
Fig. 1 |. Renal and extrarenal manifestations in polycystic kidney disease.
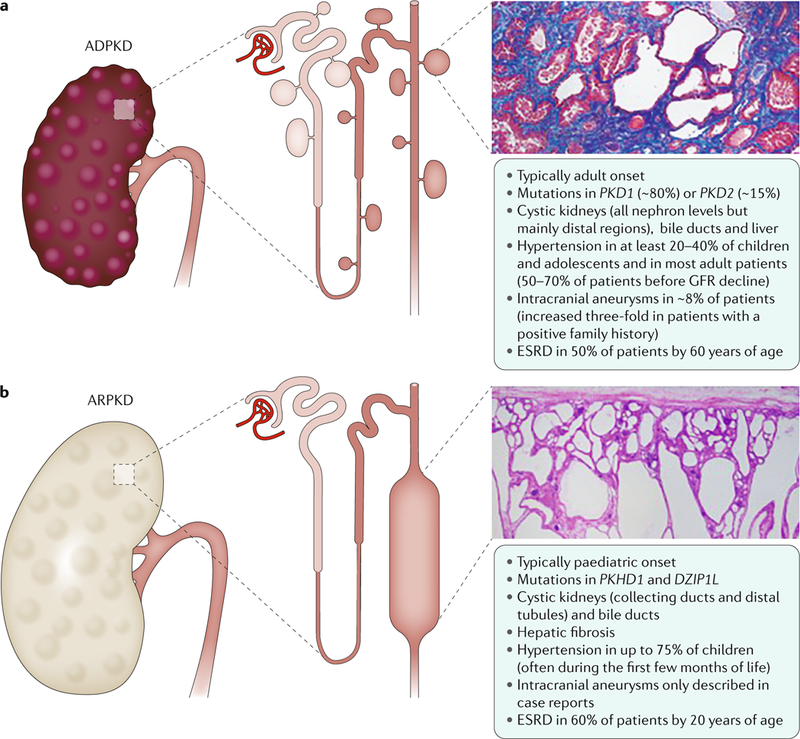
a | Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of polycystic kidney disease (PKD) and is mainly caused by mutations in PKD1 and PKD2, which encode polycystin 1 and polycystin 2, respectively. ADPKD is usually an adult-onset disease that is characterized by the formation of fluid-filled cysts in various locations in the kidneys, but mostly in the distal regions. The histology image is of the kidney (stained with Malloy trichrome stain) of a 49-year-old patient with end-stage ADPKD. Small cysts and extensive fibrosis (blue) are visible. b | Autosomal recessive PKD (ARPKD) is rarer and more severe than ADPKD and is caused by mutations in polycystic kidney and hepatic disease 1 (PKHD1; which encodes fibrocystin) and DZIP1L (which encodes DAZ-interacting protein 1-like protein (DZIP1L)). ARPKD usually presents in utero, perinatally or in infancy and is characterized by the formation of cysts from the renal distal tubules and collecting ducts. Microscopically, cysts are fusiform dilatations of the aforementioned distal parts of the nephron, which are lined by a columnar or cuboidal epithelium. Respective dilated collecting ducts run perpendicular to the renal capsule (renal section is stained with haematoxylin and eosin). Both diseases often progress to end-stage renal disease (ESRD) that requires renal replacement therapy. GFR,glomerular filtration rate.