

Abstract
Doublecortin (DCX) mutations cause abnormal development of the DCX protein that normally aids in neuronal migration during fetal development. These mutations lead to lissencephaly, or the appearance of a “smooth brain,” which is varying levels of pachygyria or agyria in severe cases. Many genetic variants of the mutation have been identified, and an even greater range of phenotypic presentations have been described in the literature. The X-linked lissencephaly (DCX) mutation leads to an X-linked gender-dependent condition that causes subcortical heterotopia in females and lissencephaly in males. The authors report the case of a 13-year-old male who presented to our clinic for new-onset seizure disorder. He had a past medical history of developmental delay and features of autism spectrum disorder which was diagnosed at age 5 years at an outside clinic. Magnetic resonance imaging (MRI) brain at age 5 years showed pachygyria of the frontal and temporal lobes. After extensive genetic testing over the course of over a decade, the patient was found to have a de novo mutation in the DCX gene diagnosed via whole-exome sequencing. Specifically, he was found to have a mosaic mutation of the DCX gene as a c.30-31 deletion. His previous MRI findings were consistent with a diagnosis of X-linked sporadic lissencephaly sequence and included mainly a diffuse bilateral pachygyria (isolated lissencephaly sequence X chromosome). Thickening of the cortex and pachygyria or agyria are classic findings of lissencephaly, but do not help specify any mutation in the gene, of which there are over 70 possibilities. Our patient is unique in that most individuals with DCX mutation have infantile seizures, severe intellectual disability, orthopedic complications, and postnatal microcephaly, which our patient does not have.
Keywords: neurodevelopment, lissencephaly, doublecortin, neuronal migration
The case involves the delay in diagnoses of lissencephaly etiology in a patient with doublecortin (DCX) mutation. This delay was due to changing availability and utilization of genetic testing. The authors also seek to highlight the ability for an individual with a severely abnormal neurological physiology to be successful in society depending on genetic mosaicism lending residual function and parental involvement in individual success.1 Genetic variants resulting in lissencephaly are varied in both their specific karyotype location and their phenotypic presentations.2 The dominant mechanisms by which cortical lamination is disrupted can be due to a failure of neuronal migration, a failure of neuronal path finding, or an imbalance in DNA replication in utero.3 The 2 main classifications of lissencephaly are therefore defined as either type 1, which is a failure to differentiate the cortex into all of the appropriate 6 layers (instead resulting in 4 layers), or type 2, which is a complete failure to layer the cortex at all. The latter is referred to as a “cobblestone” phenotype, while the former is known as “classical” lissencephaly.4 The DCX gene codes for the DCX protein that binds microtubules during neural development. Mutations in the gene cause the neuronal migration disorders subcortical band heterotopia in females and classic lissencephaly in males.2 Although lissencephaly can be diagnosed in utero, patients who presented before the popularization of in utero testing and genetic diagnosis will often go undiagnosed until presenting to clinic with behavioral or developmental abnormalities. Although the clinical manifestations of lissencephaly normally present in individuals during early development, the diagnosis of a DCX mutation specifically causing the lissencephaly can be delayed for years or even decades. Here, the authors present a case of a patient who was initially diagnosed with lissencephaly after presenting to clinic with behavior and developmental abnormalities, but whose specific genetic mutation causing the disorder was not diagnosed until over a decade later. The final diagnosis was made in the setting of new-onset seizure and an evolving clinical approach to genetic testing based on accumulated test results throughout the patient’s clinical history.
Case Report
A 13-year-old male presented to outside clinic with autistic features including lack of social interaction, repetitive behaviors, and sound hypersensitivity. Patient reached most of his appropriate early developmental milestones, walked at 8 months and said his first words at 14 months. The patient was reported to have an age-appropriate vocabulary with what his mother said was nearly 20 words by the time he was 18 months, but he could not use words to name items correctly. Magnetic resonance imaging (MRI) of the brain was completed and showed decreased topographic development of the temporal, parietal, and frontal lobes. In addition to diffuse pachygyria, there was involvement of the cerebellar vermis in repeat MRI imaging in both 2010 and 2011 (Figures 1 and 2).
Figure 1.
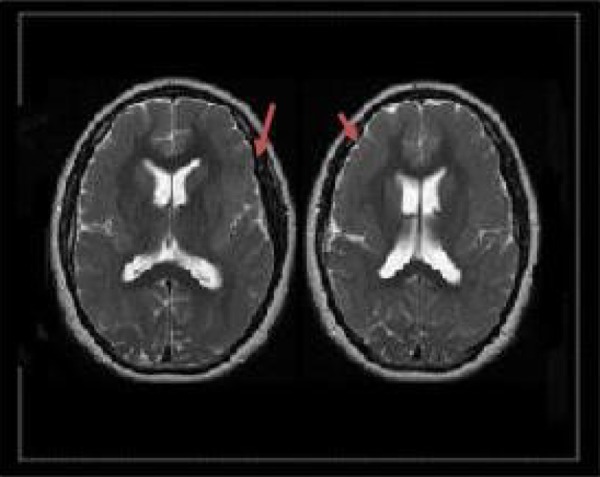
T1-weighted magnetic resonance imaging (MRI) showing pachygyria of the bilateral frontal lobes.
Figure 2.
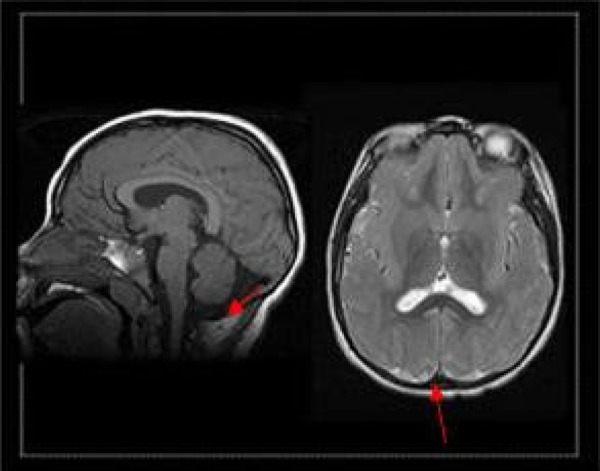
T1-weighted magnetic resonance imaging (MRI) showing cerebellar vermis involvement.
Although these findings were consistent with the patient’s eventual diagnosis of DCX mutation,5 they were not reflective of the full set of typical MRI findings in patients with the DCX mutation in males (Figure 3). The MRI done when the patient was 7-years old suggested a partial lissencephaly of unknown genetic origin.
Figure 3.

Magnetic resonance imaging (MRI) findings in patients with isolated lissencephaly sequence X chromosome (ILSX).
This is consistent with the patient’s proposed genetic etiology being de novo, since family members had no significant developmental or neurological history to suggest possible sources of the patient’s disorder. Because of mild facial dysmorphisms including large low-set ears and widely spaced eyes, the patient was tested for Fragile-X at the time which was negative. Karyotype was also performed at the age of 5 years but yielded no positive findings. No further genetic workup was done at the time. The patient was then able to live a relatively normal adolescent life, attending public school despite having mild Intellectual disability disorder. His mother requested speech and behavioral therapy 7 times a week for him to improve his skills in social interaction. At the age of 13 years, he had his first generalized tonic–clonic seizure in the setting of gastroenteritis. A repeat MRI again demonstrated lissencephaly and a 23-hour electroencephalography revealed frontal predominant intermittent slowing, consistent with his structural brain abnormalities.6 At the age of 13 years, the patient was started on Keppra with excellent seizure control from 2010 to 2015. Comparative Genomic Hybridization Assay performed at age 13 years was negative as well. At this point in adolescence, there was an increase in seizure frequency due to noncompliance and bouts of illness, and patient was brought to clinic once again for further management of his seizures. At the age of 19 years, methylation studies for Angelman syndrome and Prader-Willi syndrome were found to be normal. He then had whole-exome sequencing which resulted in the discovery of a mutation in the DCX gene on chromosome Xq22-23, specifically a c.30-31 deletion.
Discussion
The DCX mutations cause abnormal function of DCX, a protein responsible for microtubule migration in DNA replication during neural development of the fetus.7 Doublecortin binds and stabilizes microtubules (Figure 4) that normally aid in neuronal migration during fetal development and is also present in perilesional cortical8 cells of patients with stroke.
Figure 4.

Protein structure of doublecortin (DCX) isolated (L) and attached to tubulin molecule (R).9
This involvement in newly regenerating neural function suggests a role in adult neural migration as well.9 Patients with mutant DCX genes vary in their presentation based on the location of the10 mutation on the affected chromosome as well as the gender of the patient, and somatic versus germ line origin.11 Our patient had a de novo mutation of the isolated lissencephaly sequence X chromosome, which has its own specific subset of findings on MRI that typically includes involvement of the cerebellar vermis and corpus callosum.12 In patients with DCX mutation, treatment is symptomatic and supportive rather than curative. However, this does not mean that genetic etiology of each patient’s specific DCX mutation is irrelevant in clinical care. Expected complications of lissencephaly differ somewhat by etiology. For patients with DCX mutation mosaicism, complications such as seizure and orthopedic abnormality13 can be expected in early childhood and adolescence. In our patient’s case, prognosis was more guarded in the early stages after his diagnosis of lissencephaly, likely at least in part because outcomes are dependent on many factors. The degree of lissencephaly largely determines outcomes, as completely “smooth-brained” patients often die before the age of 10 years, and many others never reach a developmental stage of greater than 6 months of age. Our patient likely had a DCX mutation that was mosaic in nature, and there was likely some level of functional DCX protein available to the developing neural network during fetal stages. Patients such as ours with a partial lissencephaly can reach varied levels of social functionality. Our patient who is now 23-years old functions at approximately a 9-year-old level. Because behavioral intervention in patients with intellectual disability is more effective with earlier onset of therapy, it is significant to note onset of therapy when parental involvement in this case resulted in early aggressive speech and behavioral therapy for the patient.14 It also led to a push for our patient to be involved in public school classes with abled students. Integrative education programs such as these have been shown to improve students’ social outcomes in the long term.1 Because of a combination of the mildness of our patient’s lissencephaly and these early interventions, he has graduated high school, is able to maintain employment, and is a functioning member of society despite his medical diagnosis. The authors compare this patient profile, for example, to a different mutation on chromosome 17, which also causes congenital lissencephaly.15 This condition is coined Miller-Dieker syndrome, named after 2 separate physicians who identified the disorder in the 1960s. Instead of having orthopedic complications, these patients have associated cyanotic congenital heart malformations and kidney abnormalities, as well as early onset of visual impairment.15 In such patients, the genetic etiology of their gene mutation gives medical direction. In a patient in whom you expect to have complications of the heart and kidney, medications normally contraindicated in patients with heart or kidney disease should be viewed as suboptimal. Screening for these malformations would also be pertinent in the patient’s routine follow-up care. Although treatment at this point is symptomatic, patients with lissencephaly deserve a thorough genetic workup to identify the etiology of their own “smooth-brain” syndrome. Not only can it allow better guidance to patient families about what to expect regarding prognosis but knowing associated symptomology can help physicians anticipate and screen for complications more likely in specific patients. Pioneering research into gene replacement in utero is still in the animal model stages for DCX mutation therapy. However, if these treatments, which at the animal level, seem to have some level of success were to come to fruition, they will only be helpful if the mutation itself is diagnosed incredibly early in development.16 Our patient had an extensive genetic workup throughout the course of his clinical care and was able to receive support from his family and community in order to have the positive outcome he did.
Footnotes
Author Contributions: IZ and DP contributed to conception and design. IZ and DP drafted the manuscripts. All authors SM, DP, and IZ contributed to acquisition and editing of the manuscripts. SM gave final approval. IZ agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial funding provided by the McGovern Medical School.
Ethical Approval: Consent was taken from the care giver of the patient.
References
- 1. Cooney P, Tunney C, O’Reilly G. A systematic review of the evidence regarding cognitive therapy skills that assist cognitive behavioural therapy in adults who have an intellectual disability. J Appl Res Intellect Disabil. 2018;31(1):23–42. [DOI] [PubMed] [Google Scholar]
- 2. Friocourt G, Marcorelles P, Saugier-Veber P, Quille M, Marret S, Laquerrière A. Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type I lissencephaly. Acta Neuropathol. 2011;121(2):149–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marcorelles P, Laquerrière A, Adde-Michel C, et al. Evidence for tangential migration disturbances in human lissencephaly resulting from a defect in LIS1, DCX and ARX genes. Acta Neuropathol. 2010;120(4):503–515. [DOI] [PubMed] [Google Scholar]
- 4. Devisme L, Bouchet C, Gonzalès M, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain. 2012;135(pt 2):469–482. [DOI] [PubMed] [Google Scholar]
- 5. Leger P, Souville I, Boddaert N, et al. The location of DCX mutations predicts malformation severity in X-linked lissencephaly. Neurogenetics. 2008;9(4):277–285. [DOI] [PubMed] [Google Scholar]
- 6. Accolla EA, Kaplan PW, Maeder-Ingvar M, Jukopila S, Rossetti AO. Clinical correlates of frontal intermittent rhythmic delta activity (FIRDA). Clin Neurophysiol. 2011;122(1):27–31. [DOI] [PubMed] [Google Scholar]
- 7. Bahi-Buisson N, Guerrini R. Diffuse malformations of cortical development. Handb Clin Neurol. 2013;111:653–665. [DOI] [PubMed] [Google Scholar]
- 8. Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17-linked and Xlinked lissencephaly. Neurology. 1999;53(2):270–277. [DOI] [PubMed] [Google Scholar]
- 9. Jin K, Sun Y, Xie L, et al. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. doi:10.1016/S1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 10. UniProtKB - 043602 (DCX_HUMAN). 2002-2018. https://www.uniprot.org/uniprot/O43602. Accessed March 4, 2018.
- 11. Lissencephaly Information Page. 2017. https://www.ninds.nih.gov/Disorders/All-Disorders/Lissencephaly-Information-Page. Accessed March, 2019.
- 12. Fu X, Brown KJ, Yap CC, Winckler B, Jaiswal JK, Liu JS. Doublecortin (Dcx) family proteins regulate filamentous actin structure in developing neurons. J Neurosci. 2013;33(2):709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu JS, Schubert CR, Fu X, et al. Molecular basis for specific regulation of neuronal kinesin-3 motors by doublecortin family proteins. Mol Cell. http://www.rcsb.org/structure/4ATU. Accessed April 3, 2018. [DOI] [PMC free article] [PubMed]
- 14. Sanders MR, Markie-Dadds C, Tully LA, Bor W. The triple P-positive parenting program: a comparison of enhanced, standard, and self-directed behavioral family intervention for parents of children with early onset conduct problems. J Consult Clin Psychol. 2000;68(4):624–640. [PubMed] [Google Scholar]
- 15. Macura S, Freeman S. Miller-Dieker Syndrome In: Narins B, ed. The Gale Encyclopedia of Genetic Disorders. Detroit, MI: Cengage Gale; 2016:1174–1177. [Google Scholar]
- 16. Manent JB, Wang Y, Chang Y, Paramasivam M, LoTurco JJ. Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat Med. 2009;15(1):84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]