

Abstract
Misfolded protein toxicity and failure of protein quality control underlie neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Here, we identified Lethal(3)malignant brain tumor-like protein 1 (L3MBTL1) as a previously unknown regulator of protein quality control, the loss of which protected against the proteotoxicity of mutant SOD1 or C9orf72 dipeptide repeat proteins. L3MBTL1 acts by regulating p53-dependent quality control systems that degrade misfolded proteins. SET domain-containing protein 8 (SETD8), a L3MBTL1-associatd p53-binding protein, also regulated clearance of misfolded proteins and was increased by proteotoxicity-associated stresses in mammalian cells. Both L3MBTL1 and SETD8 were up-regulated in the central nervous systems of mouse models of ALS and human ALS/FTD patients. The role of L3MBTL1 in protein quality control is conserved from C. elegans to mammalian neurons. These results indicate a previously unrecognized pathway in both normal stress response and proteotoxicity-associated neurodegenerative diseases.
Keywords: ALS, FTD, L3MBTL1, Neurodegeneration, Proteotoxicity, SETD8
Introduction
Protein homeostasis is vital for cellular fitness and is a crucial factor in human diseases including cancer and neurodegenerative diseases such as ALS and FTD1, 2. ALS is characterized by progressive motor neuron loss that leads to muscular atrophy, paralysis, and respiratory failure. FTD is the second most common cause of dementia after Alzheimer’s disease in patients younger than 65. Genetic and pathological studies have suggested that ALS and FTD are two related diseases in a continuous clinical spectrum3. Perturbation in protein homeostasis is a common theme for ALS-associated proteins, including Cu/Zn superoxide dismutase (SOD1)4–6. The most common form of ALS/FTD identified to date is linked to a hexanucleotide repeat expansion (HRE), (GGGGCC)n, in a noncoding region of C9orf727, 8. RNAs containing the expanded HRE repeat can be toxic9, 10. In addition to loss of C9orf72 protein function and accumulation of RNAs containing the expanded HRE repeat that have been implicated in the disease9–11, the expanded repeat can initiate translation in the absence of the ATG start codon through a poorly understood mechanism, producing toxic polydipeptide proteins12–14.
To guard against proteotoxicity, the cell coordinates several major quality control machineries, including molecular chaperones, the ubiquitin-proteasome system, and autophagy, through exquisite regulatory systems. Previously, we observed that inactivation of both a ubiquitin ligase, ubiquitination factor E4B (UBE4B), and lysine-specific demethylase 1 (LSD1) promotes degradation of misfolded proteins by activating a p53-dependent pathway15. The p53 transcription factor, best known for its role in DNA damage responses, was found to promote protein quality control under these experimental conditions15. An independent study reported that protein misfolding stress activates p53-mediated transcription that is distinct from HSF1-mediated transcription16. However, the regulatory mechanisms within the network of protein quality control are not fully understood, and new key players are yet to be uncovered. Here we report the findings of previously unrecognized regulators of protein quality control for ALS/FTD-associated proteins including mutant SOD1 and C9orf72 dipeptide repeat proteins as well as dysregulated proteotoxic stress responses in relevant neurodegenerative diseases.
Results
To gain insight into the genes and pathways involved in neurodegeneration, we screened mutagenized C. elegans for suppressors of locomotion phenotypes induced by mutant SOD1G85R and selected the animals with rare salient improvement in the locomotion against a background of poorly moving non-suppressed progeny using a previously established model and screening method15, 17 (Fig. 1a). We identified one particular strain showing potent suppression of the locomotion defect when compared to the parental SOD1G85R transgene line, reaching >80% of the locomotion robustness of the SOD1WT line (Fig. 1b), without reducing the SOD1G85R transgene mRNA level (Supplementary Fig. 1a). We mapped the suppressor mutation and identified one missense mutation, G1539A, in the gene lin-61, that resulted in a single amino acid change (H358Y) in one of the encoded protein’s malignant brain tumor (MBT) repeats (Fig. 1d). LIN-61 protein is broadly expressed in C. elegans and has reported involvement in transcription and DNA repair18. To demonstrate that loss of lin-61 was responsible for the suppressor phenotype, we examined the effects of an independent allele, lin-61(n3809), which has a C->T substitution in codon 159 that generates an ochre stop codon, resulting in a truncated LIN-61 protein lacking the MBT2, MBT3, and MBT4 domains18. When crossed to the mutant SOD1 strain, lin-61(n3809) recapitulated the strong locomotor defect-suppressing phenotype observed in the original suppressor strain (Fig. 1b). These results confirmed that loss of function of lin-61 suppresses neurodegenerative phenotypes in the SOD1G85R C. elegans model of ALS.
Figure 1. Identification of lin-61 as a robust suppressor that ameliorates the proteotoxicity in the C. elegans model of SOD1-associated ALS.
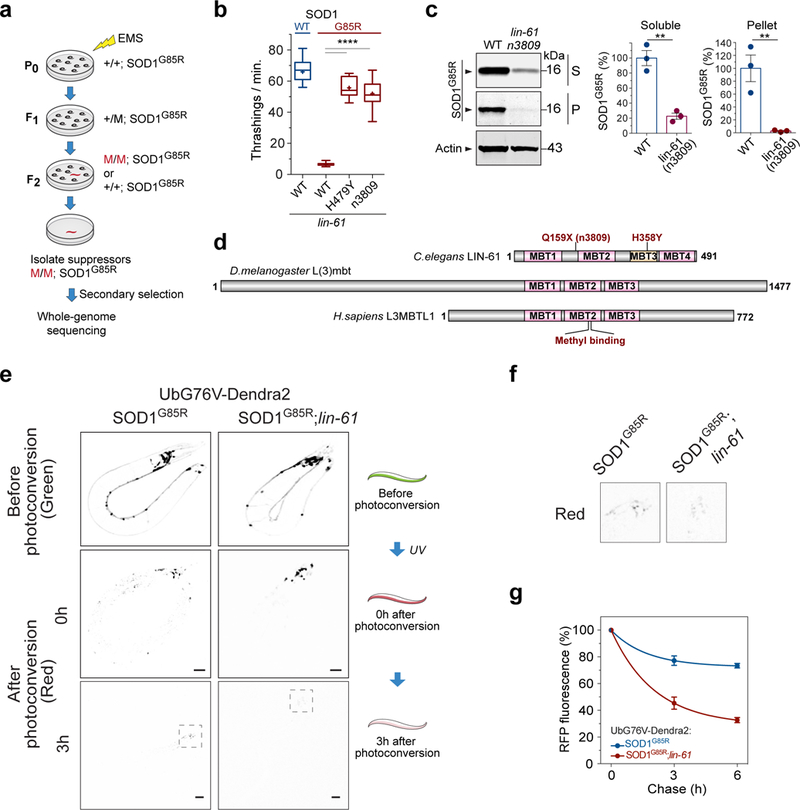
(a) Work flow of the suppressor screen that identified mutant C. elegans (red) with saliently improved locomotion. (b) Locomotor behavior of C. elegans with neuronal expression of the human, ALS-linked mutant SOD1G85R, or the wild type SOD1WT in the presence of the suppressor mutation lin-61(H358Y) or a loss-of-function mutation lin-61(n3809) as compared with the control background (WT). (“+” = mean; whiskers = min. to max.; n=15 worms; **** P<0.0001). (c) Western blot analysis of soluble (S) and pellet (P) protein fractions from the WT and lin-61(n3809) mutant worms. (n=3 independent cultures; ** P=0.002 for the soluble fraction; ** P=0.009 for the pellet fraction). (d) MBT domain proteins LIN-61, L(3)mbt, and L(3)MBTL1 across different species. (e) C. elegans with neuronal expression of UbG76V-Dendra2 reporter were crossed to SOD1G85R worms in the presence or absence of loss-of-function mutation lin-61(n3809) to examine the rate of protein degradation in vivo. Representative fluorescence images of live C. elegans before and after photoconversion of the green Dendra2 (Green) into photo-switched red Dendra2 (Red). The level of red fluorescence after photoconversion decreases faster in the presence of lin-61(n3809) than in its absence, indicating a faster degradation of the Dendra2-UbG76V protein. Scale bar: 100 μm. (f) Enlarged panel sections from the red channel at the 3-h time point in (e), enclosing worm head areas. (g) Quantification of red (RFP) fluorescence during the chase following the conversion, showing significant difference with or without lin-61(n3809) at both the 3-h and 6-h time points (n=3 independent groups of worms, P≤0.014 for each time point). Error bars represent ± SEM.
The solubility of SOD1G85R was measured by detergent extraction of the C. elegans, with large SOD1G85R protein aggregates being sedimented in the insoluble pellet (~3% of the total SOD1G85R), and soluble SOD1G85R including its misfolded monomers and small oligomers remaining in the supernatant (Supplementary Fig. 1b). The presence of lin-61(n3809) resulted in a significant reduction in the amount of SOD1G85R protein in both the supernatant and the pellet (Fig. 1c). We then examined a different strain that expressed the SOD1G85R-YFP fusion protein, which forms visible aggregates and displays strong neurotoxicity in C. elegans17. Again, the null allele lin-61(n3809) significantly reduced the amount of aggregated SOD1G85R-YFP protein in the C. elegans neurons, while not decreasing the level of SOD1G85R-YFP mRNAs (Supplementary Fig. 1c,d). As controls, lin-61(n3809) did not affect the protein levels of C. elegans endogenous WT SOD-1, neuronally expressed human WT SOD1, or a poly-glutamine reporter (Supplementary Fig. 1e-g), suggesting that the regulation of lin-61 is selective to certain misfolded proteins.
To examine the general protein degradation capacity of the lin-61(n3809) strain, we employed a neuronally expressed reporter, UbG76V-Dendra2, which consists of the photoconvertible fluorescent protein Dendra2 fused to the G76V mutant form of ubiquitin (UbG76V), a degradation signal that cannot be cleaved by a ubiquitin hydrolase19. Since the photoconversion of Dendra2 from green to red fluorescent protein is an irreversible post-translational event, the red fluorescence of the UbG76V-Dendra2 reporter allows for quantitation of the rate of protein degradation independently of protein synthesis. We crossed the strain carrying the neuronal UbG76V-Dendra2 reporter to the SOD1G85R transgenic strain and measured the turnover rate of the UbG76V-Dendra2 protein in the presence or absence of the suppressor mutation lin-61(n3809) (Fig. 1e,f, and Supplementary Fig. 2). The half-life of photoconverted UbG76V-Dendra2, as measured with the red fluorescence, was approximately 2.5 h in the presence of lin-61(n3809) but was significantly longer (>6 h) in the SOD1G85R parental controls (Fig. 1g). The accelerated degradation of UbG76V-Dendra2 indicated a generally enhanced proteasomal degradation capacity in the worm neurons, as conferred by lin-61(n3809).
Loss of L3MBTL1 enhances the clearance of misfolded proteins
LIN-61 is a protein with malignant brain tumor (MBT) repeat domains, and its homologs including are present in most metazoans (Fig. 1d, Supplementary Fig. 1h)20. To determine whether the human homolog L3MBTL1 affects protein aggregation in mammalian cells, we used a cell-based protein aggregation assay that utilizes aggregation-prone human SOD1G85R protein as a reporter. Mammalian cell lysates were subjected to detergent extraction and fractionation; the endogenous SOD1WT protein remained in the soluble supernatant, but a portion of the SOD1G85R protein (~30%) was enriched in the insoluble, aggregate-containing pellet fraction (Fig. 2a). The SOD1G85R protein migrates faster than SOD1WT on SDS-PAGE gels, making it amenable to quantification6, 21. SOD1G85R was transfected into HEK293 cells treated with either non-targeting control shRNAs or L3MBTL1 shRNAs to reduce the expression of L3MBTL1 (Fig. 2a). The reduction of L3MBTL1 significantly decreased the levels of misfolded and aggregated SOD1G85R in both the soluble and insoluble fractions (Fig. 2a), with similar changes in the levels of the total SOD1G85R protein (Supplementary Fig. 3a) but no effect on the level of endogenous SOD1WT protein (Fig. 2a,b and Supplementary Fig. 3b). The effect of reducing L3MBTL1 on SOD1G85R protein was confirmed by independent shRNAs against L3MBTL1. Conversely, over-expression of L3MBTL1 in the same assay increased the level of mutant SOD1 proteins but not SOD1WT protein (Fig. 2c,d), confirming that loss of L3MBTL1 is positively associated with reduction of the misfolded mutant SOD1 proteins.
Figure 2. Reduction of human L3MBTL1 accelerates proteasome-mediated protein degradation.
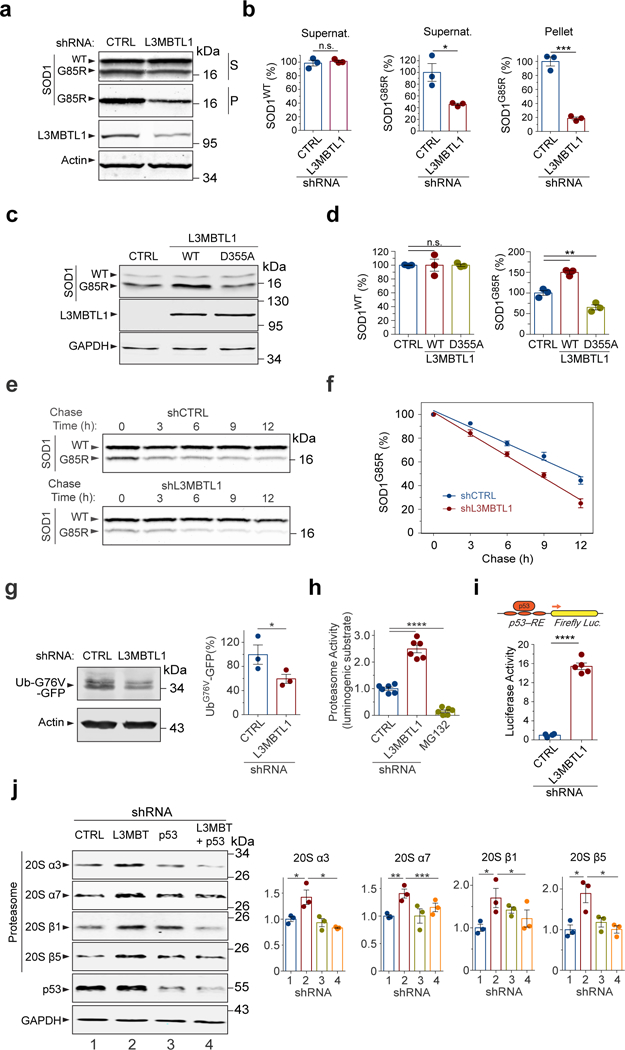
(a) Western blotting analysis of supernatant (S) and pellet (P) fractions from HEK293 cells treated with control (CTRL) or L3MBTL1 shRNAs. (b) Quantification of WT and G85R mutant SOD1 protein levels by western blotting is shown (n=3 independent cultures; n.s., non-significant, P=0.577; * P(S)=0.0235, *** P(P)=0.0003). (c) Overexpression of Flag-tagged human WT L3MBTL1 increased SOD1G85R proteins compared to the control expression of β-glucuronidase (GUS) in HEK293 cells, while the D355A mutation abolished the effects of WT L3MBTL1. (d) Quantification of WT and G85R mutant SOD1 protein levels by western blotting in (c) (n=3 independent cultures; n.s., P>0.999; ** P=0.002). (e) Western blots of a representative cycloheximide chase experiment to determine the SOD1 protein half-life in the L3MBTL1 shRNA-treated cells versus controls. (f) Quantification of the SOD1G85R clearance, as analyzed by western blotting in (e). (n=3 independent experiments; P≤0.01 for each time point). (g) Proteasome degradation assay using destabilized fusion protein UbG76V-GFP as a readout. Western blots and quantification of full length UbG76V-GFP in HEK293 cell treated with L3MBTL1 or control shRNAs (n=3 independent cultures; * P=0.041). (h) Proteasome activities in HEK293 cells treated with L3MBTL1 or control shRNAs, as measured by a luciferase-based assay (n=6 independent cultures; **** P<0.0001). (i) p53 transcriptional activity is increased in L3MBTL1 shRNA-treated cells, as measured by the p53 response element (p53-RE)-mediated luciferase activity assay (n=4 independent cultures; **** P<0.0001). (j) Western blots of proteasome subunits 20S-α3, -α7, -β1, and -β5, and quantification of subunit protein levels in the cells treated with control (1), L3MBTL1 (2) or p53 (3), and double (4) shRNAs (n=3 independent experiments; * P(α3,1vs2)=0.045; * P(α3,2vs4)=0.013; ** P(α7,1vs2)=0.0089, *** P(α7,2vs4)=0.0008, * P(β1,1vs2)=0.0391, * P(β1,2vs4)=0.011, * P(β5,1vs2)=0.026, * P(β5,2vs4)=0.023). Error bars represent ± SEM.
Since L3MBTL1 was reported to bind p5322, a positive regulator of protein quality control15, 16, we asked if the interaction between L3MBTL1 and p53 is required for the effects of L3MBTL1 on the regulation of mutant protein levels. Consistent with a previous report22, we observed that a point mutation in the second MBT domain of L3MBTL1, D355A, decreased the binding between L3MBTL1 and endogenous p53 in co-immunoprecipitation assays in HEK293 cells (Supplementary Fig. 3c). Notably, the introduction of D355A mutation abolished the effects of WT L3MBTL1 on the regulation of mutant SOD1 protein levels (Fig. 2c,d), indicating that the residue of L3MBTL1 that enables it to bind methylated p53 is required for its activity in regulating SOD1 protein quality control. Furthermore, the reduction of L3MBTL1 did not decrease the level of a polyQ reporter protein in HEK293 cells (Supplementary Fig. 3d), suggesting that the regulation of L3MBTL1 on protein degradation is selective.
To determine whether the decrease in the steady-state levels of misfolded and aggregated proteins was a consequence of increased protein degradation, we performed cycloheximide chase experiments to measure the turnover rate of the misfolded proteins. HEK293 cells were transfected with SOD1G85R together with either non-targeting control shRNAs or L3MBTL1 shRNAs, and the clearance of the SOD1G85R protein was quantified. The reduction of L3MBTL1 decreased the half-life of SOD1G85R from 11.5 h to 8.4 h, indicating that accelerated degradation of the mutant protein underlies the reduction in the SOD1G85R protein (Fig. 2e,f). As a control, the half-life of endogenous SOD1WT protein was not affected (Fig. 2e and Supplementary Fig. 3e), suggesting that loss of L3MBTL1 enhanced the quality control and clearance of misfolded proteins.
Next we asked whether the proteasomal activity was affected by L3MBTL1 reduction. A mammalian reporter UbG76V-GFP, which is degraded by the proteasome, was used as a signal for the proteasomal activity (Supplementary Fig. 3f)23. The reduction of L3MBTL1 significantly decreased the level of UbG76V-GFP protein in HEK293 cells, suggesting that the enhanced clearance of misfolded proteins associated with the reduction of L3MBTL1 is due to increased proteasomal activity (Fig. 2g). To confirm that the proteasomal activity is increased upon reduction of L3MBTL1, we employed Proteasome-Glo luminogenic proteasome substrates to measure Chymotrypsin-like proteasome activity (Supplementary Fig. 3g). When HEK293 cells were treated with L3MBTL1 shRNAs, the measured proteasome activity was increased more than twofold compared with cells treated with control shRNAs (Fig. 2h). Treatment with the proteasome-specific inhibitor MG132 completely abolished proteasome activity, confirming that the effect of reduction of L3MBTL1 was dependent on the proteasome (Fig. 2h).
To examine if the increase in proteasomal activity induced by reduction of L3MBTL1 is a result of upregulation of the proteasome levels, we measured the amounts of representative proteasome subunits in cells treated with L3MBTL1 or control shRNAs. The levels of proteasome subunits were generally increased in HEK293 cells treated with L3MBTL1 shRNAs, as shown by the western blot analysis and quantification of the core subunits (20S-α3, -α7, -β1, and -β5) (Fig. 2j). Since L3MBTL1 binds p5322, which can act as a transcription factor positively regulating protein quality control machineries15, 16, we asked whether the regulation of L3MBTL1 on proteasomal subunit levels depends on p53. Reducing p53 by shRNAs ablated the increase in the levels of proteasome subunits induced by the reduction of L3MBTL1 (Fig. 2j). Consistent with the observation of p53 acting downstream of L3MBTL1, the reduction of L3MBTL1 led to activation of p53 activity, as measured by the p53 response element (p53-RE)-mediated luciferase activity assay (Fig. 2i). These results suggest that L3MBTL1 modulates protein quality control, at least in part, via p53-dependent upregulation of the proteasome machinery.
SETD8 is up-regulated in neurodegeneration and regulates protein quality control
SETD8 is a methyltransferase that methylates K382 of p5324, which is then bound by L3MBTL1 via the methylated lysine22. We observed that the endogenous SETD8 protein level was significantly increased in HEK293 cells overexpressing SOD1G85R when compared with the SOD1WT-overexpressing cells (Fig. 3a). Furthermore, the expression of SOD1G85R but not SOD1WT increased the level of p53 protein (Fig. 3a).
Figure 3. SETD8 is up-regulated in response to proteotoxicity-associated neurodegeneration and regulates protein quality control.
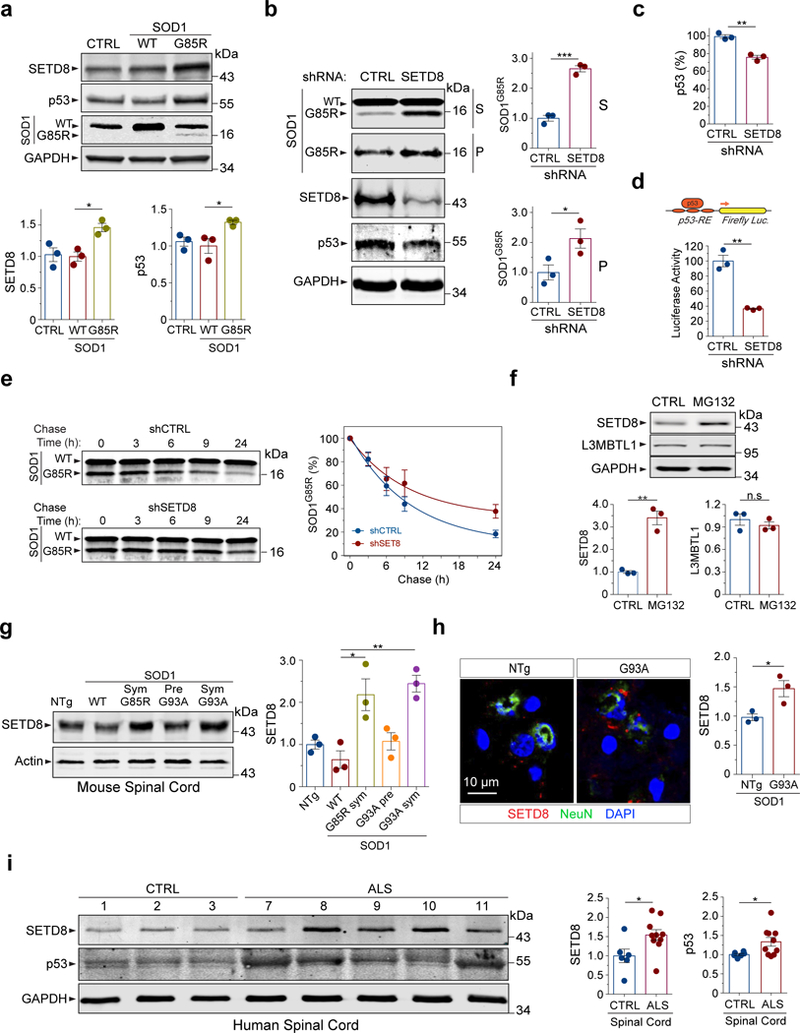
(a) Expression of mutant SOD1G85R but not SOD1WT in HEK293 cells increases the levels of both SETD8 and p53 proteins (n=3 independent cultures; * P(SETD8)=0.035, * P(p53)=0.03). (b) Reduction of SETD8 leads to accumulation of SOD1G85R protein in both supernatant (S) and pellet (P) fractions, while not affecting endogenous SOD1WT protein levels in HEK293 cells (n=3 independent cultures; *** P(S)=0.0003, * P(P)=0.049). Reduction of SETD8 also decreases the levels of p53 protein. (c) Quantification of the p53 levels in SETD8 shRNA-treated cells is shown (n=3 independent cultures; ** P=0.0015). (d) p53 transcriptional activity is reduced in SETD8 shRNA-treated cells (n=3 independent cultures; ** P=0.0012). (e) Western blots of a representative cycloheximide chase experiment to determine the SOD1 protein half-life in the SETD8 shRNA-treated cells versus controls (n=3 independent cultures; P=0.044). (f) Treatments with the proteasome inhibitor MG132 (20 µM for 1 h) increase the level of SETD8 protein but not L3MBTL1 in HEK293 cells (n=3 independent cultures; ** P(SETD8)=0.0014; n.s., P(L3MBTL1)=0.71). (g) The SETD8 protein is up-regulated in the spinal cord tissues from the mouse models of SOD1-linked ALS in a mutant SOD1- and symptom-dependent manner (n=3 sets of male mice; * P=0.023; ** P=0.0033). NTg (non-transgenic, 6 months old); WT (SOD1WT-YFP transgenic, 9–15 months old); Sym G85R (symptomatic SOD1G85R-YFP transgenic, 10–15 months old); Pre G93A (presymptomatic SOD1G93A, 2–4 months old); Sym G93A (symptomatic SOD1G93A, 4–6 months old). (h) Representative immunofluorescence staining shows increased levels of SETD8 in spinal cord neurons of symptomatic SOD1G93A transgenic mice compared to age- and gender-controlled non-transgenic mice (n=3 pairs of mice; * P=0.03). (i) Western blotting of human spinal cord tissue lysates derived from ALS/FTD patients and healthy controls show up-regulation of SETD8 and p53 in the patients’ tissues (n=6 for control and n=10 for patients; * P(SETD8)=0.03; * P(p53)=0.047). Error bars represent ± SEM.
To determine whether SETD8 regulates the clearance of misfolded proteins, we evaluated the turnover of the SOD1G85R protein in HEK293 cells when SETD8 was inactivated. SETD8 was reduced by specific shRNAs, and its inactivation was verified by the reduction of H4K20me1 as one of the enzymatic products of SETD8 (Supplementary Fig. 4a). When SETD8 was inactivated, western blots showed a nearly three-fold increase in the level of soluble SOD1G85R and a two-fold increase in the level of insoluble SOD1G85R (Fig. 3b), while not changing the level of endogenous SOD1WT protein (Supplementary Fig. 3b). Interestingly, the reduction of SETD8 led to a decrease in the level of p53 protein (Fig. 3b,c), suggesting that SETD8 is a positive regulator of p53. The reduction of SETD8 significantly decreased the transcriptional activity of p53 as measured by the p53-luciferase transcriptional assay (Fig. 3d). Furthermore, consistent with the regulation of SETD8 on p53, the interaction between the two proteins was confirmed by co-immunoprecipitation experiments (Supplementary Fig. 4b).
To investigate whether the increase in the steady-state level of SOD1G85R upon SETD8 reduction was due to a less efficient clearance, we examined the half-life of the protein following cycloheximide-induced blockade of translation. The post-translational clearance of misfolded SOD1G85R, but not the SOD1WT protein, was less efficient under the SETD8 shRNA-treated conditions when compared with the control (Fig. 3e). The half-life of the SOD1G85R protein was increased from ~10 h to ~22 h when SETD8 was reduced. As a control, the half-life of endogenous SOD1WT protein was not affected by the reduction of SETD8 (Fig. 3e, Supplementary Fig. 4c).
Further evidence suggests that the SETD8-mediated methylation promotes the stability of p53 (Supplementary Fig. 4d,e). When WT p53 or its methylation-null mutant K382R was expressed in HEK293 cells, the steady-state level of K382R mutant was consistently lower than its WT counterpart (Supplementary Fig. 4d,e). Overexpression of SETD8 increased the level of WT p53 protein but not that of K382R mutant (Supplementary Fig. 4d), suggesting that SETD8 positively regulates p53 protein levels through its methylation. Notably, when the level of p53 protein decreased with the K382R mutation, there was less co-expressed SETD8 protein accumulated in the cell (Supplementary Fig. 4d), consistent with the notion that p53 interacts with SETD8 (Supplementary Fig. 4b) and that their interaction is associated with their stability. Treatment with the proteasomal inhibitor MG132 increased the levels of K382R mutant p53 and SETD8 (Supplementary Fig. 4d), suggesting that the reduction in their protein levels was due to their instability and degradation. The faster turnover of K382R mutant p53, compared to WT p53, was confirmed in cycloheximide chase experiments (Supplementary Fig. 4e). Thus, these data suggest that SETD8 methylates p53 protein and in turn regulates its stability. Furthermore, the regulation of p53 level by SETD8 was dependent on L3MBTL1. When SETD8 was reduced by its specific shRNA, the p53 protein level was consistently decreased; however, when both L3MBTL1 and SETD8 were simultaneously reduced by their double shRNAs, the effect of SETD8 on p53 was blocked, and the p53 protein returned to control levels (Supplementary Fig. 4f). Accordingly, the increase in the SOD1G85R protein level upon the reduction of SETD8 alone was reversed when simultaneous reduction of both L3MBTL1 and SETD8 were implemented (Supplementary Fig. 4f). Together, these data suggest that the regulations of p53 and SOD1G85R proteins by SETD8 were at least partially influenced by L3BMTL1, consistent with the notion that the interactions of p53 with both SETD8 and L3MBTL1 mediate the turnover of the misfolded SOD1G85R protein.
Interestingly, the SETD8 protein level was significantly increased upon proteotoxic stress induced by the proteasome inhibitor MG132 in HEK293 or mouse embryonic fibroblast cells (Fig. 3f, Supplementary Fig. 5a,b), while the L3MBTL1 protein level was not changed. The increase in the SETD8 protein upon the MG132 treatment was associated with a significant increase in its mRNA levels, suggesting that SETD8 was transcriptionally up-regulated in response to proteotoxic stress (Supplementary Fig. 5c).
To investigate whether the regulation of SETD8 in response to protein misfolding stress is relevant to neurodegeneration in vivo, we examined endogenous SETD8 levels in several mouse models of SOD1-linked ALS. We analyzed spinal cord tissues from transgenic mice expressing SOD1G93A, SOD1G85R-YFP, and SOD1WT-YFP as well as non-transgenic control mice25, 26. Western blot analysis indicated a significant increase in the levels of SETD8 protein in the whole spinal cords of symptomatic SOD1G93A and SOD1G85R-YFP mice but not in non-transgenic, SOD1WT-YFP transgenic, or presymptomatic SOD1G93A mice (Fig. 3g). Consistent with the changes of SETD8 protein levels, the level of methylated histone H4K20me1, an enzymatic product of SETD8-mediated methylation, was increased in the spinal cords in a mutant SOD1- and symptom-dependent manner (Supplementary Fig. 5d). Furthermore, the upregulation of SETD8 in neurons was confirmed by immunofluorescence staining in the spinal cords of symptomatic SOD1G93A mice (Fig. 3h).
To extend our findings to human patients, we analyzed extracts of tissues from the central nervous systems of ALS/FTD patients. When spinal cords were examined by western blotting for ten patients and six healthy controls, we found that the level of SETD8 protein was consistently elevated in the patients, including two harboring the C9orf72 hexanucleotide repeat expansion, one carrying the SOD1 L145F mutation, and one carrying the profilin 1 G118V mutation (Fig. 3i, Supplementary Fig. 5e, and Supplementary Table 1). Moreover, p53 protein levels were increased in the spinal cords from the patients compared to the healthy controls (Fig. 3i). The unaffected brain regions of the occipital lobes in these patients were examined as controls and they did not show any change in the level of SETD8 (Supplementary Fig. 5f), suggesting that the upregulation of SETD8 was associated with ALS pathology in the spinal cords in these patients. As disease-specific controls, inferior parietal lobules of brain cortexes from seven patients with Alzheimer’s disease and four healthy controls were examined and the levels of SETD8 proteins were found to be decreased (Supplementary Fig. 5g). Interestingly, L3MBTL1 protein levels were also increased in the spinal cords of symptomatic SOD1G93A mice and a subset of the ALS/FTD patients (Supplementary Fig. 6a-d), suggesting that L3MBTL1-mediated protein quality control pathways are compromised in ALS/FTD-associated neurodegenerative conditions.
L3MBTL1 and SETD8 homologs modulate proteotoxicity in Drosophila
To evaluate the conserved effects of L3MBTL1 and SETD8 in an in vivo system of proteotoxicity-associated neurodegeneration, we studied their homologs in Drosophila. A Drosophila model expressing ALS-associated human mutant SOD1-A4V in motor neurons under the D42 driver developed deficits in climbing assays measuring the locomotor function27. Using a transgenic strain with stable RNAi-induced reduction of the Drosophila homolog of human L3MBTL1, l(3)mbt, we found that the reduction of l(3)mbt produced a significant rescue of the mutant SOD1-induced climbing deficits (Fig. 4a). The reduction in neurotoxicity was consistent with decreased levels of mutant SOD1 proteins as measured by western blot assays, suggestive of enhanced degradation of the mutant SOD1 protein (Fig. 4b). Next, we asked whether the Drosophila homolog of human SETD8, Pr-set7, modulates the mutant SOD1-induced neurotoxicity by crossing the strain with stable RNAi-induced reduction of Pr-set7 to the fly models expressing mutant SOD1 in motor neurons. The reduction of Pr-set7 led to a significant deterioration of the climbing deficits of the mutant SOD1 flies (Fig. 4a), which is consistent with increased accumulation of the mutant SOD1 proteins (Fig. 4b), suggestive of impaired clearance. To test whether dp53, the Drosophila homolog of human p5328, 29, also affects SOD1 proteotoxicity, we crossed a stable dp53 RNAi strain to the mutant SOD1 flies, and observed that the reduction of dp53 significantly worsened the climbing deficits as well as increased the accumulation of mutant SOD1 protein (Fig. 4a,b), consistent with the role of p53 as a positive regulator of protein quality control.
Figure 4. Effects of protein quality control factors in Drosophila.
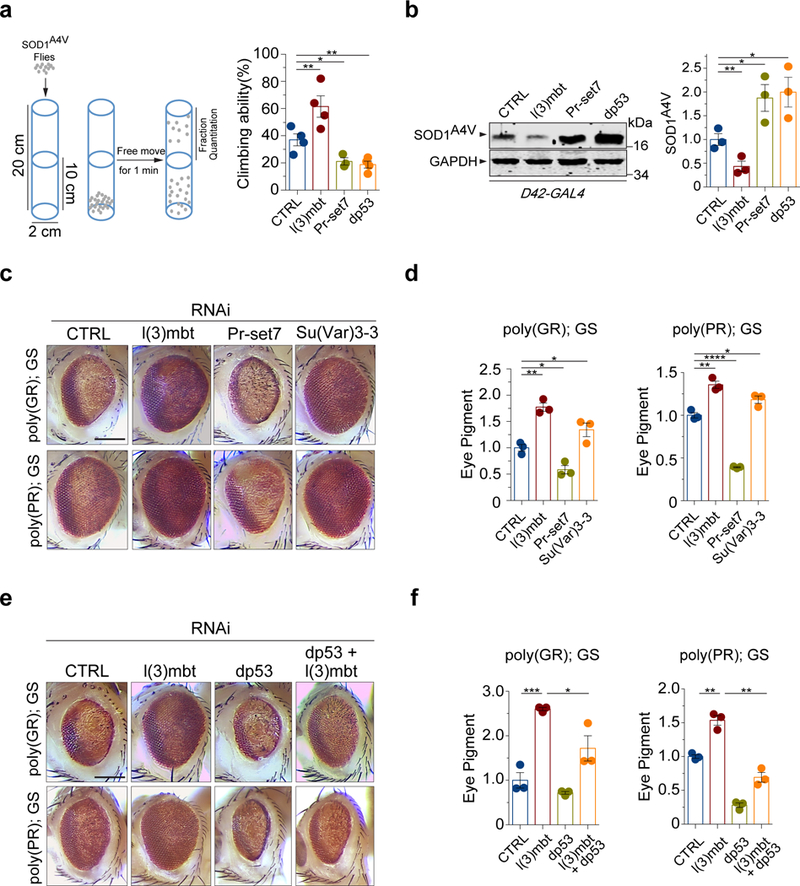
(a) Scheme of the climbing (negative geotaxis) assay in adult Drosophila expressing human SOD1A4V in motor neurons (left). Quantification of the climbing ability of Drosophila expressing human SOD1A4V in motor neurons together with gene-specific or control RNAi is shown (right; n=4 independent groups; ** P(l3mbt)=0.007, * P(Pr-set7)=0.025, ** P(dp53)=0.002). (b) Protein levels of mutant SOD1A4V in adult Drosophila motor neurons, upon treatment with gene specific or control RNAi (n=3 independent groups; ** P(l3mbt)=0.002, * P(Pr-set7)=0.036, * P(dp53)=0.036). (c) The reduction of l(3)mbt by RNAi strongly suppressed the eye degeneration phenotypes in both poly-GR or poly-PR strains when compared with the control Luc RNAi strain. Scale Bar: 100 μm. (d) Quantification of pigment content in adult eyes confirms the protection against degeneration by loss of l(3)mbt in both poly-GR or poly-PR strains. The measurements represent three independent groups with each containing fly heads from four males and four females (n=3; ** P(l3mbt)=0.0016, * P(Su(Var)3–3)=0.046, * P(Pr-set7)=0.015 with poly-GR. ** P(l3mbt)=0.0025, * P(Su(Var)3–3)=0.027, **** P(Pr-set7)<0.0001 with poly-PR). (e) The degenerative rough-eye phenotype of adult flies is alleviated by the reduction of protein demethylase Su(Var)3–3, but aggravated by the reduction of Drosophila homolog of SETD8, Pr-set7, or dp53. Scale Bar: 100 μm. (f) Quantification of the pigment content from degenerating eyes confirms the observation of the rough-eye phenotypes in the suppressor and enhancer strains. The measurements represent three independent groups with each containing fly heads from four males and four females (n=3; *** P(l3mbt)=0.0008, * P(l3mbt+dp53)=0.036 with poly-GR. ** P(l3mbt)=0.002, ** P(l3mbt+dp53)=0.0011 with poly-PR). Error bars represent ± SEM.
Additionally, we tested how L3MBTL1 and SETD8 influence phenotypes of transgenic Drosophila models expressing the ALS/FTD-related proteotoxic dipeptide repeats (DPR) encoded by the C9orf72 HRE30, 31. Flies expressing the polypeptide repeats of proline-arginine (poly-PR) and glycine-arginine (poly-GR) develop various degrees of rough-eye phenotype as a result of the loss of photoreceptors and retinal degeneration in adult eyes32, 33. To start with a moderate phenotype that enables assessment of genetic modifiers, flies were raised on regular food containing 300 μM RU486 that induced relatively low leves of DPR protein expression via the GMR-Gal4 GeneSwitch system. Reduction of the Drosophila homolog of human L3MBTL1, l(3)mbt, produced a significant rescue of the poly-PR- and poly-GR-induced rough-eye phenotypes, resulting in a smoother eye appearance and increased pigmentation (Fig. 4c). Accordingly, the loss of l(3)mbt significantly reversed the pigmentation loss and ommatidial disarray induced by each of the two reporter genes (Fig. 4d). Next, we asked whether the Drosophila homolog of human SETD8, Pr-set7, modulates the poly-PR- and poly-GR-induced proteotoxicity. The loss of Pr-set7 resulted in worsened eye roughness and robustly augmented the loss of pigmentation (Fig. 4c,d). As a control, the RNAi treatments on flies without poly(GR) or poly(PR) showed unperturbed, normal eye phenotypes (Supplementary Fig. 7). The Drosophila demethylase Su(Var)3–3, with opposing enzymatic function to the methyltransferase Pr-set7, is the fly homolog of human lysine-specific demethylase 1, which we have previously shown to negatively regulate protein quality control via p5315. The reduction of demethylase Su(Var)3–3 strongly suppressed neurodegeneration and loss of pigmentation in both the poly(GR) and poly(PR) fly models (Fig. 4c,d), supporting the notion that Pr-set7-mediated methylation is required for neuroprotection in fly eyes.
To test whether Drosophila dp53 mediates the protective effect of the loss of l(3)mbt on the eye degeneration, we crossed the strain with stable RNAi-induced reduction of dp53 to the fly models in which poly(GR) or poly(PR) dipeptides resulted in Drosophila eye neurodegeneration. The loss of dp53 resulted in worsened eye roughness and the loss of pigmentation, and completely ablated the protective effect of loss of l(3)mbt (Fig. 4e,f), suggesting that p53 acts downstream of l(3)mbt in the regulation of proteotoxicity. The dp53 RNAi treatment on flies without poly(GR) or poly(PR) showed normal eye phenotypes (Supplementary Fig. 7). Taken together, these results suggest that the regulatory effects of L3MBTL1 on proteotoxicity-associated neurodegeneration are mediated at least in part by p53.
L3MBTL1 p53-binding antagonist promotes degradation of misfolded proteins
L3MBTL1 recognizes and binds mono- and di-methylated residues on target proteins, thereby acting as a “reader” in a network of post-translational modifications34. The second MBT domain of L3MBTL1 has been shown to have methyllysine-binding activity35. This domain also shares the greatest homology to one of the MBT domains in LIN-61 (Fig. 1d)36. A specific small-molecule antagonist of L3MBTL1, UNC669, binds to the methyllysine recognition site in the second MBT domain of L3MBTL1, thereby blocking the interaction between L3MBTL1 and its methylated histone substrates37. We performed co-immunoprecipitation experiments and found that treatment of HEK293 cells with UNC669 significantly reduced the amounts of endogenous p53 pulled down by L3MBTL1 (Fig. 5a), establishing that UNC669 reduces the binding of L3MBTL1 to p53. Consistent with the negative regulation of L3MBTL1 on p53, UNC669 exhibited a dose-dependent effect on the p53 transcriptional activity, as measured by the p53-RE luciferase reporter (Fig. 5b).
Figure 5. L3MBT1 antagonist UNC669 protects against proteotoxicity in mammalian cells.
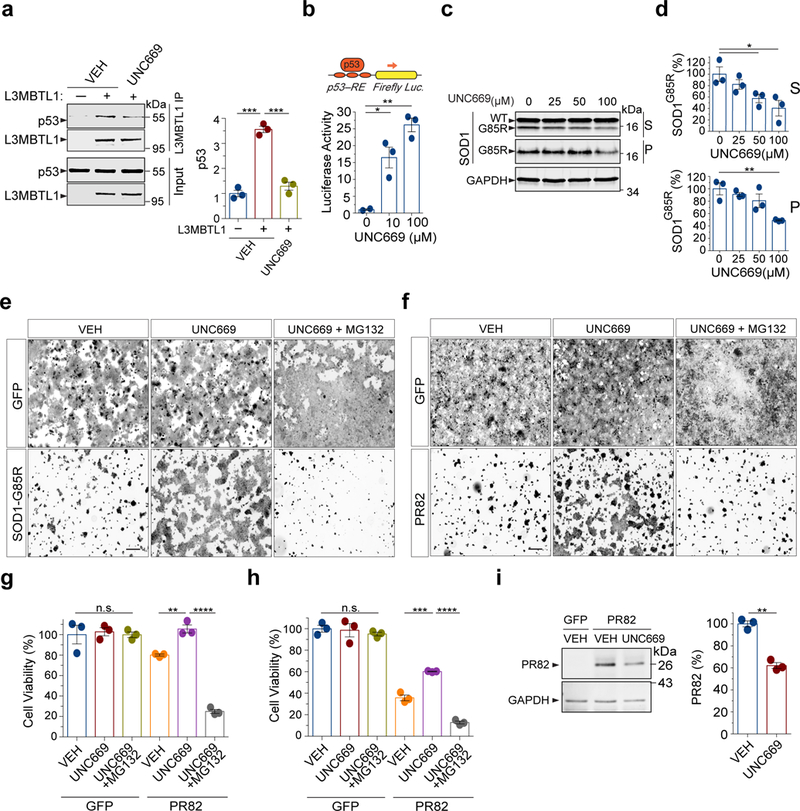
(a) Co-immunoprecipitation analysis of Flag-tagged L3MBTL1 and endogenous p53 in HEK293 cells treated with 12.5 µM UNC669 or vehicle controls (n=3 independent experiments; *** P(VEH)=0.0002, *** P(UNC669)=0.0004). (b) HEK293 cells treated with increased concentrations of UNC669 show increased p53 protein transcriptional activity, as measured by the p53 response element (p53-RE)-mediated luciferase activity assay (n=3 independent experiments; * P=0.03; ** P=0.002). (c) HEK293 cells expressing SOD1G85R were treated with increasing concentrations of UNC669 (top) and the S and P fractions were analyzed by western blotting and probed with antibodies against SOD1 and GAPDH. (d) Quantification of SOD1G85R western blot analyses in (c) (n=3 independent experiments; * P≤0.044 for S and n=3; ** P=0.006 for P). (e) HEK293 cells were transfected with SOD1G85R, treated with 100 µM UNC669 or vehicle (VEH) controls with or without 0.5 µM MG132, and measured for survival using calcein AM fluorescence. (f) HEK293 cells expressing PR82 were treated with 12.5 µM UNC669 or vehicle controls with or without 0.5 µM MG132 and measured for survival using calcein AM fluorescence. (g) Quantification of fluorescence intensity from (e), as a readout of cell viability (n=3 independent cultures; ** P=0.0033; **** P<0.0001). (h) Quantification of fluorescence intensity from (f), as a readout of cell viability (n=3 independent cultures; *** P=0.0007; **** P<0.0001). (i) Western blotting analysis of HEK293 cells expressing C9orf72-related proline-arginine (PR82) dipeptide repeats and treated with 12.5 µM UNC669 (n=3 independent experiments; ** P=0.0011). Scale Bar: 300 μm. Error bars represent ± SEM.
Next, to determine the effect of the L3MBTL1 antagonist UNC669 on the clearance of the misfolded SOD1G85R protein, we analyzed the solubility of SOD1G85R after HEK293 cells were treated with UNC669. Treatment with UNC669 led to a significant, dose-dependent decrease in the levels of misfolded SOD1G85R in both the soluble and insoluble fractions (Fig. 5c,d), while not affecting the level of endogenous SOD1WT protein (Supplementary Fig. 8a). Treatment of cells with 100 μM UNC669 significantly improved the survival of the SOD1G85R-expressing cells (Fig. 5e,g), as measured by calcein AM, a fluorescent dye that stains live cells. Moreover, compromising the proteasome activity with a low concentration of the inhibitor MG132 (0.5 μM) completely abolished the protective effects of UNC669 on the SOD1G85R-expressing cells, indicating that the proteotoxicity of SOD1G85R and the rescue by UNC669 were specific to alterations in proteasomal activity (Fig. 5e,g).
To ask whether the L3MBTL1 antagonist UNC669 affects the proteotoxicity of C9orf72 poly-DPR, we used a cell-based model using the proline-arginine dipeptide repeat PR82. Treatment with 12.5 μM UNC669 significantly improved the survival of the PR82-expressing cells (Fig. 5f,h). Thirty hours after the UNC669 treatment, the levels of PR82 in the cells were significantly reduced (Fig. 5i). Moreover, compromising the proteasome activity with a low concentration of the inhibitor MG132 (0.5 μM) blocked the protection of UNC669 on PR82-induced toxicity (Fig. 5f,h). Together, these results validated that the loss of L3MBTL1 function reduces proteotoxicity of the tested misfolded proteins and suggested that L3MBTL1 modulates protein quality control through its regulation of p53 activity.
L3MBTL1 antagonist UNC669 protects against proteotoxicity in neurons and mice
We further tested whether L3MBTL1 inhibition would alleviate proteotoxicity-induced neurotoxicity in mammalian neurons. We first developed a neurotoxicity assay using motor neurons differentiated in vitro from mouse embryonic stem cells. The lentivirus expression of SOD1G85R exerted a significant toxicity on the motor neurons, and six days after transduction, ~70% of motor neurons expressing SOD1G85R survived as measured by the calcein AM staining (Fig. 6a,b). However, a treatment of 100 μM UNC669 significantly decreased the SOD1G85R-induced toxicity with ~90% surviving motor neurons compared with GFP control (Fig. 6a,b), without affecting the survival of the GFP-transduced mouse motor neurons (Supplementary Fig. 8b).
Figure 6. L3MBTL1 antagonist UNC669 protects against proteotoxicity in neurons and mice.
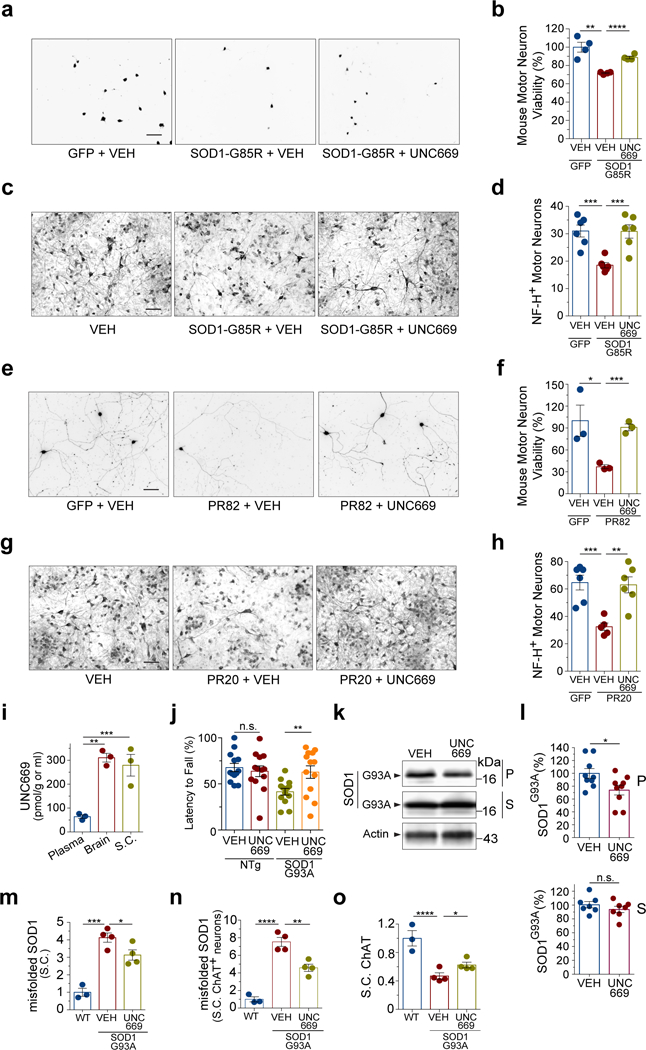
(a) Mouse embryonic stem cell-derived motor neurons were transduced with GFP or SOD1G85R viral expression vectors, treated with vehicle (VEH) or 100 µM UNC669, and measured for survival based on the calcein AM fluorescence as shown. (b) Quantification of the calcein AM fluorescence intensity from (a) (n=4 independent cultures; ** P=0.0021; **** P<0.0001). (c) Toxicity assays on primary rat spinal cord neuronal cultures infected with of HSV-SOD1G85R and treated with 100 µM UNC669 or vehicle (EtOH, VEH). SMI-32 immunostaining is shown. (d) Quantification of surviving rat motor neurons from (c) in four random fields (n=6 independent cultures, *** P(VEH GFP vs VEH SOD1G85R)=0.0004; *** P(VEH SOD1G85R vs UNC669 SOD1G85R)=0.0009). (e) Mouse embryonic stem cell-derived motor neurons were transduced with lentiviruses expressing proline-arginine (PR82) dipeptide repeats or GFP, treated with 6.25 µM UNC669 or vehicle control, and measured for survival based on the calcein AM fluorescence. (f) Quantification of the calcein AM fluorescence intensity from (e) (n=3 independent cultures; * P=0.0435; *** P=0.0006). (g) Primary rat spinal cord neuronal cultures were treated with exogenously added C9orf72-related proline-arginine (PR20) dipeptide repeats and treated with 100 µM UNC669 or vehicle controls. Images show representative fields of motor neurons in each condition after six days of treatment. (h) Quantification of rat motor neuron survival from (g) in four random fields (n=5 independent cultures; *** P(VEH GFP vs VEH PR20)=0.0008; ** P(VEH PR20 vs UNC669 PR20)=0.002). (i) Biodistribution of UNC669 in mice, 3h after intraperitoneal (IP) administration of UNC669, shows accumulation of UNC669 in brain and spinal cord (n=3 mice; *** P=0.0002; ** P=0.01). (j) IP administration of UNC669 to an ALS transgenic mouse model (SOD1G93A, 18 males and 8 females; NTg, 14 males and 12 females) improves the motor ability of SOD1G93A mice as measured by the Rotarod performance test (n=13 pairs of gender-matched SOD1G93A littermates and 13 pairs of gender-matched non-transgenic mice; n.s., P=0.59; ** P=0.0013). (k) Representative western blots of brain protein fractions from VEH- or UNC669-treated SOD1G93A transgenic mice. The S and P fractions were probed with an antibody against SOD1. (l) Quantification of SOD1G93A in S- and P-fractions by western blot analysis. A total of 7 (S) and 9 (P) pairs of VEH- or UNC669-treated brains were analyzed (n.s., P=0.31; * P=0.03). (m) Quantification of immunostaining, using an antibody that recognizes the misfolded SOD1 protein, on spinal cord sections. Y-axis represents average relative fluorescence intensity in six to nine spinal cord regions from four pairs of SOD1G93A mice treated with UNC669 or controls and three non-transgenic WT mice without treatment (n=4 for SOD1G93A and 3 for WT; * P=0.0455; ***P=0.004). (n) Quantification of immunostaining, using the same misfolded SOD1 antibody and the spinal cords as in (m), in ChAT-positive spinal cord motor neurons (n=4 for SOD1G93A and 3 for WT; ** P=0.0042; ****P<0.0001). (o) Quantification of ChAT immunostaining signals in spinal cord neurons from mice treated with UNC669 or controls (n=4 for SOD1G93A and 3 for WT; * P=0.0418; **** P<0.0001). Scale Bar: 300 μm. Error bars represent ± SEM.
We also employed a previously characterized SOD1 neurotoxicity assay38, in which spinal cord primary motor neurons were prepared from rat embryos and maintained on astrocyte monolayers. Expression of SOD1G85R via a neuron-specific herpes simplex virus (HSV) vector induced a significant loss of motor neurons over 5 days as compared with the control (Fig. 6c,d). When treated with 100 µM of L3MBTL1 inhibitor UNC669, the motor neurons showed protection from SOD1G85R-induced proteotoxicity (Fig. 6c,d), without changes on the survival of normal rat spinal motor neurons (Supplementary Fig. 8c). These results showed that the inhibition of L3MBTL1 protects against the toxicity of misfolded SOD1 proteins in neurons.
Next, we transduced mouse motor neurons with lentivirus expressing the C9orf72 proline-arginine dipeptide repeat PR82, which led to a decreased cell viability of ~40% as compared with a GFP control (Fig. 6e,f). Treatment of the motor neurons with UNC669 at the concentration of 6.25 µM resulted in substantial improvement in cell viability with ~90% of motor neurons surviving (Fig. 6e,f). Since the synthesized proline-arginine dipeptide repeat PR20 was recently shown to exert toxicity to mammalian neurons via the inhibition of proteasomal activity30, 39, we asked whether L3MBTL1 inhibition would ameliorate the neurotoxicity induced by exogenously provided PR20 peptide repeat. The primary rat spinal cord neurons were grown on astrocytes, and while PR20 killed approximately 50% of motor neurons, co-incubation with 100 µM UNC669 completely protected the neurons against this insult (Fig. 6g,h). The protection against both SOD1G85R- and C9orf72 DPR-induced neurotoxicity suggests that L3MBTL1 acts through protein quality control systems.
To test the effects of L3MBTL1 inhibition on proteotoxicity in vivo, we applied the inhibitor UNC669 to the SOD1G93A mouse model that develops paralytic symptoms. We treated the SOD1G93A animals with UNC669 through intraperitoneal injection, which led to the drug accumulation in the brain and spinal cord (Fig. 6i), starting around the age of 5 weeks. Although the treatment did not significantly alter the overall survival of the animals possibly due to the severity of the disease in this model, there was an improvement in the motor functions of the treated animals. In the rotarod assay used to study neuromuscular function, SOD1G93A mice treated with UNC669 showed significantly better locomotor activity than their vehicle-treated gender-matched littermates around the age of 12 weeks (Fig. 6j). In a separate control experiment, the UNC669 treatment did not promote the locomotor activity of non-transgenic mice, indicating that its effect is dependent on mutant SOD1-induced toxicity (Fig. 6j). Western blotting of the brain tissue showed that UNC669-treated mice had slightly less SOD1G93A protein in the P2 fraction than controls while the S1 fraction remained unchanged (Fig. 6k,l). Immunofluorescence staining analysis of the lumbar regions of spinal cords, using an antibody that specifically recognizes the misfolding conformation of SOD1 protein but not the correctly folded proteins, showed a significant decrease in the levels of misfolded SOD1 in UNC669-treated animals compared to the controls (Fig. 6m and Supplementary Fig. 8d,e). The levels of misfolded SOD1 in the motor neurons from UNC669-treated animals was significantly decreased compared to vehicle-only controls (Fig. 6n and Supplementary Fig. 8e). The intensity of choline acetyltransferase (ChAT) staining as a motor neuron marker in the spinal cords was consistently increased in the UNC669-treated animals compared to vehicle controls (Fig. 6o and Supplementary Fig. 8e), indicating an enhancement of functional gene expression in motor neurons as a result of UNC669 treatment and consistent with the improvement of motor neuron function and locomotor behaviors (Fig. 6j).
Discussion
Recognition of misfolded proteins and their selective degradation by the protein quality control system are crucial for protein homeostasis under stress. The present study identified L3MBTL1 and SETD8 as new regulators of protein quality control and suggests that their dysregulation is an integral part of the disease pathogenesis (Supplementary Fig. 9). We found that the loss of lin-61 robustly suppressed the toxicity of misfolded protein in C. elegans and that this function appears to be evolutionarily conserved in Drosophila l(3)mbt and human L3MBTL1. The enhanced degradation of misfolded proteins was consistent with up-regulation of the proteasomal subunits upon reduction of L3MBTL1 in human cells, which was dependent on p53, consistent with previous findings that p53 controls the transcription of a protein quality control network15, 16. In support of the role of p53 as a downstream effector, we found that the small molecule compound UNC669, which interferes with the binding of L3MBTL1 to methylated histones, can block the interaction of L3MBTL1 with p53 and exert protective effects against proteotoxicity in neuronal cultures and mouse models. It was reported that SETD8 could methylate p53 and promote its interaction with L3MBTL122, and in the present study, we found reduction of SETD8 decreased the level of p53 protein and impaired the turnover of misfolded SOD1 protein accordingly. It is notable that our previously identified regulators of proteotoxicity, UBE4B and LSD115, as well as L3MBTL1 and SETD8, converge on the regulation of lysine modification of p53. The data collectively suggest that p53 is a master regulatory switch for protein quality control, and as a transcription factor, it can act as a switchboard that integrates combinatorial input signals and output functions in its regulatory control.
Our finding that perturbation of protein homeostasis leads to an elevation of SETD8 protein suggests that SETD8 is a molecular effector for natural proteotoxic stress responses. In response to proteotoxic stresses induced by proteasomal inhibition, the level of SETD8 increases in different cell types. Furthermore, the accumulation of both SETD8 and L3MBTL1 in mutant SOD1 mouse models and human ALS/FTD patients suggests that they are up-regulated by pathologic insults during the disease process. Our identification of the regulators of protein quality control could lead to new strategies in therapeutic interventions.
Materials and Methods
DNA Plasmids
For mammalian expression, SOD1 variants were expressed in pEF-BOS as previously described6. Gateway Donor vectors encoding human L3MBTL1 (HsCD00398731) and SETD8 (HsCD00352315) were purchased from the Arizona State University Plasmid Repository (DNASU) and were recombined into Destination vector pDEST40 containing V5 or Flag tags. Site-directed mutagenesis was done using primer-encoded nucleotide replacements and the HiFi Assembly kit (NEB). The polyQ82 expression vector (mHTT-N171–82Q) was described previously40. The following shRNA sequences were cloned into the pRFP-C-RS vector (Origene) or obtained in pLKO-1: 5’gctggagtcatggctatgatt3’ (L3MBTL1, #1); 5’gcagtcactcacaacaagaat3’ (L3MBTL1, #2); 5’ccgaggaacagaagatcaaag3’ (SETD8, #1); 5’cgcaacagaatcgcaaactta3’ (SETD8, #2); 5’gcgcgatagcgctaataattt3’ (non-targeting control). A HSV vector expressing mutant SOD1 was previously described38. The PR82 lentiviral expression construct contained 82 proline-arginine dipeptide repeats in the pLenti-puro-CMV (w118) vector. The PR82 coding sequence was subcloned from a previous sequence with randomized codons designed to produce only the proline-arginine dipeptide repeat32.
C. elegans Strains, Suppressor Screen, and Mutation Identification
The Bristol N2 C. elegans strain was used in all experiments unless otherwise specified. A list of C. elegans strains is given in Supplementary Table 2. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which was funded by NIH Office of Research Infrastructure Programs (P40 OD010440). For the suppressor screen, worms were mutagenized with 47 mM ethyl methanesulfonate, and a semi-clonal strategy was used, with five P0 worms in one plate. Suppressors were visually selected on the basis of strong recovery in the movement phenotype in the F2 generation. The suppressor strains were backcrossed to the parent SOD1G85R line more than five times for further characterization. The suppressor mutations were mapped by using single nucleotide polymorphism markers in the Hawaiian strain and then identified by whole-genome deep sequencing, followed by Sanger-sequencing validations. In brief, C. elegans genomic DNA was purified using the DNeasy Blood and Tissue Kit (Qiagen). The deep sequencing was carried out on an Illumina HiSeq platform. Bowtie 2 was used to index the C. elegans reference genome and to align the Illumina sequencing reads to this reference41. Using Bowtie 2, a Sequence Alignment/Map (SAM) output was obtained and used in subsequent analyses. The SAMtools software package was used to identify variants and call SNPs and INDELS based on the SAM alignment files42. Variants were called and written to a VCF (variant call format) file. SnpEff was then used to annotate the effects of the variants on protein coding43. Finally, the IGV browser was used to view the variants and the underlying sequence reads44.
C. elegans Locomotor Assay
The C. elegans strains were observed stereoscopically, and their motility was quantified by the thrashing assay45: Animals were transferred from the feeding plate into M9 buffer (3 mg/ml KH2PO4, 6 mg/ml Na2HPO4, 5 mg/ml NaCl and 1 mM MgSO4). After 1 min of adaptation, the number of body bends or thrashes was counted for 1 min as an index of the locomotor phenotype. A thrash was counted when both the head and the tail bent away from the anteroposterior axis by more than 45˚. Videos of C. elegans locomotion were recorded using a Leica MZ165 fluorescence stereoscope.
C. elegans UbG76V-Dendra2 Degradation Assay
Live C. elegans at the early L4 stage were mounted onto microscope slides by immersion in a 1.5-μl drop of M9 on a 5% Noble agar (Sigma) pad. One worm was placed on one pad and covered with a standard glass coverslip. UbG76V-Dendra2 in the whole worm was converted from green to red by using the 488-nm laser at 100% power and a high number (2 min) of iterations on a Leica SP8 laser scanning confocal microscope. Both green and red fluorescence signals were imaged by a sequential mode immediately following photoconversion. Then the worm was allowed to recover on a fresh food plate. At different time points post-photoconversion, the same worm was mounted and re-imaged using the same acquisition settings. NIH ImageJ was used to quantify changes in the red fluorescence intensities of the worms over time.
Drosophila Strains and Assays
Flies were reared and crossed on standard yeast-agar-cornmeal medium at 25°C. The Drosophila transgenic strain carrying GAL4-inducible human SOD1A4V (hSOD1A4V) was recombined with D42-GAL4 (w*; P{w+mW.hs=GawB}D42;), which drives expression in motor neurons, as previously described27. The D42-GAL4, hSOD1A4V/TM3 flies strains were then crossed to RNAi transgenic strains. The following Drosophila strains were obtained from the Bloomington Stock Center or Vienna Stock Center: GAL4-inducible expression hSOD1A4V (P{UAS-hSOD1.A4V}); RNAi strains including Luc (y1 v1; P{TRiP.JF01355}attP2), l(3)mbt (y1 sc* v1; P{TRiP.HMS01466}attP2/TM3, Sb1), Pr-set7 (w1118; P{GD10930}v34590), and p53 (y1 v1; P{TRiP. GL01220}attP40). They were transferred to freshly made food every 2–3 days. Flies were aged for 35 days, and their climbing behaviors and SOD1 protein levels were examined. For the climbing assay, Drosophila were transferred to new empty vials, and the number of flies that can climb from the bottom to the top half of the vials during 1 min intervals was counted and then quantified. For western blot assays, Drosophila were lysed in RIPA lysis buffer [50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 100 mM NaF, 17.5mM β-glycerophosphate, 0.5% sodium deoxycholate, 10% glycerol, 1% NP-40, 0.2mM sodium orthovanadate and 1mM PMSF, 1/100th protease inhibitor cocktail (Sigma, P8340)], and then homogenized in a Diagenode Bioruptor. The lysates were centrifuged 10 min (4˚C, ~10,000 g) and the supernatants were used for western blot analysis.
Drosophila transgenic strains carrying the Gal4-inducible human ALS gene C9orf72-associated dipeptide repeat DPR proteins poly(PR) (w1118; P{UAS-polyPR.PO-100}att40) or poly(GR) (w1118; P{UAS-polyGR.PO-100}att40), each containing 100 repeats, were previously described32. For DPR proteins poly(PR) or poly(GR) expression, we recombined GMR-GAL4 GeneSwitch with the poly(PR) or poly(GR) lines and crossed the resulting balanced lines, GMR-Gal4 GeneSwitch, poly(PR)/Cyo and GMR-Gal4 GeneSwitch, poly(GR)/Cyo, to RNAi transgenic strains. To induce DPR protein expression using GMR-Gal4 GeneSwitch, flies were raised at 25°C on regular food containing 300 μM RU486 (Cayman Chemical). Flies were aged for 21 days, and their eye morphology was examined. Changes in pigmentation, ommatidial structure, and glossiness phenotypes were monitored for enhancement or suppression. The pigment content was detected at 488 nm and background-corrected at 600 nm using fly head protein extracts as previously described15.
Mammalian Cell Lines, Transfections, and Drug Treatments
HEK293 and mouse embryonic cell lines were grown at 37˚C / 5% CO2 in standard DMEM medium supplemented with 10% FBS, 2 mM L-glutamine, and 1x non-essential amino acids (DMEM/10). Transfections of mammalian cells were performed using Lipofectamine 2000 or 3000 (Invitrogen) according to the manufacturer’s recommendations. For shRNAs, transfections of HEK293 cells were performed by plating 3.2×105 cells in 60-mm poly(ethyleneimine) (PEI, 10 µg/ml in PBS, Sigma)-pretreated dishes one day before the transfection. For transfection, 4 µg of shRNA-encoding plasmids, 350 ng of SOD1 reporter (BOS-SOD1-G85R), and 10 µl of Lipofectamine 2000 (Invitrogen) were mixed in 500 µl Opti-MEM I (Invitrogen) and applied to cells in 2.5 ml Opti-MEM I. One day post-transfection, the medium was replaced with DMEM/10. The cells were lysed for analysis 72–96 h after the start of the transfections. UNC669 (Millipore) was dissolved at 200 mM in ethanol, while MG132 (Sigma) at 20 mM was dissolved in DMSO. All drugs were diluted in DMEM/10 prior to the cell treatments.
Western Blotting, Immunoprecipitation, Immunofluorescence Staining, and Antibodies
For western blotting, equal amounts of proteins were electrophoresed on 15% or 4–20% Tris-Cl gels (Bio-Rad). Proteins were transferred to nitrocellulose membranes and immuno-probed with the following antibodies: anti-SOD1–100 (Enzo, P00441, 1:3,000), anti-L3MBTL1 (Abcam, 1:500), anti-SETD8 (Cell Signaling Technology, C18B7, 1:1,000, and Genetex GTX119440, 1:1,000), anti-p53 7F5 (Cell Signaling Technology, #9282, 1:1,000), anti-C. elegans-SOD-1 (JH766, 1:5,000)46, anti-histone H4 (Active Motif, #61300, 1:1,000), anti-H4K20me1 (Active Motif, #39728, 1:1,000), anti-YFP (Takara, #632381, 1:3,000), anti-beta-actin (Santa Cruz, sc-47778B, 1:3,000), and anti-GAPDH (Thermo Fisher, TAB1001, 1:3,000). Proteins were visualized using Li-COR anti-mouse and anti-rabbit 680 and 800 fluorescent antibodies, then visualized and quantified using an Odyssey scanner and Image Studio 2.0 software (Li-Cor). The original scans of western blotting images are provided in the supplementary information (Supplementary Fig. 10–12). Protein quantification is normalized to loading control actin or GAPDH, with the exception of insoluble fractions normalized to total fraction protein amounts.
For co-immunoprecipitation experiments, HEK293 cells were transfected with specific plasmids using Lipofectamine 2000 for 6 hours and then maintained in serum-containing medium for 24–48 hours. For drug treatment, cells were cultured with 100 uM UNC669 for 24 hours after the transfection. Cells were then harvested in lysis buffer (50mm Tris-HCl, Ph7.4, 250mm NaCl, 0.5% Triton x-100, 10% glycerol, 1mm DTT) containing a proteasome inhibitor (Sigma) and PMSF. After sonication, cell lysates were centrifuged at 16,000g for 10 minutes at 4˚C, and the supernatant was collected for further analysis. The cell lysates were incubated with V5 antibody (Invitrogen) overnight at 4˚C and then immunoprecipitated with protein A/G magnetic beads at 4˚C for 2 hours. For Flag immunoprecipitation, cell lysates were incubated with washed magnetic Flag-beads and rotated at 4˚C for 2 hours. Beads were washed with cell lysis buffer 5 times, incubated in elusion buffer at room temperature for 15 minutes, and then neutralized with 1M Tris-HCl (pH 8.0). The eluents were then collected for western blotting analysis.
For immunofluorescence staining analysis, mouse spinal cords were fixed with 4% PFA and sliced with a Cryostat (Leica). Slices were treated with blocking buffer (10% Goat serum, 0.2% tween 20) at room temperature for 1 hour. Blocked slices were incubated with primary antibodies (anti-NeuN, Millipore, MAB377, 1:20; anti-L3MBTL1, GeneTex, GTX103793, 1:100; anti-SETD8, Cell Signaling Technology, 2996, 1:100; anti-misfolded human SOD1, MediMabs, B8H10, 1:20; anti-ChAT, AB144P, 1:100) at 4˚C overnight. After washing with PBS, slices were incubated with immunofluorescence-conjunct secondary antibodies at room temperature for 2 hours. After washing with PBS, slices were sealed with mounting medium containing DAPI (Invitrogen). Images were captured using an SP8 confocal microscope (Leica) and analyzed using ImageJ software.
Protein Aggregation Assay
The protein aggregation assay for C. elegans and mammalian cells was modified from a previously described protocol6. C. elegans strains were collected from NGM feeding plates into M9 buffer and washed five times. Mammalian cells grown on 60-mm plate were washed two times with cold PBS. C. elegans and mammalian cells were lysed in 200–300 µl of lysis buffer (50 mM Tris–HCl, pH 8.0, 1 mM ethylenediaminetetraacetic acid [EDTA],100 mM NaCl, and 0.5% NP-40, 1/100th protease inhibitor cocktail (Sigma, P8340), and 25 mM iodoacetamide (Sigma, I6125); they were then sonicated on ice in a Diagenode Bioruptor (High, 30 sec pulse, 30 sec pause, 5 min total). The lysates were centrifuged 5–10 min at ~130,000 g (25 psi) in an Airfuge (Coulter-Beckman) to separate larger pelleted aggregates (P1) from soluble proteins and smaller aggregates (S1). The P1 pellet was resuspended in lysis buffer and sonicated as described above, except for 10 min. After centrifugation (Airfuge, ~130,000 g, 5–10 min), the pellet (P2) was resuspended in 100 µl urea/SDS buffer (8 M urea, 5% SDS, 40 mM Tris-Cl pH 6.8, 0.1 mM EDTA), followed by 5 min of sonication.
Transcriptional Activity Luciferase Assay
HEK293 cells were transfected with the firefly luciferase p53 reporter plasmid containing the p53-binding consensus sequence (PG13-luc)47, together with a thymidine kinase promoter Renilla luciferase (tk-Rluc) reporter for normalization. At 72 h post-transfection, the cells were lysed in passive lysis buffer (Promega) and analyzed with the Dual Luciferase Reporter System according to the manufacturer’s recommendations (Promega), using an injector-equipped Synergy H1 microplate reader (Bio-Tek).
Proteasomal Activity Assay
HEK293 cells were plated in white-wall clear-bottom 96-well plates (Corning). After the experimental step such as a 72-h shRNA treatment, the Proteasome-Glo™ Cell-Based Reagent including the luminogenic substrate, Suc-LLVY-aminoluciferin (Promega), was added to the cell samples (1:1). The samples were mixed using a plate shaker at 700rpm for 2 minutes, incubated at room temperature for 10 minutes, and measured in a plate-reading luminometer (Synergy H1, Bio-Tek). Treatment with the proteasome-specific inhibitor MG132 (20 µM, 1h) was used as a control.
Quantitative RT-qPCR
Total RNA was isolated from C. elegans or mouse embryonic fibroblast cells with the RNeasy Mini kit. For quantitative RT-qPCR, cDNAs were synthesized with the QuantiTect reverse transcription kit using RNA samples from both (Qiagen). Primers for quantitative RT-qPCR were as follows: human SOD1 (F: 5’GGGAAGCTGTTGTCCCAAG3’; R: 5’CAAGGGGAGGTAAAAGAGAGC3’); C. elegans GAPDH (F: 5’CGGTATCATCGAAGGACTCAT3’; R: 5’GTCCATCTCTCCACAGCTTT3’); mouse SETD8 (F: 5’TCTATGCCTACATGAGTCCGAA3’; R: 5’GATTCCAGCTAATGGTTTCCCC3’); mouse actin (F: 5’GGCTGTATTCCCCTCCATCG3’; R: 5’CCAGTTGGTAACGCCATGT3’). RT-qPCRs were performed on a Bio-Rad thermal cycler with iQ SYBER Green PCR mix (Bio-Rad).
Mouse and Human Tissues
The SOD1 transgenic mice used in this study have been previously characterized: the SOD1G93A line [B6.Cg-Tg (SOD1*G93A)1Gur/J; Jackson Laboratory]25 and the SOD1G85R-YFP and SOD1WT-YFP lines26. Transgenic mice were identified by PCR amplification of DNA extracted from tail biopsies. Mice were euthanized in a CO2 chamber, and fresh tissues were harvested by flash-freezing in liquid nitrogen and then stored at −80°C. For western blot analysis, spinal cords were rinsed with cold PBS and homogenized with cold RIPA buffer using glass tissue grinders. The homogenates were then centrifuged at 4°C at 1,000 g for 10 min, and the supernatants were centrifuged again at 16,000 g for 10 min, with the final supernatant used for western blot analysis. The animal protocol was approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions. Human post-mortem brain and spinal cord tissues used in this study are provided by the Johns Hopkins Brain Bank and the Department of Veterans Affairs Biorepository Brain Bank (Supplementary Table 1).
Cell Survival Assay
HEK293 cells were plated in 12-well plates at 2 ×105 cells per well. After adherence, the cells were transfected with constructs expressing SOD1G85R, the proline-arginine (PR82) peptide, or equal amounts of a GFP control construct (1.5 μg/well). Six hours after the transfection, UNC669 (100 μM for SOD1G85R and 12.5 μM for PR82) was added and the medium and the drug were refreshed every 2 days. The cells were kept for 4 days and then gently washed with 1× DPBS (+ Mg2+, Ca2+) and stained with 3 μM calcein AM (Invitrogen), followed by measurement of fluorescence with a plate reader (Synergy H1 Hybrid, excitation/emission at 485 nm/535 nm). Images of randomly selected fields were captured under a fluorescence microscope (Nikon Eclipse TS100 equipped with lumencor).
Mouse Motor Neuron Survival Assay
Mouse embryonic stem (mES) cells were cultured, differentiated, and selected for Hb9-expressing motor neurons as described previously48. Briefly, mES cells (1×106) with integrated Hb9-Puromycin transgene were cultured in suspension, in 10 ml DFK5 media for two days, followed by a 4-day incubation in DFK5 media containing 2 μM retinoic acid and 0.6 µM smoothened agonist. Differentiating Embryonic Bodies were dissociated, plated in individual laminin-coated wells of a 12-well plate (1.5×105 cells/cm2), and incubated in DFK5 media with GDNF, BDNF, NT-3 (5 ng/ml each), and 4 µg/ml puromycin for 24 h to select for Hb9-Puromycin-expressing motor neurons. Twenty-four hours later, the medium was replaced with DFKNB media supplemented with 1× B27, GDNF, BDNF and NT-348. Before changing the media, cells were infected with an HSV construct expressing SOD1G85R, a lentiviral construct expressing PR82, or GFP expression cassettes for 6 h in 4 μg/ml polybrene. Motor neurons infected with SOD1G85R or PR82 dipeptide constructs were treated in triplicates, with vehicle (ethanol), or UNC669 (100 μM for SOD1G85R and 6.25 μM for PR82), whereas motor neurons transduced with GFP control in triplicate were treated only with the vehicle. After infection and start of drug/vehicle treatment, motor neurons were kept for additional 6 days and the drug was changed every 3 days. On the last day, motor neurons were subjected to cell survival assay using the calcein AM method as described above.
Spinal Motor Neuron Survival Assay
Embryonic Sprague Dawley rat spinal cord cultures and neuronal survival assays were previously described38. The pregnant rat dams were euthanized by overdosing with nembutal. Spinal cords from embryonic day 15 rats were dissociated and grown on cortical astrocyte monolayers established from 1–3-day-old rats. The spinal cord neurons were maintained in astrocyte-conditioned medium with neurotrophic factors, including ciliary neurotrophic factor, cardiotrophin-1, brain-derived neurotrophic factor, neurotrophin 4, and glial-derived neurotrophic factor (10 ng/ml, Alomone Labs). The spinal cord culture was treated with UNC669 or vehicle ~1 h prior to infection with a neuron-specific HSV vector expressing mutant SOD1. For the alternative PR20 toxicity assay, the protocol was described previously30, 39. The medium was replenished with fresh media three times per week. At day 5 post-infection, motor neuron number was determined by cell fixation, immunostaining with a motor neuron-specific antibody against neurofilament H (NF-H), SMI-32 (Covance), and counting stained cells with cell body diameter of ≥ 20 microns. In prior work we established that this protocol identifies motor neurons in our culture system38. The motor neurons were counted in four random fields of view for each condition with the viewer blinded to the experimental manipulation. Quadruplicate coverslips per experimental manipulation were quantified in this way and group differences assessed by ANOVA. In the post hoc analysis a P value of ≤ 0.05 was set as significant. All experiments were repeated with independently created cultures at least 3 times.
Mouse UNC669 Treatment and Rotarod Assay
To assess the bioavailability of UNC669 in the brain and spinal cord, non-transgenic C57BL/6J mice (aged 8–12 weeks) were intraperitoneally injected with UNC669 (12 mM dissolved in DPBS containing 10% DMSO) at 5 ml/kg. Three hours post intraperitoneal administration, tissues were harvested, snap frozen and analyzed for UNC669 using mass spectrometry.
To test the drug effects, gender- and litter-matched SOD1G93A transgenic mice [B6.Cg-Tg(SOD1*G93A)1Gur/J; Jackson Laboratory] and non-transgenic C57BL/6J mice were intraperitoneally injected with either UNC669 (10 mM) at 5 ml/kg mouse or vehicle. The injections were subsequently repeated every five days. Mice were trained on the Rotarod apparatus (4–32 rpm over 5 minutes) (Harvard Apparatus). Mice were given three attempts, after which they were returned to their cages. Experimental mice were tested a day after training. The time taken for mice to fall off the Rotarod was recorded. Data were collected in triplicates and mice were given 1 minute to recover before each trial. Both training and testing were performed at a fixed time to prevent confounding factors such as the circadian rhythm.
Microscopy
C. elegans and mouse spinal cord imaging were performed on a Leica SP8 laser scanning confocal microscope. Fly imaging was performed on a Leica M80 microscope with the IC80 HD camera, and images were taken and analyzed with Leica application suite V4 software.
Bioinformatic and Statistical Analysis
The evolutionary history for L3MBTL1 related proteins was inferred by using the Maximum Likelihood method based on the JTT matrix-based model49. The phylogenic tree with the highest log likelihood is used. Evolutionary analyses were conducted in MEGA750. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported previously15. Data distribution was assumed to be normal but this was not formally tested. Samples such as C. elegans, Drosophila, tissue cultures and mice were randomly selected into experimental or control groups. Data collection and analysis were not performed blind to the conditions of the experiments. The experiments were repeated in two or more independent trials. The P-values for all analyses were obtained using two-tailed Student’s t-tests performed in Excel or GraphPad Prism 6 unless otherwise indicated.
Supplementary Material
Acknowledgements
This work was supported by grants from NIH (NS074324, NS089616, and NS110098), Packard Center for ALS Research at Johns Hopkins, Muscular Dystrophy Association, ALS Association, and Maryland Stem Cell Research Fund (J.W.). We thank Ho-Yon Hwang for assisting in C. elegans genome deep-sequencing and data analysis, Carina Holmberg of University of Helsinki for the Dendra2 C. elegans strain, Valeria Culotta of Johns Hopkins University for the C. elegans SOD-1 antibody, Adrian Isaacs of University College London for the proline-arginine repeat plasmid template, and members of the Wang laboratory for discussion.
Footnotes
Competing Interests
The authors declare no competing interests.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
- 1.Balch WE, Morimoto RI, Dillin A & Kelly JW Adapting Proteostasis for Disease Intervention. Science 319, 916–919 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336, 1511–1513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Langenhove T, van der Zee J & Van Broeckhoven C The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Annals of medicine 44, 817–828 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosco DA et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13, 1396–1403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Xu G & Borchelt D High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9, 139–148 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Wang J et al. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12, 2753–2764 (2003). [DOI] [PubMed] [Google Scholar]
- 7.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renton Alan E. et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron (2011). [DOI] [PMC free article] [PubMed]
- 9.Donnelly CJ et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haeusler AR et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ugolino J et al. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet 12, e1006443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ash PE et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori K et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Zu T et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110, E4968–4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Periz G et al. Regulation of Protein Quality Control by UBE4B and LSD1 through p53-Mediated Transcription. PLoS Biol 13, e1002114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazaki Y, Chen LC, Chu BW, Swigut T & Wandless TJ Distinct transcriptional responses elicited by unfolded nuclear or cytoplasmic protein in mammalian cells. Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet 5, e1000350 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison MM, Lu X & Horvitz HR LIN-61, one of two Caenorhabditis elegans malignant-brain-tumor-repeat-containing proteins, acts with the DRM and NuRD-like protein complexes in vulval development but not in certain other biological processes. Genetics 176, 255–271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamer G, Matilainen O & Holmberg CI A photoconvertible reporter of the ubiquitin-proteasome system in vivo. Nature methods 7, 473–478 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Bonasio R, Lecona E & Reinberg D MBT domain proteins in development and disease. Semin Cell Dev Biol 21, 221–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Xu G & Borchelt DR Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J Neurochem 96, 1277–1288 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West LE et al. The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J Biol Chem 285, 37725–37732 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dantuma NP, Lindsten K, Glas R, Jellne M & Masucci MG Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 18, 538–543 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Shi X et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 27, 636–646 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurney ME et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Wang J et al. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A 106, 1392–1397 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watson MR, Lagow RD, Xu K, Zhang B & Bonini NM A Drosophila Model for Amyotrophic Lateral Sclerosis Reveals Motor Neuron Damage by Human SOD1. J. Biol. Chem 283, 24972–24981 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ollmann M et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101, 91–101 (2000). [DOI] [PubMed] [Google Scholar]
- 29.D’Brot A, Kurtz P, Regan E, Jakubowski B & Abrams JM A platform for interrogating cancer-associated p53 alleles. Oncogene 36, 286–291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta R et al. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. eNeuro 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer NJ et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet 50, 603–612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizielinska S et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110, 7778–7783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min J et al. L3MBTL1 recognition of mono- and dimethylated histones. Nat Struct Mol Biol 14, 1229–1230 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Trojer P et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129, 915–928 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Koester-Eiserfunke N & Fischle W H3K9me2/3 binding of the MBT domain protein LIN-61 is essential for Caenorhabditis elegans vulva development. PLoS Genet 7, e1002017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herold JM et al. Small-molecule ligands of methyl-lysine binding proteins. J Med Chem 54, 2504–2511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mojsilovic-Petrovic J et al. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci 26, 9250–9263 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon I et al. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science (2014). [DOI] [PMC free article] [PubMed]
- 40.Subramaniam S, Sixt KM, Barrow R & Snyder SH Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang T, Mullane PC, Periz G & Wang J TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum.Mol.Genet 20, 1952–1965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen LT & Culotta VC Activation of CuZn superoxide dismutases from Caenorhabditis elegans does not require the copper chaperone CCS. J Biol Chem 280, 41373–41379 (2005). [DOI] [PubMed] [Google Scholar]
- 47.el-Deiry WS et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993). [DOI] [PubMed] [Google Scholar]
- 48.McCreedy DA et al. A new method for generating high purity motoneurons from mouse embryonic stem cells. Biotechnol Bioeng 111, 2041–2055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones DT, Taylor WR & Thornton JM The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8, 275–282 (1992). [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, Stecher G & Tamura K MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.