. Chem. 281, 24934–24944 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambert L. A., Perri H., Halbrooks P. J., Mason A. B., Evolution of the transferrin family: Conservation of residues associated with iron and anion binding. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 142, 129–141 (2005). [DOI] [PubMed] [Google Scholar]
- 34.MacGillivray R. T. A., Moore S. A., Chen J., Anderson B. F., Baker H., Luo Y., Bewley M., Smith C. A., Murphy M. E. P., Wang Y., Mason A. B., Woodworth R. C., Brayer G. D., Baker E. N., Two high-resolution crystal structures of the recombinant N-lobe of human transferrin reveal a structural change implicated in iron release. Biochemistry 37, 7919–7928 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Li Y., Llewellyn L., Moczydlowski E., Biochemical and immunochemical comparison of saxiphilin and transferrin, two structurally related plasma proteins from Rana catesbeiana. Mol. Pharmacol. 44, 742–748 (1993). [PubMed] [Google Scholar]
- 36.Malthiéry Y., Lissitzky S., Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur. J. Biochem. 165, 491–498 (1987). [DOI] [PubMed] [Google Scholar]
- 37.Lenarčič B., Bevec T., Thyropins—New structurally related proteinase inhibitors. Biol. Chem. 379, 105–111 (1998). [PubMed] [Google Scholar]
- 38.Gunčar G., Pungerčič G., Klemenčič I., Turk V., Turk D., Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 18, 793–803 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molina F., Bouanani M., Pau B., Granier C., Characterization of the type-1 repeat from thyroglobulin, a cysteine-rich module found in proteins from different families. Eur. J. Biochem. 240, 125–133 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Novinec M., Kordiš D., Turk V., Lenarčič B., Diversity and evolution of the thyroglobulin type-1 domain superfamily. Mol. Biol. Evol. 23, 744–755 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Hall D. R., Hadden J. M., Leonard G. A., Bailey S., Neu M., Winn M., Lindley P. F., The crystal and molecular structures of diferric porcine and rabbit serum transferrins at resolutions of 2.15 and 2.60 Å, respectively. Acta Crystallogr. D Biol. Crystallogr. 58, 70–80 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Parsons W. H., Du Bois J., Maleimide conjugates of saxitoxin as covalent inhibitors of voltage-gated sodium channels. J. Am. Chem. Soc. 135, 10582–10585 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Kao C. Y., Kao P. N., James-Krace M. R., Koehn F. E., Wichmann C. F., Schnoes H. K., Actions of epimers of 12-(OH)-reduced saxitoxin and of 11-(Oso3)-saxitoxin on squid axon. Toxicon 23, 647–655 (1985). [DOI] [PubMed] [Google Scholar]
- 44.Penzotti J. L., Fozzard H. A., Lipkind G. M., Dudley S. C. Jr., Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys. J. 75, 2647–2657 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas-Tran R., Du Bois J., Mutant cycle analysis with modified saxitoxins reveals specific interactions critical to attaining high-affinity inhibition of hNaV1.7. Proc. Natl. Acad. Sci. U.S.A. 113, 5856–5861 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satin J., Kyle J. W., Chen M., Bell P., Cribbs L. L., Fozzard H. A., Rogart R. B., A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 256, 1202–1205 (1992). [DOI] [PubMed] [Google Scholar]
- 47.Shen H., Liu D., Wu K., Lei J., Yan N., Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Barad B. A., Echols N., Wang R. Y.-R., Cheng Y., DiMaio F., Adams P. D., Fraser J. S., EMRinger: Side chain–directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartesaghi A., Matthies D., Banerjee S., Merk A., Subramaniam S., Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 11709–11714 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vonck J., Mills D. J., Advances in high-resolution cryo-EM of oligomeric enzymes. Curr. Opin. Struct. Biol. 46, 48–54 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Laskowski R. A., Luscombe N. M., Swindells M. B., Thornton J. M., Protein clefts in molecular recognition and function. Protein Sci. 5, 2438–2452 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao M., Skolnick J., The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc. Natl. Acad. Sci. U.S.A. 109, 3784–3789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geffeney S. L., Fujimoto E., Brodie E. D. III, Brodie E. D. Jr., Ruben P. C., Evolutionary diversification of TTX-resistant sodium channels in a predator-prey interaction. Nature 434, 759–763 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Llewellyn L. E., Doyle J., Jellett J., Barrett R., Alison C., Bentz C., Quilliam M., Measurement of paralytic shellfish toxins in molluscan extracts: Comparison of the microtitre plate saxiphilin and sodium channel radioreceptor assays with mouse bioassay, HPLC analysis and a commercially available cell culture assay. Food Addit. Contam. 18, 970–980 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Lolicato M., Arrigoni C., Mori T., Sekioka Y., Bryant C., Clark K. A., Minor D. L. Jr., K2P2.1(TREK-1)–activator complexes reveal a cryptic selectivity filter binding site. Nature 547, 364–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fridy P. C., Li Y., Keegan S., Thompson M. K., Nudelman I., Scheid J. F., Oeffinger M., Nussenzweig M. C., Fenyö D., Chait B. T., Rout M. P., A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 11, 1253–1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaya D., Kreir M., Robbins R. A., Wong S., Hammon J., Bruggemann A., Minor D. L. Jr., Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 12313–12318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans P. R., Murshudov G. N., How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang A. H. W., MacGillivray R. T. A., Chen J., Luo Y., Wang Y., Brayer G. D., Mason A. B., Woodworth R. C., Murphy M. E., Crystal structures of two mutants (K206Q, H207E) of the N-lobe of human transferrin with increased affinity for iron. Protein Sci. 9, 49–52 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terwilliger T., SOLVE and RESOLVE: Automated structure solution, density modification and model building. J. Synchrotron Radiat. 11, 49–52 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Terwilliger T. C., Read R. J., Adams P. D., Brunger A. T., Afonine P. V., Hung L. W., Model morphing and sequence assignment after molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 69, 2244–2250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Afonine P. V., Moriarty N. W., Mustyakimov M., Sobolev O. V., Terwilliger T. C., Turk D., Urzhumtsev A., Adams P. D., FEM: Feature-enhanced map. Acta Crystallogr. D Biol. Crystallogr. 71, 646–666 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walker J. R., Merit J. E., Thomas-Tran R., Tang D. T. Y., Du Bois J., Divergent synthesis of natural derivatives of (+)-saxitoxin including 11-saxitoxinethanoic acid. Angew. Chem. Int. Ed. Engl. 58, 1689–1693 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wallace A. C., Laskowski R. A., Thornton J. M., LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Schröder E., Phillips C., Garman E., Harlos K., Crawford C., X-ray crystallographic structure of a papain-leupeptin complex. FEBS Lett. 315, 38–42 (1993). [DOI] [PubMed] [Google Scholar]
- 69.Sala A., Capaldi S., Campagnoli M., Faggion B., Labò S., Perduca M., Romano A., Carrizo M. E., Valli M., Visai L., Minchiotti L., Galliano M., Monaco H. L., Structure and properties of the C-terminal domain of insulin-like growth factor-binding protein-1 isolated from human amniotic fluid. J. Biol. Chem. 280, 29812–29819 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Sitar T., Popowicz G. M., Siwanowicz I., Huber R., Holak T. A., Structural basis for the inhibition of insulin-like growth factors by insulin-like growth factor-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 13028–13033 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pavšič M., Gunčar G., Djinović-Carugo K., Lenarčič B., Crystal structure and its bearing towards an understanding of key biological functions of EpCAM. Nat. Commun. 5, 4764 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaax2650/DC1
Table S1. Crystallographic data collection and refinement statistics.
Fig. S1. Sxph structural analysis.
Fig. S2. Sxph sequence, secondary structure, and disulfide map.
Fig. S3. Comparison of Sxph and representative transferrin family member sequences.
Fig. S4. Sxph thyroglobulin domain structural analysis.
Fig. S5. STX-binding site and STX derivatives.
Fig. S6. Structural comparison of Sxph N1 and C1 domains.
Fig. S7. Sequence comparison of Sxph and putative Sxph homologs.
Fig. S8. NaVPaS:STX and NaV1.7:STX interactions.
Movie S1. Sxph conformational changes upon STX binding.