

Abstract
Niclosamide, an antiparasitic, has been repositioned as a potential therapeutic drug for systemic diseases based on its antiviral, anticancer, and anti-infection properties. However, low bioavailability limits its in vivo efficacy. Our aim was to determine whether metabolic disposition by microsomal P450 enzymes in liver and intestine influences niclosamide’s bioavailability in vivo, by comparing niclosamide metabolism in wild-type, liver-Cpr-null (LCN), and intestinal epithelium-Cpr-null (IECN) mice. In vitro stability of niclosamide in microsomal incubations was greater in the intestine than in liver in the presence of NADPH, but it was much greater in liver than in intestine in the presence of UDPGA. NADPH-dependent niclosamide metabolism and hydroxy–niclosamide formation were inhibited in hepatic microsomes of LCN mice, but not IECN mice, compared with wild-type mice. In intestinal microsomal reactions, hydroxy–niclosamide formation was not detected, but rates of niclosamide-glucuronide formation were ∼10-fold greater than in liver, in wild-type, LCN, and IECN mice. Apparent Km and Vmax values for microsomal niclosamide–glucuronide formation showed large differences between the two tissues, with the intestine having higher Km (0.47 μM) and higher Vmax (15.8) than the liver (0.09 μM and 0.75, respectively). In vivo studies in LCN mice confirmed the essential role of hepatic P450 in hydroxy–niclosamide formation; however, pharmacokinetic profiles of oral niclosamide were only minimally changed in LCN mice, compared with wild-type mice, and the changes seem to reflect the compensatory increase in hepatic UDP-glucuronosyltransferase activity.
SIGNIFICANCE STATEMENT
These results suggest that efforts to increase the bioavailability of niclosamide by blocking its metabolism by P450 enzymes will unlikely be fruitful. In contrast, inhibition of niclosamide glucuronidation in both liver and intestine may prove effective for increasing niclosamide’s bioavailability, thereby making it practical to repurpose this drug for treating systemic diseases.
Introduction
Niclosamide (2′,5-dichloro-4′-nitrosalicylanilide) has been used as a piscicide and molluscicide to treat water since the 1970s, to control, for example, vectors for schistosomiasis (Andrews et al., 1982; Schreier et al., 2000; Chimbari, 2012). In addition, niclosamide is used as an anthelmintic drug for the treatment of tapeworm infections in both humans and animals (Pearson and Hewlett, 1985). The therapeutic effects were reported to be via the uncoupling of the electron transport chain from the adenosine triphosphate synthase, resulting in blockage of the synthesis of adenosine triphosphate, which is an essential source of energy for cellular metabolism (Al-Hadiya, 2005). Recently, niclosamide has emerged as a strong candidate for drug repurposing and repositioning, as this antiparasitic drug has been shown to have therapeutic effects against viruses, inflammation, and cancer, and can also regulate glucose metabolism in obese subjects (Chang et al., 2006; Piccaro et al., 2013; Li et al., 2014; Chowdhury et al., 2017). Niclosamide is in clinical trials for treating colon cancer and prostate cancer (Clinicaltrials.gov Identifier, NCT02532114; NCT03123978; NCT02687009).
Niclosamide, with a hydrophobic structure, is a Biopharmaceutical Classification System class II drug (Pardhi et al., 2017), which has limited absorption from the gastrointestinal tract when administered orally. Efforts have been made to increase niclosamide bioavailability, including modification of its physicochemical properties and use of drug carriers (van Tonder et al., 2004; Lin et al., 2016; Pardhi et al., 2017; Xie and Yao, 2018). The low bioavailability may also be due to rapid metabolism, i.e., first-pass extraction by the intestine and liver. Early studies detected hydroxylated niclosamide in the mouse liver (Douch and Gahagan, 1977) and niclosamide glucuronides (NIC-G) and sulfate esters in fishes (Dawson et al., 1999). A more recent report of in vitro assays using human recombinant enzymes identified CYP1A2 and UGT1A1 as the most active enzymes for hydroxylation and glucuronidation, respectively, of niclosamide (Lu et al., 2016). CYP1A2 is mainly expressed in the liver with low expression and activity in the extrahepatic tissues (Gad, 2008). UGT1A1 is highly expressed in both liver and small intestine (Fisher et al., 2000; Tukey and Strassburg, 2000; Buckley and Klaassen, 2007). Several studies have investigated the relative importance of intestinal and hepatic glucuronidation to the metabolism of various drugs (but not for niclosamide), and the results were substrate-dependent (Bowalgaha and Miners, 2001; Watanabe et al., 2002; Bernard and Guillemette, 2004; Cubitt et al., 2009).
In the present study, we aimed to gain a better understanding of the roles of liver and intestinal microsomal cytochrome P450 (P450 or CYP) enzymes in niclosamide disposition, to guide efforts to improve niclosamide bioavailability through metabolic modulation. We used two engineered mouse models with tissue-specific deletion of the P450 reductase (Cpr or Por, essential for the activities of all microsomal P450 enzymes) in either liver hepatocytes [liver-Cpr-null (LCN)] (Gu et al., 2003) or intestinal enterocytes [intestinal epithelium-Cpr-null (IECN)] (Zhang et al., 2009). We compared hepatic and intestinal microsomes from wild-type, LCN, and IECN mice for niclosamide metabolic stability and rates of formation of hydroxy-niclosamide (NIC-OH) and NIC-G in vitro. Based on an initial finding that hepatic, but not intestinal, microsomes were active in NADPH-dependent niclosamide metabolism and NIC-OH formation, we further compared wild-type and LCN mice for pharmacokinetic profiles of oral niclosamide and in vivo formation of niclosamide metabolites. Our results indicate that P450-mediated biotransformation is unlikely important for in vivo disposition of oral niclosamide and imply that both hepatic and intestinal glucuronidation may be more promising targets for increasing niclosamide’s bioavailability.
Materials and Methods
Chemicals and Reagents.
Niclosamide (purity ≥98%), uridine diphosphate glucuronic acid, trisodium salt (UDPGA) (purity ≥98%), β-nicotinamide adenine dinucleotide phosphate, reduced tetra (cyclohexyl ammonium) salt (NADPH) (purity ≥97%), and p-nitrophenol (purity ≥99%) were purchased from Sigma-Aldrich (St. Louis, MO). All solvents (acetonitrile, methanol, and water) were of liquid chromatography–mass spectrometry grade (Fisher Scientific, Houston, TX), and all other chemicals were of reagent grade.
Animals and Treatments.
Animals were housed at 22°C with 12-hour on-off light cycle and given food and water ad libitum. Two- to 3-month-old male LCN mice (Gu et al., 2003), IECN mice (Zhang et al., 2009), and their corresponding wild-type littermates were used for the study. For in vivo clearance studies, LCN and wild-type mice were given niclosamide (Sigma-Aldrich) at 120 mg/kg by oral gavage. Animal studies were approved by the Wadsworth Center Institutional Animal Care and Use Committee (Albany, NY) and University of Arizona Animal Care and Use Committee.
Preparation of Microsomes and In Vitro Assays.
Hepatic and intestinal microsomes were prepared according to previously described procedures (Zhang et al., 2009); the epithelium of the entire small intestine from two to three mice was pooled for each microsomal preparation. For assaying P450-mediated niclosamide metabolism, the incubation mixtures contained 0.1 mg microsomal protein, 0.1 M potassium phosphate buffer (pH 7.4), 1.0 mM NADPH, and 3 mM MgCl2 in a final volume of 200 µl. For assaying UDP-glucuronosyl transferase (UGT)–mediated niclosamide metabolism, the incubation mixtures were the same as for P450 assays, except that 5.0 mM UDPGA was used in place of NADPH. The substrate concentration was 100 µM for measuring the formation of niclosamide metabolites, and 1 µM for determining the rates of niclosamide disappearance. The reactions were initiated after a 3-minute preincubation, by the addition of NADPH and/or UDPGA into the reaction mixture, followed by a 30-minute incubation at 37°C in a shaking water bath. The reactions were terminated by the addition of 400 µl cold acetonitrile. Control groups were included, in which NADPH and/or UDPGA were absent. p-Nitrophenol was added (in methanol, at 250 pmol per sample) as an internal standard. Extraction efficiency for niclosamide was >95%. The intrinsic clearance (CLint) of niclosamide was calculated using the following equation: CLint = [volume of reaction (V) × 0.693]/half-life of substrate disappearance estimated by regression analysis of semilogarithmic plots (Suzuki et al., 2003).
Sample Preparation for In Vivo Clearance of Oral Niclosamide.
Blood samples were collected through the tail vein using heparinized capillary tubes (Thermo Fisher Scientific), at 0.25, 0.5, 1, 2, 4, 10, and 24 hours after dosing, and stored as plasma at −80°C until use. Plasma samples (10 µl each) were each mixed with 10 µl 25-µM methanolic solution of p-nitrophenol (as an internal standard), 10 µl methanol, and 270 µl acetonitrile. The mixtures were vortexed and then centrifuged at 4000g for 10 minutes; the organic layer (top) was transferred to a new tube, and the centrifugation step was repeated. The recovery of the standards in blank plasma was >85%. A 2-µl aliquot of the supernatant was used for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
LC-MS/MS Analysis of Niclosamide, NIC-OH, and NIC-G.
Niclosamide and niclosamide metabolites were detected using LC-MS/MS. The LC-MS/MS system consisted of an Agilent model 1200 high-performance liquid chromatography (Agilent Technologies, Santa Clara, CA) and a Sciex 4000 Q-Trap mass spectrometer (AB SCIEX, Framingham, MA). Analytes were separated on a ZORBAX SB-C18 column (2.1 × 150 mm, 3.5 µm; Agilent) at room temperature, with mobile phase A containing 0.1% formic acid (v/v) in water and mobile phase B containing 100% methanol. Isocratic elution of the analytes was conducted using 20%A/80%B at a flow rate of 0.4 ml/min. The mass spectrometer was operated in negative mode, using electrospray ionization. The ion spray voltage and temperature were set at −4500 V and 450°C, respectively. Curtain gas, ion source gas 1, and ion source gas 2 were set at 20, 40, and 20 psi, respectively. Niclosamide, NIC-OH, NIC-G, and the internal standard p-nitrophenol were detected using multiple reaction monitoring, with a dwell time of 150 milliseconds per transition, at m/z 325/171, 341/171, 501/325, and 138/108, respectively; the mass transitions were adapted from a protocol previously described by Lu et al. (2016). The optimized parameters for declustering potential, entrance potential, collision energy, and collision cell exit potential, used for analyte identification and quantification, were as follows: −50, −12, −38, and −7 V, respectively, for niclosamide; −50, −12, −40, and −10 V, respectively, for NIC-OH; −50, −12, −25, and −10 V, respectively, for NIC-G; and −50, −12, −25, and −10 V, respectively, for p-nitrophenol. Retention times for niclosamide, NIC-OH, NIC-G, and p-nitrophenol were 4.95, 3.38, 1.49, and 1.11 minutes, respectively. For quantitative analysis of niclosamide, standards (0.1–40 µM in 10 µl methanol), along with 10 µl p-nitrophenol (at 25 µM in methanol), were added to 10 µl blank mouse plasma to construct the calibration curve. Standards for NIC-OH and NIC-G were not available; therefore, the ratios of metabolite peak area to internal standard peak area were taken for estimating the relative levels of the metabolites in different samples.
RNA Extraction and Quantitative Polymerase Chain Reaction for UGT1A1.
Total RNA was isolated from liver using TRIzol reagent (Invitrogen, Carlsbad, CA) and subjected to RNA polymerase chain reaction essentially as previously described (Richardson et al., 2006). RNA concentration and purity were determined spectrally. First-strand cDNA was prepared using 2 µg RNA and SuperScript III First-Strand Synthesis System (Invitrogen). The primers used for UGT1A1 determination were as follows: forward, 5′-ccagcagaaggggcacgaagttg-3′, and reverse, 5′-tgaccacgcgcagcagaaaagaat-3′; those for glyceraldehyde 3-phosphate dehydrogenase determination were as follows: forward, 5′-agaacatcatccctgcatcca-3′, and reverse, 5′-ccgttcagctctgggatgac-3′.
Immunoblot Analysis.
Hepatic microsomal proteins (30 µg) were separated on 10% NuPAGE Bis-Tris gels (Life Technologies, Grand Island, NY) and subjected to immunoblot analysis, as described previously (Wen et al., 2013). For immunodetection, a rabbit polyclonal anti-UGT1A1 (Abcam, Cambridge, MA) and a rabbit polyclonal anti-calnexin (Abcam) (as loading control) were used. Peroxidase-conjugated goat anti-rabbit IgG (Sigma-Aldrich) was used as secondary antibody. The immunoreactive signal was detected with a chemiluminescence kit (GE Healthcare, Piscataway, NJ) and quantified using a GS-710 Calibrated Imaging densitometer or a ChemiDoc XRS1 System (Bio-Rad, Hercules, CA).
Enzyme Kinetic Analysis for Microsomal NIC-G Formation.
Contents of reaction mixtures were the same as described above for determination of rates of NIC-G formation, except that a range of substrate concentrations was used (0.03–100 µM) and the amount of microsomal protein was reduced to 0.01 mg. Reactions were carried out for 2.5 minutes for intestinal microsomes, and 10 minutes for hepatic microsomes, during which the rates were linear with incubation time. To increase sensitivity, a new LC-MS/MS system was used, which consisted of a SCIEX Q-Trap 6500+ mass spectrometer (AB SCIEX), an Infinity II Series model 1290 ultraperformance liquid chromatography system (Agilent), and an Agilent ZORBAX Elipse Plus C18 column (2.1 × 50 mm; 1.8 μm). The column was equilibrated with 75%B (methanol containing 0.1% formic acid). Samples were eluted with a 3-minute linear gradient from 75%B to 95%B, at room temperature, at a flow rate of 0.3 ml/min. The mass spectrometer was operated in negative ion mode using electrospray ionization with the following settings: ion spray voltage, −4500 V; temperature, 450°C; curtain gas, 25 psi; ion source gas 1, 40 psi; and ion source gas 2, 20 psi. NIC-G and a new internal standard nitrendipine (added at 100 pmol per sample in methanol) were detected using multiple reaction monitoring at m/z 501/325 and 359/122, respectively, with the dwell time of 150 milliseconds per transition. The optimized parameters for declustering potential, entrance potential, collision energy, and collision cell exit potential were as follows: −41, −10, −25, and −15 V, respectively, for NIC-G; and −91, −10, −35, and −12 V, respectively, for nitrendipine. For sample preparation, solid-phase extraction was performed by using solid-phase extraction C18 cartridges (Biotage, Charlottesville, VA). All samples were analyzed in triplicate. Retention times for NIC-G and nitrendipine were 1.23 and 1.26 minutes, respectively.
Data Analysis.
Pharmacokinetic parameters were calculated using PK solver (Microsoft, Redmond, WA), by assuming a noncompartmental model. Half-life values for calculation of intrinsic clearance and statistical significance of various data comparisons, as well as enzyme kinetic parameters (from Lineweaver–Burk plots), were determined with the use of GraphPad Prism (GraphPad Software, La Jolla, CA). One-way or two-way analysis of variance, followed by a post hoc test for pairwise comparisons, or Student’s t test, was used. P values <0.05 were considered statistically significant.
Results
In Vitro Metabolic Stability of Niclosamide.
The amounts of niclosamide remaining at various times after initiation of reaction are plotted in Fig. 1. With hepatic microsomes from wild-type mice (Fig. 1A), niclosamide levels decreased over time upon incubation with either NADPH or UDPGA (added at saturating amounts), whereas little metabolism occurred in the absence of a cofactor. The rate of niclosamide disappearance was slightly greater in the presence of NADPH than in the presence of UDPGA. Coincubation with both cofactors led to a further increase in niclosamide disappearance. With intestinal microsomes (Fig. 1B), all the niclosamide disappeared within 5 minutes in the presence of UDPGA, but only ∼10% was lost in that time frame when NADPH was present. The rate of niclosamide disappearance was also greater in the intestine than in the liver, as niclosamide metabolism was not complete in the liver microsomal incubations until at least 30 minutes, with either cofactor (Fig. 1A).
Fig. 1.

In vitro metabolic stability of niclosamide. Levels of niclosamide (NIC) remaining after incubation with pooled hepatic (A, C, and E) or intestinal microsomes (B, D, and F) of adult male mice for various times are shown as a percentage of the starting amount at time zero. Reaction mixtures contained either no cofactor (negative control), 1.0 mM NADPH (for P450 activity), 5.0 mM UDPGA (for UGT activity), or 1.0 mM NADPH plus 5.0 mM UDPGA (for combined CYP and UGT reactions), in addition to a common mixture of 0.1 mg hepatic or intestinal microsomal protein from wild-type, LCN, or IECN mice, 1 µM niclosamide, 0.1 M potassium phosphate buffer (pH 7.4), and 3 mM MgCl2, in a final volume of 200 µl. Relevant wild-type data from (A and B) were replotted in (C–F) for comparison with data from null mice. The values represent means ± S.D. (n = 3–5). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 compared with NADPH group (A and B) or wild-type group (C–F); one-way analysis of variance with Bonferroni’s multiple comparisons test. The aggregate values for no-cofactor control (Control) in wild-type, LCN, and IECN groups at the 60-minute time point are shown for liver (C and E) and intestine (D and F).
The intrinsic clearance of niclosamide was calculated based on data in Fig. 1, A and B. In the liver, there was no significant difference between the contributions from CYP-mediated intrinsic clearance (0.21 ± 0.04 ml/min per milligram) and UGT-mediated intrinsic clearance (0.21 ± 0.01 ml/min per milligram); in the intestine, UGT-mediated intrinsic clearance (1.33 ± 0.25 ml/min per milligram) was significantly higher (P < 0.001; one-way analysis of variance) than those mediated by either CYP or UGT enzymes in the liver, or that mediated by CYPs in the intestine (0.05 ± 0.02 ml/min per milligram).
The relative contributions of hepatic and intestinal CYP enzymes to microsomal niclosamide metabolism were further examined by comparing microsomes from LCN and IECN mouse models with those of wild-type mice. As shown in Fig. 1, C and D, hepatic microsomal NADPH-supported metabolism of niclosamide was significantly decreased in the LCN mice, but not in the IECN mice, whereas intestinal microsomal NADPH-mediated metabolism of niclosamide was not different among the three mouse genotype groups. These data are consistent with a role for CYPs in hepatic, but not intestinal, microsomal metabolism of niclosamide.
In control experiments, as expected, a notable decrease in hepatic (Fig. 1E) or intestinal (Fig. 1F) microsomal UDPGA-supported niclosamide metabolism was not observed in either LCN mice or IECN mice, compared with wild-type mice. Quite the reverse, small increases in rates of hepatic microsomal UDPGA-supported niclosamide metabolism were observed in LCN and, to a lesser degree, IECN mice (Fig. 1E). In that connection, compensatory increases in hepatic UGT1A1 mRNA (by ∼100%) and protein (by ∼50%) levels in the LCN mice, compared with wild-type mice, were observed (Fig. 2), although a significant increase in UGT1A1 mRNA or protein level was not observed in the livers of the IECN mice (data not shown). In contrast, an increase in rates of intestinal microsomal UDPGA-supported niclosamide metabolism was not observed in the null mice (Fig. 1F).
Fig. 2.
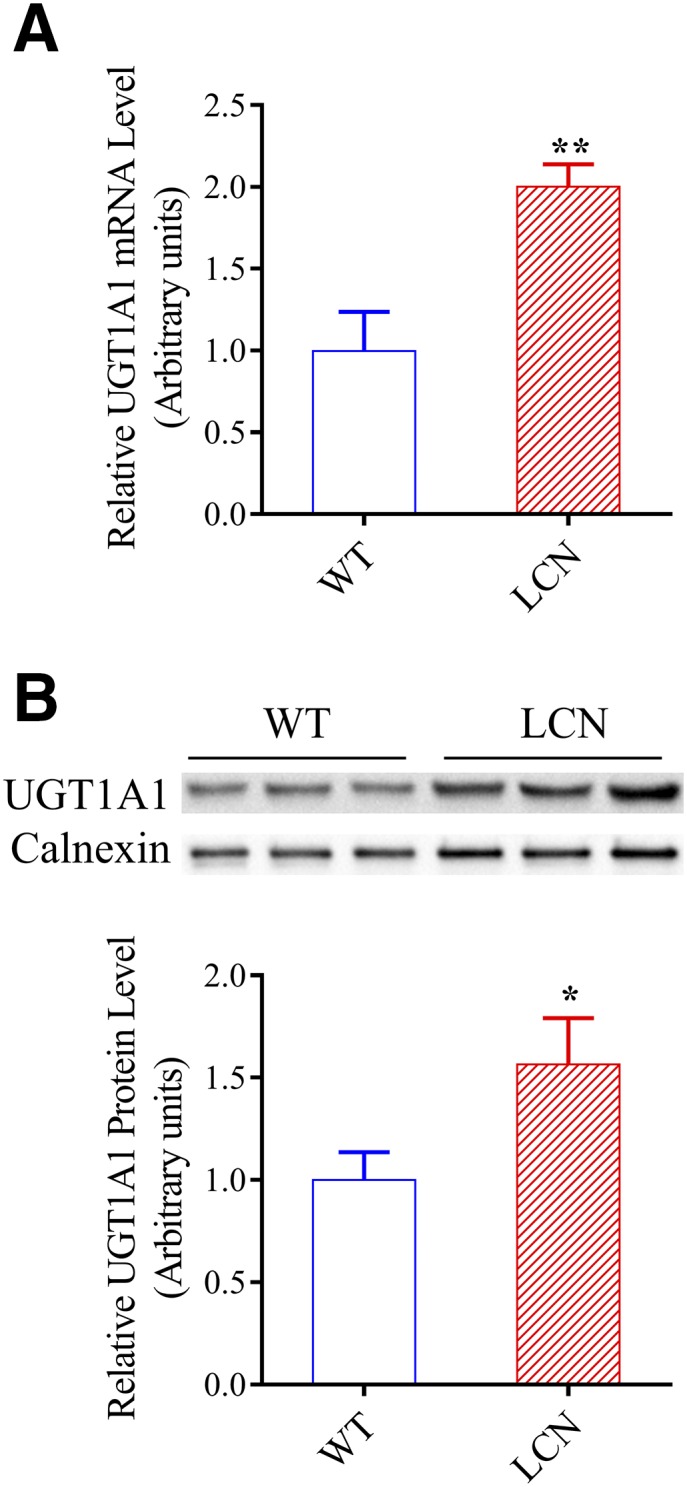
UGT1A1 expression in the liver of wild-type and LCN mice. (A) Hepatic UGT1A1 mRNA level was determined in adult male mice using RNA polymerase chain reaction, as described in Materials and Methods. The values represent means ± S.D., n = 3. (B) UGT1A1 protein was detected on immunoblots, as described in Materials and Methods. Microsomal proteins (30 µg) from liver of adult male mice were analyzed. Calnexin level was determined as a loading control. Each microsomal sample was prepared from tissues pooled from two to three mice. Results of densitometric analysis (bar graphs) are normalized by calnexin levels in each sample and are shown in arbitrary units. The values represent means ± S.D., n = 3. **P < 0.01; *P < 0.05; compared with wild-type group, Student’s t test.
Niclosamide Metabolite Formation In Vitro.
In addition to measuring overall niclosamide metabolism, the rates of NIC-OH and NIC-G formation in vitro by hepatic or intestinal microsomes were also compared among wild-type, LCN, and IECN mice. Authentic metabolite standards for NIC-OH and NIC-G were not available; thus, relative rates of metabolite formation were compared among the genotype groups.
NIC-OH formation was detected in incubations with hepatic (Fig. 3A), but not intestinal (data not shown), microsomes, in the presence of NADPH. As expected, liver microsomal NIC-OH formation rates were significantly decreased in LCN mice than wild-type mice, reflecting the loss of all hepatic microsomal P450 activity, whereas the rates were slightly increased in IECN mice, consistent with the known induction of CYP1A expression in the livers of these mice (Zhang et al., 2009).
Fig. 3.

Relative rates of NIC-OH and NIC-G formation in liver and intestinal microsomes of wild-type, LCN, and IECN mice. The relative rates of NIC-OH (A) and NIC-G (B, C) formation in hepatic (A and B) and intestinal (C) microsomes were derived from chromatographic peak area ratios of the metabolites to that of an internal standard. Formation of NIC-OH was detected with hepatic microsomes (A), but not intestinal microsomes (data not shown). The reaction mixtures contained 0.1 mg microsomal protein, 100 µM niclosamide, 0.1 M potassium phosphate buffer (pH 7.4), 1.0 mM NADPH or 5.0 mM UDPGA, and 3 mM MgCl2 in a final volume of 200 µl. The incubation was carried out at 37°C for 30 minutes. The values represented means ± S.D., n = 3–5; *P < 0.05; **P < 0.01; ****P < 0.0001, compared with wild-type group; one-way analysis of variance with Bonferroni’s multiple comparisons test.
Rates of formation of NIC-G were much lower in hepatic (Fig. 3B) than in intestinal microsomes (Fig. 3C), in the presence of UDPGA, which agreed with the tissue differences in rates of UDPGA-supported niclosamide metabolism observed in Fig. 1. Consistent with data in Fig. 1E on UDPGA-supported niclosamide metabolism in hepatic microsomes, rates of NIC-G formation were elevated in hepatic microsomes from both LCN (1.5-fold) and IECN (1.3-fold) mice, compared with wild-type mice (Fig. 3B). In contrast to the situation in the liver, there was no genotype difference in rates of NIC-G formation in intestinal microsomes (Fig. 3C). This result agreed with data in Fig. 1F for intestinal microsomal UDPGA-supported niclosamide metabolism and reflects absence of compensatory increases in UGT expression in the intestine of LCN or IECN mice.
The enzyme kinetic profiles of NIC-G formation in intestinal and hepatic microsomes of wild-type mice were compared. For both tissues, the Michaelis–Menten plot (Fig. 4, left panels) showed substrate saturation of the enzyme. Apparent kinetic parameters calculated from the transformed Linewaever–Burk plots (Fig. 4, right panels) showed large differences between the two tissues in both Km and Vmax, with the intestine having higher Km (0.47 μM) and higher Vmax (15.8) values than the liver (0.09 μM and 0.75, respectively).
Fig. 4.

Enzyme kinetic profiles of NIC-G formation in intestinal and hepatic microsomes. Reaction mixtures contained 0.1 M potassium phosphate buffer (pH 7.4), 0.01 mg intestinal or hepatic microsomal protein, 0.03–100 µM niclosamide, 5.0 mM UDPGA, and 3 mM MgCl2 in a final volume of 200 µl. The reaction was carried out at 37°C for 2.5 minutes for intestinal microsomes. The values represent means ± S.D. of triplicate determinations using pooled intestinal (A) and hepatic (B) microsomes of six adult male wild-type mice. Rates of NIC-G formation are shown as relative amounts of NIC-G formed in arbitrary unit (determined by the ratio of NIC-G peak area over peak area of the internal standard nitrendipine) per milligram of microsomal protein, per minute. The apparent Km and Vmax values, calculated from the Lineweaver–Burk blots (right panel), are shown.
Niclosamide Metabolism and Disposition in Wild-Type and LCN Mice In Vivo.
Given that both CYP and UGT enzymes can metabolize niclosamide in the liver, we further determined whether P450-catalyzed niclosamide hydroxylation contributes significantly to the in vivo disposition of oral niclosamide, by comparing between wild-type and LCN mice. It was anticipated that, if hepatic P450 enzymes contributed significantly to systemic clearance of oral niclosamide, we would observe a substantial decrease in niclosamide clearance in the LCN mice, compared with wild-type mice, as reflected by higher Cmax and/or area under the curve values. However, as shown in Fig. 5A, clearance of niclosamide from plasma following oral administration (the apparent clearance, CL/F) was not slower in LCN mice, compared with wild-type mice, with a significant decrease in Cmax (by 32%, Table 1). NIC-OH formation was nearly completely inhibited (Fig. 5B; Table 1), which is consistent with the loss of hepatic CYP1A2 activity in LCN mice, and indicates that the lower systemic exposure of niclosamide in the LCN mice was not due to a compensatory increase in niclosamide hydroxylation by P450s in other tissues. Consistent with in vitro data showing upregulation of hepatic UGT1A1 expression and UGT activity (Figs. 1–3), in vivo formation of NIC-G was increased in the LCN mice (Fig. 5C; Table 1).
Fig. 5.

In vivo clearance of niclosamide in wild-type and LCN mice. Adult male wild-type and LCN littermates were given a single oral dose of niclosamide at 120 mg/kg. Plasma was obtained at various times after dosing for determination of niclosamide (A), NIC-OH (B), and NIC-G (C). Relative levels of NIC-OH or NIC-G (in arbitrary units) are compared between wild-type and LCN mice. The values represent means ± S.D., n = 5; *P < 0.05; **P < 0.01; ****P < 0.0001, compared with corresponding wild-type group, two-way analysis of variance with Bonferroni’s multiple comparisons test.
TABLE 1.
Pharmacokinetic parameters for orally administered niclosamide and its metabolites in wild-type and LCN mice
Data in Fig. 5 were used to calculate pharmacokinetic parameters, as described in Materials and Methods. The values represent means ± S.D., n = 5.
| Analyte | Strain | Tmax | Cmax | T1/2 | AUC0–24 h | CL/F |
|---|---|---|---|---|---|---|
| NIC | h | μM | h | nmol·h/ml | l/h per kilogram | |
| Wild-type | 0.80 ± 0.27 | 1.18 ± 0.17 | 12.2 ± 3.8 | 8.80 ± 2.13 | 30.98 ± 15.35 | |
| LCN | 0.75 ± 0.71 | 0.76 ± 0.20a | 17.2 ± 5.1 | 5.90 ± 2.18 | 45.67 ± 23.98 | |
| NIC-OH | h | Arbitrary unit | h | Arbitrary unit | Arbitrary unit | |
| Wild-type | 0.80 ± 0.27 | 0.75 ± 0.39 | 4.10 ± 1.95 | 3.52 ± 1.77 | 93.20 ± 79.16 | |
| LCN | 1.00 ± 0.61 | 0.07 ± 0.03a | 6.31 ± 3.74 | 0.37 ± 0.19a | 847.6 ± 622.3b | |
| NIC-G | h | Arbitrary unit | h | Arbitrary unit | Arbitrary unit | |
| Wild-type | 0.63 ± 0.25 | 0.59 ± 0.23 | 8.17 ± 3.70 | 2.70 ± 1.12 | 62.44 ± 48.51 | |
| LCN | 0.50 ± 0.31 | 1.35 ± 0.41b | 10.4 ± 6.2 | 5.09 ± 0.85a | 31.50 ± 13.90 |
AUC, area under the curve; T1/2, half-life.
P < 0.01, compared with corresponding wild-type group; Student’s t test.
P < 0.05, compared with corresponding wild-type group; Student’s t test.
Discussion
Although it has been reported that niclosamide could be metabolized by both CYP1A2 and UGT1A1 (Lu et al., 2016), the relative contributions of the CYPs and UGTs to microsomal metabolism of niclosamide in the liver and intestine were unclear. In this work, we measured the CYP-dependent and UGT-dependent metabolic disposition of niclosamide by incubating niclosamide with mouse hepatic or intestinal microsomes in the presence or absence of the respective cofactors for the two groups of enzymes, NADPH and UDPGA. The in vitro metabolic stability results indicated that, whereas CYP and UGT enzymes both contribute to niclosamide disposition in the liver, UGT plays a major role in microsomal niclosamide metabolism in the intestine. Furthermore, intestinal microsomal metabolism of niclosamide is more rapid than hepatic microsomal niclosamide metabolism, which implies that first-pass metabolism in the gut may play an important role for in vivo disposition of oral niclosamide.
A comparison among wild-type, LCN, and IECN mice provided strong support to the notion of a tissue-specific contribution of CYPs to niclosamide disposition in the liver, as a remarkable decrease in niclosamide disappearance was only observed in the LCN mice, but not in the IECN mice (Fig. 1). Results from metabolite formation analysis are also consistent with those from measurement of substrate disappearance, given that NIC-OH formation was observed in hepatic, but not intestinal, microsomal reactions. This apparent liver-specific contribution of CYPs to niclosamide disposition is consistent with the fact that CYP1A2, the major CYP isoform reported to be active toward niclosamide hydroxylation (Lu et al., 2016), is expressed in the liver, but very little in intestine (Kaminsky and Zhang, 2003). Nonetheless, the in vivo data for comparisons of niclosamide pharmacokinetics between wild-type and LCN mice (Fig. 5) indicated that P450-mediated niclosamide hydroxylation is unlikely a major pathway for in vivo disposition of oral niclosamide.
Multiple lines of evidence indicate greater activity of intestinal UGTs than hepatic UGTs toward niclosamide, including comparisons of intrinsic clearance (Fig. 1), relative rates of NIC-G formation at a high substrate concentration (Fig. 3), and enzyme kinetic profiles (Fig. 4). Notably, the apparent Km value for hepatic microsomes determined in this study (0.09 μM) is lower than the previously reported value (1.67 ± 0.06 μM) (Lu et al., 2016). This discrepancy may be explained by the difference in mouse strain (B6 mice in this study; CD-1 mice in the previous study) and assay conditions, such as range of substrates (0.03–100 μM in this study; 0.195–37.5 μM in Supplemental Table 2 of the previous study) or microsomal protein (50 μg/ml in this study; 265 μg/ml previously) used. Remarkably, although hepatic NIC-G formation had a lower Km than intestinal NIC-G formation, the relative rates of NIC-G formation were higher in intestinal than in hepatic microsomal reactions, at all substrate concentrations tested, which included a concentration (0.03 μM) lower than the apparent Km for hepatic microsomes (0.09 μM).
The finding that intestinal microsomes have much higher apparent Km and Vmax values than hepatic microsomes suggests involvement of different UGT enzymes in these two tissues. In that regard, Lu et al. (2016) reported that other UGT isoforms, including some that are highly expressed in the intestine (UGT1A10 and UGT2B7), are also active in the in vitro metabolism of niclosamide, although they were not as active as UGT1A1 based on Km values and calculated intrinsic clearance of heterologously expressed UGT enzymes. Thus, further studies to identify the responsible UGT enzymes for NIC-G formation in the intestine, via UGT isoform-specific inhibition study and/or UGT isoform-specific knockout mouse models, such as the Ugt1-null mouse (Chen et al., 2013), would be valuable.
The reason for the apparent increase in hepatic UGT activity of LCN and IECN mice is unclear, but it is possibly due to a compensatory increase in the hepatic expression level of the niclosamide-metabolizing UGT enzyme(s) in these null mouse models, compared with wild-type mice, which was confirmed for hepatic UGT1A1 mRNA and protein expression in the LCN mice (Fig. 2). A previous study has also reported that there was a small increase in UGT1A1 mRNA expression in the livers of the LCN mice (Cheng et al., 2014). It remains to be determined whether other niclosamide-metabolizing UGT enzymes are induced in the livers of the IECN mice and would explain the observed increases in microsomal niclosamide glucuronidation (Fig. 3) and UDPGA-supported niclosamide disappearance (Fig. 1).
The relative contributrions of hepatic and intestinal UGTs to in vivo disposition of oral niclosamide remain unclear. The in vivo result showing lower systemic exposure to niclosamide in the LCN mice, compared with wild-type mice (Fig. 5), is not necessarily predicted from the in vitro data, given that 1) intestinal UGT activity was much higher than hepatic UGT activity toward niclosamide, and thus more likely to play a dominant role in the first-pass clearance of oral niclosamide; and 2) there was no increase in the UGT activity toward niclosamide in the intestine of LCN mice, compared with wild-type mice. Nonetheless, the finding that, in wild-type mice, UGT activity toward niclosamide had a much lower apparent Km value in liver than in intestine, in addition to the greater tissue mass in liver than in intestine, suggests that the hepatic UGT contribution to first-pass niclosamide metabolism is greater at relatively low drug concentrations. In that regard, maximal plasma level of niclosamide following the 120-mg/kg oral dose was ∼1 μM (Table 1). Although liver and intestinal tissue levels of niclosamide are expected to be higher than plasama levels, the concentration of niclosamide that is available for metabolism by UGT enzymes in these tissues may be similar to or somewhat above the Km value for hepatic UGT, but much lower than the Km value for intestinal UGT. Thus, the intestinal UGT may be underutilized at this dose. Notably, in other experiments not shown, the genotype differences between wild-type and LCN, as illustrated in Fig. 5, for the dose of 120 mg/kg were also observed when mice were treated with niclosamide at 40 mg/kg, a dose at which the intestinal UGTs are even less likely to be fully used.
In conclusion, our results indicate that P450-mediated biotransformation is not important for in vivo disposition of oral niclosamide. In contrast, both hepatic and intestinal glucuronidation, which appears to be catalyzed by UGT enzymes with differing kinetic properties, may significantly contribute to in vivo disposition of oral niclosamide. Thus, efforts to increase the bioavailability of niclosamide should focus on inhibition of niclosamide glucuronidation in both liver and intestine.
Acknowledgments
We thank Weizhu Yang for assistance with mouse breeding.
Abbreviations
- IECN
intestinal epithelium-Cpr-null
- LCN
liver-Cpr-null
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- NIC-G
niclosamide glucuronide
- NIC-OH
hydroxy niclosamide
- UDPGA
uridine diphosphate glucuronic acid
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Fan, Li, Ding, Zhang.
Conducted experiments: Fan.
Performed data analysis: Fan, Zhang.
Wrote or contributed to the writing of the manuscript: Fan, Li, Ding, Zhang.
Footnotes
This work was supported in part by the National Institutes of Health [Grants GM082978, ES006694, AI134568, and AI131669].
References
- Al-Hadiya BMH. (2005) Niclosamide: comprehensive profile. Profiles Drug Subst Excip Relat Methodol 32:67–96. [DOI] [PubMed] [Google Scholar]
- Andrews P, Thyssen J, Lorke D. (1982) The biology and toxicology of molluscicides, Bayluscide. Pharmacol Ther 19:245–295. [DOI] [PubMed] [Google Scholar]
- Bernard O, Guillemette C. (2004) The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug Metab Dispos 32:775–778. [DOI] [PubMed] [Google Scholar]
- Bowalgaha K, Miners JO. (2001) The glucuronidation of mycophenolic acid by human liver, kidney and jejunum microsomes. Br J Clin Pharmacol 52:605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DB, Klaassen CD. (2007) Tissue- and gender-specific mRNA expression of UDP-glucuronosyltransferases (UGTs) in mice. Drug Metab Dispos 35:121–127. [DOI] [PubMed] [Google Scholar]
- Chang Y-W, Yeh T-K, Lin K-T, Chen W-C, Yao H-T, Lan S-J, Wu Y-S, Hsieh H-P, Chen C-M, Chen C-T. (2006) Pharmacokinetics of anti-SARS-CoV agent niclosamide and its analogs in rats. J Food Drug Anal 14:329–333. [Google Scholar]
- Chen S, Yueh MF, Bigo C, Barbier O, Wang K, Karin M, Nguyen N, Tukey RH. (2013) Intestinal glucuronidation protects against chemotherapy-induced toxicity by irinotecan (CPT-11). Proc Natl Acad Sci U S A 110:19143–19148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Gu J, Klaassen CD. (2014) Adaptive hepatic and intestinal alterations in mice after deletion of NADPH-cytochrome P450 oxidoreductase (Cpr) in hepatocytes. Drug Metab Dispos 42:1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimbari MJ. (2012) Enhancing schistosomiasis control strategy for zimbabwe: building on past experiences. J Parasitol Res 2012:353768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury MKH, Turner N, Bentley NL, Das A, Wu LE, Richani D, Bustamante S, Gilchrist RB, Morris MJ, Shepherd PR, et al. (2017) Niclosamide reduces glucagon sensitivity via hepatic PKA inhibition in obese mice: implications for glucose metabolism improvements in type 2 diabetes. Sci Rep 7:40159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubitt HE, Houston JB, Galetin A. (2009) Relative importance of intestinal and hepatic glucuronidation-impact on the prediction of drug clearance. Pharm Res 26:1073–1083. [DOI] [PubMed] [Google Scholar]
- Dawson VK, Schreier TM, Boogaard MA, Gingerich WH. (1999) Uptake, metabolism, and elimination of niclosamide by fish, in Xenobiotics in Fish (Smith DJ, Gingerich WH, Beconi-Barker MG. eds) pp 167–176, Springer, Boston. [Google Scholar]
- Douch PG, Gahagan HM. (1977) The metabolism of niclosamide and related compounds by Moniezia expansa, Ascaris lumbricoides var suum, and mouse- and sheep-liver enzymes. Xenobiotica 7:301–307. [DOI] [PubMed] [Google Scholar]
- Fisher MB, Vandenbranden M, Findlay K, Burchell B, Thummel KE, Hall SD, Wrighton SA. (2000) Tissue distribution and interindividual variation in human UDP-glucuronosyltransferase activity: relationship between UGT1A1 promoter genotype and variability in a liver bank. Pharmacogenetics 10:727–739. [DOI] [PubMed] [Google Scholar]
- Gad SC. (2008) Preclinical Development Handbook: ADME and Biopharmaceutical Properties, John Wiley & Sons, Inc., Hoboken, New Jersey. [Google Scholar]
- Gu J, Weng Y, Zhang Q-Y, Cui H, Behr M, Wu L, Yang W, Zhang L, Ding X. (2003) Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem 278:25895–25901. [DOI] [PubMed] [Google Scholar]
- Kaminsky LS, Zhang QY. (2003) The small intestine as a xenobiotic-metabolizing organ. Drug Metab Dispos 31:1520–1525. [DOI] [PubMed] [Google Scholar]
- Li Y, Li P-K, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. (2014) Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett 349:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-K, Bai M-Y, Hu T-M, Wang Y-C, Chao T-K, Weng S-J, Huang R-L, Su P-H, Lai H-C. (2016) Preclinical evaluation of a nanoformulated antihelminthic, niclosamide, in ovarian cancer. Oncotarget 7:8993–9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Ma Z, Zhang T, Zhang X, Wu B. (2016) Metabolism of the anthelmintic drug niclosamide by cytochrome P450 enzymes and UDP-glucuronosyltransferases: metabolite elucidation and main contributions from CYP1A2 and UGT1A1. Xenobiotica 46:1–13. [DOI] [PubMed] [Google Scholar]
- Pardhi V, Chavan RB, Thipparaboina R, Thatikonda S, Naidu V, Shastri NR. (2017) Preparation, characterization, and cytotoxicity studies of niclosamide loaded mesoporous drug delivery systems. Int J Pharm 528:202–214. [DOI] [PubMed] [Google Scholar]
- Pearson RD, Hewlett EL. (1985) Niclosamide therapy for tapeworm infections. Ann Intern Med 102:550–551. [DOI] [PubMed] [Google Scholar]
- Piccaro G, Giannoni F, Filippini P, Mustazzolu A, Fattorini L. (2013) Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob Agents Chemother 57:1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson TA, Sherman M, Kalman D, Morgan ET. (2006) Expression of UDP-glucuronosyltransferase isoform mRNAs during inflammation and infection in mouse liver and kidney. Drug Metab Dispos 34:351–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier TM, Dawson VK, Choi Y, Spanjers NJ, Boogaard MA. (2000) Determination of niclosamide residues in rainbow trout (Oncorhynchus mykiss) and channel catfish (Ictalurus punctatus) fillet tissue by high-performance liquid chromatography. J Agric Food Chem 48:2212–2215. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Iida I, Hirota M, Akimoto M, Higuchi S, Suwa T, Tani M, Ishizaki T, Chiba K. (2003) CYP isoforms involved in the metabolism of clarithromycin in vitro: comparison between the identification from disappearance rate and that from formation rate of metabolites. Drug Metab Pharmacokinet 18:104–113. [DOI] [PubMed] [Google Scholar]
- Tukey RH, Strassburg CP. (2000) Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol 40:581–616. [DOI] [PubMed] [Google Scholar]
- van Tonder EC, Maleka TS, Liebenberg W, Song M, Wurster DE, de Villiers MM. (2004) Preparation and physicochemical properties of niclosamide anhydrate and two monohydrates. Int J Pharm 269:417–432. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Nakajima M, Yokoi T. (2002) Troglitazone glucuronidation in human liver and intestine microsomes: high catalytic activity of UGT1A8 and UGT1A10. Drug Metab Dispos 30:1462–1469. [DOI] [PubMed] [Google Scholar]
- Wen X, Donepudi AC, Thomas PE, Slitt AL, King RS, Aleksunes LM. (2013) Regulation of hepatic phase II metabolism in pregnant mice. J Pharmacol Exp Ther 344:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Yao Y. (2018) Octenylsuccinate hydroxypropyl phytoglycogen enhances the solubility and in-vitro antitumor efficacy of niclosamide. Int J Pharm 535:157–163. [DOI] [PubMed] [Google Scholar]
- Zhang Q-Y, Fang C, Zhang J, Dunbar D, Kaminsky L, Ding X. (2009) An intestinal epithelium-specific cytochrome P450 (P450) reductase-knockout mouse model: direct evidence for a role of intestinal p450s in first-pass clearance of oral nifedipine. Drug Metab Dispos 37:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]