

Abstract
GABA synapses in the brain undergo depolarization-induced suppression of inhibition (DSI) that requires activation of presynaptic cannabinoid type 1 receptors (CB1Rs). The brevity of DSI, lasting ∼1 min in most brain regions, has been ascribed to the transient production of 2-arachidonoylglycerol (2-AG). Here, we propose that the duration of DSI is controlled by heterologous interactions between presynaptic mGluRs and CB1Rs. By examining GABA synapses on parvocellular corticotropin-releasing hormone-expressing neurons in the paraventricular nucleus of the hypothalamus (PVN) of male and female mice, we show that DSI decays quickly in experimental conditions in which both GABA and glutamate are released from adjacent nerve terminals. Pharmacological inhibition of group I mGluRs prolongs DSI, whereas prior activation of mGluRs inhibits DSI, collectively suggesting that group I mGluRs quench presynaptic CB1R signaling. When photostimulation of genetically identified terminals is used to release only GABA, CB1R-dependent DSI persists for many minutes. Under the same conditions, activation of group I mGluRs reestablishes classical, transient DSI. The long-lasting DSI observed when GABA synapses are independently recruited functionally uncouples inhibitory input to PVN neurons. These observations suggest that heterologous interactions between mGluRs and CB1Rs control the temporal window of DSI at GABA synapses, providing evidence for a powerful new way to affect functional circuit connectivity in the brain.
SIGNIFICANCE STATEMENT Postsynaptic depolarization liberates endocannabinoids, resulting in a rapid and transient decrease in release probability at GABA synapses. We discovered that mGluRs control the duration of depolarization-induced suppression of inhibition (DSI), most likely through heterologous desensitization of cannabinoid type 1 receptors by presynaptic mGluR5. By shortening the duration of DSI, mGluRs control the temporal window for retrograde signaling at GABA synapses. Physiological or pathological changes that affect glutamate spillover may profoundly affect network excitability by shifting the duration of cannabinoid inhibition at GABA synapses.
Keywords: CB1, DSI, endocannabinoid, heterologous desensitization, hypothalamus, mGluR
Introduction
Endocannabinoids (eCBs) are retrograde signaling molecules that decrease release probability at central synapses through activation of presynaptic cannabinoid type 1 receptors (CB1Rs) (Kreitzer and Regehr, 2001a,b; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Although eCB signaling has been implicated in the regulation of several behavioral states including stress, the short-lived actions of eCBs, when released in response to postsynaptic depolarization, are difficult to reconcile with their profound contributions to behavioral outputs. For example, eCBs have a dramatic effect on the output of stress circuits (Patel et al., 2004; Steiner et al., 2008; Morena et al., 2016), yet GABA synapses on corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the hypothalamus (PVN) show depolarization-induced suppression of inhibition (DSI) that is transient, decaying in <1 min (Wamsteeker et al., 2010; Wamsteeker Cusulin et al., 2014). This disconnect may be a consequence of upstream actions of eCBs in key stress centers such as the amygdala (Hill et al., 2009) or bed nucleus of the stria terminalis (BNST) (Glangetas et al., 2013), but it is also possible that local signals in the PVN control eCB signaling in ways that are not yet fully understood. For example, interactions between CB1Rs and other signaling systems may affect eCB efficacy.
In the PVN, more than half of the synaptic afferents onto CRH neurons are GABAergic, but there is little spatial segregation between glutamate and GABA synapses (Decavel and van den Pol, 1992; Miklós and Kovács, 2002, 2012). Further, GABA release probability can be modulated by activation of G-protein-coupled mGluRs (Schrader and Tasker, 1997; Zhou and Hablitz, 1997; Govindaiah and Cox, 2004; Errington et al., 2011), raising the possibility that spillover-induced activation of presynaptic mGluRs may have nuanced effects on GABA terminals. This may occur through direct alterations to release probability or result from heterologous desensitization of other G-protein-coupled receptors (GPCRs) (Gordon and Bains, 2003; Zhang et al., 2004; Straiker and Mackie, 2007). In the PVN, presynaptic terminals exhibit heterologous desensitization following coincident GPCR activation (Gordon and Bains, 2003; Zhang et al., 2004). Specifically, the activation of protein kinase C by a Gq-coupled adrenoreceptor impairs the ability of Gi/o-coupled mGluRs to decrease neurotransmitter release (Gordon and Bains, 2003). This interaction between GPCRs from major signaling modalities raises the possibility that glutamatergic and GABAergic transmission may interact in the PVN as well. Although heterologous desensitization of CB1Rs has been described (Kouznetsova et al., 2002; Chu et al., 2010), the synaptic consequences of this process are unknown.
Here, we hypothesized that recruitment of presynaptic mGluRs on GABA terminals may impact DSI through heterologous desensitization of CB1Rs. To test this possibility, we examined DSI under conditions that either recruited both GABA and glutamate release (electrical stimulation) or just GABA release (photostimulation of vGAT-positive nerve terminals expressing channel rhodopsin 2). Our findings indicate that activation of presynaptic group I mGluRs promotes heterologous desensitization of CB1Rs in GABA terminals of the PVN. This does not affect the amplitude of DSI, but does shorten the duration. These findings suggest that, in the absence of coincident glutamate release, postsynaptic depolarization can have long-lasting consequences on release at GABA synapses.
Materials and Methods
Animal handling.
All protocols received approval from the University of Calgary Animal Care and Use Committee in accordance with the guidelines of the Canadian Council on Animal Care. Experiments were performed on 8- to 10-week-old vGAT-IRES-Cre transgenic mice (Stock Slc32a1tm2(cre)Lowl, backcrossed to C57BL/6J, The Jackson Laboratory stock #016962, RRID:IMSR_JAX:016962) weaned at 3 weeks from a homozygous colony. Mutants were screened for using PCR protocols provided by the supplier. The following primers were used to identify vGAT mutants: 5′-CTT CGT CAT CGG CGG CAT CTG-3′ and 5′-CCA AAA GAC GGC AAT ATG GT-3′ (200 bp band). Mice were housed individually on a 12 h/12 h light/dark cycle and were given ad libitum access to food and water.
Slice preparation and electrophysiology.
Male and female vGAT-IRES-Cre mice were anesthetized with isoflurane and decapitated. Brains were removed and placed into slicing solution (0°C, saturated with 95% O2/5%CO2) containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 d-glucose, and 75 sucrose before coronal sectioning to 250 μm on a vibratome (Leica). Slices were transferred to aCSF (30°C, saturated with 95% O2/5% CO2) containing the following (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 d-glucose. Slices recovered for 1 h before being brought to room temperature and then transferred to a recording chamber superfused with aCSF (1 ml/min, 30–32°C saturated with 95% O2/5% CO2) and visualized with an upright microscope (BX51WI, Olympus) fitted with infrared DIC optics, metal halide fluorescence lamp (X-Cite Series 120, EXFO), and digital camera (OLY-150, Olympus). In the indicated experiments, the following compounds were added via syringe pump (R-99, Razel): 100 μm 6-Imino-3-(4-methoxyphenyl)-1(6)-pyridazinebutanoic acid hydrobromide (GABAzine, Tocris Bioscience), 50 μm (RS)-3,5-dihydroxyphenylglycine (DHPG, Tocris Bioscience), 10 μm 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP, Tocris Bioscience), 10 μm 6,7-dinitroquinoxaline-2,3-dione (DNQX, Tocris Bioscience) and 5 μm N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamine (AM251, Tocris Bioscience), 750 nm (3,4 -dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone (JNJ 16259685, Tocris Bioscience), 1 μm guanosine 5′-(β-thio)diphosphate trilithium salt (GDPβs, Sigma-Aldrich), and 10 μm Alexa Fluor 568 dye (Alexa Fluor 568, Thermo Fisher Scientific).
Parvocellular neurosecretory cells were identified based on location (Biag et al., 2012), morphology under differential interference contrast, and distinct characteristic membrane voltage response to depolarizing current steps (Wamsteeker et al., 2010). Borosilicate glass micropipettes (BF150-86-10, Sutter Instruments) pulled on a horizontal pipette puller (p-97, Sutter Instruments; 3–5 MΩ) and filled with a KGluc internal solution containing the following (in mm): 108 K-gluconate, 8 KCl, 8 Na-gluconate, 1 K2EGTA, 10 HEPES, 2 MgCl2, 4 K2ATP, and 0.3 Na3GTP were used for whole-cell recordings. Astrocyte-free [Ca2+] was clamped to 100 nm based on calculations obtained via WebMaxC (RRID:SCR_003165) and Lenntech activity coefficient calculator (RRID:SCR_016475) with a modified KGluc internal containing the following (in mm): 96 K-gluconate, 4.3 CaCl2, 8 Na-gluconate, 10 HEPES, 2 MgCl2, 4 K2ATP, 0.3 Na3GTP, 10 K4BAPTA, 1 GDPβs, and Alexa Fluor 568. PNCs were voltage clamped (−80 mV) under constant perfusion of DNQX. To induce DSI, PNCs were given depolarizing voltage command to +20 mV for 5 s. Optically evoked IPSCs (oIPSCs) and electrically evoked IPSCs (eIPSCs) were elicited at a rate of 0.2 Hz. eIPSCs were elicited through a monopolar aCSF-filled micropipette (4–6 MΩ) 40–60 μm medioventral to the PNC and stimulating afferents in the neuropil (100–150 μs, 10–50 V; S88 stimulator, Grass Instruments). Experiments in which oIPSCs interrupted firing were conducted by injecting a postsynaptic depolarizing current step (40–100 pA, 2.5 s) to elicit cell-autonomous firing while afferents from the fusiform nucleus of the BNST (BNSTFu) were stimulated (10 Hz, 1 s) before and after long-lasting DSI (LDSI) induction. This protocol ran 10 sweeps at 0.2 Hz during both conditions and sweeps with <6 Hz steady-state firing at baseline were excluded from analysis. Firing during the initial 500 ms was omitted due to frequency adaptation, which is characteristic of PNCs (Tasker and Dudek, 1991). Firing frequency during the 1 s BNSTFu activation was normalized to the preceding 500 ms of steady-state firing for the percentage change in firing. Access resistance (Ra) was monitored throughout the experiment; cells were excluded from analysis if Ra exceeded 20 MΩ or a 15% change.
Optogenetics.
Surgeries were performed on 6-week-old mice anesthetized with isoflurane via nose cone (induced at 5% v/v, maintained at 2%) and thermoregulated by heating pad. Mice were head-fixed on a stereotaxic apparatus (Kopf Instruments) with bregma and lambda sutures forming a horizontal plane. Glass capillaries lowered through burr holes targeted the BNSTFu (AP: +1.2 mm; ML: ±0.6 mm; DV: −4.5 mm from dura). A Nanoject II apparatus (Drummond Scientific) bilaterally delivered 207 nl of rAAV2 (3 × 1012 genome copy per milliliter, Canadian Neurophotonics Platform Viral Vector Facility, RRID:SCR_016477; 3 × 1012 genome copy per milliliter, UNC vector core, RRID:SCR_002448) carrying channelrhodopsin 2 (ChR2)-enhanced yellow fluorescent protein (eYFP) (pAAV-EF1a-double floxed-hChR2(H134R)-eYFP-WPRE-HGHpA, a gift from Karl Deisseroth, Addgene #20298, RRID:SCR_002037). Mice were given at least 14 d of recovery before being killed. A fiber-optic cable (150 μm diameter) was positioned 1–2 mm above the slice and delivered light (2 ms, 3.0–5.0 mW, 473 nm) through a fiber-coupled laser (IKE-473-100-op, IkeCool) triggered via Digidata 1440A (Molecular Devices).
Data analysis and statistics.
Signals were amplified (Multiclamp 700B, Molecular Devices), low-pass filtered at 1 kHz, digitized at 10 kHz (Digidata 1440A, Molecular Devices), and recorded (pClamp 10.4, Molecular Devices, RRID:SCR_011323) for offline analysis (Prism 6, GraphPad, RRID:SCR_000306). oIPSC and eIPSC amplitudes were measured by subtracting the peak synaptic current from baseline. Spontaneous IPSC (sIPSC) event amplitude and frequency were detected using automated search parameters and confirmed by eye (MiniAnalysis, Synaptosoft, RRID:SCR_002184). Data were normalized and expressed as a percentage of the baseline values (−2 to 0 min before the initial DSI step) in each cell, with baseline measurements taken from the 30 s epoch before postsynaptic depolarization (gray graph regions). Peak DSI values (red graph regions) were obtained by averaging evoked amplitude, sIPSC frequency, or amplitude 30 s following the postsynaptic depolarization for each individual DSI. LDSI values (blue graph regions) were measured in the 30 s epoch 3–3.5 min following a postsynaptic depolarization for each individual DSI except for the data shown in Figure 5, B and C, in which it was measured at the 7.5–8 min epoch. Although no power analyses were performed, sample sizes were determined based on previously reported studies. Male and female mice were used, except for the optogenetic interrupt experiment, in which all mice were male. The number of neurons recorded from is indicated in the figures and the number of mice used is reported in the figure legends. Unless otherwise noted, repeated-measures one-way ANOVA with Geisser–Greenhouse correction and Dunnett's post hoc correction were run to compare the 30 s averaged baseline before postsynaptic depolarization with the subsequent DSI and LDSI values for each individual postsynaptic depolarization or to compare repeated DSI or LDSI epochs with the initial baseline. A paired two-tailed Student's t test was used to compare the effects of acute DHPG application on sIPSC amplitude and frequency and a paired one-tailed Student's t test was used to compare the effects of GABAzine on oIPSC amplitude.
Figure 5.
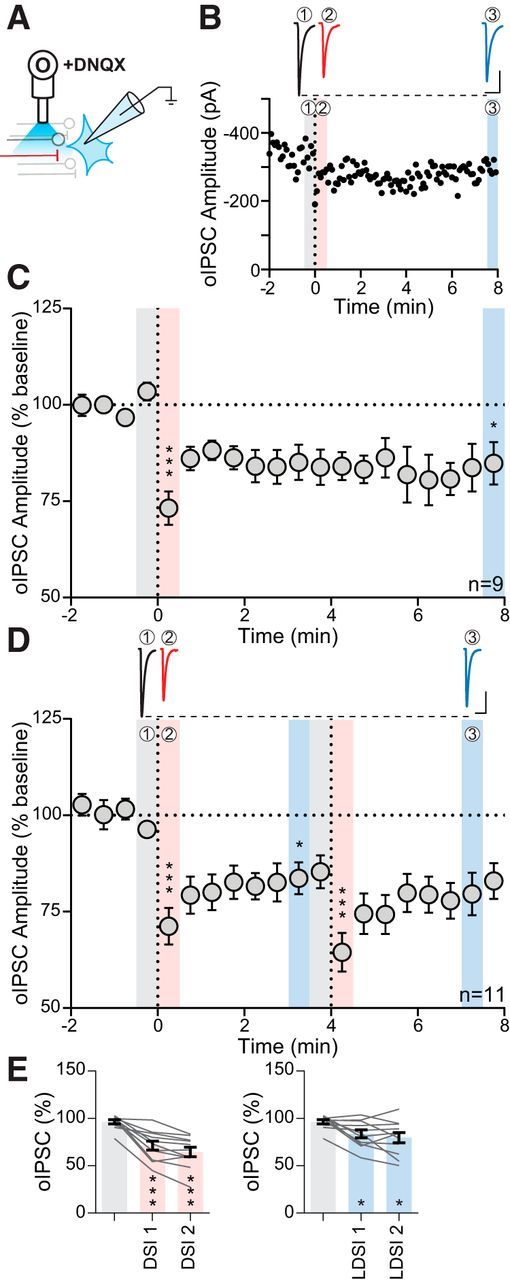
oIPSCs exhibit LDSI. A, Experimental setup of optical fiber placement for BNSTFu stimulation. B, Representative oIPSC traces (top) and amplitude time course (bottom) from an individual cell following a single postsynaptic depolarization. C, Summary time course showing that LDSI in 9 cells from 7 mice (6 male, 1 female) persists for many minutes following a single postsynaptic depolarization (F(2,8) = 13.52, p = 0.0012). D, Top, Traces of oIPSCs corresponding to shaded regions below. Bottom, Summary time course showing that LDSI in 11 cells from 11 mice (2 male, 9 female) is not enhanced by repeated postsynaptic steps (PD1: F(2,10) = 18.60, p = 0.0001, PD2: F(2,10) = 10.70, p = 0.0016). E, Summary graphs data corresponding to shaded regions in D showing DSI (F(2,10) = 39.38, p = 0.0001) and LDSI (F(2,10) = 7.94, p = 0.0055) values compared with the initial baseline indicating that LDSI2 remains significantly depressed compared with the initial baseline. Scale bars, 100 pA, 20 ms. Post hoc versus baseline p-values are shown as follows: *p < 0.05, ***p < 0.001. Data are shown as mean ± SEM.
Results
Rapid, transient inhibition through retrograde transmission of eCBs has been shown to affect GABAergic synapses in the PVN (Wamsteeker et al., 2010; Wamsteeker Cusulin et al., 2014). Here, we investigated whether electrical stimulation of the neuropil may release both glutamate and GABA to affect DSI (Fig. 1A, inset). In brain slices maintained at 30°C, postsynaptic depolarization (−80 mV to +20 mV for 5 s; vertical dashed line) resulted in the immediate depression of eIPSCs (DSI1: 69.7 ± 5.4% of baseline: 96.3 ± 2.1%; p = 0.0013; n = 10), which recovered within 4 min (recovery 1: 96.0 ± 4.7% of baseline; p = 0.93; n = 10). A second postsynaptic depolarization induced a similar DSI and recovery (DSI2: 67.6 ± 5.3% of baseline; p = 0.0001; n = 10; recovery 2: 97.2 ± 6.1% of baseline; p = 0.96; n = 10) (Fig. 1A). sIPSC frequency is manipulated by eCB-dependent reductions in GABA release probability (Diana and Marty, 2003; Wamsteeker et al., 2010). Post hoc analysis revealed that sIPSC frequency showed a similar decrease immediately following postsynaptic depolarization (DSI1: 67.2 ± 6.1% of baseline: 94.1 ± 4.5%; p = 0.0066; n = 10), which also recovered (recovery 2: 84.9 ± 7.1% of baseline; p = 0.34; n = 10) (Fig. 1B). As anticipated, the amplitude of sIPSCs was unaltered by postsynaptic depolarization (DSI1: 98.9 ± 4.4% of baseline: 99.0 ± 1.2%; p = 0.91; n = 10; recovery 2: 109.4 ± 6.9% of baseline; p = 0.25; n = 10) (Fig. 1C).
Figure 1.
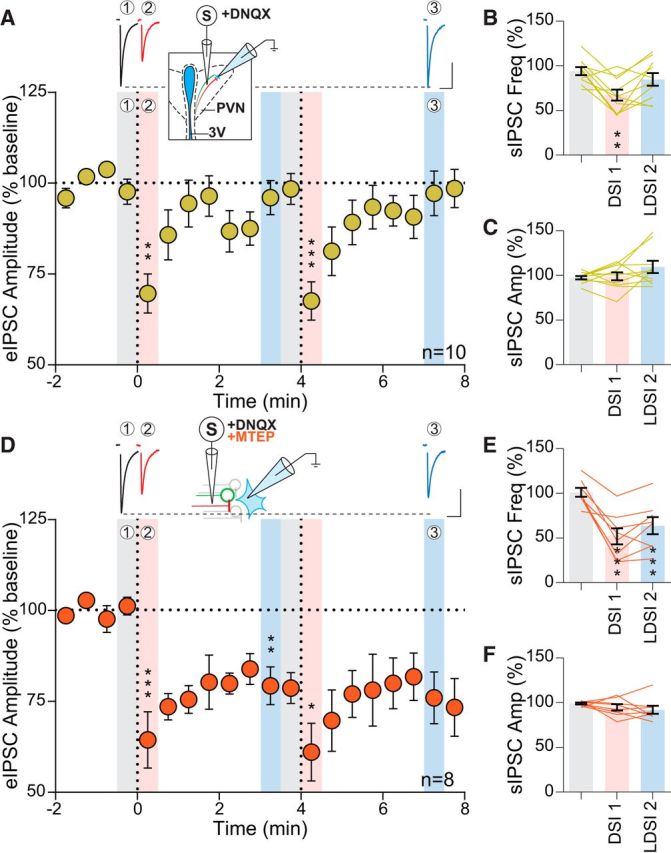
Inhibition of mGluR5 prolongs DSI. A, Top, Representative traces of eIPSCs from baseline (1, gray) immediately after postsynaptic depolarization (2, red) and 3 min later (3, blue) corresponding to the indicated regions below. Inset, Schematic of experimental setup. Bottom, Time course of eIPSC amplitude of 10 cells from 9 animals (8 male, 1 female) following repeated postsynaptic depolarization (PD) to +20 mV for 5 s (vertical dashed line) (PD1: F(2,9) = 17.06, p = 0.0001, PD2: F(2,9) = 34.15, p = 0.0001). Significance is reported with respect to the preceding shaded baseline region. B, Summary data corresponding to shaded regions in A showing that sIPSC frequency undergoes a transient reduction that returns to baseline following PD (F(2,9) = 7.89, p = 0.0035), whereas sIPSC amplitude (C) is unaffected (F(2,9) = 2.13 p = 0.15). D, Top, Sample eIPSC traces in the presence of MTEP (10 μm). Inset, Experimental setup. Bottom, Time course showing eIPSC amplitude of 8 cells from 7 mice (4 male, 3 female) undergoes a transient and persistent reduction in the presence of a mGluR5 antagonist (PD1: F(2,7) = 17.81, p = 0.0009, PD2: F(2,7) = 6.85, p = 0.018). E, MTEP causes a transient and persistent reduction in sIPSC frequency (F(2,7) = 23.33, p = 0.0001), whereas sIPSC amplitude (F) is unaltered (F(2,7) = 1.43, p = 0.27). Scale bars, 100 pA, 20 ms. Post hoc versus baseline p-values are shown as follows: *p < 0.05, **p < 0.01, ***p < 0.001. Data are shown as mean ± SEM.
Activation of group I mGluRs in several regions (Zhou and Hablitz, 1997; Lafourcade et al., 2009), including the PVN (Schrader and Tasker, 1997), can modulate synaptic GABA release. Given the proximity of glutamate and GABA terminals in the PVN (Miklós and Kovács, 2012), we hypothesized that electrical stimulation may depolarize adjacent afferents and thereby release both glutamate and GABA in close proximity. To determine the possible interactions between endogenous glutamate and DSI, we investigated whether the mGluR5-specific antagonist MTEP would affect DSI at GABA synapses (Fig. 1D, inset). There was a depression of eIPSC amplitude following postsynaptic depolarization (DSI1: 64.4 ± 7.8% of baseline: 101.2 ± 2.4%; p = 0.0010; n = 8; DSI2: 61.0 ± 8.0% of baseline; p = 0.0009; n = 8); eIPSC amplitude, however, failed to recover to baseline during the recording period (4 min). This was termed LDSI (LDSI1: 79.3 ± 5.2% of baseline; p = 0.0023; n = 8; LDSI2: 76.0 ± 7.1% of baseline; p = 0.0088; n = 8) (Fig. 1D). These lasting effects on the eIPSC were mirrored by persistent decreases in sIPSC frequency (DSI1: 51.9 ± 9% of baseline: 101.1 ± 5.0%; p = 0.0012; n = 8) and LDSI (LDSI2: 63.8 ± 9.5% of baseline; p = 0.0049; n = 8) (Fig. 1E). The amplitude of sIPSCs in MTEP was unaltered (DSI1: 94.8 ± 3.5% of baseline: 99.0 ± 1.0%; p = 0.49; n = 8; LDSI2: 92.0 ± 4.5% of baseline; p = 0.23; n = 8) (Fig. 1F). We repeated the experiment with JNJ 16259685, a specific mGluR1 antagonist, to test whether both group I mGluRs could curtail the duration of eCB signaling at GABA synapses or if this temporal control over DSI was specific to mGluR5. In the presence of the mGluR1 antagonist, eIPSCs exhibited DSI (65.7 ± 8.6% of baseline 100.4 ± 2.3%; p = 0.024; n = 4), which recovered (LDSI2: 91.7 ± 6.4% of baseline; p = 0.35; n = 4), indicating that mGluR1 does not participate in curtailing DSI duration. Together, these data suggest that glutamate, released coincidentally during electrical stimulation of the neuropil, acts heterosynaptically at mGluR5 on GABA terminals to curtail the duration of DSI.
mGluRs that modulate the duration of eCB-mediated DSI may be present in the postsynaptic neuron (van den Pol et al., 1995; Li et al., 2014), neighboring astrocytes (van den Pol et al., 1995), or presynaptic terminals. To determine the synaptic location of the mGluR5 in the findings reported above, we specifically disrupted GPCR signaling in either the postsynaptic neuron or in nearby astrocytes. First, we electrically stimulated afferents and recorded from postsynaptic neurons loaded with GDPβs. We observed transient DSI (DSI1: 63.2 ± 6.0% of baseline: 106.1 ± 4.4%; p = 0.0012; n = 7), but not LDSI (LDSI2: 119.6 ± 6.8% of baseline; p = 0.15; n = 7) (Fig. 2A). To test for the involvement of astrocytic mGluR5, we patched and loaded astrocytes with a fluorescent reporter and an internal solution that clamped Ca2+ levels (Henneberger et al., 2010) and contained GDPβs. We obtained whole-cell recordings from neurons within the fluorescent astrocytic domain. Postsynaptic depolarization as above elicited DSI (DSI1: 66.3 ± 7.7% of baseline: 101.8 ± 3.4%; p = 0.014; n = 6), but not LDSI (LDSI2: 104.1 ± 9.2% of baseline; p = 0.94; n = 6) (Fig. 2B). Together, these data suggest that the mGluR5 responsible for curtailing the duration DSI is not located in postsynaptic neuron or in adjacent astrocytes.
Figure 2.

No role for astrocytic or postsynaptic GPCR signaling in LDSI. A, Left, Schematic showing postsynaptic loading of GDPβs. Right, Time course summary of 7 cells from 6 animals (4 male, 2 female) showing rapid recovery from DSI in neurons with impaired GPCR signaling (PD1: F(2,6) = 22.89, p = 0.0001, PD2: F(2,6) = 23.87, p = 0.0012). B, Left, Schematic showing astrocytes patched and filled with an internal solution that clamps Ca2+ transients and prevents GPCR activation. Following astrocytic loading, an adjacent neuron is patched and eIPSCs are elicited. Right, Time course summary of 6 cells from 5 animals (4 male, 1 female) showing that PNCs within the field of a patched astrocyte exhibit rapid recovery from DSI (PD1: F(2,5) = 13.61, p = 0.0040, PD2: F(2,5) = 5.23, p = 0.028). Post hoc versus baseline p-values are shown as follows: *p < 0.05, **p < 0.01, ***p < 0.001. Data are shown as mean ± SEM.
Previous studies have provided functional evidence in support of mGluR5 expression on afferents to hypothalamic nuclei (Schrader and Tasker, 1997). To determine whether GABA synapses in the PVN contain functional presynaptic mGluRs, we bath applied the group I mGluR ligand DHPG and recorded spontaneous IPSCs. DHPG increased the frequency of sIPSCs, but had no effect on amplitude (frequency: 3.6 ± 0.7 Hz vs baseline: 2.3 ± 0.5 Hz; p = 0.0046; n = 12 from 7 mice: 5 male, 2 female; paired t test; amplitude: 18.5 ± 0.9 pA vs baseline: 19.2 ± 1.0 pA; p = 0.063; n = 12 from 7 mice: 5 male, 2 female; paired t test). Next, we investigated whether DSI was inhibited by prior activation of these mGluRs. We have shown previously that activation of Gq-coupled GPCRs in the PVN causes a heterologous desensitization of presynaptic Gi-coupled GPCRs (Gordon and Bains, 2003). Although the receptors in that study are different, the downstream signaling cascades are the same as those being investigated here. This raises the possibility that similar interaction may occur between presynaptic Gq-coupled mGluR5 and Gi-coupled CB1R. In the same neuron, we depolarized the postsynaptic membrane to elicit DSI and then applied DHPG; following drug washout, we depolarized the postsynaptic cell again and recorded the effect on eIPSCs (Fig. 3A). Before mGluR5 activation, postsynaptic depolarization rapidly decreased eIPSC amplitude (DSI: 82.2 ± 3.8% of baseline: 102.6 ± 2.8%; p = 0.0002; n = 11) with no LDSI (LDSI: 106.2 ± 4.2 of baseline; p = 0.66; n = 11) (Fig. 3B). Following DHPG washout, we failed to observe any suppression of inhibition (DSI: 91.3 ± 4.1% of baseline: 101.5 ± 1.3%; p = 0.13; n = 11; LDSI: 98.8 ± 4.3% of baseline; p = 0.85; n = 11) (Fig. 3C). These data suggest that activation of presynaptic group I mGluRs causes a heterologous desensitization of CB1Rs that impairs DSI.
Figure 3.
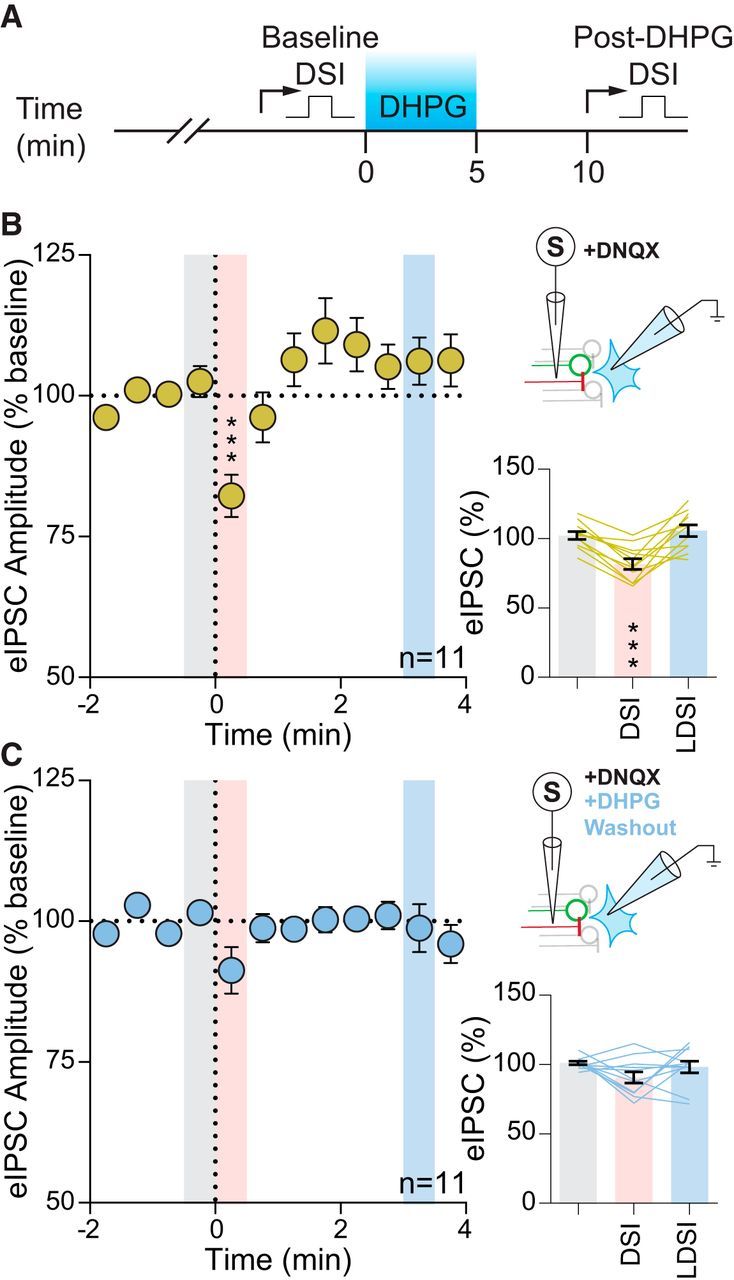
Group I mGluR activation quenches DSI. A, Temporal schematic of experiment detailing timing of recordings and drug application. B, Summary time course (left) and graphs (bottom) showing eIPSCs of 11 cells from 9 mice (3 male, 6 female) undergo DSI (F(2,10) = 19.59, p = 0.001). Top, Experimental setup. C, Following DHPG washout in the same cells, DSI was absent in both the summary time course (left) and graphs (bottom) (F(2,10) = 2.21, p = 0.14). Top, Experimental setup. Post hoc versus baseline p-values are shown as follows: ***p < 0.001. Data are shown as mean ± SEM.
The findings above indicate an interaction between mGluRs recruited by synaptically released glutamate and presynaptic CB1Rs. If activation of mGluRs inhibits CB1R signaling, then experiments in which glutamate is not liberated from terminals should enhance DSI. To elicit DSI in the absence of synaptic glutamate release, we used a combined transgenic and optogenetic approach to drive bilateral viral expression of ChR2-eYFP under a vGAT promoter. Previous work has shown that the fusiform nucleus of the fusiform nucleus of the BNST (BNSTFu) provides dense GABAergic innervation to the PVN (Dong et al., 2001). Following surgical recovery, postfixed coronal slices containing the BNSTFu were examined under confocal microscopy for ChR2-eYFP expression (Fig. 4A,B). Epifluorescence identified eYFP-positive cell bodies from live BNSTFu slices before visualized-patch recordings were made. Depolarizing current steps revealed firing characteristics (Fig. 4C) corresponding to GAD67-positive anterolateral BNST neurons (Hammack et al., 2007; Dabrowska et al., 2013). Brief (5 ms) flashes of 473 nm light elicited membrane depolarizations and inward currents, whereas prolonged (200 ms) light pulses resulted in firing activity and a biphasic (peak and steady-state) photocurrent (Fig. 4D), consistent with ChR2 expression within the cell. Confocal imaging revealed eYFP-positive BNSTFu afferents to the lateral horn of the PVN (Fig. 4E), a region densely populated by PNCs (Biag et al., 2012). Light pulses were delivered through an optical fiber positioned 1–2 mm above the slice (Fig. 4F). The synaptic GABAAR antagonist GABAzine (100 μm) abolished optically-evoked postsynaptic currents (oPSC) amplitude (−136.9 ± 40.5 pA vs −2.2 ± 0.3 pA; p = 0.044; n = 4 from 4 mice: 2 male, 2 female; one-tailed t test) (Fig. 4G), confirming that oPSCs represent a pure GABAergic afferent population.
Figure 4.
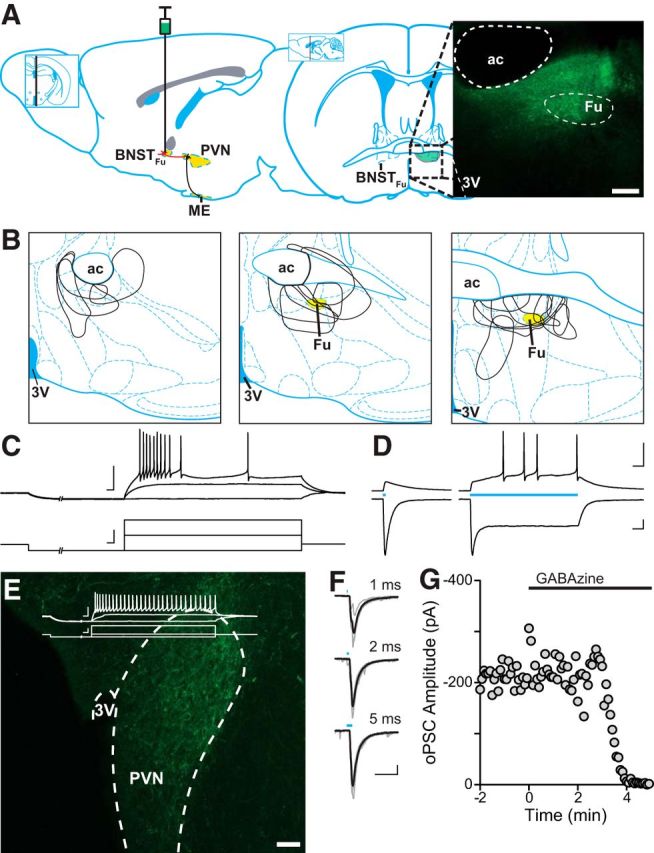
Optically evoked GABA release in the PVN of vGAT-ChR2 BNSTFu mice. A, Left, Sagittal illustration denoting the projection from the BNSTFu to the PVN, targeted for stereotaxic viral injection. Right, Coronal map through the level of the BNST, indicating the location of the BNSTFu. Inset, Confocal image (10× magnification) showing eYFP expression in the BNSTFu of a vGAT-IRES-Cre mouse. B, Injection maps outlining BNSTFu targeting. C, eYFP-positive neurons from the BNSTFu were patched and characterized by their membrane voltage response before being exposed to varying durations of 473 nm light (D). BNSTFu neurons exhibited a characteristic steady-state current in response to a sustained exposure to blue light. E, Confocal image (20× magnification) depicting ChR2-eYFP-positive BNSTFu afferent fibers innervating the PVN. Inset, Representative trace showing a PNC membrane voltage response. F, Synaptic currents evoked by 473 nm light of varying durations. A 2 ms pulse was found to elicit synchronous release and was used for all other experiments. G, oPSCs were completely inhibited in a single cell by the application of the selective GABAA antagonist GABAzine (100 μm). Scale bars: A, E, 100 μm; C–F, 50 mV/pA, 20 ms.
Next, we examined DSI and LDSI at these ChR2-expressing BNSTFu GABA synapses (Fig. 5A) following a single postsynaptic depolarization (Fig. 5B). We observed an immediate (DSI: 73.2 ± 4.3% of baseline: 103.5 ± 2.3%; p = 0.0002; n = 9) and persistent (LDSI: 84.8 ± 5.5% of baseline; p = 0.026; n = 9) depression of optically-evoked inhibitory postsynaptic current (oIPSC) amplitude (Fig. 5C). To characterize the persistent depression, we examined the effects of repeated postsynaptic depolarization. The first postsynaptic depolarization elicited both DSI and LDSI (DSI1: 71.2 ± 4.7% of baseline 1: 96.3 ± 2.1%; p = 0.0004; n = 11; LDSI1: 83.7 ± 4.2% of baseline 1; p = 0.015; n = 11) (Fig. 5D), whereas the second elicited DSI (64.5 ± 5.0% vs baseline 2: 88.4 ± 4.2%; p = 0.0005; n = 11), but did not enhance LDSI (79.6 ± 5.5% vs baseline 2; p = 0.36; n = 11) (Fig. 5D). Together, these results show that GABAergic BNSTFu afferents in the PVN undergo a LDSI that persists at least 8 min and is not enhanced by further postsynaptic depolarization.
To confirm that LDSI is CB1R dependent, we elicited postsynaptic depolarization in the presence of the CB1R antagonist AM251 (5 μm) (Fig. 6A, inset). AM251 alone did not alter basal oIPSC amplitude (AM251: −258.9 ± 47.8 pA vs control: −254.9 ± 61.0 pA; p = 0.96; n = 10, 11 respectively), sIPSC frequency (AM251: 3.2 ± 1.1 Hz vs control: 4.2 ± 0.7 Hz; p = 0.42; n = 10, 11 respectively), or sIPSC amplitude (AM251: 24.4 ± 1.2 pA vs control: 28.9 ± 2.2 pA; p = 0.091; n = 10, 11 respectively). AM251 blocked both DSI (DSI1: 91.2 ± 1.3% of baseline: 97.7 ± 2.8%; p = 0.16; n = 10) and LDSI (LDSI2: 102.4 ± 6.4% of baseline; p = 0.53; n = 10) following repeated postsynaptic depolarization (Fig. 6A), confirming that both DSI and LDSI are eCB dependent. Across all conditions tested, changes to evoked IPSCs mirror changes to sIPSC frequency. Therefore, we characterized the time course of changes to sIPSC frequency (Fig. 6C) to determine whether LDSI is a process specific to BNSTFu synapses or if LDSI globally affected GABAergic inputs onto PNCs. The amplitude of sIPSCs were unaltered by eCBs (DSI1: 92.7 ± 3.6% of baseline: 99.0 ± 1.2%; p = 0.23; n = 11; LDSI2: 96.3 ± 1.7% of baseline; p = 0.48; n = 11) (data not shown), whereas sIPSC frequency underwent a rapid, repeatable, DSI (DSI1: 61.3 ± 4.4% of baseline 1: 100.9 ± 1.6%; p = 0.0001; n = 11; DSI2: 53.2 ± 4.4% vs baseline 2: 74.5 ± 5.2%; p = 0.0002; n = 11) (Fig. 6D) and persistent LDSI (LDSI1: 76.2 ± 5.7% of baseline 1; p = 0.0023; n = 11) that was not enhanced by repeated postsynaptic depolarization (LDSI2: 76.2 ± 7.7% vs baseline 2; p = 0.95; n = 11) (Fig. 6D). AM251 blocked the effects of postsynaptic depolarization on sIPSC frequency (DSI1: 103.1 ± 2.3% of baseline: 96.4 ± 3.1%; p = 0.23; n = 10; LDSI2: 105.2 ± 6.9% of baseline; p = 0.52; n = 10) (Fig. 6D), indicating that LDSI of evoked and spontaneous GABA transmission in PNC afferents is eCB dependent.
Figure 6.
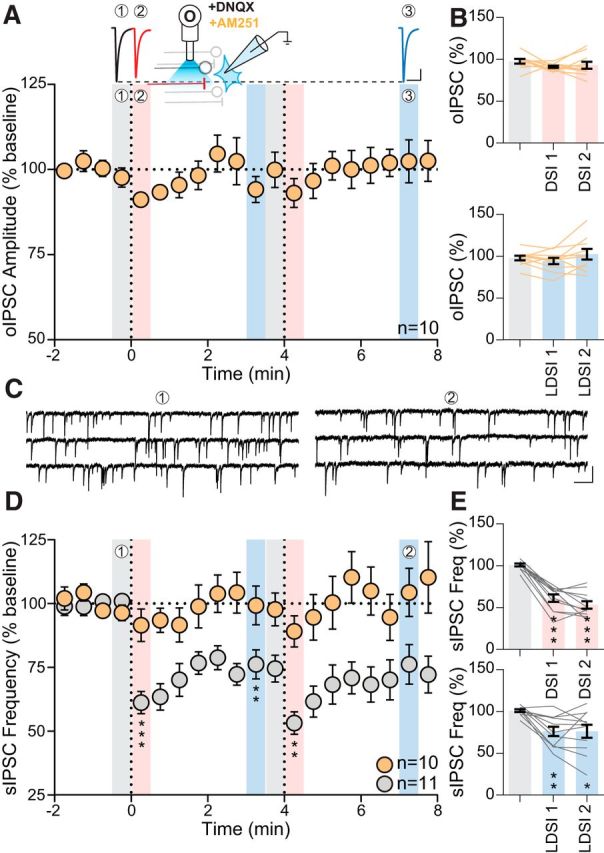
LDSI is an eCB-dependent process that affects GABA synapses globally. A, Top, Traces from recordings in the presence of AM251 (5 μm). Inset, Schematic describing experimental setup. Bottom, Summary time course of 10 cells from 7 mice (4 male, 3 female) showing that oIPSCs do not undergo DSI or LDSI with AM251 (PD1: F(2,9) = 1.89, p = 0.19, PD2: F(2,9) = 1.57, p = 0.24). B, Summary graphs showing that repeated postsynaptic depolarization affects neither DSI (F(2,9) = 1.03, p = 0.35) nor LDSI (F(2,9) = 1.92, p = 0.19). C, Representative traces of control sIPSC events during the baseline (1) and LDSI (2) time points shown in D. D, Time course summary of sIPSC frequency in control depresses following repeated postsynaptic depolarization (gray, PD1: F(2,10) = 33.59, p = 0.0001, PD2: F(2,10) = 11.44, p = 0.0025), but is unaltered in AM251 (orange, PD1: F(2,9) = 1.73, p = 0.21, PD2: F(2,9) = 0.54, p = 0.57). E, Summary graphs showing that, compared with the initial baseline, postsynaptic depolarization causes phasic (F(2,10) = 72.66, p = 0.0001) and persistent (F(2,10) = 9.52, p = 0.0039) depression of sIPSC frequency in control. Scale bars: A, 100 pA, 20 ms; C, 25 pA, 200 ms. Post hoc versus baseline p-values are shown as follows: *p < 0.05, **p < 0.01, ***p < 0.001. Data are shown as mean ± SEM.
Given that eIPSCs undergo LDSI in the presence of an mGluR5 antagonist, we tested whether LDSI in oIPSCs could be reversed to rapidly recovering DSI by exogenously activating mGluR5 with the group I mGluR agonist DHPG (Fig. 7A, inset). DHPG application did not alter DSI (DSI1: 61.4 ± 7.8% of baseline: 95.4 ± 2.5%; p = 0.0064; n = 6; DSI2: 64.8 ± 7.9% of baseline; p = 0.015; n = 6); however, LDSI was absent (LDSI1: 95.4 ± 5.2% of baseline; p = 0.99; n = 6; LDSI2: 100.5 ± 5.7% of baseline; p = 0.62; n = 6) (Fig. 7A). Similarly, sIPSC frequency showed DSI (DSI1: 47.2 ± 8.8% of baseline: 93.9 ± 5.2%; p = 0.0019; n = 6), but not LDSI (LDSI2: 82.6 ± 15.7% of baseline; p = 0.61; n = 6) (Fig. 7B). The amplitude of sIPSC events were unaltered (DSI1: 96.0 ± 1.1% of baseline: 99.2 ± 1.6%; p = 0.42; n = 6; LDSI2: 98.3 ± 5% of baseline; p = 0.98; n = 6) (Fig. 7C). Therefore, activation of group I mGluRs (mGluR5 specifically) prevents LDSI, allowing rapid recovery from DSI.
Figure 7.

mGluR5 coactivation curtails optically induced LDSI. A, Top, Representative traces during DHPG application (50 μm). Inset, Experimental setup. Bottom, Summary time course of 6 cells from 6 mice (2 male, 4 female) showing postsynaptic depolarizations in DHPG repeatedly elicit DSI, but not LDSI (PD1: F(2,5) = 19.59, p = 0.0010, PD2: F(2,5) = 16.63, p = 0.0038). B, C, Summary graphs of a phasic suppression of sIPSC frequency (F(2,5) = 10.30, p = 0.0037; B), whereas sIPSC amplitude is unaffected by postsynaptic depolarization (F(2,5) = 0.29, p = 0.67; C). Scale bars, 100 pA, 20 ms. Post hoc versus baseline p-values are shown as follows: *p < 0.05, **p < 0.01. Data are shown as mean ± SEM.
Inhibition of GABAergic tone in the PVN results in HPA axis activation (Hewitt et al., 2009). The BNSTFu relays stressor information from higher-order structures to the PVN (Dong et al., 2001), suggesting that BNSTFu synapses should have functional control over CRH neuron firing. We found that a single oIPSC could transiently impair firing activity in PNCs (Fig. 8A). Next, we tested whether inhibition from repeated BNSTFu stimulation (10 Hz, 1 s) could be ameliorated by LDSI (Fig. 8B). Before LDSI induction, BNSTFu stimulation inhibited PNC firing (gray: 61.7 ± 9.3% of baseline; p = 0.0091; n = 6) (Fig. 8C,E). Following LDSI induction, inhibition from BNSTFu stimulation was weaker, resulting in impaired control over PNC firing (blue: 86.0 ± 8.4% of baseline; p = 0.16; n = 6; gray vs blue: 61.7 ± 9.3% vs 86.0 ± 8.4%; p = 0.045, n = 6) (Fig. 8D,E). These data demonstrate that, in the absence of desensitization of CB1R by mGluR5, eCBs exert control over the functional inhibition of PNC activity for many minutes.
Figure 8.
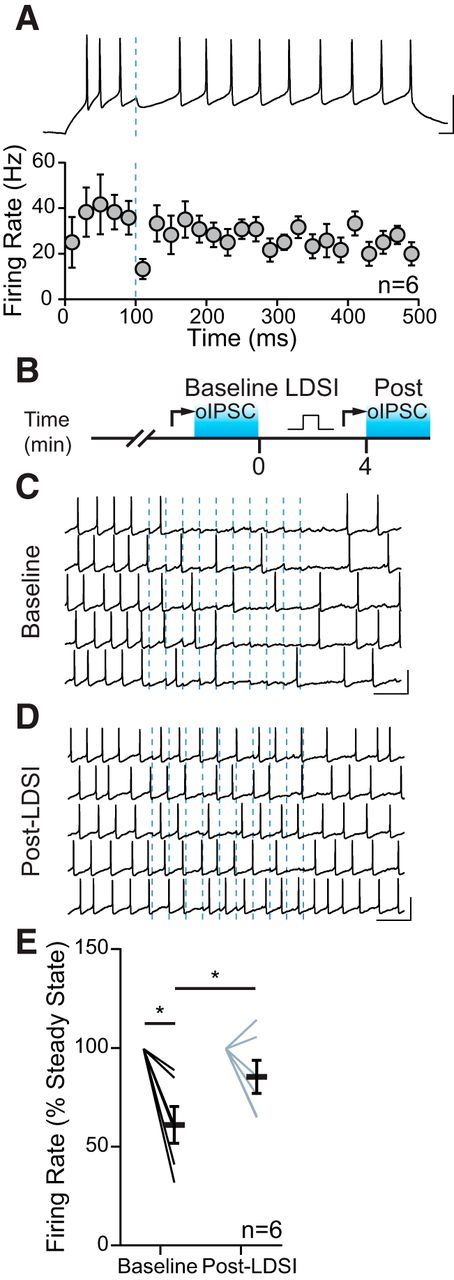
LDSI impairs afferent control of firing. A, Spiking is reliably inhibited by a single oIPSC from BNSTFu afferents (blue line), as shown in the current-clamp trace (top) and summary graph (bottom). B, Temporal schematic of experiment detailing timing of recordings, optical stimulation, and postsynaptic depolarization (square pulse). C, Action potentials in one cell are inhibited by optical stimulation at 10 Hz (dashed blue lines) before LDSI. D, Inhibition is less reliable following LDSI induction. E, Summary data of the mean changes in firing of 6 cells from 4 mice (4 male) during the 1 s, 10 Hz stimulation for both conditions (F(3,5) = 9.89, p = 0.0048). Scale bars: A, 50 mV, 20 ms; C, D, 50 mV, 200 ms. Post hoc versus baseline p-values are shown as follows: *p < 0.05. Data are shown as mean ± SEM.
Discussion
Here, we demonstrate that eCB-mediated suppression of inhibitory terminals in the PVN persists for up to 8 min, globally affecting GABAergic afferents onto PNCs, and is curtailed by heterologous desensitization through mGluR coactivation. Specifically, mGluR5 activation is necessary to curtail LDSI, whereas activation of group I mGluRs in the absence of coincident glutamate release is sufficient to curtail LDSI. LDSI represents a novel form of plasticity previously unattributed to the cannabinoid system. Together, the data illustrate that using electrical stimulation to assay DSI in the PVN and other brain regions may underestimate the duration of eCB-mediated effects at inhibitory synapses following postsynaptic depolarization.
By loading specific synaptic compartments with GDPβs, we selectively blocked GPCR function in postsynaptic and astrocytic sites. With this method, we thereby tested whether mGluR5 function in those specific sites was required to curtail LDSI by looking at the speed of eIPSC recovery from DSI. Rapid recovery from postsynaptic depolarization with either astrocytic or postsynaptic GPCR function blocked suggests that the mGluR5 responsible for controlling the duration of LDSI is not located at those sites. Our group has previously shown heterologous desensitization within presynaptic terminals of the PVN, where PKC activation by Gq receptors impaired the function of downstream Gi signaling (Gordon and Bains, 2003). Here, we show that, following a transient activation of Gq-coupled mGluR5, Gi-coupled CB1R function is impaired. Although this does not directly show that the mGluR5 responsible for curtailing LDSI is presynaptic, these data, when taken together with the absence of postsynaptic or astrocytic GPCR involvement, support our hypothesis that LDSI at inhibitory afferents to PNCs is curtailed by presynaptic mGluR5.
Interestingly, following acute DHPG application and washout (Fig. 3C), CB1R activity is impaired to the point that postsynaptic depolarization no longer causes a significant DSI. However, when DHPG is continuously applied to examine oIPSC recovery (Fig. 7), DSI remains intact. This discrepancy may be explained by differences in the duration of DHPG application, where a longer time course of mGluR5 activation may be blunted by receptor internalization or desensitization (Ribeiro et al., 2009), thereby reducing the heterologous desensitization of CB1R. This may represent a mechanism by which glutamate spillover and activation of presynaptic mGluR5 could prevent GABA synapses from undergoing DSI, but with persistent mGluR5 activation, allow for rapidly recovering DSI to occur.
GPCRs (Gyombolai et al., 2012) including mGluRs (Varma et al., 2001) can recruit eCB signaling, which, under certain conditions, impart long-term plasticity at glutamatergic and GABAergic terminals (Gerdeman et al., 2002; Gerdeman and Lovinger, 2003). GPCR-mediated eCB signaling paired to patterned activity can induce LTD at GABA synapses (Marsicano et al., 2002; Heifets and Castillo, 2009). In contrast to DSI or LDSI, this eCB-LTD requires persistent activation of presynaptic terminals through high-frequency, theta burst, or spike-timing dependent stimulation paired with prolonged postsynaptic depolarization (Chevaleyre and Castillo, 2003; Crozier et al., 2007; Zhu and Lovinger, 2007). Mechanistically, these eCB-LTD induction methods require either presynaptic Ca2+ elevations, or afferent activity coupled to GPCR-mediated eCB production (Chevaleyre et al., 2006; Heifets and Castillo, 2009). However, the methods for inducing eCB-LTD are distinct from LDSI; coordinated activity in presynaptic terminals is not required for LDSI and the timeframe of eCB production and release is much shorter for LDSI than for eCB-LTD (Chevaleyre et al., 2006). We suggest that LDSI is a novel form of fast eCB action distinct from short-acting DSI/DSE and the prolonged actions of eCB-LTD, adding to the complexity of eCB function at the synaptic level.
It is unclear whether LDSI is specific to the neuron from which eCBs were released or if neighboring neurons might also undergo a suppression of inhibitory inputs due to eCB diffusion (Kreitzer et al., 2002). Due to the global afferent effects of LDSI within a single neuron, as shown by the LDSI of sIPSC frequency, it is possible that eCBs could impart LDSI to neighboring neurons when released in an environment containing little free circulating glutamate. This raises the possibility of a cluster of PNCs with inhibitory constraint removed by a globally diminished GABAergic drive primed to fire in response to signals that were previously subthreshold. This scenario may represent a mechanism by which the ensemble activity of PNCs could be coupled to recruit a larger or more reliable hormonal release to the median eminence and may be a site for dysregulation in stress disorders.
Selective activation of GABA synapses allows for careful examination of DSI in the absence of neurotransmitters coincidentally released by electrical stimulation. Here, the data suggest that DSI has remarkably different recovery kinetics when examined in this manner. Enhancing the recovery from persistent eCB-dependent depression is a mechanism by which glutamate can effectively reset the strength of GABA terminals. Physiologically, this may occur through several means: the coordinated recruitment of glutamatergic afferents from one or more stress-associated principal projection site; release of glutamate as a gliotransmitter; or lateral diffusion of glutamate to heterosynaptic mGluRs, the likelihood of which could be increased through state-dependent network remodeling of astrocytic coverage (Panatier et al., 2006). LDSI may also act as a state-switching mechanism in which depressed GABAergic afferents can be brought back to full strength after an excitatory afferent population reached an activity threshold permissive for glutamate spillover. Conversely, afferents undergoing DSI during periods of high glutamatergic activity would be only transiently suppressed, allowing the inhibitory tone to maintain a higher dynamic range.
eCB signaling can either rapidly and transiently inhibit GABA transmission through DSI or, through activation of postsynaptic metabotropic receptors, induce eCB-LTD at GABA synapses. Here, we describe a novel action of eCBs in suppressing the activity of synapses. We report a role for Gq-coupled mGluRs in curtailing the actions of Gi-coupled CB1Rs at presynaptic GABA terminals through heterologous desensitization. These findings indicate that heterosynaptic signaling during coincident glutamate and GABA release accelerates recovery from eCB-mediated depression and that conditions favoring glutamate spillover may limit the temporal window of eCB action at GABA synapses. Therefore, presynaptic mGluR5-mediated quenching of DSI is a third mechanism of control over the temporal window of eCBs.
Footnotes
This work was supported by the Canadian Institutes for Health Research (CIHR Grant 86501, CIHR Foundation Scheme to J.S.B.). P.L.W.C. is supported by a studentship from Alberta Innovates Health Solutions (AIHS). We thank Cheryl Breiteneder, Mio Tsui, and Rodney Barasi for technical assistance and mouse colony maintenance.
The authors declare no competing financial interests.
References
- Biag J, Huang Y, Gou L, Hintiryan H, Askarinam A, Hahn JD, Toga AW, Dong HW (2012) Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J Comp Neurol 520:6–33. 10.1002/cne.22698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. 10.1016/S0896-6273(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76. 10.1146/annurev.neuro.29.051605.112834 [DOI] [PubMed] [Google Scholar]
- Chu J, Zheng H, Zhang Y, Loh HH, Law PY (2010) Agonist-dependent μ-opioid receptor signaling can lead to heterologous desensitization. Cell Signal 22:684–696. 10.1016/j.cellsig.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF (2007) Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci U S A 104:1383–1388. 10.1073/pnas.0609596104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska J, Hazra R, Guo JD, Dewitt S, Rainnie DG (2013) Central CRF neurons are not created equal: phenotypic differences in CRF-containing neurons of the rat paraventricular hypothalamus and the bed nucleus of the stria terminalis. Front Neurosci 7:156. 10.3389/fnins.2013.00156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decavel C, van den Pol AN (1992) Converging GABA- and glutamate-immunoreactive axons make synaptic contact with identified hypothalamic neurosecretory neurons. J Comp Neurol 316:104–116. 10.1002/cne.903160109 [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A (2003) Characterization of depolarization-induced suppression of inhibition using paired interneuron–purkinje cell recordings. J Neurosci 23:5906–5918. 10.1523/JNEUROSCI.23-13-05906.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW (2001) Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol 436:430–455. 10.1002/cne.1079 [DOI] [PubMed] [Google Scholar]
- Errington AC, Di Giovanni G, Crunelli V, Cope DW (2011) mGluR control of interneuron output regulates feedforward tonic GABAA inhibition in the visual thalamus. J Neurosci 31:8669–8680. 10.1523/JNEUROSCI.0317-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM (2003) Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol 140:781–789. 10.1038/sj.bjp.0705466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. 10.1038/nn832 [DOI] [PubMed] [Google Scholar]
- Glangetas C, Girard D, Groc L, Marsicano G, Chaouloff F, Georges F (2013) Stress switches cannabinoid type-1 (CB1) receptor-dependent plasticity from LTD to LTP in the bed nucleus of the stria terminalis. J Neurosci 33:19657–19663. 10.1523/JNEUROSCI.3175-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Bains JS (2003) Priming of excitatory synapses by alpha1 adrenoceptor-mediated inhibition of group III metabotropic glutamate receptors. J Neurosci 23:6223–6231. 10.1523/JNEUROSCI.23-15-06223.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindaiah, Cox CL (2004) Synaptic activation of metabotropic glutamate receptors regulates dendritic outputs of thalamic interneurons. Neuron 41:611–623. 10.1016/S0896-6273(04)00013-3 [DOI] [PubMed] [Google Scholar]
- Gyombolai P, Pap D, Turu G, Catt KJ, Bagdy G, Hunyady L (2012) Regulation of endocannabinoid release by G proteins: a paracrine mechanism of G protein-coupled receptor action. Mol Cell Endocrinol 353:29–36. 10.1016/j.mce.2011.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammack SE, Mania I, Rainnie DG (2007) Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. J Neurophysiol 98:638–656. 10.1152/jn.00382.2007 [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. 10.1146/annurev.physiol.010908.163149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463:232–236. 10.1038/nature08673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS (2009) Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci 12:438–443. 10.1038/nn.2274 [DOI] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB (2009) Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 34:2733–2745. 10.1038/npp.2009.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA (2002) Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol 61:477–485. 10.1124/mol.61.3.477 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001a) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. 10.1016/S0896-6273(01)00246-X [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001b) Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21:RC174. 10.1523/JNEUROSCI.21-20-j0005.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG (2002) Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron 34:787–796. 10.1016/S0896-6273(02)00695-5 [DOI] [PubMed] [Google Scholar]
- Lafourcade CA, Zhang L, Alger BE (2009) Novel mGluR- and CB1R-independent suppression of GABA release caused by a contaminant of the group I metabotropic glutamate receptor agonist, DHPG. PLoS One 4:e6122. 10.1371/journal.pone.0006122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhu LH, Pachuau J, Lee HA, Pan HL (2014) mGluR5 upregulation increases excitability of hypothalamic presympathetic neurons through NMDA receptor trafficking in spontaneously hypertensive rats. J Neurosci 34:4309–4317. 10.1523/JNEUROSCI.4295-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418:530–534. 10.1038/nature00839 [DOI] [PubMed] [Google Scholar]
- Miklós IH, Kovács KJ (2002) GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience 113:581–592. 10.1016/S0306-4522(02)00147-1 [DOI] [PubMed] [Google Scholar]
- Miklós IH, Kovács KJ (2012) Reorganization of synaptic inputs to the hypothalamic paraventricular nucleus during chronic psychogenic stress in rats. Biol Psychiatry 71:301–308. 10.1016/j.biopsych.2011.10.027 [DOI] [PubMed] [Google Scholar]
- Morena M, Patel S, Bains JS, Hill MN (2016) Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 41:80–102. 10.1038/npp.2015.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. 10.1016/S0896-6273(01)00247-1 [DOI] [PubMed] [Google Scholar]
- Panatier A, Gentles SJ, Bourque CW, Oliet SH (2006) Activity-dependent synaptic plasticity in the supraoptic nucleus of the rat hypothalamus. J Physiol 573:711–721. 10.1113/jphysiol.2006.109447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ (2004) Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 145:5431–5438. 10.1210/en.2004-0638 [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Ferreira LT, Paquet M, Cregan T, Ding Q, Gros R, Ferguson SS (2009) Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J Biol Chem 284:23444–23453. 10.1074/jbc.M109.000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG (1997) Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J Neurophysiol 77:527–536. 10.1152/jn.1997.77.2.527 [DOI] [PubMed] [Google Scholar]
- Steiner MA, Marsicano G, Nestler EJ, Holsboer F, Lutz B, Wotjak CT (2008) Antidepressant-like behavioral effects of impaired cannabinoid receptor type 1 signaling coincide with exaggerated corticosterone secretion in mice. Psychoneuroendocrinology 33:54–67. 10.1016/j.psyneuen.2007.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K (2007) Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J Physiol 578:773–785. 10.1113/jphysiol.2006.117499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE (1991) Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol 434:271–293. 10.1113/jphysiol.1991.sp018469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Romano C, Ghosh P (1995) Metabotropic glutamate receptor mGluR5 subcellular distribution and developmental expression in hypothalamus. J Comp Neurol 362:134–150. 10.1002/cne.903620108 [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC188. 10.1523/JNEUROSCI.21-24-j0003.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker Cusulin JI, Senst L, Teskey GC, Bains JS (2014) Experience salience gates endocannabinoid signaling at hypothalamic synapses. J Neurosci 34:6177–6181. 10.1523/JNEUROSCI.0163-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS (2010) Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci 30:11188–11196. 10.1523/JNEUROSCI.1046-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. 10.1038/35069076 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gray TS, D'Souza DN, Carrasco GA, Damjanoska KJ, Dudas B, Garcia F, Zainelli GM, Sullivan Hanley NR, Battaglia G, Muma NA, Van de Kar LD (2004) Desensitization of 5-HT1A receptors by 5-HT2A receptors in neuroendocrine neurons in vivo. J Pharmacol Exp Ther 310:59–66. 10.1124/jpet.103.062224 [DOI] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ (1997) Metabotropic glutamate receptor enhancement of spontaneous IPSCs in neocortical interneurons. J Neurophysiol 78:2287–2295. 10.1152/jn.1997.78.5.2287 [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM (2007) Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol 97:4386–4389. 10.1152/jn.01228.2006 [DOI] [PubMed] [Google Scholar]