

ABSTRACT
There have been few new therapies for patients with chronic kidney disease in the last decade. However, the management of patients affected by genetic kidney disease is rapidly evolving. Inherited or genetic kidney disease affects around 10% of adults with end‐stage kidney disease and up to 70% of children with early onset kidney disease. Advances in next‐generation sequencing have enabled rapid and cost‐effective sequencing of large amounts of DNA. Next‐generation sequencing‐based diagnostic tests now enable identification of a monogenic cause in around 20% of patients with early‐onset chronic kidney disease. A definitive diagnosis through genomic testing may negate the need for prolonged diagnostic investigations and surveillance, facilitate reproductive planning and provide accurate counselling for at‐risk relatives. Genomics has allowed the better understanding of disease pathogenesis, providing prognostic information and facilitating development of targeted treatments for patients with inherited or genetic kidney disease. Although genomic testing is becoming more readily available, there are many challenges to implementation in clinical practice. Multidisciplinary renal genetics clinics serve as a model of how some of these challenges may be overcome. Such clinics are already well established in most parts of Australia, with more to follow in future. With the rapid pace of new technology and gene discovery, collaboration between expert clinicians, laboratory and research scientists is of increasing importance to maximize benefits to patients and health‐care systems.
Keywords: genetic kidney disease, genetic testing, genomic testing, inherited kidney disease
Summary at a Glance
The authors reckoned the importance of genomic testing as it allows better understanding of disease pathogenesis, provides prognostic information and facilitates development of targeted treatment, particularly for patients with inherited or genetic kidney disease.
Chronic kidney disease (CKD) has a significant impact on morbidity and mortality in Australia, affecting up to 10–16% of the adult population.1, 2 Despite decades of research, there have been few new therapies for patients with CKD. In contrast, the management of patients affected by inherited or genetic kidney disease (GKD) is rapidly evolving. GKD affects around 10% of adults with end‐stage kidney disease (ESKD)3 and up to 70% of children with early onset CKD,4 highlighting opportunities to optimize the care of a significant proportion of CKD patients through genomics. The most common cause of GKD in adult patients receiving renal replacement therapy is autosomal dominant polycystic kidney disease, which represented 9% of patients in Australia and New Zealand receiving dialysis or transplantation in 2016.5 The prevalence of GKD in the Australian paediatric population is at least 70.6/million aged <20 years, with congenital abnormalities of the kidney and urinary tract (CAKUT) and steroid‐resistant nephrotic syndrome being the most frequent.6 This article aims to provide an overview of the clinical utility, service delivery models and challenges and opportunities relating to the translation of genomics in patients with GKD in an Australian context. To provide an evidence‐based review, we searched PubMed and MEDLINE for original and review articles up until 28th of February 2018.
Next‐generation sequencing (NGS) involves simultaneous sequencing of multiple DNA segments, and may also be referred to as massively parallel sequencing.7 NGS is able to sequence vast quantities of data compared to traditional Sanger Sequencing techniques, although this is associated with higher error rates.8 Advances in NGS in the last decade has enabled rapid and cost‐effective sequencing of large regions of the genome.9 The unit cost price of NGS has reduced faster than other comparator disruptive technologies. For example, the current cost of sequencing a whole human genome using NGS being in the region of $1–2000.10 This does not include the cost of data analysis and interpretation, which remains considerable.
In the research setting, genomic technologies have enabled the identification of new causative genes in GKD,11 improved delineation of conditions12 and elucidated novel targets for therapy.13 Genomic testing technologies are rapidly transitioning from the research to the clinical environment, and it is estimated that genomic data from over 60 000 000 individuals will be generated within healthcare in the next 7 years, worldwide.14 However, many implementation challenges remain, not least demonstrating clinical utility and cost‐effectiveness of genomic testing compared to standard diagnostic care for specific indications, such as renal disease, as well as the development of sustainable models for service delivery.
GENOMIC SEQUENCING AS A DIAGNOSTIC TEST
Most GKD is classified according to their broad phenotypes, such as cystic kidney disease, nephrotic syndrome and immune‐mediated or thrombotic glomerulopathies; however, currently, there are no consensus guidelines which systematically classify these groups. For the purposes of diagnostic testing, classification based on the likely underlying molecular cause allows prioritization of the most relevant genes for analysis. These include glomerular diseases, renal tubular diseases and metabolic diseases, nephrolithiasis, ciliopathies, CAKUT, and disorders of complement.3, 15Monogenic renal disorders are phenotypically diverse, and the number of causative genes is continually expanding. NGS‐based testing now enables identification of a monogenic cause in around 20% of patients with early onset CKD.4 There are several NGS testing modalities currently used, which are summarized in Table 1. NGS panel tests to target a pre‐determined set of genes and detect single‐nucleotide variants (SNV) and small insertions or deletions (indels). Targeted testing reduces the risk of incidental findings; however, it relies on the correct gene panel being selected, and the panel content being regularly updated in light of new gene discoveries.7 In 2013, an expert team of nephrologists, clinical geneticists and molecular geneticists developed an exome‐based panel approach to provide a comprehensive national diagnostic service in Australia. This involved the establishment of 10 ‘virtual’ multi‐gene panels, encompassing 207 known disease‐causing genes, sequenced on a single exome‐based platform. The results of this laboratory service were recently published, demonstrating a diagnostic rate of 43% in 135 families referred over a two‐year period.15 By contrast, whole‐exome sequencing (WES) targets all the coding regions of the genome, and allows more flexible analysis compared with panel sequencing, particularly, for those with non‐specific, complex or overlapping phenotypes. WES data can also be stored and reanalyzed over time in light of new gene discoveries, without the need for additional sequencing.
Table 1.
Testing modalities
| Test | Description | Indications | Example |
|---|---|---|---|
| Chromosomal microarray | Detects unbalanced chromosome abnormalities, Genome wide | Suspect genomic disorder (multi‐organ anomalies) | CAKUT |
| Single Gene Sanger | Detects SNV and small indels (<10 bp) within a DNA segment. Detects conditions associated with variants in one gene | Suspect single‐gene disorder. Confirm NGS findings | Fabry disease |
| Targeted NGS panel | Detection of SNV and small indels (<1 kb) within specified sample of genes. Unable to reanalyze at later date | Suspect condition that affects several discrete genes | Alport syndrome |
| Targeted WES | ‘Virtual panel’ which also detects SNV and small indels (<1 kb) within specified sample of genes. Able to go back and reanalyze as new genes are discovered/ of interest | Suspect condition that affects several discrete genes | Alport syndrome – |
| WES | Detects SNV and small indels (<1 kb) within coding regions of the exome | Suspect condition associated that affects moderate‐large number of genes. Inconclusive phenotype | Nephronophthisis |
| WGS | Detects SNV and small indels within coding and non‐coding regions of the genome | Suspect condition which involves pseudogenes. Inconclusive phenotype | ADPKD |
ADPKD, autosomal dominant polycystic kidney disease; CAKUT, Congenital anomalies of the kidney and urinary tract; Indels: insertions or deletions; NGS: next‐generation sequencing; SNV: single nucleotide variant; WES, whole exome sequencing; WGS: Whole genome sequencing.
Although it provides more comprehensive testing compared with targeted panels, there are potential pitfalls of WES that must be mentioned. These mainly relate to variant interpretation. As there is a large degree of sequence variation within a human exome or genome, there is risk of attributing causality to benign rare variants.16 Although organisations, such as the American College of Medical Genetics and Genomics have well established guidelines for diagnostic interpretation,17, 18, 19 the accuracy of results heavily rely on the phenotypic information and family history provided by the ordering physician and on genotype–phenotype correlation during reporting. Currently, no standards exist for the quality of clinical information that is given prior to testing and who should provide this.20 In addition, genomic tests, such as WES have the potential to identify incidental findings, which are variants unrelated to the primary indication for testing but may have health implications for patients and extended family members. While this is unlikely to occur when there is a narrow phenotypic spectrum, and only limited analysis of the WES data is undertaken, incidental findings are more likely to arise where broader analysis is undertaken in complex cases, depending on the level of consent obtained pre‐test.
Until now, the diagnostic utility of WES in a broad cohort with suspected GKD has only been assessed in a small number of pilot studies. Recently, results of a cohort study demonstrated that WES provided a diagnosis in 22 of 94 (24%) adults referred for suspected inherited CKD or hypertension. This is one of the few studies to date that have also explored the clinical utility of genomic testing in a CKD cohort. The authors highlighted cases where genetic diagnoses lead to direct changes in clinical managements, such as the avoidance of immunosuppression, carrier screening of at‐risk relatives and introduction of auditory and ophthalmologic screening in patients with an initial diagnosis of familial Focal Segmental Glomerular Sclerosis (FSGS) who were found to have COLA3/4/5 mutations.21 In a North American paediatric cohort of 79 consanguineous or familial cases of suspected nephronophthisis, WES found causative mutation(s) in 50 families (63%). While the suspected diagnosis of nephronophthisis was confirmed in most of these cases, 18/50 (36%) were found to have a different molecular diagnosis, such as renal tubulopathies, Alport syndrome and CAKUT.22
Although there is a paucity of data on utility in a broad CKD cohort, several studies have investigated the frequency of mutations in specific renal phenotypes within a research setting. Within a cohort of 1783 unrelated families with SRNS, exon sequencing identified a single gene cause in 29.5%.23 The advent of WES may have improved the diagnostic yield, with a recent study demonstrating a monogenic causative mutation in 15 out of 51 families who presented with suspected nephrolithiasis or nephrocalcinosis before the age of 25 years.24 Studies are ongoing in Australia and internationally. The 100 000 Genomes Project25 in the United Kingdom is expected to complete recruitment and sequencing later this year, and includes a large sub‐cohort of patients with suspected inherited renal diseases who have remained unsolved using standard testing. The data from the project is expected to offer new insights into the pathogenesis of IHD, including the contribution of structural and non‐coding variants.
While WES is currently costly compared with single gene or panel testing, it is a more cost‐effective approach compared to WGS, which interrogates both coding and non‐coding regions, although this difference in cost is likely to change in future. WGS has the advantage of being able to identify copy number and structural variation and provides more uniform coverage of the coding region.26, 27 Currently, several Australian laboratories have accreditation to perform WES as a clinical test. One laboratory has accreditation to perform WGS, with more expected to follow.
WHO SHOULD BE REFERRED FOR GENETIC/GENOMIC TESTING?
Patients should be referred for genetic or genomic testing if it is necessary to confirm a suspected genetic diagnosis, or to clarify or exclude other differential diagnoses.28, 29 A definitive diagnosis may negate the need for prolonged diagnostic investigations and surveillance.7 In addition, it may provide prognostic information, including informing targeted surveillance of extra‐renal manifestations.30, 31 Indications for genetic testing are outlined in Table 2.
Table 2.
Indications for testing
| Indications | Benefits | Cautions/limitations* |
|---|---|---|
| Confirm a suspected diagnosis (e.g. Alport syndrome) | Targeted management of disease (e.g. aHUS) | Is the renal disease likely to be of genetic origin? |
| Clarify/exclude differential diagnoses (e.g. ARTKD/ADTKD, cystic renal disease) | Avoidance of therapies which will not provide benefit (e.g. SRNS) | What is the best test? Consider disease mechanism (e.g. chromosome microarray – HNF1B deletions) |
| Facilitate reproductive options | Avoidance of renal biopsy in proband/relatives (e.g. Alport syndrome) | Identify accredited laboratory to perform test |
| Clarify inheritance in family (e.g. Alport syndrome) | Active surveillance of extra‐renal manifestations (e.g. ADTKD‐HNF1B, syndromic NPHP) | Consider cost of test and identify appropriate funding mechanism |
| Provide prognostic information (e.g. ADPKD) | Obtain appropriate consent including limitations of test, incidental findings, family implications | |
| Reproductive planning (e.g. prenatal genetic diagnosis, preimplantation genetic diagnosis) | Correct clinical interpretation of laboratory results (e.g. variants of unknown significance) | |
| Early identification of at‐risk for relatives | – | |
| Identification of live related kidney donors | – |
ARTKD, autosomal recessive tubulointerstitial kidney disease; ADTKD, autosomal dominant tubulointerstitial kidney disease; aHUS, atypical haemolytic‐uraemic syndrome; HNF1B: hepatocyte nuclear factor 1 beta; NPHP: nephronophthisis; SRNS: steroid‐resistant nephrotic syndrome.
These factors are considered at the multidisciplinary renal genetics clinic.
Confirming or clarifying a genetic diagnosis has demonstrated clinical utility in a variety of situations, especially as GKD can be phenotypically diverse. FSGS is a primary glomerular disease, which is associated with a 50% risk of progressing to ESKD within 5 years of diagnosis if patients do not achieve at least partial remission.32, 33 COL4A3–5 variants causing Alport syndrome have been found in around 10% of families with a clinical diagnosis of hereditary FSGS,34, 35 which highlights the importance of molecular testing in establishing an accurate diagnosis. Confirmation of a genetic diagnosis is also important in the management of atypical haemolytic uremic syndrome, particularly surrounding transplantation. The risk of post‐transplant recurrence is especially high in patients with mutations in complement genes,36 with up to 90% risk if recurrence with those with a CFH mutation.37, 38 Therefore, a genetic diagnosis will assist to inform the decision about when to use prophylactic complement inhibitors in this situation.39 Furthermore, with new treatments, such as tolvaptan emerging for autosomal dominant polycystic kidney disease, it may be necessary to have a precise molecular diagnosis, especially for those participating in therapeutic trials and those without a positive family history to demonstrate accurate results.40
A definitive diagnosis may negate the need for prolonged diagnostic investigations and surveillance in addition to guiding management. For example, confirming a diagnosis of Alport syndrome may negate the need for a renal biopsy for some individuals as well as at‐risk relatives. Accurate and timely diagnosis along with treatment with angiotensin‐converting enzyme inhibitors, when indicated has been shown to improve the long‐term prognosis of Alport syndrome.41, 42 Alport syndrome is traditionally thought of as affecting men, and therefore women are likely to be underdiagnosed. Although women may often have a milder disease course, up to one‐third will develop renal failure.43 Therefore, it is recommended that all women with suspected Alport syndrome should be offered genetic testing to confirm a molecular diagnosis, even if asymptomatic. This will allow prognostic information for surveillance for proteinuria and hypertension and allow accurate reproductive risk counselling.44
Genetic/genomic testing can provide prognostic information, including informing targeted surveillance of extra‐renal manifestations. For example, it is important to screen for diabetes and liver function in patients with an HNF1B mutation,31 which is a disease with a variable multisystem phenotype that can be commonly misdiagnosed.45 Recent data indicates that impaired neurocognitive function in some children with CKD is independent of the severity of kidney disease. This suggests that the genetic lesions have an impact on both kidney and neurocognitive development,46 further highlighting the opportunity for early diagnosis and individual interventions to reduce this effect.
Another important indication for genetic/genomic testing is to facilitate reproductive options. Pre‐implantation genetic testing can be performed during in‐vitro fertilization to select embryos unaffected by a genetic disorder. Furthermore, genetic testing may be used to clarify inheritance patterns in a family. This will allow early identification of at‐risk family members, and release some family members from screening. By doing so, transplant planning can be facilitated by early identification of potential donors. Although genetic testing has been more widely used in paediatric nephrology in the past, more recently diagnostic benefits have also been demonstrated in adults with CKD, with results of an Australian cohort who underwent exome‐based gene panel testing reporting similar diagnostic rates between families with a paediatric versus adult proband (46% vs. 40%).15 The interpretation of these tests is often complex, and therefore often requires the assistance of a clinical geneticist, discussion at a multidisciplinary meeting, or referral to renal genetics service.47
RENAL GENETICS CLINICAL SERVICES AND PROJECTS
While there is an increasing body of evidence of the value of genomic tests in patients with CKD, most of the current evidence is limited to specific disease groups within a research setting .48, 49 Therefore, it is difficult to guide clinical practice until more data becomes available in the clinical setting.15 Application in routine clinical care presents many practical challenges, including but not limited to appropriate patient and test selection, result interpretation, and counselling of extended family members. Access to funded testing is highly variable. Federal funding for genetic testing is limited, and genomic testing is supported by clinical services at a state level or through research studies designed to evaluate the application of genomics in health care.50 Multidisciplinary renal genetics clinics (RGC) are one model of how some of these implementation challenges may be addressed. The current RGC model involves a patient being seen by a clinical nephrologist, clinical geneticist and a genetic counsellor within the one clinic. In 2013, the first multidisciplinary RGC in Australia was established in Brisbane with initial outcomes subsequently reported.47 Since then, 240 patients (22 paediatrics and 218 adults) have been assessed. Referral indications include diagnostic and management opinions, and genetic counselling issues. The clinic has utilized the expertise of the specialists, along with current genomic sequencing technology, to alter the prior clinical diagnosis in 33% of patients and provided them with a clear clinical and/or genetic diagnosis.
Building on the model established in Queensland, the KidGen Collaborative was formed in 2016, with the goal of providing a definitive diagnosis to patients with GKD within a multidisciplinary RGC setting across Australia. The KidGen Consortium has well‐established multidisciplinary RGC located in Queensland, New South Wales and Victoria (Fig. 1). In the last year, services have commenced in South Australia and Western Australia. Multidisciplinary RGC will soon be underway in Darwin and Tasmania, with the aim to provide access to 90% of the Australian population over the next 12 months. Over the next 3 years, KidGen will provide a new standard of care for patients with GKD, including access to a multi‐disciplinary clinic, with genomic testing and genetic counselling for the family where appropriate. KidGen, in conjunction with the Melbourne Genomics Health Alliance and the Australian Genomics Health Alliance is evaluating whether multidisciplinary clinics improve the outcome, patient experience and standard of care for patients with GKD and their families. As genomic medicine is increasingly incorporated into mainstream medical practice, more nephrologists will need to be upskilled in genomics and this multidisciplinary model is likely to evolve.
Figure 1.
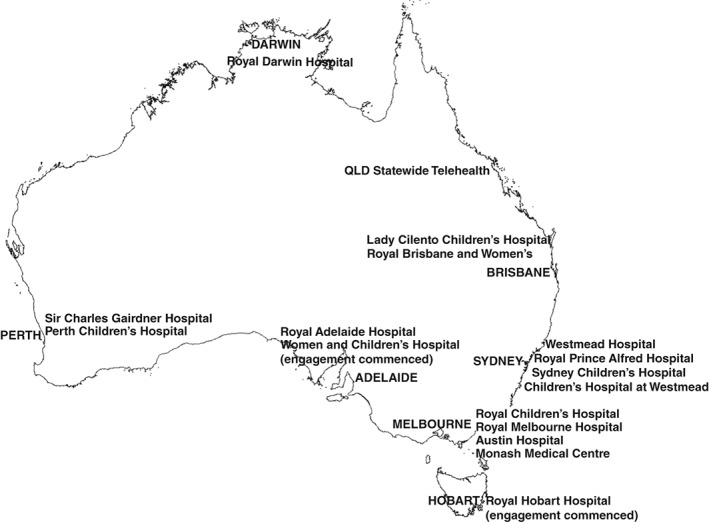
Map of Australian Renal Genetics Flagship (2018).
In Victoria, the establishment of multidisciplinary RGC has been coupled with funding from the Melbourne Genomics Health Alliance for 200 adult and paediatric patients with suspected renal genetic disease to be recruited over 2 years and undergo diagnostic WES. The Victorian cohort is part of a nation‐wide cohort funded by the Australian Genomics Health Alliance, which is expected to comprise 361 patients. Within these projects, multiple sub‐studies are underway, including an implementation science project, which will explore the attitudes and practices of nephrologists regarding genomic testing and analyze practical differences between the function of the multidisciplinary RGC across Australia. The Australian Genomics Health Alliance is a Driver project for the Global Alliance for Genomics and Health (www.ga4gh.org), and has close links with Genomics England, enabling international collaboration to accelerate the implementation of genomics in health care.
In the second half of 2018, a second Australian Genomics‐funded project (‘wHole genome Investigation to iDentify unDEtected Nephropathies (HIDDEN) flagship) will commence recruitment of renal patients with early onset unexplained CKD. The cause of ESKD in Australia is unknown in over 10% of patients.51 Earlier diagnoses may enable specific care prior to development of ESKD and/or predict and influence outcomes post‐transplantation. The HIDDEN flagship will enrol patients with ESKD and no definitive diagnosis with the aim of determining whether genomic sequencing can help to diagnose and better guide clinical management in such patients. The immediate aim is to evaluate 200 participants with unexplained ESKD over the next 24 months. The Flagship will also evaluate the role of dynamic consent and pharmacogenomics in improving management of patients with ESKD.
RESEARCH GENOMICS
While clinical testing helps to establish a definitive diagnosis in many patients with GKD, there are patients who remain undiagnosed. Patients assessed in KidGen RGC in whom genetic testing has been unsuccessful in achieving a diagnosis will be offered recruitment into the National Health and Medical Research Council (NHMRC)‐funded study “NGS and induced pluripotent stem cells (iPSC) applications in genetic renal disease.”52 This research genomics arm will undertake WES and WGS in multiple family members to allow more complete genomic analysis, coupled with functional analysis to validate novel genetic findings. Functional genomics involves the iPSC in modelling kidney disease, leveraging local expertise in the generation of kidney organoids.53 Patient‐derived iPSC will be used to generate renal and relevant extra‐renal tissue in vitro to validate novel genetic findings, understand the underlying the pathophysiology and work towards applying stem cells to cellular therapy.
SCIENTIFIC MEETINGS
The KidGen Renal Genetics Symposium is currently one of the few dedicated renal genetics meetings to be held internationally on an annual basis. In 2017, the 5th annual meeting was held in Melbourne.54 This meeting addressed clinical, diagnostic and research aspects of GKD. More than 100 clinicians, researchers and patient representatives attended the conference. The overall goal was to improve the understanding and direction of genomics in renal medicine in Australia and discuss barriers to the use of genomic testing within this area. The next meeting will be held in Sydney in 2018 in conjunction with the annual scientific meeting of the Australian and New Zealand Society of Nephrology.
CHALLENGES AND FUTURE DIRECTIONS
We face many challenges with the implementation of genomic testing. Most of these challenges apply to all types of rare genetic disease, whereas some challenges are specific to nephrology. While there has been considerable progress in the molecular causes of GKD, with the current diagnostic rate being up to 46%,15 the molecular aetiology for many rare kidney diseases remains to be elucidated. In addition, the use and clinical impact of genomic testing for patients with GKD remains limited. There are limited representative studies on genomic testing in CKD, with even fewer studies evaluating clinical utility. There are many reasons for this; first, patients with rare diseases represent limited sample sizes, which are not feasible to participate in large‐scale randomized studies. Informed consent for genomic testing is lengthier and more complicated compared with other diagnostic trials. Demonstrating clinical utility usually needs a longer duration of follow‐up, which may be unachievable in trials. In addition, the high‐cost and long‐turnaround times of several months prevent its generalized use in clinical practice, resulting in the ongoing need for traditional diagnostic investigations at present. Reassuringly however, costs of genomic tests are diminishing, and turnaround times are reducing. Rare kidney disease is now being recognized as an important issue amongst the international nephrology community, and recently an international conference dedicated to addressing issues on rare kidney disease was held by the Kidney disease: Improving Global Outcomes.55Alternative innovative trial designs are being developed to maximize the opportunities from limited cohorts56, 57 Finally, as genomic tests are becoming more acceptable and is considered as a standard diagnostic investigation, there will be increased participation in clinical trials, thereby improving evidence for efficacy. Nonetheless, even if evidence can demonstrate clinical utility, poor appreciation of genetic studies by health‐care providers remains another challenge.58 There is a lack of literature reporting nephrologists’ knowledge and practice of genomics/genetics; however, themes from other subspecialties include needs for effective education strategies and organizational support, and importance of genetic counsellors in facilitating implementation.59, 60, 61, 62 Current research is looking at some of the barriers to implementation of genomic testing within the nephrology field.
Genomic data interpretation remains a complex, labour‐intensive task, with significant risks for generating both false‐positive and false‐negative results. The identification of variants of uncertain significance, and of secondary or incidental findings unrelated to the reason for testing pose additional clinical challenges.63 Most research in this area suggests that patients wish to be informed of secondary findings, even when limited treatment options are available,64 raising important issues about how to incorporate providing this information within the RGC service delivery model. Patient preferences for genomic testing have mainly been evaluated in the oncology and obstetric setting.65, 66 There is a growing emphasis of the importance of shared decision making in genomic testing,65, 67 which results in improved patient confidence and satisfaction. Incorporating these lessons in the care of patients with GKD and evaluating impact are key priorities. Many ethical and legal issues remain unresolved, including the insurance ramifications of a genetic diagnosis. Countries, such as the United States and Canada have passed laws to protect patients from genetic discrimination,68 while such legislation is yet to be introduced in Australia. Finally, the increased technical ability to generate genomic data needs to be accompanied by the expansion and upskilling of the existing workforce of laboratory scientists, clinical geneticists, genetic counsellors and nephrologists with an interest in genomics in order to fully realise the potential of this technology to improve patient care.
DISCLOSURE
We have no conflict of interest to report.
REFERENCES
- 1. Chadban SJ. Prevalence of kidney damage in Australian adults: The AusDiab kidney study. J. Am. Soc. Nephrol. 2003; 14: 131S–138. [DOI] [PubMed] [Google Scholar]
- 2. White SL, Polkinghorne KR, Atkins RC, Chadban SJ. Comparison of the prevalence and mortality risk of CKD in Australia using the CKD epidemiology collaboration (CKD‐EPI) and modification of diet in renal disease (MDRD) study GFR estimating equations: The AusDiab (Australian diabetes, obesity and lifestyle) study. Am. J. Kidney Dis. 2010; 55: 660–70. [DOI] [PubMed] [Google Scholar]
- 3. Devuyst O, Knoers NV, Remuzzi G, Schaefer F, Board of the Working Group for Inherited Kidney Diseases of the European Renal Association and European Dialysis and Transplant Association . Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014; 383: 1844–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vivante A, Hildebrandt F. Exploring the genetic basis of early‐onset chronic kidney disease. Nat. Rev. Nephrol. 2016; 12: 133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. ANZDATA . ANZDATA Registry 40th Report 2017. Adelaide, Australia: Australia and New Zealand Dialysis and Transplant Registry, 2018. Cited XXX.] Available from URL: http://www.anzdata.org.au. [Google Scholar]
- 6. Fletcher J, McDonald S, Alexander SI, Australian and New Zealand Pediatric Nephrology Association (ANZPNA) . Prevalence of genetic renal disease in children. Pediatr. Nephrol. 2013; 28: 251–6. [DOI] [PubMed] [Google Scholar]
- 7. Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat. Rev. Nephrol. 2018; 14: 83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodwin S, McPherson JD, McCombie WR. Coming of age: Ten years of next‐generation sequencing technologies. Nat. Rev. Genet. 2016; 17: 333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next‐generation sequencing: Impact of exome sequencing in characterizing Mendelian disorders. J. Hum. Genet. 2012; 57: 621–32. [DOI] [PubMed] [Google Scholar]
- 10. Hayden EC. Technology: The $1,000 genome. Nature 2014; 507: 294–5. [DOI] [PubMed] [Google Scholar]
- 11. Bekheirnia MR, Bekheirnia N, Bainbridge MN et al Whole‐exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet. Med. 2017; 19: 412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schenk H, Müller‐Deile J, Kinast M, Schiffer M. Disease modeling in genetic kidney diseases: Zebrafish. Cell. Tissue. Res. 2017; 369: 127–41. [DOI] [PubMed] [Google Scholar]
- 13. Emma F, Nesterova G, Langman C et al Nephropathic cystinosis: An international consensus document. Nephrol. Dial. Transplant. 2014; 29: iv87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birney E, Vamathevan J, Goodhand P. Genomics in healthcare: GA4GH looks to 2022. BioRxiv. 2017. 10.1101/203554 accessed July 2018. [DOI] [Google Scholar]
- 15. Mallett AJ, McCarthy HJ, Ho G et al Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017; 92: 1493–506. [DOI] [PubMed] [Google Scholar]
- 16. MacArthur DG, Manolio TA, Dimmock DP et al Guidelines for investigating causality of sequence variants in human disease. Nature 2014; 508: 469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthijs G, Souche E, Alders M et al Guidelines for diagnostic next‐generation sequencing. Eur. J. Hum. Genet. 2016; 24: 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S et al Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17: 405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rehm HL, Bale SJ, Bayrak‐Toydemir P et al ACMG clinical laboratory standards for next‐generation sequencing. Genet. Med. 2013; 15: 733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strande NT, Berg JS. Defining the clinical value of a genomic diagnosis in the era of next‐generation sequencing. Annu. Rev. Genomics Hum. Genet. 2016; 17: 303–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lata S, Marasa M, Li Y et al Whole‐exome sequencing in adults with chronic kidney disease: A pilot study. Ann. Intern. Med. 2018; 168: 100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Braun DA, Schueler M, Halbritter J et al Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood‐onset increased renal echogenicity. Kidney Int. 2016; 89: 468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sadowski CE, Lovric S, Ashraf S et al A single‐gene cause in 29.5% of cases of steroid‐resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015; 26: 1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daga A, Majmundar AJ, Braun DA et al Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018; 93: 204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turnbull C, Scott RH, Thomas E et al The 100 000 genomes project: Bringing whole genome sequencing to the NHS. BMJ 2018; 361: k1687. [DOI] [PubMed] [Google Scholar]
- 26. Belkadi A, Bolze A, Itan Y et al Whole‐genome sequencing is more powerful than whole‐exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. 2015; 112: 5473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 2014; 15: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valencia C, Husami A, Holle J et al Clinical impact and cost‐effectiveness of whole exome sequencing as a diagnostic tool: A pediatric center's experience. Front Pediatr. 2015; 3: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haskell GT, Adams MC, Fan Z et al Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol. Genet. 2018; 4: e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ellingford JM, Sergouniotis PI, Lennon R et al Pinpointing clinical diagnosis through whole exome sequencing to direct patient care: A case of Senior‐Loken syndrome. Lancet 2015; 385: 1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B‐associated renal and extra‐renal disease‐an expanding clinical spectrum. Nat. Rev. Nephrol. 2015; 11: 102–12. [DOI] [PubMed] [Google Scholar]
- 32. Gipson DS, Chin H, Presler TP et al Differential risk of remission and ESRD in childhood FSGS. Pediatr. Nephrol. 2006; 21: 344–9. [DOI] [PubMed] [Google Scholar]
- 33. Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC, Toronto Glomerulonephritis Registry Group . Focal and segmental glomerulosclerosis: Definition and relevance of a partial remission. J. Am. Soc. Nephrol. 2005; 16: 1061–8. [DOI] [PubMed] [Google Scholar]
- 34. Malone AF, Phelan PJ, Hall G et al Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014; 86: 1253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gast C, Pengelly RJ, Lyon M et al Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016; 31: 961–70. [DOI] [PubMed] [Google Scholar]
- 36. Le Quintrec M, Zuber J, Moulin B et al Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am. J. Transplant. 2013; 13: 663–75. [DOI] [PubMed] [Google Scholar]
- 37. Bresin E, Daina E, Noris M et al Outcome of renal transplantation in patients with non‐Shiga toxin‐associated hemolytic uremic syndrome: Prognostic significance of genetic background. Clin. J. Am. Soc. Nephrol. 2006; 1: 88–99. [DOI] [PubMed] [Google Scholar]
- 38. Sellier‐Leclerc AL, Fremeaux‐Bacchi V, Dragon‐Durey MA et al Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2007; 18: 2392–400. [DOI] [PubMed] [Google Scholar]
- 39. Fox LC, Cohney SJ, Kausman JY et al Consensus opinion on diagnosis and management of thrombotic microangiopathy in Australia and New Zealand. Nephrology (Carlton) 2018; 23: 507–17. [DOI] [PubMed] [Google Scholar]
- 40. Cornec‐Le Gall E, Blais JD, Irazabal MV et al Can we further enrich autosomal dominant polycystic kidney disease clinical trials for rapidly progressive patients? Application of the PROPKD score in the TEMPO trial. Nephrol. Dial. Transplant. 2017; 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gross O, Licht C, Anders HJ et al Early angiotensin‐converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012; 81: 494–501. [DOI] [PubMed] [Google Scholar]
- 42. Temme J, Peters F, Lange K et al Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of Xchromosomal and autosomal recessive Alport mutations. Kidney Int. 2012; 81: 779–83. [DOI] [PubMed] [Google Scholar]
- 43. Jais JP, Knebelmann B, Giatras I et al X‐Linked Alport Syndrome: Natural History and Genotype‐Phenotype Correlations in Girls and Women Belonging to 195 Families: A “European Community Alport Syndrome Concerted Action” Study. Journal of the American Society of Nephrology 2003; 14: 2603–10. [DOI] [PubMed] [Google Scholar]
- 44. Savige J, Colville D, Rheault M et al Alport syndrome in women and girls. Clin. J. Am. Soc. Nephrol. 2016; 11: 1713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verhave JC, Bech AP, Wetzels JFM, Nijenhuis T. Hepatocyte nuclear factor 1beta‐associated kidney disease: More than renal cysts and diabetes. J. Am. Soc. Nephrol. 2016; 27: 345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verbitsky M, Kogon AJ, Matheson M et al Genomic disorders and neurocognitive impairment in pediatric CKD. J. Am. Soc. Nephrol. 2017; 28: 2303–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mallett A, Fowles LF, McGaughran J, Healy H, Patel C. A multidisciplinary renal genetics clinic improves patient diagnosis. Med. J. Aust. 2016; 204: 58–9. [DOI] [PubMed] [Google Scholar]
- 48. Halbritter J, Porath JD, Diaz KA et al Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis‐related ciliopathy. Hum. Genet. 2013; 132: 865–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heidet L, Morinière V, Henry C et al Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 2017; 28: 2901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gaff CL, M. Winship I, M. Forrest S et al Preparing for genomic medicine: A real world demonstration of health system change. NPJ Genom. Med. 2017; 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Australia and New Zealand Dialysis and Transplant Registry. Annual ANZDATA Report. South Adelaide, Australia: ANZDATA Registry, 2016. [Google Scholar]
- 52. Mallett A, Patel C, Maier B et al A protocol for the identification and validation of novel genetic causes of kidney disease. BMC Nephrol. 2015; 16: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takasato M, Er PX, Chiu HS, Little MH. Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 2016; 11: 1681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jayasinghe K, Quinlan C, Stark Z et al Meeting report of the 2017 KidGen Renal Genetics Symposium. Hum. Genomics 2018; 12: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aymé S, Bockenhauer D, Day S et al Common elements in rare kidney diseases: Conclusions from a kidney disease: Improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017; 92: 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Korn EL, McShane LM, Freidlin B. Statistical challenges in the evaluation of treatments for small patient populations. Sci. Transl. Med. 2013; 5: 178sr3–3. [DOI] [PubMed] [Google Scholar]
- 57. Gagne JJ, Thompson L, O'Keefe K, Kesselheim AS. Innovative research methods for studying treatments for rare diseases: Methodological review. BMJ 2014; 349: g6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Riel E, Wárlám‐Rodenhuis CC, Verhoef S, Rutgers EJTH, Ausems MGEM. BRCA testing of breast cancer patients: Medical specialists' referral patterns, knowledge and attitudes to genetic testing. Eur. J. Cancer Care (Engl) 2010; 19: 369–76. [DOI] [PubMed] [Google Scholar]
- 59. Delikurt T, Williamson GR, Anastasiadou V, Skirton H. A systematic review of factors that act as barriers to patient referral to genetic services. Eur. J. Hum. Genet. 2015; 23: 739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Martin G, Currie G, Finn R. Bringing genetics into primary care: Findings from a national evaluation of pilots in England. J. Health Serv. Res. Policy 2009; 14: 204–11. [DOI] [PubMed] [Google Scholar]
- 61. Johnson LM, Valdez JM, Quinn EA et al Integrating next‐generation sequencing into pediatric oncology practice: An assessment of physician confidence and understanding of clinical genomics. Cancer 2017; 123: 2352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chow‐White P, Ha D, Laskin J. Knowledge, attitudes, and values among physicians working with clinical genomics: A survey of medical oncologists. Hum. Resour. Health 2017; 15: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tan N, Amendola LM, O'Daniel JM et al Is “incidental finding” the best term?: A study of patients' preferences. Genet. Med. 2017; 19: 176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brothers KB, East KM, Kelley WV et al Eliciting preferences on secondary findings: The preferences instrument for genomic secondary results. Genet. Med. 2017; 19: 337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaphingst KA, Ivanovich J, Lyons S et al Preferences for learning different types of genome sequencing results among young breast cancer patients: Role of psychological and clinical factors. Transl. Behav. Med. 2018; 8: 71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Agatisa PK, Mercer MB, Mitchum A, Coleridge MB, Farrell RM. Patient‐centered obstetric Care in the age of cell‐free fetal DNA prenatal screening. J. Patient Exp. 2018; 5: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vadaparampil ST, Cragun D. Shared decision making: Implications for return of results from whole‐exome and whole‐genome sequencing. Transl. Behav. Med. 2018; 8: 80–4. [DOI] [PubMed] [Google Scholar]
- 68. Joly Y, Feze IN, Song L, Knoppers BM. Comparative approaches to genetic discrimination: Chasing shadows? Trends Genet. 2017; 33: 299–302. [DOI] [PubMed] [Google Scholar]