

Abstract
The SPECC1L protein plays a role in adherens junctions involved in cell adhesion, actin cytoskeleton organization, microtubule stabilization, spindle organization and cytokinesis. It modulates PI3K-AKT signaling and controls cranial neural crest cell delamination during facial morphogenesis. SPECC1L causative variants were first identified in individuals with oblique facial clefts. Recently, causative variants in SPECC1L were reported in a pedigree reported in 1988 as atypical Opitz GBBB syndrome. Six families with SPECC1L variants have been reported thus far. We report here eight further pedigrees with SPECC1L variants, including a three-generation family, and a further individual of a previously published family. We discuss the nosology of Teebi and GBBB, and the syndromes related to SPECC1L variants. Although the phenotype of individuals with SPECC1L mutations shows overlap with Opitz syndrome in its craniofacial anomalies, the canonical laryngeal malformations and male genital anomalies are not observed. Instead, individuals with SPECC1L variants have branchial fistulae, omphalocele, diaphragmatic hernias, and uterus didelphis. We also point to the clinical overlap of SPECC1L syndrome with mild Baraitser-Winter craniofrontofacial syndrome: they share similar dysmorphic features (wide, short nose with a large tip, cleft lip and palate, blepharoptosis, retrognathia, and craniosynostosis), although intellectual disability, neuronal migration defect, and muscular problems remain largely specific to Baraitser-Winter syndrome. In conclusion, we suggest that patients with pathogenic variants in SPECC1L should not be described as “dominant (or type 2) Opitz GBBB syndrome”, , and instead should be referred to as “SPECC1L syndrome” as both disorders show distinctive, non overlapping developmental anomalies beyond facial communalities.
Keywords: Opitz BBBG syndrome, Teebi hypertelorism syndrome, SPECC1L, MID1, nosology, omphalocele, bicornuate uterus
Introduction
John Opitz reported as distinct X-linked disorders the G syndrome (Opitz et al., 1969a) and the BBB syndrome (Opitz et al., 1969b) that shared hypertelorism and hypospadias. G syndrome was mainly distinguished by the presence of laryngeal malformations. Both disorders were tentatively merged on clinical grounds (Verloes et al., 1989) and finally shown to be the same disorder when pathogenic variants in MID1, an X-linked gene which encodes a microtubule-associated cytoskeletal protein, were identified with both phenotypes without genotype-phenotype correlation (Quaderi et al., 1997). Recently, a variant in MID2 (also X-linked) was also incriminated in a single family with mild GBBB (Geetha et al., 2014; Li et al., 2016). The distinctive facial features of the X-linked Opitz syndrome (Opitz GBBB syndrome, type I; OMIM 300000) are hypertelorism, prominent forehead, broad nasal bridge and anteverted nares. Congenital anomalies include hypospadias, cleft lip/palate, laryngeal and tracheoesophageal abnormalities (typically: cleft larynx), imperforate anus and cardiac defects. Developmental delay and intellectual disability are inconstant. Some individuals thought to have Opitz syndrome on clinical grounds were shown to carry a 22q11 deletion (Fryburg et al., 1996).
An autosomal locus was mapped on 22q11 (Robin et al., 1995), based on a large “unusual G syndrome” pedigree reported in 1988 (Allanson, 1988) (Opitz GBBB syndrome, type II: OMIM 145410). In 2014, a variant in SPECC1L was identified in Allanson’s family, still considered by the authors as Opitz syndrome (Kruszka et al., 2015), and in a second family. Both were identified among a series of 21 pedigrees selected for the presence of a craniofacial dysmorphic syndrome reminiscent of Opitz syndrome. In Kruszka’s family 1 (Kruszka et al., 2015), affected individuals spanned three generations. The proband presented with congenital diaphragmatic hernia, vesicoureteral reflux and deafness. His brother had metopic craniosynostosis, aortic stenosis and tracheomalacia. Their mother had bilateral cleft lip and palate, congenital umbilical hernia and bicornuate uterus. The three affected family members had similar facial features, including hypertelorism with slightly downslanting palpebral fissures, prominent forehead, a prominent nasal root, and a large nose with a large tip (with a median groove in the second boy), arched eyebrows and retrognathia. In Kruzska’s second family, previously described by Judith Allanson (Allanson, 1988) and reported as Family 1 in the article establishing the linkage of the autosomal dominant form of Opitz to chromosome 22q11.2 (Robin et al., 1995), the proband girl had sagittal craniosynostosis, cleft palate, swallowing difficulties and stridor, bilateral hearing loss, ventricular dilation, ureteral reflux, and umbilical hernia. Her dysmorphism included wide forehead with prominent metopic suture, widow’s peak, a marked hypertelorism, downslanting palpebral fissures, prominent nasal root and bridge, a midline groove in the tip of the nose, a wide and poorly defined philtrum, and posteriorly rotated ears. Her sister had hypertelorism and high, broad nasal bridge. Her father and his four siblings had large head circumferences, hypertelorism, down-slanting palpebral fissure, high and broad nasal bridge, wide nasal base with a hooked tip, and long columella. The father had a non-specified upper gastrointestinal malformation. One paternal uncle had a unilateral cleft lip and palate. None of the males had hypospadias.
Ahmad Teebi described in 1987 a four-generational Arabian family (Hypertelorism, Teebi type: OMIM 145420) in which 16 individuals showed a striking dysmorphism, with male-to-male transmission (Teebi, 1987). Clinical details were provided for six of them. Their phenotype included severe hypertelorism, widow’s peak, mildly downslanting palpebral fissures, proptosis, ptosis, broad nasal bridge, short nose with a large tip in some, long deep philtrum and a thin upper lip. They had mild interdigital webbing and shawl scrotum, but none of the males had hypospadias. Umbilical hernia was present in two out of six patients. Intelligence and stature were normal in all of them. Thirteen other patients were subsequently reported with a purported clinical diagnosis of Teebi syndrome (reviewed in (Han et al., 2006)) A duplication of 937 kb in the X chromosome encompassing EFNB1 and two flanking genes (PJA1 and STARD8) was recently shown to segregate with marked hypertelorism and mild nail ridging, but no craniosynostosis, in three female patients from three generations. The authors described this family as X-linked Teebi hypertelorism syndrome (Babbs et al., 2011). We recently reported heterozygous variants in SPECC1L in two families clinically diagnosed with Teebi hypertelorism syndrome (Bhoj et al., 2015).
Cerebrofrontofacial syndromes were tentatively delineated by Robin Winter (Winter, 2001) for patients with frontonasal anomalies (broad nose, flared nares, hypertelorism, epicanthus), dysplastic ears and often CNS anomalies. Many of them, retrospectively, fitted a diagnosis of Baraitser-Winter cerebrofrontofacial syndrome (BWCFF). BWCFF is caused by pathogenic variants in ACTB (Baraitser-Winter syndrome 1: OMIM 243310) or ACTG1 (Baraitser-Winter syndrome 2: OMIM 614583) (Riviere et al., 2012). Individuals with causative variants in either gene show a broad and rather variable phenotype, with marked hypertelorism, sometimes ridged metopic suture, ptosis, arched eyebrows, wide, short and upturned nose and a constellation of malformations comprising cleft lip and palate, ocular coloboma, predominantly frontal pachygyria, muscle waste at shoulder girdle, sensorineural deafness, and usually intellectual deficiency (Verloes et al., 2015).
SPECC1L (Sperm antigen with calponin homology and Coiled-Coil domains 1 Like) is located in 22q11.23 (genomic coordinates GRGh38:22:24,270,816–24,417,739). The protein coded by SPECC1L shows several functional domains: a calponin-like actin-binding, a 2-oxo-acid dehydrogenase, an acyltransferase component, and a lipoyl-binding domain. SPECC1L protein is expressed broadly in many tissues, but its expression is prominent in neural crest cell populations of the developing facial primordia. In situ hybridization of mouse Specc1l showed prominent expression in the maxillary prominence and lateral nasal processes, the eyes and limbs at embryonic day E9.5-E10.5 (Saadi et al., 2011). Homozygous LOF mutants are embryonic lethal and show impaired neural tube closure and defective cranial neural crest cells delamination (Wilson et al., 2016). SPECC1L is a cytoskeletal protein that associates with both actin and microtubules to affect larger cytoskeletal function. It colocalizes with acetylated a-tubulin and F-actin, and is observed in a ring around γ-tubulin containing centrioles, possibly in the microtubule organizing center. Actin cytoskeleton reorganization is required for adequate cell adhesion and migration. SPECC1L may play a role in cytoskeleton organization and acetylated α-tubulin microtubule stabilization. Overexpression or reduction of SPECC1L results in actin reorganization defects. Endogenous SPECC1L colocalizes with filamentous actin and acetylated α-tubulin of the mitotic spindle, suggesting a role for SPECC1L in cytokinesis and spindle organization. SPECC1L is required for proper cell adhesion and migration: in vitro, SPECC1L deficiency causes altered adhesion of cultured cells to the growth substrate (Saadi et al., 2011). In a mouse model, loss of Specc1l increases the staining of adherens junctions in the neural folds, whereas their dissolution is expected to allow neural crest cell delamination. This function appears to be mediated through PI3K-AKT signaling (Wilson et al., 2016). Thus, SPECC1L may mediate transduction of signals required to remodel the actin cytoskeleton during cell adhesion and migration and is a regulator of adherens junction stability. Dysfunction of SPECC1L disrupts the cell adhesion-based signaling necessary for correct neural crest cell behavior during craniofacial morphogenesis, including the expression of CHD1 and CTTNNB1. MID1 is expressed in a subset of cranial neural crest cells that migrate to populate the branchial arches and frontonasal process. MID1 is an RBCC (RING-finger, B-boxes and Coiled-coil) scaffold protein. It forms a large microtubule-associated protein complex that stabilizes microtubules. It also forms a translational activator complex. Finally, it plays a role as ubiquitin E3 ligase degrading the microtubule protein phosphatase 2A and thus activating mTOR pathway. Variants in MID1, SPECC1L and ubiquitous actins express overlapping phenotypes that may indicate convergent functions, but the real interactions between those proteins in neural crest biology remain to be described (Latta and Golding, 2012; Winter et al., 2016).
In this paper, we report 16 patients from eight unreported families with SPECC1L pathogenic variants and a further individual of a previously published family and revisit the nosology of GBBB and Teebi syndromes.
Methods
All patients were gathered through a clinical multicentric collaboration. Authors obtained written consents of the patient or their parents (for patients < 16 years). Patients were ascertained both through targeted Sanger testing of SPECC1L based on suggestive phenotypic features or through exome sequencing (see table 1 for details). Exome capture was carried out using Agilent SureSelect Human All Exon kit (Agilent Technologies, Santa Clara, CA, USA), guided by the manufacturer’s protocols. Parental samples, where available, were also tested. Variant annotation was performed using ACMG variant classification rules (Richards et al., 2015). Variants were declared to ClinVar.
Table 1:
Molecular investigation of the families. SPECC1L variants based on RefSeq NM_015330.4. WES: whole exome sequencing; CES: clinical exome sequencing; TSS: targeted Sanger sequencing. ACMG evidences: PS1: same amino acid change as a previously established pathogenic variant; PS2: de novo with parentage confirmed; PS3: well established in vitro or in vivo functional studies supportive of a damaging effect; PM2: absent from population databases; PM5: novel missense change at an amino acid residue where a different pathogenic missense change has been seen before; PP1: segregation with disease in multiple affected individuals; PP3: multiple computational evidences support deleterious effect
| DNA variant | Protein amino acid change |
ACMG PP3 evidences | ACMG evidence levels |
Family # |
Familial investigation |
Mode of detection |
|---|---|---|---|---|---|---|
| c.1189A>C | p.(Thr397Pro) | Functional assay reported previously | PS3, PM2, PP1, PP3 | 8 | Familial | TSS |
| c.1246G>A | p.(Ala416Thr) | SIFT, Polyphen2, LRT, and MutationTaster: deleterious or disease-causing variant; CADD=28 | PS2, PM2, PP3 | 7 | De novo | TSS |
| c.1258G>A | p.(Glu420Lys) | SIFT, Polyphen2, LRT, and MutationTaster: deleterious or disease-causing variant; CADD=29.1. A variant in the same codon (c.1260G>C:p.Glu420Asp) previously published |
PS1, PS2, PM2, PM5, PP3 | 2 | De novo | WES |
| 4 | De novo in pt 4–1. Present in the 2 fetuses. Absent in a normal sib. | TSS | ||||
| c.1258G>A | p.(Glu420Lys) | SIFT, Polyphen2, LRT, and MutationTaster: deleterious or disease-causing variant; CADD=26.4 | PM2, PP1, PP3 | 5 | Shared by affected sisters, parents unavailable for testing | TSS |
| c.1382G>A | p.(Arg461 Gln) | Mutation Taster, SIFT and PolyPhen2: potentially deleterious variant | PS2, PM2, PP3 | 3 | De novo | WES |
| c.3026A>C | p.(Tyr1009Ser) | SIFT, Polyphen2, LRT, and MutationTaster: deleterious or disease-causing variant; CADD=24.9 | PS2, PM2, PP1, PP3 | 6 | Shared by proband, affected sister, and affected father. Paternal grandparents unavailable for testing. | CES |
| c.3293G>A | p.(Arg1098Gln) | SIFT, Polyphen2, LRT, and MutationTaster: deleterious or disease-causing variant; CADD=35 | PS1, PM2, PP1 PP3 |
1 | Present in the 4 affected patients | ES in patients 1–4 and 4–1; TSS in Relatives |
| PS1, PS2, PM2, PP3 | 9 | De novo | CES |
Results
Family 1 (Figure 1)
Figure 1:
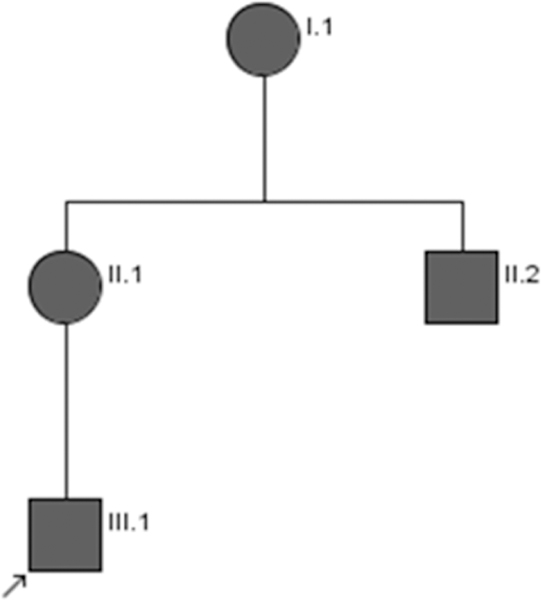
Pedigree of Family 1
Individual 1-III-1.
This individual is the first child of non-consanguineous parents, born at 36 weeks of gestation (WG). He had bilateral cleft lip and palate, left diaphragmatic hernia that was operated at the first day of life, bilateral inguinal hernia (operated in infancy) and normal genitalia. He sat at eighteen months and was able to walk at age 3. He was first seen in the genetic clinics at age 4 (Fig 2a,b), where a tentative diagnosis of Teebi hypertelorism syndrome was suggested. He had a triangular face, mild hypertelorism with intercanthal distance (ICD) of 35 mm, long, slightly downslanted palpebral fissures and mild ptosis, prominent orbital ridges with arched eyebrows, prominent nasal root, a short and large nose, with a large, bulbous tip, flared alae nasi, tick columella, a long philtrum with surgical scars, and a receding chin. Fingers and feet were relatively short. Head circumference was 51 cm (+0.18 SD). At that time, skeletal survey excluded craniosynostosis. MRI disclosed short corpus callosum. EEG showed an irritative wave pattern on the occipital area, but he never had seizures. Heart and kidneys ultrasound scans were normal. His development was severely delayed: He has severe intellectual disability, no speech and major behavioral problems in relation with an autism spectrum disorder (ASD), treated by risperidone and cyamemazine. At his last assessment, at age 19 (Fig 2c,d), he was still non-verbal, eating pureed foods and was not toilet trained at night.
Figure 2:

Craniofacial morphology in individuals with SPECC1L variants. a-d: Individual 3–1 aged 4 years and 15 years; e-g: Individual 2.1 in infancy and aged 30; h,i: individual 1–3; j,k: individual 1–4; l: individual 2–1; m,n: individual 4–1 in infancy and adult; o-q: individual 3–1 aged 4 months and 2 ½ years; r: individual 5–1; s: individual 5–2; t: individual 91
Individual 1-II-1.
This woman (Fig 2 e–g), the mother of 1-III-1, was born at term to non-consanguineous parents. She followed normal schooling, with support. She had a triangular face, mild hypertelorism (ICD 35 mm) with long, slightly downslanted palpebral fissures and mild ptosis, prominent orbital ridges with arched eyebrows, prominent, flat nasal root, short and large nose, with a large bulbous tip, small alae nasi, tick columella, a long philtrum with smooth grooves, a large mouth, low set ears with long earlobes and preauricular pits and a receding chin. Umbilical hernia was operated at age 4. She had bicornuate uterus and normal kidneys.
Individual 1-II-2.
This man was individual 1-II-1’s brother. He was born at 33 WG. He underwent surgery for inguinal and umbilical hernias and had hyperopia (+3 d). He had a normal development and followed normal schooling, albeit with difficulties. In early adulthood, a single, painful tumor of the right common fibular nerve was diagnosed. His right leg was amyotrophic compared to the left. After extensive neuroimaging (including brain and medullar MRI) that excluded NF1, a diagnosis of solitary schwannoma or neurofibroma was proposed. He had a triangular face, major hypertelorism (ICD 45 mm) with long palpebral fissures and mild ptosis, prominent orbital ridges with thick, arched eyebrows, prominent nasal root with short and large nose, with a large, flat tip, small alae nasi, thick columella, a long, smooth philtrum with a thin vermillion border, and a receding chin (Fig 2j,k). The ears were small, triangular shaped, with preauricular pits and tags.
Individual 1-I-1.
This woman, the mother of individuals 1-II-1 and 1-II-2, was born prematurely with a reported birth weight of 750 g. She had midline cleft palate (surgically repaired during infancy). She developed a secondary hydrocephalus. She had bicornuate uterus and normal kidneys. She underwent surgery for intestinal obstruction of imprecise etiology at age 46 years. In her fifties, she developed non-insulin-resistant diabetes mellitus (despite absence of obesity) and glaucoma. She was illiterate but nevertheless had a normal, independent social life. She had marked hypertelorism (ICD 40 mm) with long, slightly downslanted palpebral fissures and mild ptosis, prominent orbital ridges, arched eyebrows, prominent, flat nasal root with marked lateral edges, a short and large nose, with a bulbous tip, small alae nasi, tick columella, a long philtrum with smooth grooves, thin vermillion border, a large mouth, low set ears with long earlobe and preauricular pits, and a receding chin (Fig 2h,i). She had unilateral, probably congenital deafness.
All affected family members were found to carry the SPECC1L c.3293G>A p.(Arg1098Gln) variant (Table 1).
Family 2
Individual 2–1.
This man was born at term to healthy parents. He had a 4 cm omphalocele containing gut, surgically repaired. Large anterior fontanel and hypertelorism were also noted. He underwent surgery for cryptorchidism and obstruction of the ureteropelvic junction at 3 months and 12 years of age respectively. He had normal intelligence and normal academic achievement. His dysmorphism (Fig 2I) included hypertelorism (ICD: 5 cm), downslanting palpebral fissures, wide nasal bridge, prominent supraorbital ridges, and pterygium colli (Fig 2o). In addition, he had a mild left hemihypertrophy (circumferences of 37cm vs 34 cm for left and right calves respectively), already reported in infancy. A SPECC1L c.1258G>A p.(Glu420Lys) variant was found to be de novo in him (Table 1).
Family 3
Individual 3–1.
This boy was born to healthy non-consanguineous parents. He was referred to genetic clinics at the age of 4 months. He had hypertelorism, and a broad prominent nasal root and bridge (Fig 2 o–q), swallowing difficulties. Initial psychomotor development was normal. Follow up in the third year showed mild language delay. A de novo SPECC1L c.1382G>A p.(Arg461Gln) variant was identified in him (Table 1).
Family 4
Individual 4-I-1.
This female individual was born by cesarean section at term to healthy, unrelated parents after an uneventful pregnancy. At birth, she was noted to have prominent ocular hypertelorism, a natal tooth, atrial septum defect and umbilical hernia. She had normal development. She was first seen in genetics after the termination of pregnancy for Patient 4–2. At that time (Fig 2m, n), she was noted to have short stature, short limbs, short fingers and toes, arched eyebrows, high and broad nasal bridge, anteverted nares, long philtrum, micrognathia (surgically repaired), and bicornuate uterus. Before thorough syndromic assessment and molecular diagnostics established the diagnosis, the patient became pregnant again. Ultrasound examination and subsequent fetal autopsy revealed a phenotype similar to that of the first fetus.
Individual 4-II-1.
The routine ultrasound scan at 20 WG showed that this female fetus of Individual 4–1 had a severe diaphragmatic hernia. Pregnancy was terminated at 22 WG. Birth length, weight and HC were in the normal range. Necropsy revealed ocular hypertelorism, high and broad nasal bridge, anteverted nares, long philtrum, large leftsided posterior diaphragmatic hernia with the spleen, pancreas, ventricle and left liver lobe in the thoracic cavity (leading to pulmonary hypoplasia), very large ovaries and a bicornuate uterus.
Individual 4-II-2.
Ultrasound scan at 15+6 WG showed that this second female fetus of Invidiual 4–1 had a severe diaphragmatic hernia and abnormal brain imaging: inward scalloping of the frontal bones (“lemon sign”), and a “banana shaped” cerebellum (due to a shallow posterior fossa). Pregnancy was terminated at 17+3 WG. Birth length, weigh and HC were in the normal range. Autopsy revealed prominent forehead, low set and posteriorly rotated ears, ocular hypertelorism, high and broad nasal bridge, anteverted nares, median groove of the tip of the nose, long philtrum, wide mouth, micrognathia, muscular ventricular septum defect of the heart, complete absence of the left side of the diaphragm with spleen, pancreas, ventricle, and left liver lobe in the thoracic cavity (leading to pulmonary hypoplasia), enlarged ovaries and bicornuate uterus. No dysraphism was observed despite the abnormal head imaging.
A c.1258G>A p.(Glu420Lys)variant in SPECC1L was found to be de novo in 4–1 and inherited in the two fetuses (Table 1).
Family 5
Individual 5–1.
The proband of family 5 was clinically diagnosed with Teebi hypertelorism syndrome after referral to ophthalmogenetics at age 23 years (Fig 2r). She has no major medical issues other than her dysmorphic facial features. These include ocular hypertelorism, prominent forehead, arched eyebrows, bilateral ptosis (L>R), a broad nasal bridge, anteverted nares, long philtrum, and mild micrognathia.
Individual 5–2.
The full sister of individual 5–1 (Fig 2s) was found to have similar facial features with no additional medical issues. At age 25 years, her facial features included ocular hypertelorism, prominent forehead, a broad nasal bridge, and a long philtrum.
A SPECC1L variant, c.1258G>A p.(Glu420Lys) was found in both affected sisters (Table 1). The parents of both sibs were not available for testing.
Family 6
Individual 6-II-1.
This individual is the second child of consanguineous parents (coefficient of relationship 1/8). He was born at 40 weeks of gestation, with normal birth parameters. He had cryptorchidism, umbilical and unilateral inguinal hernia that were operated at 21 months. The growth and weight gain were normal until five months when he started failure to thrive. At 12 months, steroid-resistant nephrotic syndrome was diagnosed. By the age of 3 years 10 months, he developed stage 5 chronic kidney disease, and peritoneal dialysis was started. The renal biopsy revealed minimal change disease. Renal failure caused arterial hypertension, anemia, secondary hyperparathyroidism, and growth delay with short stature. He also has mild developmental delay. Craniofacial anomalies included large forehead, thick eyebrows, hypertelorism, downslanted palpebral fissures, upturned nostrils with a wide tip and short columella, wide mouth with a tented upper lip and a marked cupid’s bow, and large ears. Brain MRI and cardiac evaluation were normal. Before WES, an analysis of a nephrotic syndrome NGS gene panel analysis was performed: no clinically relevant variant was found in ACTN4, ADCK4, ARHGDIA, DGKE, EMP2, LAMB2, NPHS1, NPHS2, PLCE1, PTPRO, WT1. His renal disorder was felt to be unrelated to SPECC1L.
Individual 6-II-2.
This nine-year-old girl is the sister of individual 6-II-1. She was referred to the genetics clinics at age three years, for etiological investigation of developmental delay, poor growth and congenital heart defect (atrial septal defect and subaortic ventricular septal defect). Karyotype, MLPA for 22q11 and 10p14 regions, and metabolic tests (plasma amino acids, urine organic acids and carbohydrate-deficient transferrin) were normal. At nine-years-old she has learning difficulties. Her growth parameters (length and weight) where at −2SD. She has no renal problems. She had a triangular face with thick eyebrows, hypertelorism, downslanted palpebral fissures, upturned tip of the nose, wide mouth with everted lower lip.
Individual 6-I-1.
This 35-year-old man is the father of individuals 6-II-1 and 6-II-2. He has a history of learning difficulties. He has neither renal problems nor cardiac anomalies. He had a widow’s peak, thick eyebrows, bulbous tip of the nose, a long philtrum, and thick lips. His palpebral fissures were normal.
All three affected family members carry the SPECC1L c.3026A>C p.(Tyr1009Ser) variant (Table 1).
Family 7
Individual 7–1.
This individual is a two-month-old female born to a 38-year-old mother and 45-year-old father at 40 weeks of gestation. Prenatally there were concerns for a small ventral septal defect, two vessel umbilical cord, pelviectasis, and large placenta, but karyotype and SNP microarray from amniocytes were normal. She weighed 3385 g (50th centile) and was 53cm long (95th centile), and her head circumference was 34cm (50th centile). There were no complications with the delivery and her APGAR scores were 9 and 9. Abnormal facial features prompted a genetics consult, which noted a prominent widow’s peak, hypertelorism with a interpupillary distance of 5.25 cm (>97th%, 50th% for 9 years), a short nasal bridge, a thin upper lip, but no other congenital anomalies. On further post-natal imaging, she was noted to have uterus didelphys, but no other urogenital abnormalities. Brain ultrasound was normal except for possible thinning of the corpus callosum. A post-natal echocardiogram was normal with no evidence of a ventral septal defect. She has normal tone and development. There is no family history of hypertelorism or congenital anomalies. She was found to carry a de novo c.1246G>A p.(Ala416Thr) variant in SPECC1L (Table 1).
Family 8
Individual 8–1.
This individual is the maternal cousin of Kruzska’s Family A (Kruszka et al., 2015). She is currently 32-years-old and has developed multiple medical conditions. She was born with hypertelorism, ear pits, an umbilical hernia, bicornuate uterus, simple sacral dimple, and a thyroglossal duct cyst. She had normal growth and development and was able to graduate from secondary school. While in her second decade of life she developed pulmonary hypertension, diastolic heart failure, microvascular disease, and vasospastic angina. She has a hiatal hernia with gastroparesis and gastroesophageal reflux disease. She has astigmatism, amblyopia, and dry corneas. There has also been a concern for a mitochondrial myopathy based on her cardiac condition and skeletal muscle weakness. She was found to carry the same familial SPECC1L c.1189A>C p.(Thr397Pro) variant (Table 1).
Family 9
Individual 9–1.
This individual is a 12 months old male born to a 29-year-old mother and 41-year-old father at 38 weeks of gestation by cesarean section. Pregnancy was complicated by polyhydramnios. At birth, he presented with congenital diaphragmatic hernia and dysmorphic features including frontal bossing and wide fontanel. At 4-month physical exam showed prominent forehead, large fontanelle, wide open metopic suture, wide set eyes, wide nasal bridge, small bilateral ear pits in the medial end of upper helix, long and deep philtrum, pectus excavatum, umbilical hernia, bilateral undescended testicles, and brachyclinodactyly of 5th fingers (Fig 2t). He was intubated after birth and placed on ECMO. The diaphragmatic hernia was repaired on day of life 3 and ECMO decannulation was done on day of life 5. He remained in the NICU for two months due to poor feeding and some residual oral aversion, and was eventually discharged home with a G-tube. His hearing and echocardiogram were normal. He wears glasses for significant hyperopia. FISH for 12p tetrasomy to rule out Pallister Killian syndrome and whole-genome microarray analysis were non-diagnostic. He was found to carry a SPECC1L c.3293G>A p.(Arg1098Gln)variant (Table 1).
Discussion
SPECC1L associated syndromes
Heterozygous loss of function of SPECC1L causes Tessier IV oblique facial cleft (ObFC) : disruption of intron 14 of SPECC1L by a de novo t(1;22)(q21.3;q11.23) translocation leading to SPECC1L haploinsufficiency was observed in a girl with Tessier IV ObFC, cleft palate, microphthalmia, palpebral coloboma and talipes deformity; a de novo functionally defective variant (NM_015330.2 (SPECC1L_v001) c.1244A>C (p.Gln415Pro) ) was identified by the same group in an individual with isolated Tessier IV ObFC and normal eyes (facial clefting, oblique, type 1 - OMIM 600251). (Dasouki et al., 1988; Saadi et al., 2011).
More recently, missense variants (c.1189A>C - p.(Thr397Pro) - and c.3247G>A - p.(Gly1083Ser)) were reported in two large pedigrees considered as Opitz GBBB syndrome (Kruszka et al., 2015), one of them previously described by Judith Allanson (Allanson, 1988) (a diagnosis that we question below). We also reported heterozygous missense variants in SPECC1L (Bhoj et al., 2015) in a patient and his affected mother, previously-proposed as a new syndrome resembling Teebi hypertelorism and Aarskog syndromes (Hoffman et al., 2007), and in a single patient with typical Teebi phenotype.
A total of 31 patients (14 males and 17 females) from 12 families are recorded with SPECC1L missense variants (including 2 recurrent mutations) and a clinical diagnosis of GBBB or Teebi syndrome. The phenotype that emerges from this series is summarized in Table 2 and supplementary Table 1. None of them has Tessier IV cleft, suggesting that the syndrome discussed here occurs by another mechanism than haploinsufficency.
Table 2:
Clinical spectrum of 31 individuals with SPECC1L variants
| Patients | |||
| Number | 31 | ||
| Sex | 14 males | 17 females | |
| Mean age at last examination (for those seen after age 1) | 23 years (n=16) | ||
| Growth | |||
| Birth parameters (excluding TOP) | Number | Mean | |
| Birth head circumference (SD) | 6 | −0,17 | |
| Birth length (SD) | 7 | 0,08 | |
| Birth weight (SD) | 9 | −0,34 | |
| Latest growth parameter (mean age: 21 years) | |||
| Head circumference (SD) | 15 | −0,25 | |
| Length (SD) | 16 | −1,56 | |
| Weight (SD) | 10 | −1,55 | |
| Clinical features | Present | Absent | % |
| Short stature | 9 | 5 | 64 |
| Prominent forehead | 19 | 3 | 86 |
| Craniosynostosis (metopic or sagittal ± coronal sutures) | 3 | 13 | 19 |
| Ocular hypertelorism | 30 | 1 | 97 |
| Palpebral ptosis | 12 | 5 | 71 |
| Widow’s peak | 8 | 14 | 36 |
| High/broad nasal bridge | 30 | 0 | 100 |
| Anteverted nares | 19 | 4 | 83 |
| Long philtrum | 20 | 1 | 95 |
| Micrognathia | 12 | 9 | 57 |
| Downslanted palpebral fissures | 8 | 10 | 44 |
| Median groove of the tip of the nose | 3 | 16 | 16 |
| Posteriorly rotated ears | 5 | 12 | 29 |
| Preauricular or heliceal pits/tags | 7 | 13 | 35 |
| Cleft lip | 5 | 26 | 16 |
| Cleft palate | 7 | 23 | 23 |
| Laryngeal/tracheal anomaly (stridor, malacia...) | 2 | 27 | 7 |
| Laryngeal cleft | 0 | 29 | 0 |
| Feeding anomalies | 2 | 12 | 14 |
| Congenital heart defect (ASD, VSD and/or aortic root dilation) | 6 | 17 | 26 |
| Umbilical hernia / omphalocoele | 10 | 11 | 48 |
| Diaphragmatic hernia | 5 | 17 | 23 |
| Inguinal hernia | 5 | 12 | 29 |
| Bicornuate uterus | 9 | 0 | 100 |
| Enlarged ovaries | 2 | 3 | 40 |
| Anorectal malformation | 0 | 31 | 0 |
| Urinary tract malformation | 3 | 13 | 19 |
| Hypospadias | 0 | 14 | 0 |
| Shawl scrotum | 1 | 5 | 17 |
| CNS defect (ventriculomegaly, abnormal corpus callosum) | 6 | 3 | 67 |
| Hearing loss | 1 | 9 | 10 |
| Developmental delay or learning disability (with or without ID) | 11 | 7 | 61 |
| Intellectual disability (> 2 years) | 3 | 20 | 13 |
| Autism spectrum disorder | 2 | 11 | 15 |
Rare anomalies: thyreoglossal cyst, atresia of ear meatus, vertebral hypersegmentation (1 case each), natal teeth (3 cases)
SPECC1L-related syndrome clearly belongs to a common spectrum of developmental anomalies with shared dysfunction of several proteins involved in microtubule biology, particularly in neural crest cells. Despite their overlap, each of these genes has some phenotypic specificity that are underlined in Table 3: SPECC1L appears to play a specific role in branchial morphogenesis, diaphragmatic development, umbilical ring closure and fusion of the Mullerian ducts, which are seldom affected in the other syndromes.
Table 3:
Comparison of SPECC1L spectra with male MID1 and with Baraitser-Winter syndrome (BWS). MID1 and BWS frequencies based on the previously published compilations (Li et al., 2016; Verloes et al., 2015). p values were computed using Fisher exact test.
| Phenotypic trait |
MID1 males |
MID1 (females) |
SPECC1L | p value (male MID1 vs SPECC1L) |
BWS (ACTB + ACTG1, combined) |
p value (BWS vs SPECC1L) |
|---|---|---|---|---|---|---|
| Hypertelorism | 82/82 + 27/28 = 109/110 | 38/42 | 30/31 | Ns (0,39) | 39/41 | Ns (1) |
| Anteverted nares | 17/25 | ¼2 | 19/23 | Ns (0,49) | 26/37 | Ns (0,37) |
| Cleft lip/palate | 42/85 + 14/35 = 56/120 | 0/42 | 7/30 | 0.02 | 8/38 | Ns (1) |
| laryngeal defects | 40/85 + 16/35 = 56/120 | ¼2 | 2/29 (0/29 laryngeal cleft) | <0.001 | 0/42 | Ns (0,16) |
| Dysphagia, reflux and other esophageal problems | 31/85 | 2/42 | 4/21 | Ns (0,19) | 0 ? | NA |
| Ear pits | 0/120 | 7/20 | <0.001 | 0/40 | <0.001 | |
| Heart defects | 20/85 + 7/35 = 27/120 | 0/42 | 6/23 | Ns (0,79) | 12/36 | Ns (0,58) |
| Omphalocele | Not reported ? |
10/21 | <0.001 | 0/42 | <0.001 | |
| Anal defects | 18/85 + 4/35 = 22/120 | 0/42 | 0/31 | <0.001 | 0/18 | Ns (1) |
| Hypospadias | 68/85 + 27/35 = 95/120 | NA | 0/14 | <0.001 | 0/19 | Ns (1) |
| Uterine anomalies | NA | NA | 9/9 | Likely significant | NA | - |
| CNS malformation | 3 ACC 8 hypoplasia of the vermis |
6/9 (no pachygyria) | NA | 37pachygyria 9 ACC | <0.001 | |
| ID /DD | 28/85 + 5/35 = 33/120 | 0/42 | 3/23 | Ns (0,19) | 42/42 | <0.001 |
Comparing SPECC1L- and MID1-related disorders
Inheritance of Opitz syndrome has been subject of a long debate: more severe expression in males was initially suggesting X-linked inheritance. Multiple reports of male-to-male inheritance lead subsequently to the conclusion that GBBB syndrome was autosomal dominant. In 1995, Robin showed that at least one autosomal and one X-linked forms of Opitz could be distinguished (Robin et al., 1995). Retrospective review of the other older reports showing male-to-male is disappointing: :a convincing phenotype was reported in a father and son with hypertelorism, and hypospadias; the son further had severe manifestations, including stridor, hoarse voice and feeding difficulties (Farndon and Donnai, 1983). But the diagnosis of GBBB appears uncertain or too poorly documented in other reports (Chemke et al., 1984; Funderburk and Stewart, 1978; Tolmie et al., 1987; Wilson nd Oliver, 1988).
Robin’s segregation analysis was performed in nine pedigrees, and showed linkage to DXS987 on X chromosome in three multigenerational pedigrees and to D22S345 in 22q in six further multigenerational pedigrees (four of them with male-to-male transmission of the phenotype). They compared the two forms (Robin et al., 1996) and stated that anorectal malformations, posterior laryngeal cleft and anteverted nares were not seen in 22q-linked families, and that the craniofacial appearance was different, with broader forehead and coarser traits in the 22q group. In 22q-linked families, 25% of the carriers had ID. “Structural laryngotracheal anomalies” (but not cleft larynx) were mentioned in 2/13 males and 1/7 females within the 22q-linked pedigrees. Compared to our data, the main discordance was the presence of hypospadias in 5/16 males of 22q-linked families. Retrospectively, using present criteria, the diagnosis of GBBB syndrome may not be perfectly convincing in some of these families. For instance, the Gestalt of X-linked family 2 (with male-to-male transmission of hypertelorism, and hypospadias in a son (Robin et al., 1996)) appears surprisingly mild to fit a diagnosis of Teebi or GBBB. Hence, the contradiction between our observation and Robin’s series may reflect the use of looser clinical criteria than what would be accepted now to consider Opitz or specc1l-related syndrome. Because of these uncertainties in the clinical diagnosis, we decided to limit the comparison to the patients with proven causative variants. In Table 2, we tabulated the most striking features of 31 individuals with SPECC1L syndrome and compared them to the phenotypic profile of males with MID1 syndrome and to the full cohort of MID1 individuals. For MID1, we used the tables published by Li (Li et al., 2016) who updated two previous reviews (Ferrentino et al., 2007; Fontanella et al., 2008) encompassing a total of 184 GBBB patients.
Older clinical reports of autosomal dominant Teebi and Opitz syndromes
We reviewed nine reports published between 1987 and 2006 as Teebi syndrome, for which no molecular nor cytogenetic investigations are available. They total 19 individuals, including the six well-described patients from Teebi’s article. Among those patients, in our opinion, four reports beside the original one (Teebi, 1987) fit the clinical spectrum of SPECC1L (Stratton, 1991; Koenig, 2003; Toriello and Delp, 1994; Han et al., 2006). Male to male transmission was observed in Teebi’s and Stratton’s report. Umbilical hernia and preauricular pits were reported repeatedly. Two other reports are more likely dealing with BWCFF (Tsai et al., 2002; Machado-Paula and Guion- Almeida, 2003). Three have unclassifiable syndromes with hypertelorism (Nakagawa et al., 1998; Tsukahara et al., 1995).
Overlap with Baraitser-Winter cerebrofrontofacial syndrome
The facial dysmorphism of BWCFF (hypertelorism, broad nose with large tip, ptosis, and highly arched eyebrows, and ridged metopic suture) share similarities with both Opitz and Teebi syndrome. Indeed, some of the individuals in our paper were originally thought to have BWCFF syndrome. Table 3 points out the dissimilarities between the three syndromes.
Conclusion
Despite the resemblances between MID1-, SPECC1L- and actin-linked syndromes (which are explained by the pathophysiological commonalities in cell biology), it is important for living nosology to continue to analyze separately phenotypes of genotyped cohorts, in order to build gene-specific natural histories and to allow more accurate genetic counseling. The use of type 1 to type n is usually justified in syndromology when the phenotypes caused by variation in different genes are not distinguishable, such as in Bardet-Biedl syndrome. Beyond facial similarities, the general pattern of developmental anomalies caused by SPECC1L (with defects of the abdominal walls and diaphragm, uterine and branchial defects, sometimes craniosynostosis and aortic root dilation) is different enough from the pattern associated with MID1 (with urogenital malformations in males and laryngeal cleft) to justify to discard “Opitz GBBB type 2” from the nosology and to replace it by SPECC1L-related syndrome - or Teebi syndrome. A clear distinction between specc1l-related syndrome and MID1-related syndrome is also important for the genetic counseling of affected patients, as the medical or surgical complications are not superimposable.
At the time when exome sequencing enters clinical practice, reanalyzing published data keeping in mind the historical perspective, the history and evolution of syndrome delineation and the possible approximations made by our mentors in a time where syndrome recognition was a matter of art, without biological substrate, remains an important task for dysmorphologists in the era of genomic medicine.
Supplementary Material
Footnotes
Disclosure
the authors have no conflict of interest to disclose
Supplementary material
Tabulated individual record of the 31 patients with SPECC1L variants (xls file)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Allanson JE, 1988. G syndrome: an unusual family. Am. J. Med. Genet. 31, 637–642. [DOI] [PubMed] [Google Scholar]
- Babbs C, Stewart HS, Williams LJ, Connell L, Goriely A, Twigg SRF, Smith K, Lester T, Wilkie AOM, 2011. Duplication of the EFNB1 gene in familial hypertelorism: imbalance in ephrin-B1 expression and abnormal phenotypes in humans and mice. Hum. Mutat. 32, 930–938. 10.1002/humu.21521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj EJ, Li D, Harr MH, Tian L, Wang T, Zhao Y, Qiu H, Kim C, Hoffman JD, Hakonarson H, Zackai EH, 2015. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. Am. J. Med. Genet. A. 167A, 2497–2502. 10.1002/ajmg.a37217 [DOI] [PubMed] [Google Scholar]
- Chemke J, Shor E, Ankori-Cohen H, Kazuni E, 1984. Male to male transmission of the G syndrome. Clin. Genet. 26, 164. [DOI] [PubMed] [Google Scholar]
- Dasouki M, Barr M, Erickson RP, Cox B, 1988. Translocation (1;22) in a child with bilateral oblique facial clefts. J. Med. Genet. 25, 427–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farndon PA, Donnai D, 1983. Male to male transmission of the G syndrome. Clin. Genet. 24, 446–448. [DOI] [PubMed] [Google Scholar]
- Ferrentino R, Bassi MT, Chitayat D, Tabolacci E, Meroni G, 2007. MID1 mutation screening in a large cohort of Opitz G/BBB syndrome patients: twenty-nine novel mutations identified. Hum. Mutat. 28, 206–207. 10.1002/humu.9480 [DOI] [PubMed] [Google Scholar]
- Fontanella B, Russolillo G, Meroni G, 2008. MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum. Mutat. 29, 584–594. 10.1002/humu.20706 [DOI] [PubMed] [Google Scholar]
- Fryburg JS, Lin KY,Golden WL, 1996. Chromosome 22q11.2 deletion in a boy with Opitz (G/BBB) syndrome. Am. J. Med. Genet. 62, 274–275. [DOI] [PubMed] [Google Scholar]
- Funderburk SJ, Stewart R, 1978. The G and BBB syndromes: case presentations, genetics, and nosology. Am. J. Med. Genet. 2, 131–144. [DOI] [PubMed] [Google Scholar]
- Geetha TS, Michealraj KA, Kabra M, Kaur G, Juyal RC, Thelma BK, 2014. Targeted deep resequencing identifies MID2 mutation for X-linked intellectual disability with varied disease severity in a large kindred from India. Hum. Mutat. 35, 41–44. 10.1002/humu.22453 [DOI] [PubMed] [Google Scholar]
- Han X-D, Cox V, Slavotinek A, 2006. Atrioventricular block and wiry hair in Teebi hypertelorism syndrome. Am. J. Med. Genet. A. 140, 1960–1964. 10.1002/ajmg.a31439 [DOI] [PubMed] [Google Scholar]
- Hoffman JD, Irons M, Schwartz CE, Medne L, Zackai EH, 2007. A newly recognized craniosynostosis syndrome with features of Aarskog-Scott and Teebi syndromes. Am. J. Med. Genet. A. 143A, 1282–1286. 10.1002/ajmg.a31780 [DOI] [PubMed] [Google Scholar]
- Koenig R, 2003. Teebi hypertelorism syndrome. Clin. Dysmorphol. 12, 187–189. 10.1097/01.mcd.0000077563.66911.c4 [DOI] [PubMed] [Google Scholar]
- Kruszka P, Li D, Harr MH, Wilson NR, Swarr D, McCormick EM, Chiavacci RM, Li M, Martinez AF, Hart RA, McDonald-McGinn DM, Deardorff MA, Falk MJ, Allanson JE, Hudson C, Johnson JP, Saadi I, Hakonarson H, Muenke M, Zackai EH, 2015. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J. Med. Genet. 52, 104–110. 10.1136/jmedgenet-2014-102677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latta EJ, Golding JP, 2012. Regulation of PP2A activity by Mid1 controls cranial neural crest speed and gangliogenesis. Mech. Dev. 128, 560–576. https://doi.org/10.1016Zj.mod.2012.01.002 [DOI] [PubMed] [Google Scholar]
- Li B, Zhou T, Zou Y, 2016. Mid1/Mid2 expression in craniofacial development and a literature review of X-linked opitz syndrome. Mol. Genet. Genomic Med. 4, 95–105. 10.1002/mgg3.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Paula LA, Guion-Almeida ML, 2003. Teebi hypertelorism syndrome: additional cases. Am. J. Med. Genet. 117A, 181–183. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Kondo M, Matsui A, 1998. Teebi hypertelorism syndrome with tetralogy of Fallot. Am. J. Med. Genet. 77, 345–347. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Frias JL, Gutenberger JE, Pellett JR, 1969a. The G syndrome of multiple congenital anomalies. Birth Defects Orig. Artic. Ser. V(2), 95–101. [Google Scholar]
- Opitz JM, Summitt RL, Smith DW, 1969b. The BBB syndrome: familial telecanthus with associated congenital anomalies. Birth Defects Orig. Artic. Ser. V(2), 86–94. [Google Scholar]
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A, 1997. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 17, 285–291. 10.1038/ng1197-285 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee, 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, van Bon BW, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, Gijsen S, Sullivan CT, Christian SL, Abdul-Rahman OA, Atkin JF, Chassaing N, Drouin-Garraud V, Fry AE, Fryns JP, Gripp KW, Kempers M, Kleefstra T, Mancini GM, Nowaczyk MJ, van Ravenswaaij-Arts CM, Roscioli T, Marble M, Rosenfeld JA, Siu VM, de Vries BB, Shendure J, Verloes A, Veltman JA, Brunner HG, Ross ME, Pilz DT, Dobyns WB, 2012. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 44, 440–4, S1–2. 10.1038/ng.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Feldman GJ, Aronson AL, Mitchell AF, Weksberg R, Leonard CO, Burton BK, Josephson KD, Laxova R, Aleck KA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M, 1995. Opitz syndrome is genetically heterogeneous, with one locus on Xp22 and a second locus on 22q11.2. Nat. Genet. 11, 459–461. [DOI] [PubMed] [Google Scholar]
- Robin NH,Opitz JM,Muenke M, 1996. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am. J. Med. Genet. 62, 305–317. [DOI] [PubMed] [Google Scholar]
- Saadi I, Alkuraya FS, Gisselbrecht SS, Goessling W, Cavallesco R, Turbe-Doan A, Petrin AL, Harris J, Siddiqui U, Grix AW, Hove HD, Leboulch P, Glover TW, Morton CC, Richieri-Costa A, Murray JC,Erickson RP, Maas RL, 2011. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am. J. Hum. Genet. 89, 44–55. https://doi.Org/10.1016/j.ajhg.2011.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton RF, 1991. Teebi hypertelorism syndrome. Am. J. Med. Genet. 39, 78–80. [DOI] [PubMed] [Google Scholar]
- Teebi AS, 1987. New autosomal dominant syndrome resembling craniofrontonasal dysplasia. Am. J. Med. Genet. 28, 581–591. 10.1002/ajmg.1320280306 [DOI] [PubMed] [Google Scholar]
- Tolmie JL, Coutts N, Drainer IK, 1987. Congenital anal anomalies in two families with the Opitz G syndrome. J. Med. Genet. 24, 688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello HV, Delp K, 1994. Teebi hypertelorism syndrome: report of a third family. Clin. Dysmorphol. 3, 335–339. [PubMed] [Google Scholar]
- Tsai AC-H, Robertson JR, Teebi AS, 2002. Teebi hypertelorism syndrome: report of a family with previously unrecognized findings. Am. J. Med. Genet. 113, 302–306. 10.1002/ajmg.10870 [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Uchida M, Shinohara T, 1995. Teebi hypertelorism syndrome: further observations. Am. J. Med. Genet. 59, 59–61. 10.1002/ajmg.1320590113 [DOI] [PubMed] [Google Scholar]
- Verloes A, Di Donato N, Masliah-Planchon J, Jongmans M, Abdul-Raman OA, Albrecht B, Allanson J, Brunner H, Bertola D, Chassaing N, David A, Devriendt K, Eftekhari P, Drouin-Garraud V, Faravelli F, Faivre L, Giuliano F, Guion Almeida L, Juncos J, Kempers M, Eker HK, Lacombe D, Lin A, Mancini G, Melis D, Lourengo CM, Siu VM, Morin G, Nezarati M, Nowaczyk MJM, Ramer JC, Osimani S, Philip N, Pierpont ME, Procaccio V, Roseli Z-S, Rossi M, Rusu C, Sznajer Y, Templin L, Uliana V, Klaus M, Van Bon B, Van Ravenswaaij C, Wainer B, Fry AE, Rump A, Hoischen A, Drunat S, Rivière J-B, Dobyns WB, Pilz DT, 2015. Baraitser-Winter cerebrofrontofacial syndrome: delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. EJHG 23, 292–301. 10.1038/ejhg.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloes A,Le Merrer M,Briard ML, 1989. BBBG syndrome or Opitz syndrome: new family. Am. J. Med. Genet. 34, 313–6. 10.1002/ajmg.1320340303 [DOI] [PubMed] [Google Scholar]
- Wilson GN,Oliver WJ,1988Further delineation of the G syndrome:a manageable genetic cause of infantile dysphagia. J. Med. Genet. 25, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Olm-Shipman AJ, Acevedo DS, Palaniyandi K, Hall EG, Kosa E, Stumpff KM, Smith GJ, Pitstick L, Liao EC, Bjork BC, Czirok A, Saadi I, 2016. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci. Rep. 6, 17735 10.1038/srep17735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Basilicata MF, Stemmler MP, Krauss S, 2016. The MID1 protein is a central player during development and in disease. Front. Biosci. Landmark Ed. 21, 664–682. [DOI] [PubMed] [Google Scholar]
- Winter RM, 2001. Cerebro-fronto-facial syndrome: three types? Clin. Dysmorphol. 10, 79–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.