

Methanotrophs are bacteria that sequester methane, a significant greenhouse gas, and thereby perform an important ecosystem function. Understanding the mechanisms by which these organisms interact in the environment may ultimately allow us to manipulate and to optimize this activity. Here we show that members of a genus of methane-oxidizing bacteria can be influenced by a chemical signal produced by a possibly competing species. This provides insight into how gene expression can be controlled in these bacterial communities via an exogenous chemical signal.
KEYWORDS: acyl-homoserine lactone, LuxR solo, methane, methanotroph, orphan LuxR, quorum sensing, sociomicrobiology
ABSTRACT
Multiple species of bacteria oxidize methane in the environment after it is produced by anaerobic ecosystems. These organisms provide reduced carbon substrates for species that cannot oxidize methane themselves, thereby serving a key role in these niches while also sequestering this potent greenhouse gas before it enters the atmosphere. Deciphering the molecular details of how methane-oxidizing bacteria interact in the environment enables us to understand an important aspect that shapes the structures and functions of these communities. Here we show that many members of the Methylomonas genus possess a LuxR-type acyl-homoserine lactone (acyl-HSL) receptor/transcription factor that is highly homologous to MbaR from the quorum-sensing (QS) system of Methylobacter tundripaludum, another methane oxidizer that has been isolated from the same environment. We reconstitute this detection system in Escherichia coli and use mutant and transcriptomic analysis to show that the receptor/transcription factor from Methylomonas sp. strain LW13 is active and alters LW13 gene expression in response to the acyl-HSL produced by M. tundripaludum. These findings provide a molecular mechanism for how two species of bacteria that may compete for resources in the environment can interact in a specific manner through a chemical signal.
IMPORTANCE Methanotrophs are bacteria that sequester methane, a significant greenhouse gas, and thereby perform an important ecosystem function. Understanding the mechanisms by which these organisms interact in the environment may ultimately allow us to manipulate and to optimize this activity. Here we show that members of a genus of methane-oxidizing bacteria can be influenced by a chemical signal produced by a possibly competing species. This provides insight into how gene expression can be controlled in these bacterial communities via an exogenous chemical signal.
INTRODUCTION
Methane-oxidizing bacterial communities sequester this potent greenhouse gas after it is produced by anaerobic ecosystems (1, 2). The aerobic methanotrophs involved in this process have been extensively studied in the sediment communities in Lake Washington (Seattle, WA, USA) (3–6). Evidence shows that several methane-oxidizing bacterial species are present in these environments and compete under different compositions of oxygen and methane gas (5–8). These species also provide a carbon and energy source for organisms that cannot oxidize methane themselves (9–11), thereby serving a foundational role in these ecosystems.
We previously identified and characterized a quorum-sensing (QS) system in Methylobacter tundripaludum (12), a dominant member of enrichments of these methane-oxidizing sediment communities (5, 13). QS is a chemical signaling mechanism that enables bacteria to control gene expression in a cell-density-dependent manner (reviewed in references 14 and 15). One well-studied form of QS uses as chemical signals acyl-homoserine lactones (acyl-HSLs), which are produced by LuxI-type acyl-HSL synthases and are detected by LuxR-type receptors that also serve as transcription factors to regulate gene expression. In M. tundripaludum, the acyl-HSL synthase MbaI produces N-3-hydroxydecanoyl-l-homoserine lactone (3-OH-C10-HSL), which is detected by the transcription factor/receptor MbaR (12). MbaR in turn activates a biosynthetic gene cluster that produces the specialized metabolite tundrenone (16).
Although QS was first described as an intraspecies communication system, there is precedence for its use in interspecies signaling as well. Genes encoding LuxR-type receptors in many bacterial genomes are found with no cognate signal synthase and are known as orphans or solos (17, 18). These receptors can be found in species that do not produce any acyl-HSL signals, such as SdiA in Escherichia coli and Salmonella enterica, which regulates adhesion and resistance to the innate immune system in S. enterica in response to exogenous acyl-HSLs (19). Orphan receptors can also be found in bacteria that possess luxI/luxR-type pairs in their genomes, such as QscR in Pseudomonas aeruginosa, which controls acyl-HSL and virulence factor production in response to both endogenously and exogenously produced signals (20, 21). Bioinformatic surveys (22–24) show that orphan LuxR-type receptors are widespread in bacterial genomes, which suggests that they could play a prominent role in interspecies chemical signaling. To date, however, relatively few of these systems have been characterized experimentally.
In this work, we identify an orphan QS receptor in the genome of many members of the methane-oxidizing genus Methylomonas, including strains isolated from the same environment as the QS-active M. tundripaludum. We show that the orphan receptor/transcription factor from Methylomonas species strain LW13 (13) is active in response to multiple acyl-HSL signals and controls transcription of a neighboring gene cluster that is partially conserved in all orphan-containing members of the Methylomonas genus. Finally, we demonstrate that this gene cluster is also transcribed during coculture with M. tundripaludum but this eavesdropping is not a dominant influence on the dynamics of competition between these two organisms when they are grown planktonically under methane gas in the laboratory. These results identify a molecular mechanism for interspecies chemical communication between two members of a methane-oxidizing bacterial community, an important ecological system.
RESULTS
Identification of an orphan QS receptor in LW13.
We recently characterized a QS system used by the methane-oxidizing bacterium M. tundripaludum 21/22, a strain isolated from sediment from Lake Washington (Seattle, WA, USA). Many other bacterial isolates from the same environment have sequenced genomes (13, 25), and we hypothesized that other organisms that may compete for the same niche might be able to detect the QS signal produced by M. tundripaludum. We performed a BLAST search with the amino acid sequence of the M. tundripaludum LuxR-type receptor/transcription factor MbaR. Methylomonas species strain LW13 possesses an ortholog with 67% amino acid identity to MbaR, which we have named MmsR (Fig. 1A). The amino acid sequence of MmsR retains all of the residues conserved among LuxR-type transcription factors/receptors that bind acyl-HSLs (Fig. 1B) (26, 27).
FIG 1.

Orphan LuxR-type receptor/transcription factor in members of the Methylomonas genus. (A) LW13 genomic region containing the orphan receptor gene mmsR and the predicted MmsR binding site (green arrow) (also see Fig. 2A) upstream of a neighboring gene cluster. Locus tags listed below ORFs are preceded by U737_12 and correspond to the LW13 genome described in this work. (B) Amino acid sequence comparison of the LuxR-type receptor MbaR from M. tundripaludum 21/22 (IMG locus tag T451DRAFT_0820) and homologs from Methylomonas strains, including Methylomonas sp. strain LW13 (U737_12750), Methylomonas denitrificans FJG1 (IMG locus tag Ga0213656_112931), Methylomonas methanica S1/NCIMB 11130 (IMG locus tag Ga0133021_101333), and Methylomonas sp. strain MK1 (IMG locus tag G006DRAFT_0604). Black boxes indicate amino acids conserved in LuxR-type transcription factors/receptors that bind acyl-HSLs (26, 27).
We were unable to locate a cognate luxI-type acyl-HSL synthase gene in the LW13 genome, implicating MmsR as an orphan (or solo) LuxR-type receptor/transcription factor. However, the LW13 draft genome is made up of 42 scaffolds (13), suggesting that it may be missing several open reading frames (ORFs). We used nanopore long read sequencing to close the genome, resulting in one 5,233,521-bp circular chromosome (see Fig. S1 in the supplemental material). This revised genome sequence confirmed that the LW13 genome does not contain an acyl-HSL synthase gene, and it also provided genomic context for the region around mmsR, which was originally near the end of one of the scaffolds.
When we analyzed published genomes, we discovered that many other Methylomonas strains also possess MmsR, including the type strain Methylomonas methanica S1/NCIMB 11130 (28), the well-studied organism Methylomonas denitrificans FJG1 (29), and another isolate from Lake Washington sediment, Methylomonas sp. strain MK1 (13) (Fig. 1; also see Fig. S2). Therefore, we sought to determine the function of MmsR.
MmsR responds to acyl-HSL signals via a neighboring binding site in the genome.
In each Methylomonas genome that contains mmsR, it is neighbored by a cluster of genes downstream of a predicted LuxR-type transcription factor binding site (“lux box”) (30, 31) (Fig. 1A and Fig. 2A). We hypothesized that the orphan receptor activates gene expression via this site upon acyl-HSL signal binding. To test this hypothesis experimentally, we constructed a two-plasmid E. coli reporter strain (P12745-gfp) in which one plasmid expresses mmsR via its native promoter and the other contains the putative MmsR binding site upstream of gene U737_12745 fused to a green fluorescent protein (GFP) gene (gfp), as has been done for other QS systems (12, 32).
FIG 2.
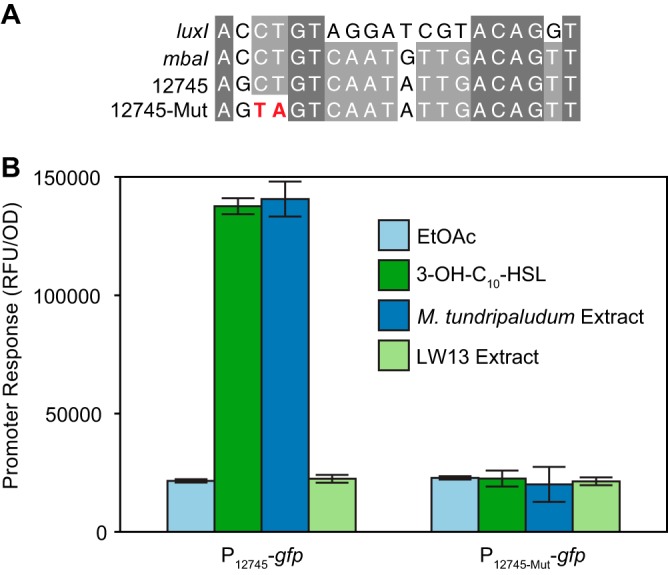
Direct MmsR activation of the promoter of the neighboring gene U737_12745 via a predicted binding site. (A) Comparison of the wild-type and mutated genes for the predicted MmsR binding site upstream of ORF U737_12745 with known lux boxes in the promoter sequences of Vibrio fischeri luxI and M. tundripaludum mbaI. (B) Response of an E. coli reporter strain containing gfp fused to the promoter region of ORF U737_12745, in addition to mmsR on a separate plasmid, to an acyl-HSL signal. EtOAc, ethyl acetate (solvent control); 3-OH-C10-HSL, 10 nM commercially available signal; M. tundripaludum and LW13 extract, ethyl acetate extracts from the supernatants of stationary-phase cultures of M. tundripaludum and LW13, respectively. Data are the mean ± standard deviation of three cultures.
When we grew this reporter strain in the presence of an ethyl acetate extract of supernatant from wild-type M. tundripaludum, an approximately 5-fold increase in normalized green fluorescence was observed (Fig. 2B). Commercially available 3-OH-C10-HSL, the QS signal produced by M. tundripaludum, also activated the reporter strain. To confirm that the acyl-HSL is the only factor produced by M. tundripaludum that activates the reporter strain, we also tested supernatant extract from an M. tundripaludum ΔmbaI strain that does not produce 3-OH-C10-HSL (12), which resulted in no increase in normalized green fluorescence (Fig. S3).
We also did not see an increase in fluorescence when supernatant extract from LW13 itself was tested (Fig. 2B), confirming that LW13 does not produce an acyl-HSL. We then constructed another version of the strain (P12745mut-gfp), containing a mutation of CT to TA in the predicted MmsR binding site (Fig. 2A). This mutation resulted in a loss of responses to both M. tundripaludum supernatant extract and 3-OH-C10-HSL (Fig. 2B). These results demonstrate that the orphan MmsR from LW13 is functional and this transcription factor can directly activate a gene downstream of an MmsR binding site in response to the acyl-HSL signal 3-OH-C10-HSL.
MmsR has broader acyl-HSL specificity than MbaR in E. coli reporter assays.
We tested the difference in acyl-HSL signal specificity of the orphan MmsR and the highly homologous MbaR from M. tundripaludum. We used a previously constructed reporter strain containing mbaR and the MbaR binding site (12) for comparison with the P12745-gfp reporter strain, using several commercially available acyl-HSLs with different acyl chain lengths and/or substituents at the third carbon of the acyl chain. MbaR was most responsive to its cognate signal, 3-OH-C10-HSL, but also was sensitive (saturation at <150 nM) to the other signals with a hydroxyl at the third carbon of the acyl chain (3-OH-C8-HSL and 3-OH-C12-HSL) tested in this assay (Fig. 3). MbaR showed very little response to the other signals, even at the maximum concentration tested (300 nM). In contrast, MmsR showed some response to all acyl-HSLs tested at a concentration of 50 nM in this assay. Among the panel of signals tested, MmsR also appeared most responsive to 3-OH-C10-HSL. Together, these results show that the orphan transcription factor MmsR responds to a broader range of acyl-HSL signals than MbaR under the conditions tested.
FIG 3.

Broader acyl-HSL signal specificity of MmsR than MbaR. E. coli reporter strains assessing MbaR and MmsR activity were assayed with several commercially available acyl-HSL signals. Data are the mean and range of two cultures and are representative of two independent experiments.
MmsR activates the expression of a neighboring gene cluster in the LW13 genome in response to 3-OH-C10-HSL.
In order to determine which genes MmsR regulates in response to an acyl-HSL signal, we compared the transcriptome of exponentially growing LW13 cells in the presence versus the absence of 2 µM 3-OH-C10-HSL. To assess whether any detected expression changes are not dependent on signal binding to MmsR, we constructed an unmarked, in-frame deletion of mmsR (ΔmmsR) and compared the transcriptome of that strain in the presence versus the absence of an acyl-HSL signal. The doubling time of the mutant was not significantly different from that of the wild-type LW13 strain, and there were no significant differences in the doubling times in the presence versus the absence of acyl-HSL for either strain (Table S1).
The neighboring cluster downstream of the identified MmsR binding site was the only group of genes found to be activated in the presence of acyl-HSL (Table 1). The first ORF (U737_12745), which was predicted to encode an NAD(P)H-dependent oxidoreductase, was upregulated approximately 6-fold in the presence of acyl-HSL. The results were very similar for comparisons of wild-type LW13 in the presence versus the absence of acyl-HSL or LW13 versus ΔmmsR in the presence of acyl-HSL (Table 1).
TABLE 1.
Genes with increased expression in LW13 in the presence of 2 µM 3-OH-C10-HSL, compared to the absence of an acyl-HSL signal or compared to the ΔmmsR strain also in the presence of an acyl-HSL signala
| Gene locus | Predicted productb | With vs without acyl-HSL |
Wild type with acyl-HSL vs ΔmmsR with acyl-HSL |
||
|---|---|---|---|---|---|
| Fold change | Adjusted P | Fold change | Adjusted P | ||
| U737_12595 | Hypothetical protein | 1.6 | 1.0E−03 | 1.6 | 1.5E−04 |
| U737_12600 | Antibiotic biosynthesis monooxygenase | 1.9 | 3.0E−08 | 1.8 | 1.5E−06 |
| U737_12605 | Glutathione S-transferase | 1.9 | 4.3E−09 | 2.0 | 2.7E−10 |
| U737_12610 | Phosphotyrosine protein phosphatase | 2.1 | 1.5E−11 | 2.1 | 1.5E−11 |
| U737_12615 | GFA family protein | 2.4 | 1.6E−17 | 2.6 | 1.3E−19 |
| U737_12620 | Glutathione S-transferase family protein | 2.2 | 7.9E−15 | 2.5 | 2.5E−19 |
| U737_12625 | α/β-Hydrolase | 3.0 | 7.6E−26 | 3.2 | 4.0E−30 |
| U737_12630 | Hypothetical protein | 3.4 | 9.8E−34 | 3.5 | 2.7E−34 |
| U737_12635 | PAP2 family protein | 3.2 | 3.8E−32 | 3.6 | 1.6E−39 |
| U737_12640 | GNAT family N-acetyltransferase | 3.5 | 1.7E−33 | 4.0 | 5.3E−40 |
| U737_12645 | FMN-binding negative transcriptional regulator | 4.2 | 3.7E−45 | 4.8 | 8.4E−53 |
| U737_12650 | Tautomerase family protein | 3.4 | 7.6E−31 | 3.2 | 1.6E−27 |
| U737_12655 | DUF1330 domain-containing protein | 3.3 | 3.0E−30 | 3.2 | 1.1E−28 |
| U737_12660 | CTP synthase | 4.6 | 5.4E−58 | 4.8 | 3.7E−61 |
| U737_12665 | α/β Hydrolase | 4.5 | 4.9E−50 | 4.7 | 3.5E−53 |
| U737_12670 | Amidase | 3.3 | 6.8E−29 | 3.3 | 3.8E−30 |
| U737_12675 | Signal peptidase II | 3.2 | 1.4E−28 | 3.5 | 4.7E−33 |
| U737_12680 | GFA family protein | 3.1 | 7.9E−26 | 3.3 | 6.4E−29 |
| U737_12685 | Methyltransferase domain-containing protein | 4.1 | 5.7E−49 | 4.4 | 4.2E−53 |
| U737_12690 | HAD family hydrolase | 6.6 | 5.3E−86 | 7.2 | 2.7E−93 |
| U737_12695 | Hypothetical protein | 9.4 | 1.5E−118 | 9.9 | 3.4E−123 |
| U737_12700 | GNAT family N-acetyltransferase | 7.6 | 3.2E−86 | 8.7 | 8.2E−99 |
| U737_12705 | SAM-dependent methyltransferase | 9.0 | 4.0E−107 | 9.8 | 6.4E−116 |
| U737_12710 | DUF4260 family protein | 7.6 | 5.6E−88 | 8.7 | 4.0E−100 |
| U737_12715 | Hypothetical protein | 2.9 | 7.9E−24 | 3.1 | 4.4E−28 |
| U737_12720 | Cupin domain-containing protein | 2.9 | 4.9E−23 | 2.5 | 1.2E−16 |
| U737_12725 | DUF4442 domain-containing protein | 2.4 | 5.5E−16 | 2.6 | 1.4E−18 |
| U737_12730 | Hypothetical protein | 1.7 | 8.5E−06 | 1.9 | 1.7E−08 |
| U737_12735 | CYTH domain-containing protein | 4.5 | 5.7E−49 | 4.4 | 8.9E−47 |
| U737_12740 | Transglutaminase family protein | 5.1 | 4.6E−55 | 5.6 | 2.5E−62 |
| U737_12745 | NAD(P)H-dependent oxidoreductase | 6.2 | 1.4E−68 | 6.8 | 1.2E−76 |
All genes with adjusted P values (48) of <0.1 are shown.
GFA, glutathione-dependent formaldehyde-activating enzyme; SAM, S-adenosylmethionine; PAP2, phosphatidic acid phosphatase 2; GNAT, Gcn5-related N-acetyltransferase; FMN, flavin mononucleotide; HAD, haloacid dehydrogenase.
In order to confirm that MmsR is the cause of the response to the acyl-HSL, we complemented the ΔmmsR mutation by inserting mmsR, under its native promoter, into a distal site in the genome. This strain (ΔmmsR::mmsR) regained sensitivity to the acyl-HSL signal, as demonstrated by real-time quantitative reverse transcription-PCR (qRT-PCR) (Fig. 4A). These results confirm that MmsR activates expression of the neighboring gene cluster in the LW13 genome in response to an acyl-HSL signal.
FIG 4.
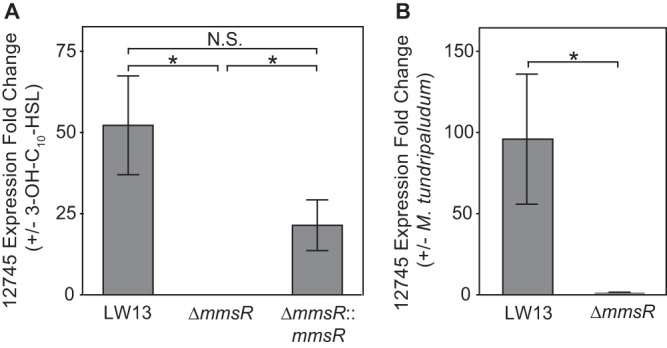
MmsR activation of U737_12745 transcription in response to 3-OH-C10-HSL or coculture with M. tundripaludum. Real-time qRT-PCR results showing relative expression of U737_12745, the first ORF in the neighboring gene cluster, for the wild-type LW13, ΔmmsR, and complemented ΔmmsR::mmsR strains in the presence versus the absence of 2 µM 3-OH-C10-HSL (A) or the wild-type LW13 and ΔmmsR strains with versus without coculture with M. tundripaludum (B). Data are the mean ± standard deviation of three technical replicates and are representative of two independent experiments. *, significantly different (one-tailed homoscedastic t test, P value of <0.01); N.S., not significant.
LW13 gene expression is affected during coculture with M. tundripaludum in an mmsR-dependent manner.
Several studies have examined the dynamics of competition between different methane-oxidizing species, including LW13 and M. tundripaludum, during growth on methane gas (5, 6). Because these two bacteria were isolated from the same environment (13) and 3-OH-C10-HSL is produced by M. tundripaludum and detected by LW13, we tested whether LW13 gene expression was affected during coculture of these two organisms. We isolated RNA from an exponentially growing coculture of these strains and confirmed that U737_12745, the first ORF in the MmsR-regulated cluster, was upregulated compared to a coculture of M. tundripaludum and the ΔmmsR strain (Fig. 4B).
Researchers previously observed that LW13 outcompetes M. tundripaludum in laboratory cocultures (33). We tested whether this result is due to eavesdropping of the M. tundripaludum acyl-HSL signal by LW13. However, a planktonic coculture of M. tundripaludum and the ΔmmsR strain showed no difference in dynamics, compared to a planktonic coculture of M. tundripaludum and wild-type LW13 (Fig. S3). This suggests that, under these conditions, gene regulation by MmsR is not the dominant factor in LW13 outcompeting M. tundripaludum in laboratory cocultures. Together, these results show that LW13 can use MmsR to alter its gene expression in response to a chemical signal produced by another methane-oxidizing species that may compete for the same ecological niche.
DISCUSSION
We have determined that several species of the methane-oxidizing bacterial genus Methylomonas, including strain LW13, possess an orphan acyl-HSL receptor/transcription factor, termed MmsR, that can detect and respond to multiple acyl-HSL signals, including 3-OH-C10-HSL, which is produced by the QS system of an M. tundripaludum strain that was isolated from the same environment. In response to a signal, MmsR activates the transcription of a neighboring gene cluster via an identified binding site.
MmsR shows broader specificity for acyl-HSL signals than does MbaR from M. tundripaludum in E. coli reporter assays. This promiscuity has been observed for other orphan LuxR-type receptor/transcription factors as well, including QscR from Pseudomonas aeruginosa, which responds to both 3-oxo-C12-HSL made by P. aeruginosa and other signals that presumably come from external environments (34). This makes intuitive sense, because orphans like MmsR do not have a cognate signal synthase, unlike MbaR with the 3-OH-C10-HSL-producing MbaI. It may also point to a different role for these orphans, related to detecting the presence of other species rather than detecting population density. There are other species in the same environment that also produce acyl-HSLs that would activate MmsR. For example, some Burkholderia and Pseudomonas species are known to produce 3-OH-C10-HSL (35, 36), and these genera have been detected in the same samples of Lake Washington sediment (5, 6, 9).
The phenotypic response of LW13 to acyl-HSL signal detection by MmsR is still under investigation. The MmsR-activated gene cluster contains several genes that are conserved in all Methylomonas strains containing mmsR, including genes predicted to encode an NAD(P)H-dependent oxidoreductase, a transglutaminase, and a phosphatase (see Fig. S2 in the supplemental material). The role of this gene cluster is currently unknown, and it is not predicted to produce a specialized metabolite (37).
There are also notable differences in the LW13 cluster, compared to the gene clusters in other Methylomonas species. For example, in all other strains the NAD(P)H-dependent oxidoreductase gene is fused with the downstream transglutaminase gene, while in LW13 these are separate ORFs. Additionally, a predicted shikimate dehydrogenase gene that is conserved in all of the other Methylomonas clusters is truncated in LW13 (Fig. S2). This raises the possibility that the MmsR-regulated cluster in LW13 is not functional, and future studies will need to be performed on other Methylomonas strains in order to determine the function of this cluster in response to acyl-HSL signals.
We investigated whether MmsR plays a role in interactions between LW13 and M. tundripaludum in laboratory cocultures. First, we demonstrated that coculturing M. tundripaludum with LW13 caused expression of the MmsR-regulated gene cluster, showing that interspecies chemical communication occurs under these conditions. Next, we assessed the dynamics of competition between M. tundripaludum and an LW13 ΔmmsR mutant. The ΔmmsR strain still outcompeted M. tundripaludum in a planktonic coculture (Fig. S3), showing that acyl-HSL eavesdropping is not the dominant reason behind LW13 outcompeting M. tundripaludum under these laboratory conditions.
These pairwise competitions may not reflect what occurs in the environment; therefore, we do not know the role of this communication system in a natural setting. When lake sediment known to contain both Methylomonas species and M. tundripaludum is enriched on methane under some conditions, it is M. tundripaludum that eventually outcompetes Methylomonas species (5, 6, 33). Further studies will be needed to determine the role of this newly identified interspecies chemical communication system in this ecologically significant group of bacteria.
MATERIALS AND METHODS
Plasmid construction.
Plasmids used in this study are listed in Table 2, and primers used in this study are listed in Table 3. All plasmids were constructed using Gibson assembly (38), with the exception of the reporter vectors pAWP169 and pAWP179. pAWP169 was constructed by amplifying the sequence containing the putative lux box from −400 bp to +21 bp of the translation start site of U737_12745 from LW13 genomic DNA. This sequence was inserted into the promoter probe plasmid pPROBE-GFP[LVA] (39) using the SacI and EcoRI restriction sites. pAWP179 was constructed using the same backbone, promoter, and restriction sites, but the 421-bp promoter sequence contains a CT-to-TA mutation in the MmsR binding sequence (Fig. 2A) and was ordered as a gBlock from Integrated DNA Technologies.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli TOP10 | F– mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu) 7697 galU galK rpsL (Smr) endA1 nupG | Invitrogen |
| E. coli S17-1 λpir | Donor strain; Tpr Smr recA thi pro hsd(r–m+) RP4-2-Tc::Mu::Km Tn7 λpir | 41 |
| mbaR E. coli reporter strain PmbaI-gfp | Acyl-HSL reporter strain; E. coli TOP10 with pAWP112 and pAWP113 | 12 |
| mmsR E. coli reporter strain P12745-gfp | Acyl-HSL reporter strain; E. coli TOP10 with pAWP134 and pAWP169 | This study |
| mmsR E. coli reporter strain with mutated lux box, P12745mut-gfp | Acyl-HSL reporter strain; E. coli TOP10 with pAWP134 and pAWP179 | This study |
| M. tundripaludum strain 21/22 | Aerobic methane-oxidizing bacterium isolated from sediment from Lake Washington (Seattle, WA, USA) | 13 |
| M. tundripaludum strain 21/22 ΔmbaI strain | Strain containing unmarked deletion of luxI-type acyl-HSL synthase gene mbaI (T451DRAFT_0796) | 12 |
| Methylomonas sp. strain LW13 | Aerobic methane-oxidizing bacterium isolated from sediment from Lake Washington | 13 |
| Methylomonas sp. strain LW13 ΔmmsR strain | Strain containing unmarked deletion of orphan luxR-type transcription factor gene mmsR (U737_12750) | This study |
| Methylomonas sp. strain LW13 ΔmmsR::mmsR strain | ΔmmsR with mmsR under its native promoter (400-bp upstream sequence) inserted between U737_06900 and U737_06905 | This study |
| Plasmids | ||
| pAWP112 | pPROBE-gfp[LVA] (39) containing gfp driven by the PmbaI promoter | 12 |
| pAWP113 | pACYC184 (49) expressing mbaR under its native promoter (400-bp upstream sequence) | 12 |
| pAWP134 | pACYC184 (49) expressing mmsR under its native promoter (400-bp upstream sequence) | This study |
| pAWP136 | pCM433kanT (42) containing flanks to create clean deletion of mmsR | This study |
| pAWP169 | pPROBE-gfp[LVA] (39) containing gfp driven by −400 bp to +21 bp of the translational start site of U737_12745 | This study |
| pAWP179 | pAWP169 with mutation of CT to TA in MmsR binding site (AGCTGTCAATATTGACAGTT to AGTAGTCAATATTGACAGTT) (see Fig. 2) | This study |
| pAWP195 | Insertion vector (site between U737_06900 and U737_06905) | This study |
| pAWP197 | pAWP195 containing mmsR under its native promoter (400-bp upstream sequence) for insertion between U737_06900 and U737_06905 | This study |
Smr, streptomycin resistance; Tpr, trimethoprim resistance.
TABLE 3.
Cloning primers used in this study
| Primer name | Sequence (5′ to 3′)a | Description |
|---|---|---|
| AP186_pCM433kanT_fwd1 | ATGTGCAGGTTGTCGGTGTC | For amplification of pCM433kanT (42) backbone |
| AP187_pCM433kanT_rev1 | TGGTAACTGTCAGACCAAGTTTACTC | |
| AP410_113V_fwd1 | ACTATGACTGAGAGTCAACG | For amplification of pACYC184 (49) vector backbone |
| AP411_113V_rev1 | TATGCGACTCCTGCATTAGG | |
| AP517_134I_fwd1 | CCTAATGCAGGAGTCGCATATAAGGGCAAGTCGCCAAGCC | For amplification of mmsR under its native promoter (400-bp upstream sequence) for insertion into pACYC184 |
| AP518_1341_rev1 | CGTTGACTCTCAGTCATAGTTTACAAAAAACCGAGCAGCG | |
| AP521_136U_fwd1 | ATATGAGTAAACTTGGTCTGACAGTTACCAAGAGTATTTCCGGCATAGCC | For amplification of flanks to create ΔmmsR clean deletion with pCM433kanT (42) |
| AP522_136U_rev1 | CCAGTTGGATGGTTGGGGTTTACCGTCGTTAGAAC | |
| AP523_136D_fwd1 | AACGACGGTAAACCCCAACCATCCAACTGGAGTCC | |
| AP524_136D_rev1 | CGTGCATCACGACACCGACAACCTGCACATTTTCGAGTCGGACATTTGGC | |
| AP658_169I_SacI_Fwd1 | GGTGGTGAGCTCCGCCAACCTAAGTCCATAGGCA | For amplification of −400 bp to +21 bp of translational start site of U737_12745 for insertion into pPROBE-gfp[LVA] (39) promoter probe vector |
| AP659_169I_EcoRI_Rev1 | GGTGGTGAATTCGGATAAAGCCAGTATTTTCATTGC | |
| AP758_195I_fwd1 | ATATGAGTAAACTTGGTCTGACAGTTACCAACCATTAACGGCGAAGTCAG | For amplification of insertion region between U737_06900 and U737_06905 in LW13 genome; region was added to pCM433kanT (42) opened with AP186 and AP187 to create insertion vector pAWP195 |
| AP759_195I_rev1 | CGTGCATCACGACACCGACAACCTGCACATAATTTCTCGGCGATTGTCAG | |
| AP764_197V_fwd1 | TACGCGATACAGCGGGCTTT | For addition of mmsR under its native promoter (400-bp upstream sequence) to insertion vector pAWP195 |
| AP765_197V_rev1 | CCGAGTACCACTACAGAGCT | |
| AP766_197I_fwd1 | ATTAGTTGTAAGCTCTGTAGTGGTACTCGGTAAGGGCAAGTCGCCAAGCC | |
| AP767_197I_rev1 | CAGGCAAAAAAAAGCCCGCTGTATCGCGTATTACAAAAAACCGAGCAGCG |
Homology regions used for Gibson assembly and restriction enzyme sites are underlined.
Strain growth.
Strains used in this study are listed in Table 2. E. coli strains were grown in Luria-Bertani (LB) medium at 37°C. M. tundripaludum 21/22 and Methylomonas sp. strain LW13 were cultured in an atmosphere of 25% methane in air. For routine culturing, plates were incubated at room temperature in sealed jars (Oxoid Ltd.), while liquid cultures were grown at 18°C in 250-ml glass serum bottles (Kimble Chase) or tubes (18 by 150 mm; Bellco Glass) sealed with rubber stoppers and aluminum seals (Wheaton), with shaking at 200 rpm. Cultures were grown in nitrate mineral salts (NMS) medium (40) containing 0.2 g/liter MgSO4·7H2O, 0.2 g/liter CaCl2·6H2O, 1 g/liter KNO3, and 30 µM LaCl3, as well as 1× trace elements (500× trace elements contains 1.0 g/liter Na2-EDTA, 2.0 g/liter FeSO4·7H2O, 0.8 g/liter ZnSO4·7H2O, 0.03 g/liter MnCl2·4H2O, 0.03 g/liter H3BO3, 0.2 g/liter CoCl2·6H2O, 0.6 g/liter CuCl2·2H2O, 0.02 g/liter NiCl2·6H2O, and 0.05 g/liter Na2MoO·2H2O). A final concentration of 5.8 mM phosphate buffer (pH 6.8) was added immediately before use.
Genetic manipulation.
Genetic manipulation of LW13 was carried out at 30°C. Plasmids were conjugated into Methylomonas sp. strain LW13 using the E. coli donor strain S17-1 (41), as described previously (42). For conjugation, LW13 biomass was spread on an NMS plate and grown under methane. The next day, an equal volume of donor biomass containing the plasmid of interest was added and the mixture was grown under methane for 2 additional days. Successful integrants (single crossovers) were selected on NMS plates containing kanamycin (50 µg/ml) and then were used to inoculate a 5-ml NMS liquid culture containing no antibiotics that was grown under methane. After one passage, this culture was plated on an NMS plate containing 1% (m/v) sucrose for counterselection, and the resulting colonies were screened for double crossovers by kanamycin sensitivity and colony PCR.
Genome resequencing, assembly, and annotation.
Short-read data for LW13 (genome identification no. 2561511042) were downloaded from the Joint Genome Institute Integrated Microbial Genomes and Microbiomes (JGI IMG/M) data management system (43). The paired-end reads were split into separate forward and reverse read files. Long reads were generated from a separate DNA isolation, using the MasterPure complete DNA and RNA purification kit (Lucigen), from 500 ml of stationary-phase culture. The sequencing library was prepared using the Oxford Nanopore rapid sequencing kit (product no. SQK-RAD004) and immediately loaded onto an Oxford Nanopore MinION long-read sequencer (see the supplemental material). Bases were called from the raw data using MinKNOW v1.15.1. Adapter sequences were removed using porechop v.0.2.3 with the command “porechop -i long_reads.fastq -o trimmed_long_reads.fastq -t 10.”
Both short and long reads were included in a hybrid assembly using Unicycler v0.4.7 (44), with the command “python unicycler-runner.py -1 short_forward.fastq -2 short_reverse.fastq -l trimmed_long_reads.fastq -o Assembly,” resulting in a single 5,233,523-bp circular contig (see Fig. S1 in the supplemental material). Final annotations were created through submission to the National Center for Biotechnology Information whole-genome sequencing (WGS) submission portal.
RNA extraction, transcriptome sequencing, and data analysis.
Growing cultures of Methylomonas sp. strain LW13 were subdiluted to an optical density at 600 nm (OD600) of 0.01 and added to bottles containing dried 3-OH-C10-HSL (or solvent control), resulting in a final concentration of 2 µM signal. RNA was extracted during log phase (OD600 of 0.4 to 0.6). Briefly, cultures were chilled on ice and then centrifuged at 5,000 rpm for 15 min at 4°C. Pellets were subsequently flash frozen in liquid nitrogen and stored at −80°C until further processing. Cell pellets were lysed by bead beating with 0.1-mm zirconia-silica beads (Biospec Products) in 1 ml TRIzol (Thermo Fisher Scientific). The lysate was then separated according to the TRIzol instructions; subsequently, 1.5 volumes of 100% ethanol was added to the aqueous phase of the extract, which was then used as the input for an RNEasy RNA isolation kit (Qiagen). The resulting eluate was then digested using Ambion DNase I (Thermo Fisher Scientific) for 30 min at 37°C before being repurified via an RNEasy RNA isolation kit with the addition of an on-column DNase (Qiagen) digestion step for 15 min at room temperature. The resulting purified RNA was checked for DNA contamination by PCR using the degenerate 16S primers 27F and 1492R.
cDNA library preparation and RNA sequencing were performed by GENEWIZ using Illumina HiSeq paired-ended reads (2 by 150 bp). The raw reads from the sequencing facility were aligned to the newly assembled Methylomonas sp. strain LW13 genome. Alignment was performed using BWA v0.7.12-r1044, using the BWA-MEM algorithm and default parameters (45). The alignments were postprocessed into sorted BAM files with SAMTools v1.2-232-g87cdc4a (46). Reads were attributed to ORFs using the htseq-count tool from HTSeq framework v0.6.1p1, in the intersection-nonempty mode (47). Differential abundance analysis was performed with DESeq2 1.2.10 (48) using R 3.3.0.
Acyl-HSL response E. coli reporter assay.
Responses to acyl-HSLs signals were detected as described previously (12, 32). Briefly, overnight cultures of E. coli reporter strains containing plasmids pAWP112 and pAWP113 (PmbaI-gfp), pAWP134 and pAWP169 (P12745-gfp), or pAWP134 and pAWP179 (P12745mut-gfp) were subcultured to an OD600 of 0.1 in LB medium with kanamycin (50 µg/ml) and chloramphenicol (35 µg/ml). Subsequently, 500 µl was added to 1.5-ml tubes containing dried signal, and the tubes were shaken at 37°C for 4 h. Cultures were then pelleted and resuspended in 500 µl of 50 mM Tris (pH 7.5), and 100 µl was measured for GFP fluorescence (excitation, 485 nm; emission, 510 nm) in a 96-well plate (Nunc black optical bottom) with a plate reader (Tecan Infinite F500). The signals N-3-hydroxydecanoyl-l-homoserine lactone (3-OH-C10-HSL), N-3-hydroxyoctanoyl-l-homoserine lactone (3-OH-C8-HSL), and N-decanoyl-l-homoserine lactone (C10-HSL) were purchased from Cayman Chemical. The signals N-3-oxodecanoyl-l-homoserine lactone (3-oxo-C10-HSL) and N-3-hydroxydodecanoyl-dl-homoserine lactone (3-OH-C12-HSL) were purchased from Sigma-Aldrich. When an ethyl acetate extract of methanotroph supernatant was used, it contained the equivalent of 100 µl of supernatant.
Real-time qRT-PCR.
One microgram of RNA, isolated as described above, was reverse transcribed using the SensiFAST cDNA synthesis kit (Bioline). PCR mixtures were prepared using the SensiFAST SYBR No-ROX kit (Bioline), containing 400 nM primers and 4 µl undiluted cDNA in a total volume of 10 µl. Reactions were performed on a PTC-200 thermal cycler with a Chromo4 continuous fluorescence detector (MJ Research), and threshold cycle (CT) values were calculated using Opticon Monitor v3.1.32, at 1 standard deviation from baseline. All gene CT values were normalized to LW13 recA (U737_07580) CT values, and primers used for all reactions are listed in Table 4.
TABLE 4.
Real-time quantitative PCR primers used in this study
| Primer name | Sequence (5′ to 3′) | Target |
|---|---|---|
| AP692_FMNred_fwd1 | GCGGCATCAATCACTTTACCG | NAD(P)H-dependent oxidoreductase gene U737_12745 |
| AP693_FMNred_rev1 | ATGGTTTCATCGCCGGTGAT | |
| AP854_LW13-recA_qPCR_fwd1 | AAGTCCGGTTCCTGGTATGC | recA gene U737_07580 |
| AP855_LW13-recA_qPCR_rev1 | CGTCGTCGTCTTCACTGAC | |
| 21/22 MtYF | GATAGTCGCCGGCTCAGGACGCAT | M. tundripaludum formaldehyde oxidation gene orfY (T451DRAFT_3399); for coculture species abundance quantification |
| 21/22 MtYR | CTACAGCAGGCGCTAAATCTTGTTC | |
| LW13YF | CCTAAGTGTTTGTTAGTGATTGCC | LW13 formaldehyde oxidation gene orfY (U737_14825); for coculture species abundance quantification |
| LW13YR | CCAATGACGGCACCTGATATGTAT |
Coculture growth.
For qRT-PCR assays, exponentially growing cultures of M. tundripaludum 21/22 and LW13 were each subdiluted to an OD600 of 0.01 in a 50-ml volume to initiate cocultures. Cocultures were grown to log phase (OD600 of 0.4 to 0.6), and RNA was then isolated and analyzed by qRT-PCR as described above.
Coculture competition assays were performed as described previously (33). All cocultures were grown in an atmosphere of 25% methane in air. Briefly, a 1:1 ratio of exponentially growing cultures of M. tundripaludum and wild-type or ΔmmsR LW13 strains were added to a total OD600 of ∼0.1 in a 50-ml volume and were grown to an OD600 of ∼0.6 before subdilution to 0.1 again in order to maintain exponential growth. This process was repeated for a total of 8 days, with samples taken every 2 days for determination of species abundance. DNA was isolated from coculture samples using a GeneJET genomic DNA purification kit (Thermo Fisher Scientific), and species abundance was determined by real-time quantitative PCR as described above, using species-specific primers (Table 4). Bacterial abundances were calculated from CT values by comparison to a standard curve.
Accession number(s).
The new LW13 genome is available at GenBank under accession no. CP033381. The transcriptome sequencing data have been submitted to the Gene Expression Omnibus (GEO) database under accession no. GSE122293.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the U.S. National Institutes of Health (grant K99 GM118762 to A.W.P. and grant R01 GM059026 to E.P.G.) and by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research (award DE-SC-0010556 to M.E.L.). M.W.P. was supported by a postdoctoral fellowship from the Mistletoe Foundation.
We thank A. H. S. Jmaileh and N. Ahmed for assistance with preliminary experiments, N. Smalley for assistance with RNA quality assessment, and members of the Lidstrom and Greenberg laboratory for helpful discussions.
A.W.P., A.L.S., E.P.G., and M.E.L. designed the experiments, A.W.P., D.L., and Z.Y. performed experiments, M.W.P. performed genome resequencing, assembly, and annotation and helped with RNA sequencing analysis, and A.W.P., A.L.S., E.P.G., and M.E.L. wrote and edited the manuscript; all authors have read and approved the final version.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02702-18.
REFERENCES
- 1.Nisbet EG, Dlugokencky EJ, Bousquet P. 2014. Atmospheric science: methane on the rise–again. Science 343:493–495. doi: 10.1126/science.1247828. [DOI] [PubMed] [Google Scholar]
- 2.Singh BK, Bardgett RD, Smith P, Reay DS. 2010. Microorganisms and climate change: terrestrial feedbacks and mitigation options. Nat Rev Microbiol 8:779–790. doi: 10.1038/nrmicro2439. [DOI] [PubMed] [Google Scholar]
- 3.Auman AJ, Stolyar S, Costello AM, Lidstrom ME. 2000. Molecular characterization of methanotrophic isolates from freshwater lake sediment. Appl Environ Microbiol 66:5259–5266. doi: 10.1128/AEM.66.12.5259-5266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L. 2008. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat Biotechnol 26:1029–1034. doi: 10.1038/nbt.1488. [DOI] [PubMed] [Google Scholar]
- 5.Oshkin IY, Beck DA, Lamb AE, Tchesnokova V, Benuska G, McTaggart TL, Kalyuzhnaya MG, Dedysh SN, Lidstrom ME, Chistoserdova L. 2015. Methane-fed microbial microcosms show differential community dynamics and pinpoint taxa involved in communal response. ISME J 9:1119–1129. doi: 10.1038/ismej.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez ME, Beck DAC, Lidstrom ME, Chistoserdova L. 2015. Oxygen availability is a major factor in determining the composition of microbial communities involved in methane oxidation. PeerJ 3:e801. doi: 10.7717/peerj.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z, Chistoserdova L. 2017. Communal metabolism of methane and the rare earth element switch. J Bacteriol 199:e00328-17. doi: 10.1128/JB.00328-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Z, Beck DAC, Chistoserdova L. 2017. Natural selection in synthetic communities highlights the roles of Methylococcaceae and Methylophilaceae and suggests differential roles for alternative methanol dehydrogenases in methane consumption. Front Microbiol 8:2392. doi: 10.3389/fmicb.2017.02392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck DAC, Kalyuzhnaya MG, Malfatti S, Tringe SG, Glavina Del Rio T, Ivanova N, Lidstrom ME, Chistoserdova L. 2013. A metagenomic insight into freshwater methane-utilizing communities and evidence for cooperation between the Methylococcaceae and the Methylophilaceae. PeerJ 1:e23. doi: 10.7717/peerj.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Ha D, Vanwonterghem I, Hoefman S, De Vos P, Boon N. 2013. Selection of associated heterotrophs by methane-oxidizing bacteria at different copper concentrations. Antonie Van Leeuwenhoek 103:527–537. doi: 10.1007/s10482-012-9835-7. [DOI] [PubMed] [Google Scholar]
- 11.Krause SMB, Johnson T, Samadhi Karunaratne Y, Fu Y, Beck DAC, Chistoserdova L, Lidstrom ME. 2017. Lanthanide-dependent cross-feeding of methane-derived carbon is linked by microbial community interactions. Proc Natl Acad Sci U S A 114:358–363. doi: 10.1073/pnas.1619871114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puri AW, Schaefer AL, Fu Y, Beck DAC, Greenberg EP, Lidstrom ME. 2017. Quorum sensing in a methane-oxidizing bacterium. J Bacteriol 199:e00773-16. doi: 10.1128/JB.00773-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalyuzhnaya MG, Lamb AE, McTaggart TL, Oshkin IY, Shapiro N, Woyke T, Chistoserdova L. 2015. Draft genome sequences of gammaproteobacterial methanotrophs isolated from Lake Washington sediment. Genome Announc 3:e00103-15. doi: 10.1128/genomeA.00103-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papenfort K, Bassler BL. 2016. Quorum sensing signal-response systems in Gram-negative bacteria. Nat Rev Microbiol 14:576–588. doi: 10.1038/nrmicro.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whiteley M, Diggle SP, Greenberg EP. 2017. Progress in and promise of bacterial quorum sensing research. Nature 551:313–320. doi: 10.1038/nature24624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puri AW, Mevers E, Ramadhar TR, Petras D, Liu D, Piel J, Dorrestein PC, Greenberg EP, Lidstrom ME, Clardy J. 2018. Tundrenone: an atypical secondary metabolite from bacteria with highly restricted primary metabolism. J Am Chem Soc 140:2002–2006. doi: 10.1021/jacs.7b12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patankar AV, González JE. 2009. Orphan LuxR regulators of quorum sensing. FEMS Microbiol Rev 33:739–756. doi: 10.1111/j.1574-6976.2009.00163.x. [DOI] [PubMed] [Google Scholar]
- 18.Subramoni S, Venturi V. 2009. LuxR-family ‘solos’: bachelor sensors/regulators of signalling molecules. Microbiology 155:1377–1385. doi: 10.1099/mic.0.026849-0. [DOI] [PubMed] [Google Scholar]
- 19.Ahmer BMM. 2004. Cell-to-cell signalling in Escherichia coli and Salmonella enterica. Mol Microbiol 52:933–945. doi: 10.1111/j.1365-2958.2004.04054.x. [DOI] [PubMed] [Google Scholar]
- 20.Chugani SA, Whiteley M, Lee KM, D'Argenio D, Manoil C, Greenberg EP. 2001. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:2752–2757. doi: 10.1073/pnas.051624298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lequette Y, Lee J-H, Ledgham F, Lazdunski A, Greenberg EP. 2006. A distinct QscR regulon in the Pseudomonas aeruginosa quorum-sensing circuit. J Bacteriol 188:3365–3370. doi: 10.1128/JB.188.9.3365-3370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudaiberdiev S, Choudhary KS, Vera Alvarez R, Gelencsér Z, Ligeti B, Lamba D, Pongor S. 2015. Census of solo LuxR genes in prokaryotic genomes. Front Cell Infect Microbiol 5:20. doi: 10.3389/fcimb.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramoni S, Florez Salcedo DV, Suarez-Moreno ZR. 2015. A bioinformatic survey of distribution, conservation, and probable functions of LuxR solo regulators in bacteria. Front Cell Infect Microbiol 5:16. doi: 10.3389/fcimb.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer AL, Lappala CR, Morlen RP, Pelletier DA, Lu T-YS, Lankford PK, Harwood CS, Greenberg EP. 2013. LuxR- and LuxI-type quorum-sensing circuits are prevalent in members of the Populus deltoides microbiome. Appl Environ Microbiol 79:5745–5752. doi: 10.1128/AEM.01417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck DAC, McTaggart TL, Setboonsarng U, Vorobev A, Goodwin L, Shapiro N, Woyke T, Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L. 2015. Multiphyletic origins of methylotrophy in Alphaproteobacteria, exemplified by comparative genomics of Lake Washington isolates. Environ Microbiol 17:547–554. doi: 10.1111/1462-2920.12736. [DOI] [PubMed] [Google Scholar]
- 26.Whitehead NA, Barnard AML, Slater H, Simpson NJL, Salmond GPC. 2001. Quorum-sensing in Gram-negative bacteria. FEMS Microbiol Rev 25:365–404. doi: 10.1111/j.1574-6976.2001.tb00583.x. [DOI] [PubMed] [Google Scholar]
- 27.Zhang R, Pappas KM, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. 2002. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature 417:971–974. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- 28.Heylen K, De Vos P, Vekeman B. 2016. Draft genome sequences of eight obligate methane oxidizers occupying distinct niches based on their nitrogen metabolism. Genome Announc 4:e00421-16. doi: 10.1128/genomeA.00421-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orata FD, Kits KD, Stein LY. 2018. Complete genome sequence of Methylomonas denitrificans strain FJG1, an obligate aerobic methanotroph that can couple methane oxidation with denitrification. Genome Announc 6:e00276-18. doi: 10.1128/genomeA.00276-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egland KA, Greenberg EP. 1999. Quorum sensing in Vibrio fischeri: elements of the luxI promoter. Mol Microbiol 31:1197–1204. doi: 10.1046/j.1365-2958.1999.01261.x. [DOI] [PubMed] [Google Scholar]
- 31.Antunes LCM, Ferreira RBR, Lostroh CP, Greenberg EP. 2008. A mutational analysis defines Vibrio fischeri LuxR binding sites. J Bacteriol 190:4392–4397. doi: 10.1128/JB.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antunes LCM, Schaefer AL, Ferreira RBR, Qin N, Stevens AM, Ruby EG, Greenberg EP. 2007. Transcriptome analysis of the Vibrio fischeri LuxR-LuxI regulon. J Bacteriol 189:8387–8391. doi: 10.1128/JB.00736-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Z, Krause SMB, Beck DAC, Chistoserdova L. 2016. A synthetic ecology perspective: how well does behavior of model organisms in the laboratory predict microbial activities in natural habitats? Front Microbiol 7:946. doi: 10.3389/fmicb.2016.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J-H, Lequette Y, Greenberg EP. 2006. Activity of purified QscR, a Pseudomonas aeruginosa orphan quorum-sensing transcription factor. Mol Microbiol 59:602–609. doi: 10.1111/j.1365-2958.2005.04960.x. [DOI] [PubMed] [Google Scholar]
- 35.Cha C, Gao P, Chen Y-C, Shaw PD, Farrand SK. 1998. Production of acyl-homoserine lactone quorum-sensing signals by Gram-negative plant-associated bacteria. Mol Plant Microbe Interact 11:1119–1129. doi: 10.1094/MPMI.1998.11.11.1119. [DOI] [PubMed] [Google Scholar]
- 36.Duerkop BA, Varga J, Chandler JR, Peterson SB, Herman JP, Churchill MEA, Parsek MR, Nierman WC, Greenberg EP. 2009. Quorum-sensing control of antibiotic synthesis in Burkholderia thailandensis. J Bacteriol 191:3909–3918. doi: 10.1128/JB.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos ELC, Kim HU, Nave M, Dickschat JS, Mitchell DA, Shelest E, Breitling R, Takano E, Lee SY, Weber T, Medema MH. 2017. antiSMASH 4.0: improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45:W36–W41. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 39.Miller WG, Leveau JH, Lindow SE. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 40.Whittenbury R, Phillips KC, Wilkinson JF. 1970. Enrichment, isolation and some properties of methane-utilizing bacteria. J Gen Microbiol 61:205–218. doi: 10.1099/00221287-61-2-205. [DOI] [PubMed] [Google Scholar]
- 41.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 42.Puri AW, Owen S, Chu F, Chavkin T, Beck DAC, Kalyuzhnaya MG, Lidstrom ME. 2015. Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl Environ Microbiol 81:1775–1781. doi: 10.1128/AEM.03795-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen I-MA, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, Huntemann M, Varghese N, White JR, Seshadri R, Smirnova T, Kirton E, Jungbluth SP, Woyke T, Eloe-Fadrosh EA, Ivanova NN, Kyrpides NC. 2019. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res 47:D666–D677. doi: 10.1093/nar/gky901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anders S, Pyl PT, Huber W. 2015. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 49.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.