

Abstract
Human blood group A and B glycosyltransferases (GTA, GTB) are retaining glycosyltransferases, requiring a catalytic mechanism that conserves the anomeric configuration of the hexopyranose moiety of the donor substrate (UDP‐GalNAc, UDP‐Gal). Previous studies have shown that GTA and GTB cycle through structurally distinct states during catalysis. Here, we link binding and release of substrates, substrate‐analogs, and products to transitions between open, semi‐closed, and closed states of the enzymes. Methyl TROSY based titration experiments in combination with zz‐exchange experiments uncover dramatic changes of binding kinetics associated with allosteric interactions between donor‐type and acceptor‐type ligands. Taken together, this highlights how allosteric control of on‐ and off‐rates correlates with conformational changes, driving catalysis to completion.
Keywords: glycosyltransferase, Methyl TROSY, chemical shift perturbation, allosteric effects, binding kinetics
Complete control: NMR experiments show that association and dissociation rate constants for binding of donor‐ and acceptor substrates to human blood group A and B glycosyltransferases are under mutual allosteric control.
Human blood group A and B glycosyltransferases (GTA, GTB) have been extensively studied in the past. Many structural and kinetic aspects are now known1 but there are also unresolved issues. It has been shown by native mass spectrometry,2 by NMR spectroscopy,3 and by isothermal titration calorimetry4 that donor‐ and acceptor‐substrate binding to GTA or GTB are under mutual allosteric control. Acceptor‐substrate binding is significantly enhanced in the presence of donor substrate or analogs and vice versa. In previous studies we have also described how the NMR exchange regime changes upon the presence of donor and acceptor substrates.5 This work proposes that changes in binding kinetics are the cause of allosteric control of binding affinities. In the meantime, we have succeeded in the complete assignment of methyl TROSY spectra of MILpr ° SVpr ° SA 13C‐methyl labeled GTA and GTB.6 Building on these assignments we studied the binding kinetics of donor‐ and acceptor substrates and analogs thereof in more detail. As reported, depending on the ligand combination, exchange may be fast, intermediate, or slow on the chemical shift time scale.5a Knowledge of the corresponding on‐ and off‐rate constants would provide a link to the conformational changes associated with substrate binding as observed by crystallography and would assist in understanding the mechanisms of allosteric control.
It needs to be emphasized that glycosylation transfer (see Figure S1 for the reactions catalyzed by GTA and GTB) is too fast for studying the mutual influence of donor‐ and acceptor substrates (Scheme 1) with NMR experiments. Furthermore, the donor substrates UDP‐Gal and UDP‐GalNAc undergo enzymatic hydrolysis in the presence of the respective glycosyltransferase, GTB or GTA. To simulate the interplay between donor‐ and acceptor substrates we used combinations of donor‐type ligands such as UDP, which is formed upon glycosyltransfer, and e. g. H‐disaccharide, a true acceptor substrate. Likewise, we employed the mock acceptor substrate 3DD (Scheme 1) as an acceptor‐type ligand. Another approximation is the use of Mg2+ instead of Mn2+, which is believed to be the relevant counter ion in the Golgi apparatus. Because of its paramagnetism, Mn2+ would significantly impede observation of NMR signals.
Scheme 1.
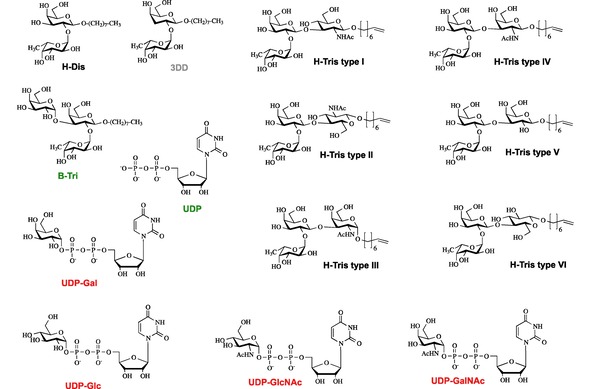
Donor (red) and acceptor (black) substrates, mock‐acceptor substrate (grey), and products (green) resulting from glycosyltransfer were used as ligands to study binding to GTA, GTB, and AAGlyB by methyl TROSY based CSP NMR experiments. The color code is also used in all tables.
NMR lineshape analysis has been used in numerous instances for the evaluation of chemical shift titration experiments, yielding quantitative descriptions of chemical exchange processes such as conformational equilibria or binding of ligands to receptor proteins. Typically, such analysis is performed recording 1D NMR spectra and fitting equations reflecting the exchange process to the experimental titration data (for a review see ref.7). This approach has been extended to 2D data sets such as 1H,15N HSQC based chemical shift perturbation (CSP) titrations by extracting 1D cross sections.8 Unfortunately, here discrimination between fast, intermediate, and slow exchange becomes complicated since CSPs of individual cross peaks differ depending on the nucleus observed, e. g. 1H or 15N. As a consequence, fitting becomes unstable and error prone. Moreover, CSP titrations based on methyl TROSY (1H,13C HMQC) experiments9 cannot be evaluated since multiple quantum coherences evolve during the indirect time dimension. This is particularly frustrating as methyl TROSY spectra have a huge potential to study binding phenomena with very large proteins or even protein complexes.10
A novel approach for analyzing CSP titration data from HSQC‐ and HMQC‐type 2D NMR spectra engages direct quantum mechanical simulation of a given pulse sequence.11 In an iterative least‐squares procedure simulated 2D spectra with varying chemical shifts, linewidths, dissociation constants, and off‐rates are compared to experimental spectra until a best fit is achieved. This new lineshape analysis procedure is implemented in the TITAN (TITration ANalysis) software package.11 Here, we have used TITAN to analyze the kinetics of ligand‐binding to GTB, GTA, and selected mutants.
Our previous qualitative studies into the kinetics of substrate binding to GTA and GTB were based on selective 15N side‐chain labeling of Trp residues and on 13C ϵ‐methyl labeling of Met residues.5 A major finding was that donor‐ and acceptor‐binding is driven into the so‐called slow‐exchange regime on the NMR chemical shift time scale when both, donor‐ and acceptor‐substrate, are present, allowing the enzyme to adopt the catalytically active state. In the present study we employed MILpr ° SVpr ° SA 13C‐methyl labeled samples of GTA and GTB, and of AAGlyB, a mutant that can transfer both Gal and GalNAc residues to acceptor substrates, to study substrate and substrate‐analog binding to these enzymes. Methyl TROSY spectra were recorded in the absence and presence of ligands (Scheme 1) yielding corresponding residue‐specific CSPs.
In a first step we obtained CSP fingerprints for each ligand at saturating or close to saturating ligand concentrations. Representative examples are shown in Figure 1a (binding of the donor‐type ligand UDP to GTB and binding of UDP to GTB saturated with H‐disaccharide, GTB:H‐Dis) and in Figure 2a (binding of the acceptor substrate H‐disaccharide to GTB and binding of H‐disaccharide to GTB saturated with UDP, GTB:UDP). A complete collection of CSP fingerprints for all ligands in Scheme 1 is found in the supplementary material (Figs. S2 to S5). For all CSP fingerprints shown in this study individual σ‐levels have been calculated for each titration to allow for a better comparison of ligands. Therefore, one also needs to consider the maximum CSPs for individual ligands (compare e. g. Figs. 1a and 2a). In general, CSPs are larger for donor‐type ligands (Figure S4) than for acceptor‐type ligands (Figs. S2 and S3). Whereas donor‐type ligands induce sizeable CSPs in the internal disordered loop (residues 173–192), the C‐term, and the β3 strand‐turn‐α2 helix motif (residues 118–130), there is almost no effect in these regions upon acceptor binding.
Figure 1.
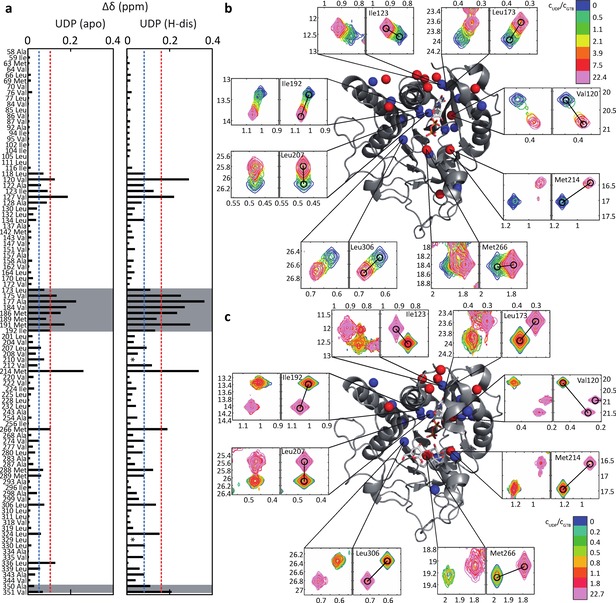
Titrations of GTB with UDP (donor‐type ligand) induce residue‐specific CSPs (Euclidean distances) in methyl TROSY spectra of MILpr ° SVpr ° SA 13C‐methyl labeled GTB (280 μM). a) Left: Endpoint of the titration GTB+UDP (donor‐type ligand). ▵δ refers to differences in chemical shifts between apo‐GTB and GTB saturated with UDP (6200 μM). Right: Endpoint of the titration GTB:H‐Dis (acceptor substrate)+UDP (donor‐type ligand). ▵δ refers to differences in chemical shifts between GTB saturated with H‐disaccharide (GTB:H‐Dis) and GTB:H‐Dis saturated with UDP (6200 μM). The dashed lines indicate 1σ (blue) and 2σ levels (red). b) and c) Selected experimental and simulated cross peaks and mapping of CSPs from a) onto GTB. The internal disordered loop and residues of the C‐terminus are highlighted in grey. Blue and red symbolize 1σ and 2σ levels as in a). b) Selection of experimental (left panels) and simulated (right panels) cross‐peaks for the titration of apo‐GTB with UDP. Relative concentrations of UDP (n‐fold excess over the GTB concentration) are color coded. c) as in b) for a titration of UDP into a solution of GTB saturated with H‐disaccharide (GTB:H‐Dis).
Figure 2.
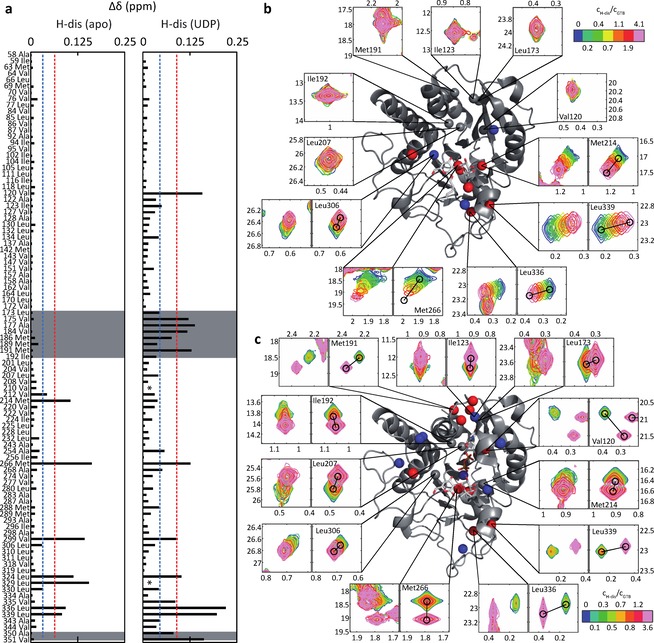
Titrations of GTB with H‐Dis (acceptor substrate) induce residue‐specific CSPs (Euclidean distances) in methyl TROSY spectra of MILpr ° SVpr ° SA 13C‐methyl labeled GTB (280 μM). a) Left: Endpoint of the titration GTB+H‐Dis (acceptor substrate). ▵δ refers to differences in chemical shifts between apo‐GTB and GTB saturated with H‐dis (1120 μM). Right: Endpoint of the titration GTB:UDP (donor‐type ligand)+H‐Dis (acceptor substrate). ▵δ refers to differences in chemical shifts between GTB:UDP and GTB:UDP saturated with H‐Dis (1120 μM). N.B.: ▵δ values comparing apo‐GTB to GTB:UDP:H‐Dis are larger as this is seen in Figure S5 of the supporting information. b) and c) Selected experimental and simulated cross peaks and mapping of CSPs from a) onto GTB. The internal disordered loop and residues of the C‐terminus are highlighted in grey. Blue and red symbolize 1σ and 2σ levels as in a). b) Selection of experimental (left panels) and simulated (right panels) cross‐peaks through titration of apo‐GTB with H‐Dis. Relative concentrations of H‐Dis (n‐fold excess over the GTB concentration) are color coded. Residues with maximal CSPs (cf. panel in a)) larger than 1σ or 2σ are colored in blue and red, respectively. c) as in b) for a titration of H‐Dis into a solution of GTB saturated with UDP (GTB:UDP).
The 13C‐methyl group of Met 214 responds strongly to binding of donor‐type as well as acceptor‐type ligands, although Met 214 is not in the immediate proximity of any ligand. This sensitivity to ligand binding has already been described before and has been linked to a close proximity of the Phe 341 side chain.5a
Binding of the acceptor substrates H‐disaccharide, H‐type II trisaccharide, and H‐type VI trisaccharide affects the chemical shifts of the 13C‐methyl groups of Met 266, Val 299, Leu 324, and Leu 329 (CSPs>2σ), which belong to the acceptor substrate binding pocket (Figure S2 and S3). Long‐range CSPs (>10 Å) are observed for Leu 336 and Leu 339. H‐type II and VI structures are characterized by a β(1,4)‐glycosidic linkage between the Gal and the reducing GlcNAc or Glc residue. CSPs for H‐type I, III, IV, and V trisaccharides are very small (Figure S2). These acceptor ligands all carry a β(1,3)‐glyosidic linkage between the Gal and the reducing GlcNAc (type I), GalNAc (type III), or Gal (type V) residue. The observation of small CSPs strongly indicates weak binding of the latter trisaccharides and is in accordance with previous studies where a rather high Km value has been observed for H‐type I trisaccharide as compared to H‐type II trisaccharide or H‐disaccharide.12
To further examine the allosteric coupling between acceptor and donor binding it would be desirable to record methyl TROSY spectra with both donor and acceptor substrates binding simultaneously. As pointed out above, glycosyltransfer is much too fast, and we had to choose combinations of donor‐ and acceptor‐type ligands that provided stable systems for hours or, in the case of titrations, for days. The combination of H‐disaccharide and UDP represents such a stable system, and although UDP is the byproduct of glycosyltransfer and inhibits glycosyltransfer this system yielded a comprehensive set of kinetic data likely reflecting the principles of mutual effects of donor‐ and acceptor‐ligands on the binding kinetics.
Crystallographic studies with GTB utilized H‐disaccharide containing a 3‐deoxy galactose moiety (3DD) in order to mimic the natural acceptor but preventing galactosyltransfer to the 3‐position of the β‐D‐Gal moiety.1a Unfortunately, this system was not an option to better mimic the natural combination of donor (UDP‐Gal) and acceptor (H‐disaccharide or H‐type trisaccharides) since 3DD accelerates hydrolysis of UDP‐Gal3 such that recording a set of methyl TROSY spectra during a titration was impossible.
In fact, hydrolysis of UDP‐Gal by GTB and of UDP‐GalNAc by GTA was much too fast to allow for NMR titrations. Therefore, we studied binding of donor‐type ligands UDP‐GalNAc, UDP‐Glc, or UDP‐GlcNAc to GTB and of UDP‐Gal, UDP‐Glc, or UDP‐GlcNAc to GTA since here hydrolysis is practically not observable. UDP served as a donor‐type ligand in most experiments probing the influence of donor substrate on acceptor‐substrate binding.
At this point, it is instructive to compare the movement of methyl TROSY cross peaks in Figs. 1 and 2 as this comparison highlights the mutual allosteric effects of donor‐ and acceptor binding. The positions of the cross peaks at the endpoint of the titration of GTB with donor‐type ligand UDP (Figure 1b) compared to the positions of the corresponding cross peaks at the endpoint of the titration of GTB:H‐Dis (GTB saturated with acceptor substrate H‐Dis) with UDP (Figure 1c) are clearly different. This excludes the possibility of a direct displacement of H‐Dis by UDP and highlights the allosteric interaction between H‐Dis and UDP (see also Fig S6, which shows individual 1H‐ and 13C‐chemical shift perturbations further substantiating this conclusion). Likewise, the positions of the cross peaks at the endpoint of the titration of GTB with acceptor substrate H‐Dis (Figure 2b) compared to the positions of the corresponding cross peaks at the endpoint of the titration of GTB:UDP (GTB saturated with donor‐type ligand UDP) with H‐Dis (Figure 2c) are also different, supporting the allosteric nature of this binding process. Furthermore, binding of H‐Dis in the presence of saturating amounts of UDP induces significant CSPs of Ala 350 and Val 351 (Figure 2a) indicating ordering of C‐terminus (Lys 346 – Pro354) into a short α‐helical segment. These effects are absent in the binding of H‐Dis or UDP alone. Finally, the positions of the cross peaks at the endpoint of the titration of GTB:H‐Dis with UDP (Figure 1c) compared to the corresponding positions of cross peaks at the endpoint of the titration of GTB:UDP with H‐Dis (Figure 2c) are identical as both titrations arrive at the ternary complex GTB : UDP : H‐Dis.
Given the large differences in CSP patterns observed for binding of H‐type trisaccharide‐acceptor substrates to GTB (Figs. S2 and S3) it was surprising that in the presence of saturating amounts of UDP the CSP signatures of all H‐type trisaccharide‐acceptor substrates were practically identical (Figure S5). This observation suggests that binding of donor substrate levels differences in acceptor binding, which is in accordance with the observation that relative rates of galactosyltransfer to H‐type I and H‐type II antigen are similar.12
Analysis of the kinetics of ligand binding proceeded using methyl TROSY based titrations. For each titration point, the corresponding methyl TROSY spectrum was imported into MATLAB and then analyzed with the TITAN algorithm, applying a two‐state ligand binding model. Donor‐type ligands are in intermediate to fast exchange as this is seen from selected cross peaks of GTB during a titration with UDP (Figure 1b) as a representative example. Peak positions shift, and in addition peak intensities vary during titration. When titrating UDP to GTB presaturated with H‐disaccharide exchange becomes slow on the chemical shift time scale, which is reflected by separate peaks for the H‐disaccharide and the UDP/H‐disaccharide bound forms of GTB, leading to varying peak intensities during titration. Quantitative lineshape analysis using TITAN yields dissociation constants KD and on‐ and off‐rate constants kon and koff for binding of donor‐type ligands UDP, UDP‐Gal, UDP‐Glc, UDP‐GalNAc, and UDP‐GlcNAc to GTB and GTA (Table 1). We have also studied binding of these ligands to a mutant, AAGlyB, which transfers Gal as well as GalNAc residues (Table S1). Dissociation constants derived from SPR and STD NMR experiments have been previously reported and are of the same order of magnitude.4 In general, the values determined in the present work are lower by a factor of two to three, but it should be noted that the values from SPR and STD NMR show a similar spread. On‐rate constants kon are around 105 M−1s−1, which is much slower than expected for a diffusion‐controlled process, indicating that binding of donor ligands involves solvent reorientation and possibly conformational changes. Dissociation rate constants koff seem to depend on the enzyme and go down into the range of a few Hz (Table S1), translating into residence times of the ligand in the binding pocket of hundreds of milliseconds.
Table 1.
Dissociation constants KD, on‐ and off‐rate constants kon and koff for donor‐type ligands (cf. Scheme 1 for the color code) from fitting a two‐state binding model to methyl TROSY titration data using the TITAN algorithm. Values in brackets are biased by slow hydrolysis of UDP‐Gal during the titration. For UDP‐Gal binding to GTB and for UDP‐GalNAc binding to GTA no titration data could be obtained due to hydrolysis.
|
|
GTA |
GTB |
||||
|---|---|---|---|---|---|---|
|
|
K D (μM) |
k on (M−1s−1) |
k off (s−1) |
K D (μM) |
k on (M−1s−1) |
k off (s−1) |
|
UDP |
– |
– |
– |
161±3 |
1.5×106 |
243±7 |
|
UDP‐Gal |
237±4 |
2.7×105 |
64±2 |
– |
– |
– |
|
UDP‐Glc |
375±6 |
2.1×105 |
78±2 |
598±7 |
3.6×105 |
219±4 |
|
UDP‐GalNAc |
– |
– |
– |
3370±60 |
1.5×105 |
493±15 |
|
UDP‐GlcNAc |
374±13 |
5.9×104 |
22±2 |
3300±110 |
1.9×105 |
610±50 |
Comparison of donor‐type ligands reveals interesting points, correlating well with known crystal structure and binding data:
UDP has the highest affinity for GTB, which is in accordance with published data.4 The present study shows that this is due to an increased on‐rate constant kon (Table 1). In contrast, the low affinities of UDP‐GalNAc and UDP‐GlcNAc binding to GTB are due to increased off‐rate constants koff, reflecting steric hindrance caused by the bulkier amino acids Met 266 and Ala 268 at the floor of the donor‐substrate binding pocket of GTB as compared to Leu 266 and Gly 268 in GTA.1c
Dissociation constants KD for UDP‐Glc and UDP‐GlcNAc binding to GTA are identical. However, on‐ and off‐rate constants differ. The smaller off‐rate constant of UDP‐Glc is compensated by a concurrent decrease of the off‐rate constant. Binding affinities of UDP‐Gal and UDP‐Glc for GTA are also rather similar, with the lower dissociation‐rate constant koff for UPD‐Gal yielding a slightly lower KD value (Table 1).
These observations highlight how subtle structural differences of ligands can have measurable influence on the binding kinetics.
Acceptor‐type ligands show a different pattern of association‐ and dissociation‐rate constants kon and koff. Except for acceptor substrates H‐type II and H‐type VI trisaccharide, which carry a β(1,4)‐glycosidic linkage between the Gal and the reducing GlcNAc or Glc residue, dissociation rate constants are of the order of a few thousand Hz. For acceptor substrates H‐type II and H‐type VI trisaccharide the values are one order of magnitude smaller (Table 2). Interestingly, this difference is compensated by a lower kon value (ca. 10x) yielding similar dissociation constants. Values for H‐type I and H‐type III trisaccharides have to be treated with care since the final ligand concentration was in the range of KD. CSPs for H‐type IV trisaccharide were too small to allow analysis (see also Figs. S1 and S2). Interestingly, the acceptor substrate H‐disaccharide and the mock‐acceptor substrate 3DD display very similar on‐ and off‐rate constants compared to B‐trisaccharide, the product of glycosylation of H‐disaccharide, suggesting that the reducing end β(1,4)‐linked GlcNAc/Glc residue in the acceptor substrates H‐type II and VI trisaccharide causes the lower dissociation rate constant.
Table 2.
Dissociation constants KD, on‐ and off‐rate constants kon and koff for acceptor substrates, mock‐acceptor substrate, and the product B‐trisaccharide (for the color code see Scheme 1) binding to GTB from fitting a two‐state binding model to methyl TROSY titration data using the TITAN algorithm. Values in brackets are to be treated with care as the final ligand concentration was in the range of KD. For H‐type IV trisaccharide CSPs were too small to allow for analysis.
|
|
K D (μM) |
k on (M−1s−1) |
k off (s−1) |
|---|---|---|---|
|
H‐Dis |
164±4 |
1.9×107 |
3100±300 |
|
3DD |
420±8 |
7.9×106 |
3300±200 |
|
B‐Tris |
264±4 |
1.1×107 |
2900±100 |
|
H‐Tris type I |
(3200±100) |
(5.0×105) |
(1600±200) |
|
H‐Tris type II |
291±6 |
7.2×105 |
210±7 |
|
H‐Tris type III |
(2900±200) |
(2.3×105) |
(700±100) |
|
H‐Tris type IV |
– |
– |
– |
|
H‐Tris type V |
1270±20 |
2.0×106 |
2600±200 |
|
H‐Tris type VI |
318±5 |
1.1×106 |
340±10 |
Binding of donor or acceptor ligands to GTB, presaturated with the respective counterpart ligand, drives the system into slow exchange as this is obvious from Figs. 1c and 2c, showing that intensities of peaks attributable to the presaturated state and the state with the second ligand bound change during titration. Interestingly, affinities and binding kinetics of the acceptor substrates H‐disaccharide, H‐type I trisaccharide, and H‐type II trisaccharide binding to GTB presaturated with UDP are very similar (Table 3), contrasting binding of these acceptors to apo‐GTB (Table 2). On‐rate constants kon are down by two orders of magnitude, from 106–107 M−1s−1 to 104–105 M−1s−1. and off‐rate constants koff are now in the single‐digit Hz range leading to micromolar dissociation constants (Table 3). Likewise, binding of UDP in the presence of saturating amounts of any type of H‐type acceptor is now characterized by slow association and dissociation, with koff of the order of few Hz and kon ranging from 104 ‐ 105 M‐1s−1, respectively yielding enhanced affinities with KD values down to 6 μM (H‐type II trisaccharide).
Table 3.
Dissociation constants KD, on‐ and off‐rate constants kon and koff for donor‐ and acceptor‐type ligands binding to GTB in the presence of saturating amounts of complementary ligands (in parentheses) from fitting a two‐state binding model to methyl TROSY titration data using the TITAN algorithm.
|
|
K D (μM) |
k on (M−1s−1) |
k off (s−1) |
|---|---|---|---|
|
Donor |
|
|
|
|
UDP (H‐Dis) |
103±2 |
2.5×104 |
2.6±0.3 |
|
UDP (B‐Tris) |
660±10 |
2.8×104 |
18±1 |
|
UDP (H‐Tris I) |
38±1 |
5.8×104 |
2.2±0.2 |
|
UDP (H‐Tris II) |
6±1 |
2.8×105 |
1.7±0.3 |
|
UDP (H‐Tris III) |
29±1 |
1.2×105 |
3.6±0.5 |
|
UDP (H‐Tris IV) |
65±2 |
1.9×105 |
12±1 |
|
UDP (H‐Tris V) |
31±1 |
1.2×105 |
3.4±0.3 |
|
UDP (H‐Tris VI) |
32±1 |
6.9×104 |
2.2±0.2 |
|
UDP‐Glc (3DD) |
580±10 |
8.6×104 |
50±2 |
|
Acceptor |
|
|
|
|---|---|---|---|
|
H‐Dis (UDP) |
28±1 |
2.5×105 |
6.9±0.4 |
|
H‐Tris I (UDP) |
44±2 |
<2.3×104 |
<1 |
|
H‐Tris II (UDP) |
10±1 |
1.7×105 |
1.7±0.5 |
|
3DD (UDP‐Glc) |
120±10 |
3.3×106 |
390±20 |
It has to be noted at this point that slow exchange as reflected by single digit off‐rate constants as reported in Table 3 likely is at the limit of TITAN analysis. The software has been validated using synthetic data for a range of off‐rate constants between 5 and 5000 Hz.11 As a plausibility check, we compared the KD values in Table 3 for UDP binding to GTB:H‐Dis and of H‐Dis binding to GTB:UDP to values obtained for the same systems using isothermal titration calorimetry (ITC).4 For UDP(H‐Dis) a value of 27 μM from ITC compares to 103 μM from NMR titration analysis, and for H‐Dis(UDP) a value of 7 μM compares to 28 μM in the present study. In both cases the values from ITC are lower by a factor of ca. four. The question, whether this discrepancy is due to off‐rate constants being at the detection limit of the method or to other factors remains open at present. Although this suggests caution in the interpretation of the kinetic data in Table 3 we think that the on‐ and off‐rate constants at least reflect trends, correlating binding kinetics with mutual allosteric interactions between donor‐ and acceptor‐type ligands. This perception is substantiated by examining the kinetic data for the ligand system UDP‐Glc (donor‐type ligand) and 3DD (mock acceptor substrate), where the off‐rate constants fall within the limits of 5 to 5000 Hz. The trend is very similar to what is observed for the UDP/H‐Dis system: The affinity of UDP‐Glc is not affected significantly by the presence of 3DD as the decrease in kon is compensated by an about equal decrease in koff. In contrast, the presence of UDP‐Glc has a significant effect on the dissociation constant of 3DD, which is caused by a decrease of koff by ca. one order of magnitude.
It is of note that koff for binding of UDP in the presence of saturating amounts of B‐trisaccharide is also substantially reduced (Table 3), but as kon is not reduced proportionally the resulting dissociation constant is increased by a factor of almost four. It appears that the presence of B‐trisaccharide triggers the same allosteric process when UDP enters its binding pocket that is also induced by the presence of e. g. H‐disaccharide. This is further substantiated by the observation that CSPs measured for the complex of GTB with H‐disaccharide and UDP bound correlate extremely well with CSPs obtained from comparing the complex of GTB with B‐trisaccharide and UDP, both with reference to apo‐GTB (Figure 3). Qualitatively, this indicates rather similar bound conformations. On the other hand, a recent crystallographic study came to the conclusion that blood group A/B trisaccharide13 and UDP cannot be bound at the same time as steric clashes between the β‐phosphate of UDP and the α‐D‐GalNAc/α‐D‐Gal unit of GTA/GTB disorder the active site and prevent UDP from being stably bound. Also, in a mass spectrometry‐based study no binding of B‐trisaccharide to GTB could be detected in the presence of UDP.2a To resolve this apparent contradiction, it has to be realized that the NMR experiments had to be performed in the presence Mg2+ instead of Mn2+, which is paramagnetic. It is well established that the affinity of UDP binding to GTB is at least one order of magnitude higher in the presence of Mn2+,4 perhaps completely blocking binding of blood group B trisaccharide. This explanation is supported by the observation that the relative affinities for UDP change in opposite directions when adding H‐disaccharide or B‐trisaccharide (Tables 2 and 3). Whereas H‐disaccharide increases the binding affinity of UDP, the presence of B‐trisaccharide leads to a ca. fourfold decrease. In the presence of Mn2+ this effect is likely more pronounced, eventually leading to a complete loss of affinity.
Figure 3.
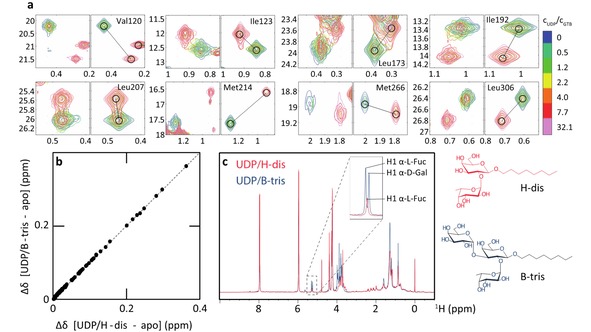
CSPs observed for a titration of GTB (193 μM), saturated with 1400 μM of the product of glycosyltransfer, B‐trisaccharide, (GTB:B‐Tri) with 6200 μM UDP (donor‐type ligand). a) Experimental (left panels) and fitted (right panels) line shapes of selected residues. The relative UDP concentrations are color‐coded. As in Figure 1c most resonances are in the limit of slow chemical exchange. However, higher UDP concentrations are needed to saturate the protein. b) Correlation of CSPs for GTB saturated with UDP and H‐disaccharide or B‐trisaccharide (CSPs are relative to the apo state). Each dot corresponds to a methyl group. An almost perfect linear correlation is obtained, indicating rather similar bound conformations. c) Proton NMR spectra of GTB in the presence of saturating amounts of UDP and H‐disaccharide (blue) or UDP and B‐trisaccharide (red), demonstrating integrity of the ligands.
To obtain further insight into allosteric coupling of donor and acceptor binding we studied two mutants of GTB, GTB Trp181Glu and GTB Trp181Met. It has been shown that a stacking interaction between Trp181 of the internal loop and Arg352 of the C‐term is important for stabilization of the closed form of GTB,1a and inhibitors have been designed that specifically block this stacking interaction.14 We hypothesized that the closed state could be artificially destabilized in the case of the Trp181Met mutant, and may be conserved or even stabilized in the case of the Trp181Glu mutant where a charged interaction can be envisioned between Glu181 and Arg352. The results of the titration analyses are given in the supporting information (see Figure S7 and Table S2). In short, data analysis shows that the allosteric interactions between UDP and H‐Dis are conserved but significantly attenuated in the mutants, which are catalytically inactive. This suggests, that the allosteric interactions between donor‐ and acceptor substrates are essential for catalytic activity.
As our studies yielded dissociation rate constants of the order of a few Hz for donor substrates in the presence of saturating amounts of acceptor substrate these systems are accessible to the direct measurement of exchange rates using so‐called ZZ‐exchange experiments.15 With such experiments one measures exchange between one or more states on time scales ranging from ∼0.5 to ∼50 s−1.16 In our case, the interchanging species were GTB:H‐Dis (state A) and GTB:H‐Dis:UDP (state B). For the ZZ‐exchange experiment we prepared a sample of ILMV 13C‐methyl labeled GTB with H‐disaccharide at saturating concentration and UDP at half‐saturating concentration, yielding an equal population of UDP‐bound and UDP‐free states. Spectra were recorded for seven mixing times in the range between 10 and 600 ms (for details see Figure S5). Only for four residues spectral overlap was moderate enough to perform an analysis based on signal intensities, yielding exchange rate constants kAB and kBA of 2.0±0.5 Hz and 2.5±0.6 Hz, respectively (Table S1). While kBA corresponds to koff from titration analysis, the second order on‐rate constant kon needs to be multiplied by the UDP concentration to obtain a value comparable to kAB. Multiplication of kon=2.5×104 M−1s−1 (Table 3) with a concentration of 250 μM (see legend of Figure S8) yields a value of ca. 6.3 Hz compared to 2.0 Hz from the ZZ‐exchange experiment. For the off‐rate constant koff (2.6 Hz, Table 3) the match with kAB (2.5 Hz) is surprisingly good, given the signal overlap (see Figure S8) in the ZZ‐exchange spectra.
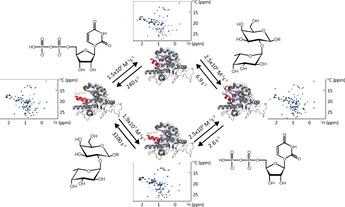
Crystallography has shown that GTB and GTA populate distinct conformational states depending on whether no ligand (open conformation), only acceptor‐type ligand (open conformation), only donor‐type ligand (semi‐closed conformation), or both types of ligand (closed conformation) are bound1a (Figure 4). Here, we have shown by dynamic NMR experiments that mutual allosteric interactions of donor‐type ligands and acceptor‐type ligands determine the exchange kinetics in binary and ternary complexes of GTA or GTB and ligands. Binary complexes of GTA or GTB with acceptor‐type ligands are characterized by fast exchange kinetics leaving the enzyme in the open conformation. Binary complexes with donor‐type ligands are in the intermediate exchange regime, inducing the semi‐closed state. For ternary complexes with donor‐ and acceptor‐type ligands exchange becomes slow with off‐rate constants koff in the range of a few Hz (Table 3). It turns out that this exchange regime matches very well catalytic rate constants kcat determined in previous studies.1a, 12 In particular, it was observed in two‐substrate kinetic experiments with the nucleotide sugar donor at a concentration of 5 mM that H‐type I antigen yields a rather high KM value of 1.3 mM, whereas H‐type II antigen is characterized by a KM value of 0.5 mM.12 This reflects the lower binding affinities of H‐type I compared to H‐type II trisaccharide in our binding experiments.
Figure 4.
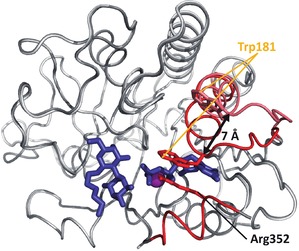
Overlay of the open (light grey) and closed (dark grey) conformation of GTA/GTB. The most flexible regions highlighted in red are only resolved in crystal structures of the AABB chimeric enzyme (PDB entries 3RIZ (open), 2RJ1 (closed)). The C‐terminal tail is only visible in the closed conformation. The internal disordered loop moves by ca. 7 Å between open and closed conformation. The closed conformation is locked via a stacking interaction between Trp181 and Arg352. Donor and acceptor substrate are represented as blue sticks. The metal ion is shown as a purple sphere.
It should be kept in mind that the Michaelis‐Menten kinetics was performed with Mn2+ occupying the metal binding site of GTA and GTB. For the present NMR study, we had to substitute Mn2+ with Mg2+ as Mn2+ is paramagnetic. It had been demonstrated before that dissociation constants for donor‐type substrates are about one order of magnitude smaller when Mn2+ is occupying the metal binding site.4 Therefore, corresponding off‐rate constants will even be smaller, implying that for ternary complexes kcat would be larger than koff, which at first sight seems contradicting, but as a matter of fact we did not measure true donor‐substrates in our experiments. For the real donor‐substrates, UDP‐Gal and UDP‐GalNAc, kcat would certainly constrain koff. At this point, it is interesting to speculate that the enzymes may have evolved to hold on to their substrates for a longer time than required for enzymatic turnover.
To visually summarize our results, Figure 5 reflects the equilibria between binary and ternary complexes for UDP and H‐disaccharide binding to GTB. Two routes are possible, one where the donor ligand binds first (“donor route”) and one where the acceptor ligand binds first (“acceptor route”). It is clear that the conformational rearrangements (Figure 5) of the enzyme have to be in tune with the exchange kinetics. Our data suggest that ligand binding dictates transition rates between open, semi‐closed, and closed states of the enzyme but it cannot be excluded that motions on faster time scales than determined by the exchange kinetics are also present. As all states of GTB are connected by equilibria the principle of detailed balancing must be fulfilled. Therefore, multiplying the two dissociation along the donor route should give the same result as multiplying the corresponding values along the acceptor route. Using the data from Tables 1 to 3 shows that there is a discrepancy of a factor of 3.6, indicating the limits of the current analysis.
Figure 5.
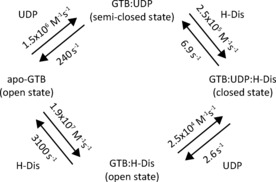
Formation of the ternary complex of GTB, UDP, and H‐disaccharide. Depending on ligand concentrations, the system will follow the donor or acceptor route.
Nevertheless, our data suggest that depending on substrate concentrations in the Golgi apparatus two different “routes”, the “acceptor route” and the “donor route”, are possible. At high local concentrations of UDP‐Gal or UDP‐GalNAc binding of the nucleotide sugar to GTB or GTA would precede binding of the H‐antigen. In cases where nucleotide sugar concentrations are low, it can be assumed that H‐antigen binds first. In this latter case, the enzymes may discriminate between different types of H‐antigens, preferring e. g. type‐II over type‐I H‐antigens (cf. Table 2). This discrimination would be switched off for the “donor route” (cf. Table 3). Current work in our laboratory is aiming at better understanding the complex allosteric mechanism behind these observations.
Experimental Section
E.coli BL21 cells, carrying the pCWΔlac plasmid with the gene of interest are inoculated into 50 mL of TB rich medium and grown until an optical density at 600 nm (OD600) >1 is reached. Carbenicillin (Carb) (100 μg/mL) is used as selecting agent throughout the expression. Cells for inoculation of 50 mL 100 % D2O‐based M9 minimal medium with a start OD600 of 0.1 are harvested by centrifugation, and excessive TB medium is removed. This starter culture is grown overnight at 37 °C with agitation. Cells from the starter culture are harvested by centrifugation (3000 x g, 2 min) and inoculated into 50 mL of M9 minimal medium for protein synthesis in D2O (CortecNet; 99.9 % 2H) containing Na2HPO4 (8.9 g/L), KH2PO4 (6.8 g/L), NaCl (0.5 g/L), (15NH4)2SO4 (CIL; 3.3 g/L), D‐glucose‐d7 (CIL; 3 g/L), trace metals and vitamins. Exchangeable protons of Na2HPO4, KH2PO4, (15NH4)2SO4, and of D‐glucose‐d7 are removed by lyophilization before use. The pH* is adjusted to 7.2. The expression culture is grown at 37 °C with agitation. When an OD600 of 0.5 is reached, the culture is brought to its final volume of 0.5 L with M9 minimal medium containing 0.4 g/L of L‐alanine‐3‐13C‐2‐d (Sigma‐Aldrich) and 2.5 g/L of succinate‐d4 (CIL) for Ala methyl labeling; 72 mg/L 2‐ketobutyric acid‐4‐13C‐3,3‐d2 (CortecNet) for Ile δ1 methyl labeling; 8 vials per liter of DLAM‐LVPr°S (NMR‐BIO) containing 2‐hydroxy‐2‐(13C‐methyl)‐3‐oxo‐4‐d3 butanoate for stereoselective labeling of leucine/valine pro‐S methyl groups; 120 mg/L of 2‐keto‐3‐(methyl‐d3)‐3‐d‐4‐13C butanoate (CIL) for labeling of leucine/valine pro‐R and pro‐S methyl groups; 100 mg/L of L‐methionine‐(methyl‐13C) (CIL) for labeling of methionine ϵ methyl groups. Protein expression is induced 1 h after addition of labeled precursors using 1 mM (f.c.) IPTG. After incubation overnight at 37 °C with agitation, the cells are harvested by centrifugation (5000 x g, 20 min, 4 °C), when the maximal cell density is reached (OD600 of 4–5).
Target proteins are purified as described earlier employing cation exchange and affinity chromatography.6 In the affinity chromatography step with uridine diphosphate (UDP)‐hexanolamine sepharose, 20 mM EDTA is used for elution of GTB. This is effective in removing all manganese ions (5 mM) that are present in the binding buffer. The elution fractions are combined and GTB is concentrated with Amicon Ultra‐4 Centrifugal Filter Units (Millipore, MWCO 10 kDa). The exchange into the desired NMR buffer is performed in Zellu/Trans Mini‐Dialyzer membranes (Roth, MWCO 12 kDa) with a sample‐to‐dialysate ratio of 1 : 15. The reservoir buffer is replaced at least five times until EDTA concentrations are <0.1 μM. The NMR buffer contains 35 mM sodium phosphate (pH* 6.8), 50 mM NaCl, 5 mM MgCl2, 1 mM Tris(2‐carboxyethyl)phosphine‐d16 (CIL, TCEP), 0.1 mM 2,2‐Dimethyl‐2‐silapentane‐5‐sulfonate‐d6 (Sigma‐Aldrich, DSS‐d6) in D2O (CIL, 99.96 %). Aggregates are removed by filtration employing 0.5 mL centrifugal filter devices (0.22 μm) at 5,000 x g for 5 min. Final protein concentrations are determined by UV absorbance at 280 nm with the NanoDrop™ spectrometer employing a theoretical ϵ=52370 M−1cm−1. Glycosyltransferase activity is checked in a radiochemical assay as described earlier.3, 17 The protein samples are stored at 4 °C until being used for NMR experiments.
UDP, UDP‐Glc, UDP‐GlcNAc, and UDP‐GalNAc were purchased from Sigma Aldrich. UDP‐Gal was a kind gift from Prof. Dr. Beat Ernst (Institute of Molecular Pharmacy, University of Basel, Switzerland). H‐disaccharide (α‐L‐Fuc‐(1, 2)‐β‐D‐Gal‐O‐(CH2)7‐CH3), 3DD (α‐L‐Fuc‐(1, 2)‐β‐D‐(3‐deoxy)‐Gal‐O‐(CH2)7‐CH3), and blood group B trisaccharide (α‐L‐Fuc‐(1, 2)‐[α‐D‐Gal‐(1, 3)]‐β‐D‐Gal‐O‐(CH2)7‐CH3) were chemically synthesized in our laboratory. H‐type I to VI trisaccharides18 were a kind gift of Prof. Dr. Todd Lowary, University of Alberta, Edmonton, Canada).
Titration experiments were performed with AILpr°SMVpr°S methyl‐labeled GTA, GTB and AAGlyB on a Bruker Avance III 500 MHz NMR spectrometer equipped with a TCI cryogenic probe. All experiments were performed at 298 K, and chemical shifts are referenced to 2,2‐dimethyl‐2‐silapentane‐d6‐5‐sulfonic acid (DSS‐d6, Sigma‐Aldrich) as external standard. Sample volumes ranged from 150 to 190 μL in a 3 mm NMR tube. The same stock of D2O‐based phosphate buffer (pH* 6.8) was used for dissolving the ligands, as well as for buffer exchange of the protein. The pH* of the ligand stock solutions was carefully readjusted to 6.8. Ligand working solutions were generated as dilutions of the ligand stock solution with protein solution in order to provide a constant protein concentration during the titration. At each titration step Methyl‐TROSY experiments9 were recorded using a slightly modified standard Bruker pulse program (hmqcphpr), yielding phase sensitive spectra with decoupling during acquisition.19 The total relaxation delay (acquisition time plus relaxation delay) was 1.64 s similar to the value used by Tugarinov et al..9a The spectra are processed with 4 Hz (1H) and 8 Hz (13C) exponential line broadening and zero filling to 512 complex points in the indirect dimension using NMRPipe.20
Peak positions are followed using CcpNmr Analysis.21 Ligand binding‐induced chemical shift perturbations (CSPs) are calculated as weighted Euclidean distances (Δδ) between peak positions in the spectrum recorded with the highest ligand concentration, and the positions in the spectrum recorded for the apo state: 22 The weighting factor of 0.18 is estimated from the observed 13C chemical shift range of ca. 17 ppm and of the 1H shift range of ca. 3 ppm. The standard deviation (σ) of all Euclidean chemical shift changes is calculated independently for each ligand titration in order to allow for a better comparison between ligands of different binding affinities.
The processed 2D [13C,1H] HMQC spectra of each titration point are imported into MATLAB 2015a (The MathWorks, Inc., Natick) for analysis with TITAN.11 All data are fitted to a two‐state ligand binding model, respectively
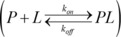 .
.
Several peaks are selected for global fitting, in order to describe the common interaction event more accurately. Chemical shifts and linewidths of the apo state are determined using only the first spectrum (absence of ligand). These parameters are hold constant for the remainder of the fitting session. Peak positions of the bound state and linewidths of all states as well as the dissociation constant (Kd) and the off‐rate (koff) of the ligand are fitted using the entire dataset. The bootstrap residual resampling procedure provided within TITAN is applied for estimation of parameter uncertainties in 200 runs.
Two sets of 2D longitudinal exchange experiments are recorded using previously described pulse sequences.16, 23 In one set of experiments the 13C frequency labeling precedes the mixing time. In the second set of experiments the order of frequency labeling and mixing time is interchanged. The experiments are performed on a Bruker Avance I spectrometer, operating at a proton frequency of 700 MHz, and equipped with a TXI cryogenic probe. The spectra are recorded with ILMV methyl‐labeled GTB (465 μM) in presence of saturating amounts of H‐dis (1.5 mM), and half‐saturating concentrations of UDP (250 μM). The mixing times range from 10 to 600 ms. For normalization, a set of reference spectra is acquired, where the mixing time is set to 0. Data are acquired with 48 scans and a recycle delay of 2.7 s, using 1024 (1H) and 256 (13C) complex data points. The spectra are processed with a cosine window function and zero filled to 2048 x 512 real points.
The peak intensities of the direct correlation signals in both sets of spectra (with/without frequency labeling) are extracted using CcpNmr analysis,21 and normalized with the peak intensities observed in the spectra with 0 mixing time, respectively. The normalized intensities are transferred to Origin (OriginLab, Northampton, MA) and are globally and simultaneously fitted to equations describing a two‐site exchanging system.16
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has been funded by the German Research Council DFG (DFG Pe494/11‐1). We acknowledge continuing support from the University of Lübeck. Professor Monica Palcic (University of Victoria, BC, Canada) is thanked for many stimulating discussions. We thank Professor Todd Lowary (University of Alberta, Edmonton, Canada) for the kind gift of synthetic H‐trisaccharides.
F. Flügge, T. Peters, ChemistryOpen 2019, 8, 760.
References
- 1.
- 1a. Alfaro J. A., Zheng R. B., Persson M., Letts J. A., Polakowski R., Bai Y., Borisova S. N., Seto N. O., Lowary T. L., Palcic M. M., Evans S. V., J. Biol. Chem. 2008, 283, 10097–10108; [DOI] [PubMed] [Google Scholar]
- 1b. Angulo J., Langpap B., Blume A., Biet T., Meyer B., Krishna N. R., Peters H., Palcic M. M., Peters T., J. Am. Chem. Soc. 2006, 128, 13529–13538; [DOI] [PubMed] [Google Scholar]
- 1c. Patenaude S. I., Seto N. O., Borisova S. N., Szpacenko A., Marcus S. L., Palcic M. M., Evans S. V., Nat. Struct. Biol. 2002, 9, 685–690. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Soya N., Shoemaker G. K., Palcic M. M., Klassen J. S., Glycobiology 2009, 19, 1224–1234; [DOI] [PubMed] [Google Scholar]; Shoemaker bG. K., Soya N., Palcic M. M., Klassen J. S., Glycobiology 2008, 18, 587–592. [DOI] [PubMed] [Google Scholar]
- 3. Sindhuwinata N., Munoz E., Munoz F. J., Palcic M. M., Peters H., Peters T., Glycobiology 2010, 20, 718–723. [DOI] [PubMed] [Google Scholar]
- 4. Sindhuwinata N., Grimm L. L., Weissbach S., Zinn S., Munoz E., Palcic M. M., Peters T., Biopolymers 2013, 99, 784–795. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Weissbach S., Flügge F., Peters T., ChemBioChem 2018, 19, 970–978; [DOI] [PubMed] [Google Scholar]
- 5b. Grimm L. L., Weissbach S., Flügge F., Begemann N., Palcic M. M., Peters T., ChemBioChem 2017, 18, 1260–1269. [DOI] [PubMed] [Google Scholar]
- 6. Flugge F., Peters T., J. Biomol. NMR 2018, 70, 245–259. [DOI] [PubMed] [Google Scholar]
- 7. Bain A. D., Prog. Nucl. Magn. Reson. Spectrosc. 2003, 43, 63–103. [Google Scholar]
- 8. Gunther U. L., Schaffhausen B., J. Biomol. NMR 2002, 22, 201–209. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Tugarinov V., Hwang P. M., Ollerenshaw J. E., Kay L. E., J. Am. Chem. Soc. 2003, 125, 10420–10428; [DOI] [PubMed] [Google Scholar]
- 9b. Ollerenshaw J. E., Tugarinov V., Kay L. E., Magn. Reson. Chem. 2003, 41, 843–852. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Wiesner S., Sprangers R., Curr. Opin. Struct. Biol. 2015, 35, 60–67; [DOI] [PubMed] [Google Scholar]
- 10b. Karagoz G. E., Duarte A. M., Akoury E., Ippel H., Biernat J., Luengo T. Moran, Radli M., Didenko T., Nordhues B. A., Veprintsev D. B., Dickey C. A., Mandelkow E., Zweckstetter M., Boelens R., Madl T., Rudiger S. G., Cell 2014, 156, 963–974; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Stoffregen M. C., Schwer M. M., Renschler F. A., Wiesner S., Structure 2012, 20, 573–581; [DOI] [PubMed] [Google Scholar]
- 10d. Sprangers R., Kay L. E., Nature 2007, 445, 618–622. [DOI] [PubMed] [Google Scholar]
- 11. Waudby C. A., Ramos A., Cabrita L. D., Christodoulou J., Sci. Rep. 2016, 6, 24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Letts J. A., Rose N. L., Fang Y. R., Barry C. H., Borisova S. N., Seto N. O., Palcic M. M., Evans S. V., J. Biol. Chem. 2006, 281, 3625–3632. [DOI] [PubMed] [Google Scholar]
- 13. Gagnon S. M. L., Legg M. S. G., Sindhuwinata N., Letts J. A., Johal A. R., Schuman B., Borisova S. N., Palcic M. M., Peters T., Evans S. V., Glycobiology 2017, 27, 966–977. [DOI] [PubMed] [Google Scholar]
- 14. Pesnot T., Jorgensen R., Palcic M. M., Wagner G. K., Nat. Chem. Biol. 2010, 6, 321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montelione G. T., Wagner G., J. Am. Chem. Soc. 1989, 111, 3096–3098. [Google Scholar]
- 16. Kloiber K., Spitzer R., Grutsch S., Kreutz C., Tollinger M., J. Biomol. NMR 2011, 51, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palcic M. M., Heerze L. D., Pierce M., Hindsgaul O., Glycoconjugate J. 1988, 5, 49–63. [Google Scholar]
- 18.
- 18a. Meloncelli P. J., Lowary T. L., Carbohydr. Res. 2010, 345, 2305–2322; [DOI] [PubMed] [Google Scholar]
- 18b. Meloncelli P. J., Lowary T. L., Aust. J. Chem. 2009, 62, 558–574; [Google Scholar]
- 18c. Meloncelli P. J., West L. J., Lowary T. L., Carbohydr. Res. 2011, 346, 1406–1426. [DOI] [PubMed] [Google Scholar]
- 19. Bax A., Griffey R. H., Hawkins B. L., J. Magn. Reson. (1969-1992) 1983, 55, 301–315. [Google Scholar]
- 20. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 21. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D., Proteins 2005, 59, 687–696. [DOI] [PubMed] [Google Scholar]
- 22. Williamson M. P., Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Farrow N. A., Zhang O., Forman-Kay J. D., Kay L. E., Biochemistry 1995, 34, 868–878; [DOI] [PubMed] [Google Scholar]
- 23b. Farrow N. A., Zhang O., Forman-Kay J. D., Kay L. E., J. Biomol. NMR 1994, 4, 727–734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary