

Abstract
The loss of presynaptic proteins Munc18-1, syntaxin-1, or SNAP-25 is known to produce cell death, but the underlying features have not been compared experimentally. Here, we investigated these features in cultured mouse CNS and DRG neurons. Side-by-side comparisons confirmed massive cell death, before synaptogenesis, within 1–4 DIV upon loss of t-SNAREs (syntaxin-1, SNAP-25) or Munc18-1, but not v-SNAREs (synaptobrevins/VAMP1/2/3 using tetanus neurotoxin (TeNT), also in TI-VAMP/VAMP7 knock-out (KO) neurons). A condensed cis-Golgi was the first abnormality observed upon Munc18-1 or SNAP-25 loss within 3 DIV. This phenotype was distinct from the Golgi fragmentation observed in apoptosis. Cell death was too rapid after syntaxin-1 loss to study Golgi abnormalities. Syntaxin-1 and Munc18-1 depend on each other for normal cellular levels. We observed that endogenous syntaxin-1 accumulates at the Golgi of Munc18-1 KO neurons. However, expression of a non-neuronal Munc18 isoform that does not bind syntaxin-1, Munc18-3, in Munc18-1 KO neurons prevented cell death and restored normal cis-Golgi morphology, but not synaptic transmission or syntaxin-1 targeting. Finally, we observed that DRG neurons are the only Munc18-1 KO neurons that do not degenerate in vivo or in vitro. In these neurons, cis-Golgi abnormalities were less severe, with no changes in Golgi shape. Together, these data demonstrate that cell death upon Munc18-1, syntaxin-1, or SNAP-25 loss occurs via a degenerative pathway unrelated to the known synapse function of these proteins and involving early cis-Golgi abnormalities, distinct from apoptosis.
SIGNIFICANCE STATEMENT This study provides new insights in a neurodegeneration pathway triggered by the absence of specific proteins involved in synaptic transmission (syntaxin-1, Munc18-1, SNAP-25), whereas other proteins involved in the same molecular process (synaptobrevins, Munc13–1/2) do not cause degeneration. Massive cell death occurs in cultured neurons upon depleting syntaxin-1, Munc18-1, and/or SNAP-25, well before synapse formation. This study characterizes several relevant cellular phenotypes, especially early cis-Golgi abnormalities, distinct from abnormalities observed during apoptosis, and rules out several other phenotypes as causal (defects in syntaxin-1 targeting and synaptic transmission). As proteins, such as syntaxin-1, Munc18-1, or SNAP-25, modulate α-synuclein neuropathy and/or are dysregulated in Alzheimer's disease, understanding this type of neurodegeneration may provide new links between synaptic defects and neurodegeneration in humans.
Keywords: exocytosis, neurodegeneration, presynaptic proteins, synaptic transmission
Introduction
Neuronal survival is supported by a complex interplay of multiple molecular pathways (Morrison et al., 2000; Dekkers et al., 2013). Loss of synapse integrity and function has received considerable attention as a putative starting point for cell loss and is considered an important early feature in many neurodegenerative disorders (Davies et al., 1987; Selkoe, 2002; Guégan and Przedborski, 2003; Venderova and Park, 2012). However, how loss of synapse integrity/function leads to cell death is unclear.
One approach to study such links between synapses and cell loss has been to exploit genetic or proteolytic depletion of synaptic proteins in mouse models and in vitro. Depletion of specific presynaptic proteins involved in exocytosis, Munc18-1, syntaxin-1, and SNAP-25, produces neuronal cell death (Kurokawa, 1987; Williamson and Neale, 1998; Verhage et al., 2000; Washbourne et al., 2002; Heeroma et al., 2003, 2004; Berliocchi et al., 2005; Delgado-Martínez et al., 2007; Zhao et al., 2010; Peng et al., 2013; Kofuji et al., 2014). Neurodegeneration has also been observed for presynaptic proteins involved in endocytosis (Cremona et al., 1999; Milosevic et al., 2011). However, depletion of other presynaptic proteins equally important for exocytosis, Munc13–1/2 or synaptobrevin-2/VAMP2, does not produce cell death (Schoch et al., 2001; Varoqueaux et al., 2002; Peng et al., 2013), nor does depletion of postsynaptic proteins. Hence, the relationship between loss of (pre-)synaptic function and neuronal viability is complex and remains poorly understood.
It is conceivable that the role of these (pre-)synaptic proteins in neuronal viability is distinct from their role in synaptic transmission. In the first study (of Munc18-1 knock-out [KO] neurons) (Verhage et al., 2000), it was proposed that neurons die due to defective synaptic transmission. However, in subsequent studies, on Munc13–1/2 double knock-out (DKO) and synaptobrevin-2/VAMP2 KO neurons (Schoch et al., 2001; Varoqueaux et al., 2002; Peng et al., 2013), synaptic transmission was also abolished but without cell death. Alternatively, it has been emphasized that some presynaptic proteins depend on each other for normal cellular levels. For instance, syntaxin-1 levels are 70% reduced in Munc18-1 KO neurons (Voets et al., 2001; Toonen et al., 2005) and Munc18-1 levels are reduced in syntaxin-1 KO (Zhou et al., 2013) and knock-in mice (Gerber et al., 2008). In addition, Munc18-1 regulates targeting of overexpressed syntaxin-1 to the plasma membrane in heterologous cells (Rowe et al., 2001; Arunachalam et al., 2008), suggesting that syntaxin-1 and Munc18-1 are cochaperones (Rowe et al., 1999, 2001; Toonen et al., 2005; McEwen and Kaplan, 2008). Such interdependencies may contribute to cell viability. However, this interdependency is unlikely to explain cell loss in SNAP-25 KO neurons. Instead, cell loss in SNAP-25 KO neurons has been attributed to impaired release of trophic factors (Delgado-Martínez et al., 2007) because high-density SNAP-25 KO cultures showed no degeneration (Bronk et al., 2007). In contrast, even in intact brain, cell-specific deletion of Munc18-1 expression in a subpopulation of neurons (i.e., within the normal network of trophic factor secreting neurons) produces loss of that population of neurons (Heeroma et al., 2004). Hence, different degenerative pathways may operate in these different models, probably unrelated to the shared role these proteins have in synaptic transmission.
To systematically compare the cell death mechanism(s) activated by depletion of presynaptic proteins, we performed a side-by-side study of Munc18-1 KO neurons, SNAP-25 KO neurons, Munc18-1/SNAP-25 DKO neurons, and syntaxin-1 depletion using botulinum neurotoxin C (BoNT/C) versus several nondegenerative models, synaptobrevin/VAMP depletion using tetanus neurotoxin (TeNT), TI-VAMP/VAMP7 KO neurons, and Munc13–1/2 DKO neurons. To monitor and compare cell loss with high cellular/time resolution, we used cultured primary mouse neurons and DRG explants and monitored viability, protein targeting, and synaptic transmission. We found that cell death upon Munc18-1, syntaxin-1, or SNAP-25 depletion occurs via a degenerative pathway unrelated to the known synaptic function of these proteins and identified early cis-Golgi abnormalities, distinct from apoptosis, as the earliest sign of neurodegeneration.
Materials and Methods
Animals.
Munc18-1 KO, SNAP-25 KO, Munc13–1/2 DKO, and TI-VAMP KO mice were generated as described previously (Verhage et al., 2000; Varoqueaux et al., 2002; Washbourne et al., 2002; Danglot et al., 2012). Munc18-1/SNAP-25 DKO mice were generated by crossing Munc18-1 HZ with SNAP-25 HZ. Embryonic day 18 (E18) or E14 embryos were obtained by caesarian section of pregnant females from timed mating. Animals were housed and bred according to the Institutional Dutch and United States governmental guidelines.
Neuronal cultures.
Hippocampi and cortices were separately collected in ice-cold Hanks buffered salt solution (Sigma) buffered with 7 mm HEPES (Invitrogen). Meninges were removed and neurons incubated in Hanks-HEPES with 0.25% trypsin (Invitrogen) for 20 min at 37°C. After washing, neurons were triturated with fire polished Pasteur pipettes and counted in a Fuchs-Rosenthal chamber. Neurons were plated in prewarmed Neurobasal medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), 1.8% HEPES, 0.25% glutamax (Invitrogen), and 0.1% Pen/Strep (Invitrogen). Hippocampal neurons were plated at a density of 6000/well on 18 mm glass coverslips on microislands of rat glia. Glial islands were created by plating 8000/well rat glia on UV-sterilized agarose-coated etched glass coverslips stamped with a 0.1 mg/ml poly-d-lysine (Sigma) and 0.2 mg/ml rat tail collagen (BD Biosciences) solution. Network cultures were created by plating 25,000–50,000/well on 18 mm glass coverslips or 250,000/well on a 35 mm glass-bottom dish of cortical neurons on a confluent layer of rat glia grown on etched glass coverslips sprayed with a 0.1 mg/ml poly-d-lysine and 0.2 mg/ml rat tail collagen (BD Biosciences) solution.
DRG cultures.
DRG from E14 Munc18-1 KO mice of were dissected (∼45 DRGs per animal) in ice-cold Hanks buffered salt solution (Sigma) buffered with 1 m HEPES (Invitrogen). Washed DRGs were transferred to coated coverslips with poly-L-lysine 0.1 mg/ml (Sigma) and Matrigel (BD Biosciences). DRGs were placed in the middle of the coverslip on top of a drop of Neurobasal (Invitrogen) supplemented with 45% d-(+)-glucose (Sigma), 1% Glutamax (Invitrogen), 2% B-27 (Invitrogen), and 50 ng/ml nerve growth factor (Sigma).
Constructs, lentiviruses, and chemicals.
Neuronal cortical cultures were infected at DIV 0 with lentiviral particles encoding SNAP-25 (S25), Munc18-1 wild-type (WT) (M18–1), Munc18-3 (M18–3), TeNT, BoNT/C expressing eGFP, mCherry, or no tagged fluorescent protein for DIV 3 and DIV 14 experiments, together with control lentiviruses expressing eGFP. Secretory vesicle marker neuropeptide Y (NPY) fused to mCherry was described previously (Farina et al., 2015) and delivered using lentiviral particles at DIV 0. For live cell image, cultures were infected with RFP as a morphological marker and ManII-GFP (gift from V. Malhotra, Centre for Genomic Regulation, Barcelona, Spain) (Villeneuve et al., 2013) as a cis-Golgi marker, which was delivered with lentiviral particles at DIV 0. F-actin was visualized live using mCherry-UtrCH (gift from William Bement, Addgene plasmid #26740) (Burkel et al., 2007), which was delivered using lentiviral particles at DIV 0. For electrophysiology recordings, hippocampal cultures were infected with Munc18-1 WT, Munc18-2, and Munc18-3 cDNAs linked to enhanced green fluorescent protein (EGFP) via an internal ribosomal entry site.
To induce apoptosis, neuronal cultures were treated with 1 μm staurosporine for 3 h or DMSO as control.
Immunocytochemistry and confocal microscopy.
Cortical neuronal cultures were fixed at DIV 3, 5, 7, 9, 11, and 14 and DRG cultures at DIV 1, 6, 13, and 21 with 4% formaldehyde (Electron Microscopy Sciences). After washing with PBS, neurons were permeabilized with 0.5% Triton X-100, followed by a 30 min incubation in PBS containing 0.1% Triton X-100 and 2% normal goat serum to block aspecific binding. The same solution was used for diluting antibodies. Neurons were then stained with primary antibodies for 2 h at room temperature, washed with PBS, and stained with AlexaFluor-conjugated secondary antibodies (1:1000; Invitrogen) for 1 h at room temperature. Phalloidin rhodamine (1:1000; Thermo Fisher Scientific) probe was stained together with the secondary antibodies. The primary antibodies and dilutions used were chicken anti-MAP2 (1:10,000; Abcam), mouse anti-GM130 (1:1000; Transduction Laboratories), rabbit anti-syntaxin-1 (1:1000; clone I379, gift from T. Südhof, Department of Molecular and Cellular Physiology and Howard Hughes Medical Institute, Stanford University School of Medicine), rat anti-LAMP1 (1:100; Abcam), rabbit anti-calnexin (1:500; ENZO Life Sciences), mouse anti-Human Transferrin Receptor (1:500; Invitrogen), chicken anti-Neurofilament (1:2000; Novus Biologicals), mouse anti-VAMP (1:1000; clone 69.1, SySy), guinea pig anti-vGlut (1:10,000; Millipore), mouse anti-SMI312 (1:1000; BioLegend), and anti-tubulin (1:500; SySy). After an additional 3 washes, coverslips were mounted on microscopic slides with Mowiol-DABCO and imaged on a NIKON Ti-Eclipse microscope, with a confocal scanner model A1R+. Image acquisition was performed using the resonant scanning mode, using a 60× oil-immersion objective (NA 1.40) with 2× zoom. z stacks were acquired with 1 μm interval for apoptotic markers. For cell survival, a 40× oil-immersion objective (NA 1.30) was used to scan an area with a radius of 1.5 mm (97 fields of view); or on a Zeiss 510 Meta Confocal microscope (Carl Zeiss) with a 63× Plan-Neofluar lens (NA 1.4, Carl Zeiss) with or without 4× zoom, z stacks were acquired with 0.63 μm interval, or 10× lens (NA 0.3, Carl Zeiss). Neuronal organelle morphology and colocalization were analyzed using ImageJ software; z stacks images were collapsed to maximal projection and analyzed. Total dendrite length, total number of presynaptic terminals, and dendrite branching (using Sholl analysis) were measured using custom written software routines in MATLAB. For Golgi fragmentation analyses, MATLAB software was used and a stack of several slices was loaded in the interface and converted to a 3D matrix of size w × h × z and thresholded using the Otsu method. The resulting binary image stack was further processed to examine the connectedness of the voxels in a 26-neighborhood fashion. The resulting objects were quantified by calculating the surface and volume of each object, and the number of objects was used as a measure for fragmentation.
Live cell imaging.
For live mitochondria experiments, cortical neuronal cultures were incubated, at DIV 3, with 50 mm of MitoTracker Green FM (Invitrogen, gift from N. Raimundo, Institute for Cellular Biochemistry, University Medical Center Göttingen, Göttingen, Germany) for 20 min at 37°C with 5% CO2. Afterward coverslips were placed in an imaging chamber with Tyrode's solution (2 mm CaCl2, 2.5 mm KCl, 119 mm NaCl, 2 mm MgCl2, 20 mm glucose, and 25 mm HEPES, pH 7.4) and imaged live at room temperature on a Zeiss 510 Meta Confocal microscope (Carl Zeiss) with a 63× Plan-Neofluar lens (NA 1.40, Carl Zeiss). For live Golgi and UtrCH experiments, cortical neurons were plated on a 35 mm glass-bottom dish, in prewarmed Neurobasal medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), 1.8% HEPES, 0.25% glutamax (Invitrogen), and 0.1% Pen/Strep (Invitrogen). Neurons were relocated at DIV 3 to a NIKON Ti-Eclipse microscope, with a confocal scanner model A1R+, and a Tokai Hit heating system with heated stage, water bath and objective, prewarmed to 37°C and regulated CO2 to 5%. For Golgi experiments, images were acquired every minute using the resonant scanning mode for 2 h 15 min, with a 60× oil-immersion objective (NA 1.4) and 2× zoom. z stacks were acquired with 1 μm interval. For live UtrCH, images were acquired using the resonant scanning mode, with a 60× oil-immersion objective (NA 1.4). z stacks were acquired with 1 μm interval.
Brain lysate and Western blot.
Brain lysate for Western blot analysis was homogenized in ice-cold PBS with protease inhibitors (Sigma) and then incubated for 2 h at 4°C in 1% Triton X-100 (Fisher Scientific) in 1× PBS with protease inhibitors. The brain lysate was then centrifuged for 20 min at 10,000 × g at 4°C; 1 ml of Laemmli Sample Buffer was added per 0.1 g of brain and boiled for 20 min at 100°C. Samples were loaded on 15% SDS-PAGE gel. Acrylamide gels were transferred onto 0.2 μm pore size Immune-Blot PVDF Membrane for Protein Blotting (Bio-Rad Laboratories). Membranes were incubated on an orbital shaker for 30 min at room temperature in 2% skim milk powder (Merck), 0.5% BSA (Thermo Fisher Scientific) containing 0.1% Tween 20 (TBS-T) (Sigma) to block nonspecific binding. Primary antibody incubation was done in 1% PBS-Tween or overnight at 4°C, followed by 3 washes with PBS-T. Membranes were stained with secondary antibody conjugated with alkaline phosphatase or Alexa-647 (1:1000) (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. After 3 washes with TBS-T, the alkaline phosphatase-conjugated antibody was visualized using an attophos AP fluorescent substrate (Promega). Membranes were scanned with a Fuji Image FLA-5000 Reader and analyzed with ImageJ software. Primary antibodies used are as follows: anti-TI-VAMP (1:1000; SySy), anti-Munc18 (1:1000, clone #610336, Transduction Laboratories Laboratories), anti-SNAP-25 (1:1000, SMI-81, Sternberger), and anti-γ-tubulin (1:2000, clone #GTU-88, Sigma).
Electrophysiological recordings.
Whole-cell voltage-clamp (Vm = −70 mV) recordings of Munc18-1 KO neurons rescued with Munc18-1-ires-EGFP or Munc18-3-ires-EGFP were performed on DIV 10–15. The patch pipette solution contained the following (in mm): 125 K+-gluconic acid, 10 NaCl, 4.6 MgCl2, 4 K2-ATP, 15 creatine phosphate, 1 EGTA, and 20 U/ml phosphocreatine kinase, pH 7.30. The external medium used contained the following components (in mm): 140 NaCl, 2.4 KCl, 4 CaCl2, 4 MgCl2, 10 HEPES, 10 glucose, pH 7.30. We used fast double-barrel application of extracellular (500 mm) hypertonic sucrose to reliably assess ready releasable pool (RRP) vesical size. Axopatch 200A was used for whole-cell recordings. All recordings were at 33°C. Signal was acquired using Digidata 1322A and Clampex 8.1. Clampfit 8.0 was used for offline analysis.
RNA isolation, cDNA synthesis, and real-time qPCR.
Cortical neuronal cultures, DRG neurons and brain tissue were lysed and scraped in TRIzol reagent (Invitrogen), and organic and aqueous phase separation was initiated by the addition of and mixing with chloroform (Merck). Subsequently, RNA isolation was performed automated with an RNeasy MiniKit on a Qiacube (QIAGEN) according to the manufacturer's protocol). RNA purity and integrity were assessed spectrophotometrically on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). cDNA synthesis was performed on 200 ng of RNA per reaction (RNA sample quantities do not vary within each experiment) using a SuperScript II Reverse Transcriptase Kit (Invitrogen). Priming of mRNA poly-A tails was performed with 125 pmol oligo(dT)12-VN primer in a final volume of 10 μl and was incubated at 70°C for 10 min. MgCl2 (2 mm), dNTPs (0.5 mm), 5 μl 5 × First-Strand Buffer, and 100 U SuperScript II reverse transcriptase were added to the primed mRNAs in a final volume of 25 μl per reaction and incubated at 42°C for 60 min. The reverse transcription reaction was stopped by incubation at 70°C for 10 min. Per sample, 1 μl cDNA was pipetted in triplicate into a 384 well plate and dried in a DNA110 SpeedVac (Thermo Fisher Scientific). Primers used are as follows: Munc18-1, forward, 5′-AACATTGCGTTTCTCCCCTA-3′, reverse, 5′-GAAAGAGTCAGCGGAGTCCA-3′; Munc18-2, forward, 5′-GCGGCACTGTCACCAACT-3′, reverse, 5′-CAGCGAGACAGCTGGTAGG-3′; Munc18-3, forward, 5′-TCTCGATGGACACCTTTTATCA-3′, reverse, 5′-CGGCCACTCTTTGGAATCTA-3′; mEEFA1 (Mouse Eukaryotic Elongation Factor 1A), forward, 5′-ACACGTAGATTCCGGCAAGT-3′, reverse, 5′-AGGAGCCCTTTCCCATCTC-3′. Roche Universal Probes used are as follows: Munc18-1 #95, Munc18-2 #19, Munc18-3 #88, and mEEFA1 #31.
Results
Rapid neuronal loss upon depletion of syntaxin-1/-2/-3, Munc18-1, or SNAP-25, but not synaptobrevin/VAMP1/2/3 and/or VAMP7/TI-VAMP
To systematically study how depletion of different presynaptic proteins relates to cell death, we monitored neuronal survival in parallel cortical cultures of Munc18-1 KO, SNAP-25 KO, Munc18-1/SNAP-25 DKO, and WT neurons infected with lentiviral vectors expressing TeNT, which cleaves synaptobrevin/VAMP1/2/3 proteins (Link et al., 1992, 1993; Schiavo et al., 1992b), and BoNT/C, which cleaves syntaxin-1/-2/-3 and SNAP-25 (Blasi et al., 1993a; Schiavo et al., 1995). BoNT/C-dependent cleavage of SNAP-25 is less efficient (Blasi et al., 1993a; Foran et al., 1996; Osen-Sand et al., 1996; Vaidyanathan et al., 1999; Berliocchi et al., 2005) and not relevant for neuronal viability (Peng et al., 2013). Therefore, we consider BoNT/C expression as a tool to study syntaxin-1/-2/-3-dependent viability. All cultures were grown on rat (WT) glia feeders to provide optimal support for neuronal viability independent of the genetic background of the neurons. TeNT and BoNT/C-expressing viral vectors used a synapsin-1 promoter, which does not induce expression in glia feeders (data not shown). Cultures were fixed at different time points (DIV 3, 5, 7, 9, 11, and 14), and immunocytochemistry was performed using MAP2 to count neurons and study their morphology. MAP2-positive cells were counted in 97 fields of view for each experimental condition. WT neurons all survived in culture during the first 5 d. A fraction of these neurons died during the second week, 39.8% at DIV 14 (Fig. 1A,B). Similarly, WT neurons treated with TeNT showed no significant cell death during the first week in culture and survival was comparable with WT neurons in the second week (Fig. 1A,B). However, WT neurons treated with BoNT/C died before DIV 3, whereas 45.8% of Munc18-1 KO neurons and 51.2% SNAP-25 KO neurons survived at DIV 3. For Munc18-1/SNAP-25 DKO, only 17.1% of neurons survived by DIV 3 and appeared underdeveloped. Beyond DIV 3, further cell loss was observed for Munc18-1 KO, SNAP-25 KO, and Munc18-1/SNAP-25 DKO neurons (Fig. 1A,B). Only in the case of SNAP-25 KO, a fraction of the neurons survived and by DIV 14, 4.1% of normally developed neurons were alive (Fig. 1A,B). Together, these data show that considerable heterogeneity exists in neuronal viability of cultured neurons lacking specific presynaptic proteins.
Figure 1.

Neuronal loss upon depletion of t-SNAREs and Munc18-1 but not v-SNAREs. A, Cortical neuronal cultures from WT, WT infected with TeNT or BoNT/C, Munc18-1 knock-out (M18–1 KO), SNAP-25 (S25) KO, and Munc18-1/SNAP-25 DKO (M18/S25 DKO) were fixed at different time points (DIV 3, 5, 7, 9, 11, and 14) and stained with a dendritic marker (MAP2). B, Quantification of number of neurons per 97 fields of view; not significant, p > 0.05. ***p < 0.001 (two-way ANOVA followed by the post hoc Bonferroni's test). C, E18 brain lysates from WT, M18–1 KO, and S25 KO were analyzed by Western blot and showed no differences in TI-VAMP levels between mutants and control animals (n = 3). D, Cortical neuronal cultures from TI-VAMP WT and TI-VAMP KO infected with TeNT or RFP (as control) were fixed at DIV 14 and stained with a dendritic marker (MAP2). E, Quantification of number of neurons per field of view. No differences in cell survival were found: not significant, p > 0.05 (one-way ANOVA followed by the post hoc Bonferroni's test). Data are mean ± SEM.
The fact that depletion of the v-SNAREs (synaptobrevins) produces such a different effect on cell survival than depletion of t-SNAREs (syntaxins and SNAP-25) or Munc18-1 might be explained by the expression of Synaptobrevin7/TI-VAMP, which is resistant to TeNT (Galli et al., 1998). TI-VAMP is expressed at normal levels in both Munc18-1 and SNAP-25 KO neurons (Fig. 1C). However, TeNT expression in neuronal cortical cultures of TI-VAMP KO mice showed no significant increase in cell loss up to DIV 14 compared with control (TI-VAMP WT) neurons (92.7% TI-VAMP WT treated with TeNT and 99.4% for TI-VAMP KO treated with TeNT; not significant, p > 0.05, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 1D,E). Together, these data show that depletion of v-SNAREs or t-SNAREs/Munc18-1 has very different effects on neuronal viability. The fact that cell death occurs before synaptogenesis points to a distinct role of t-SNAREs/Munc18-1, but not v-SNAREs in neuronal survival. Furthermore, Munc18-1/SNAP-25 DKO neurons show more severe cell loss than the individual mutants (additivity), suggesting that the underlying degenerative pathways are not the same for SNAP-25 and Munc18-1 depletion.
Abnormal cis-Golgi morphology is an early phenotype in Munc18-1 and SNAP-25 KO neurons
To investigate subcellular compartments involved in neuronal survival/death, we studied the Golgi morphology using cis-Golgi markers GM130 and Mannosidase II; the size and number of mitochondria using MitoTracker; the ER compartment using anti-calnexin antibody; lysosomes using anti-LAMP1 antibody; recycling endosomes using anti-transferrin receptor (Trf) antibody; and somata/whole neurons using MAP2 as a dendritic/soma marker. Cortical neuronal cultures were fixed at DIV 2 and DIV 3, as most neuronal loss occurs in this phase (Fig. 1A,B). Munc18-1 KO neurons showed a significant difference in cis-Golgi morphology over time (Fig. 2A,B). At DIV 2, the difference was small but significant; and although the Golgi grew and elongated substantially in control neurons between DIV 2 and 3, this was not the case in the mutant neurons (Fig. 2A,B). The size of the soma was also smaller in the mutant neurons, but this difference was minor relative to the changes in Golgi morphology and remained constant between DIV 2–3 (Fig. 2C). The differences in Golgi size were still significant after correcting for the difference in soma size (by quantifying cis-Golgi extent as a fraction of the soma; Fig. 2D). cis-Golgi structure was further analyzed using automated shape analysis (the ImageJ “round” parameter). This analysis confirmed the condensed Munc18-1 KO cis-Golgi compared with the elongated WT cis-Golgi structure (Fig. 2E). Mannosidase II (ManII), a Golgi enzyme, was used as a cis-Golgi live marker and independent approach to confirm the effects observed using GM130 staining. ManII imaging indeed showed a significant decrease in cis-Golgi area compared with WT (Fig. 2F,G).
Figure 2.
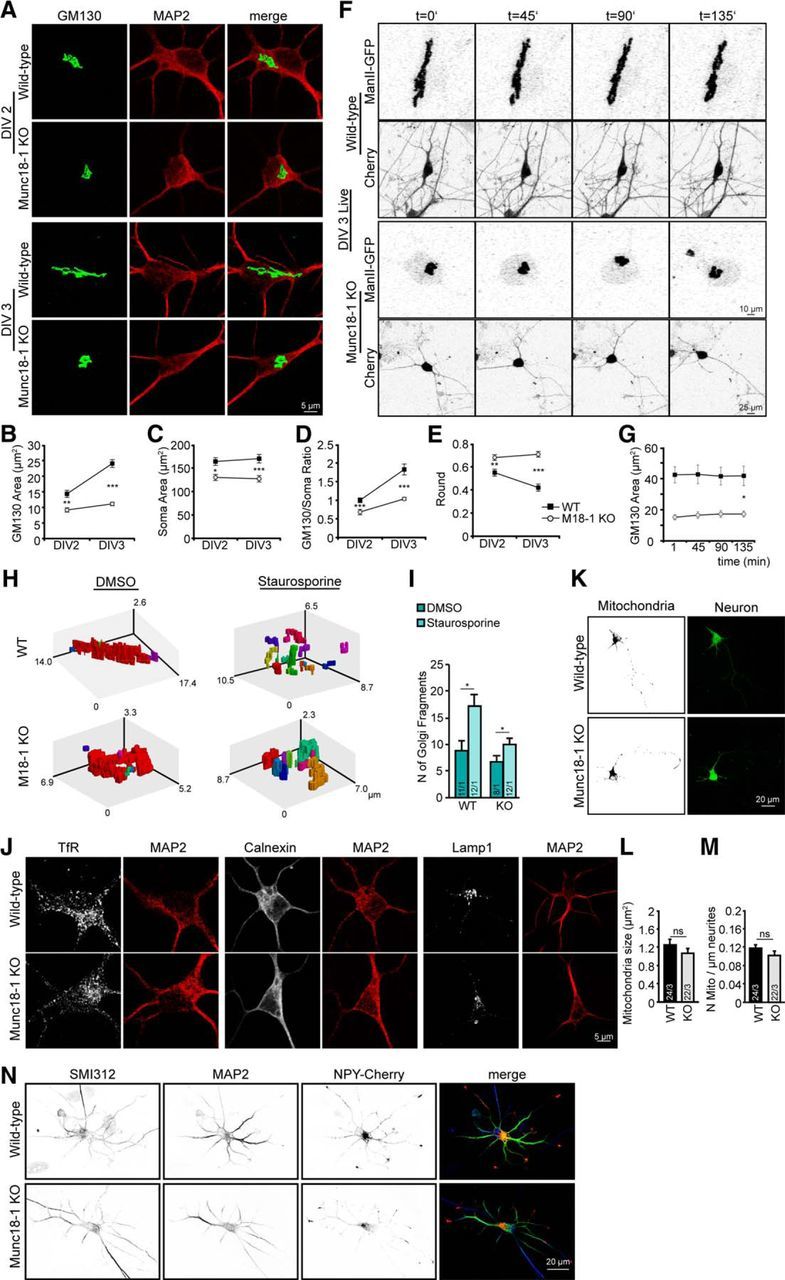
Cell loss in Munc18-1 KO neurons is preceded by abnormal cis-Golgi morphology, but no changes in other organelles. A, Cortical neurons from WT and Munc18-1 (M18–1) KO, fixed at DIV 2 and DIV 3, were stained for a cis-Golgi marker (GM130) and a dendritic marker (MAP2). B, cis-Golgi area is smaller in Munc18-1 KO. cis-Golgi (GM130) area was measured quantitatively using ImageJ software; DIV 2 WT: 14.3 ± 1.13 μm2; Munc18-1 KO: 9.2 ± 0.70 μm2. **p < 0.01 (two-way ANOVA). DIV 3 WT: 24.0 ± 1.26 μm2; Munc18-1 KO: 11.1 ± 0.58 μm2. ***p < 0.001 (two-way ANOVA). C, Munc18-1 KO neurons are smaller than WT neurons. Soma (MAP2) area was analyzed quantitatively using ImageJ software. DIV 2 WT: 163.86 ± 8.15 μm2; Munc18-1 KO: 130.02 ± 6.52 μm2. *p < 0.05 (two-way ANOVA). DIV 3 WT: 127.10 ± 9.03 μm2; Munc18-1 KO: 170.01 ± 6.81 μm2. ***p < 0.001 (two-way ANOVA). D, cis-Golgi area corrected for soma size; DIV 2 WT: 1.0 ± 0.06; Munc18-1 KO: 0.7 ± 0.07; DIV 3 WT: 1.8 ± 0.15; Munc18-1 KO: 1.0 ± 0.04. ***p < 0.001 (two-way ANOVA). E, WT cis-Golgi neurons grow and elongate over time. Shape was measured quantitatively using ImageJ software. DIV 2 WT: 0.6 ± 0.03; Munc18-1 KO: 0.7 ± 0.02. **p < 0.01 (two-way ANOVA). DIV 3 WT: 0.4 ± 0.03; Munc18-1 KO: 0.7 ± 0.02. ***p < 0.001 (two-way ANOVA). F, live cell imaging of cis-Golgi marker ManII-GFP for 2 h 15 min show again a smaller Munc18-1 KO cis-Golgi. G, Quantification over time of ManII-GFP area using ImageJ software. ManII area at 135 min WT: 41.7 ± 6.26 μm2; Munc18-1 KO: 17.23 ± 1.96 μm2. *p < 0.05 using linear mixed-effects model, auto regression correlation for time AR(1), t(9) = −4.16. H, Representation of 3D cis-Golgi WT versus Munc18-1 KO treated with 1 μm staurosporine to induce apoptosis or DMSO (control). Fragments are individually colored. I, Number of cis-Golgi (GM130) fragments were measured using MATLAB software. Number of fragments: WT DMSO, 9; WT staurosporine, 16; Munc18-1 KO DMSO, 6; Munc18-1 KO staurosporine, 10. *p < 0.05 (Student's t test). J, Cortical neurons from WT and Munc18-1 KO, fixed at DIV 3, were stained for different organelles: (TfR); ER (Calnexin); Lysosome (LAMP1). K, Cortical neurons from WT and Munc18-1 KO were incubated with MitoTracker and imaged live. L, No changes in mitochondria area measured quantitatively using ImageJ software. WT: 1.2 ± 0.12 μm2; Munc18-1 KO: 1.1 ± 0.11 μm2; not significant, p > 0.05 (Student's t test). M, Mitochondria number per total neurite length is equal in Munc18-1 KO and WT, measured quantitatively using ImageJ software. WT: 0.1 ± 0.01 μm−1; Munc18-1 KO: 0.1 ± 0.01 μm−1; not significant, p > 0.05 (Student's t test). N, WT and Munc18-1 KO cortical neurons expressing NPY-Cherry were fixed at DIV 3 and stained for SMI312 (axonal marker) and MAP2 (dendritic marker). No changes in NPY-Cherry were observed between mutant and control neurons. Merge image: blue represents SMI312; green represents MAP2; red represents NPY-Cherry. Data are mean ± SEM.
Golgi abnormalities are also a prominent feature of apoptosis (Mourelatos et al., 1990, 1996; Gonatas et al., 1992; Stieber et al., 1996; Huse et al., 2002; Fujita et al., 2006). Therefore, we tested whether the observed abnormalities in cis-Golgi morphology were similar to the abnormalities observed in apoptosis by applying 1 μm of staurosporine for 3 h, which induces apoptosis (Koh et al., 1995). Using MATLAB, a Golgi 3D structure was generated from confocal images and the number of Golgi fragments was counted. Upon apoptosis induction, a fragmented Golgi was observed in both WT and Munc18-1 KO neurons, with a significant increase in the fragment numbers (Fig. 2H,I). Hence, the changes observed in cis-Golgi morphology upon the induction of apoptosis were similar in both genotypes and distinct from the condensed cis-Golgi observed in Munc18-1 KO neurons. These two approaches, using GM130 staining and ManII imaging, indicate that Munc18-1 depletion leads to Golgi abnormalities, distinct from typical apoptotic fragmentation, before cells die.
No significant differences were observed in recycling endosomes, ER, or lysosomes between WT and Munc18-1 KO (Fig. 2J). A tendency toward bigger mitochondria was observed in WT compared with Munc18-1 KO, 1.3 μm2 versus 1.1 μm2, respectively (Fig. 2K,L), but the difference was not significant (p > 0.05, Student's t test) and the suggested effect size (<20%) was minor relative to the (significant) Golgi defects (50% difference; Fig. 2C–F). The distribution of mitochondria per neurite also showed no differences (0.1 and 0.1 μm for WT and Munc18-1 KO, respectively; not significant, p > 0.05, Student's t test; Fig. 2M). In addition, we tested the biogenesis and targeting of secretory vesicles in developing neurons. NPY-Cherry, a marker for secretory vesicles (van de Bospoort et al., 2012; Farina et al., 2015), was expressed at the time of plating, and the distribution of secretory vesicles was analyzed at DIV 3. In both control and Munc18-1 KO neurons, secretory vesicles accumulated at the tips of the outgrowing neuritis (Fig. 2N). This indicates that, during early development, trafficking of secretory vesicles to the ends of outgrowing neurites is not affected by the loss of Munc18-1; therefore, it cannot explain cell death in these neurons. Hence, Munc18-1 KO neurons show a selective defect in cis-Golgi morphology, not in other organelles and distinct from Golgi abnormalities in apoptosis, within 2 d in culture, well before synapse formation.
To study whether the condensed cis-Golgi is a specific feature of Munc18-1 KO cell death, neuronal cultures from other genotypes were also tested: SNAP-25 KO neurons, Munc13–1/2 DKO and WT neurons treated with TeNT. All these genotypes are characterized by severe defects in synaptic transmission, comparable with the Munc18-1 KO phenotype (Schiavo et al., 1992b; Varoqueaux et al., 2002; Washbourne et al., 2002; Delgado-Martínez et al., 2007). WT neurons treated with BoNT/C were not analyzed because cell death occurred too rapidly after plating, before DIV 3 (Fig. 1A,B), to allow Golgi comparisons with the other genotypes. WT neurons treated with TeNT and Munc13–1/2 DKO neurons showed no Golgi abnormalities (Fig. 3A–E). In contrast, SNAP-25 KO neurons showed a significant defect in cis-Golgi shape (Fig. 3E), similar to the Munc18-1 KO neurons (Fig. 2E), and a trend toward a smaller size (not significant; Fig. 3B). SNAP-25 KO neurons and SNAP-25 KO neurons infected with GFP showed a trend toward a smaller cis-Golgi, as observed in Munc18-1 KO neurons: 14.0 μm2, 13.7 μm2 (not significant, p = 0.208 and p = 0.474, respectively, one-way ANOVA followed by the post hoc Bonferroni's test) and a significantly smaller soma size in SNAP-25 KO neurons 125.5 μm2, but not in SNAP-25 KO infected with GFP 132.8 μm2, compared with WT neurons (**p < 0.01, ***p < 0.001, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 3A–C). While overexpression of SNAP-25 in SNAP-25 KO neurons restored the cis-Golgi shape defect observed in SNAP-25 KO neurons, overexpressing of Munc18-1 or -3 did not restore the cis-Golgi shape in SNAP-25 KO neurons (*p < 0.05, ***p < 0.001, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 3A,E). The Munc18-1/SNAP-25 DKO mutant showed a smaller cis-Golgi and also a smaller cis-Golgi/soma ratio compared with WT (Fig. 3A–D), and also compared with the single KOs. Thus, cis-Golgi abnormalities are an early characteristic of Munc18-1 KO and SNAP-25 KO neurons. The additive effect of Munc18-1 and SNAP-25 deficiency suggests that absence of these two presynaptic proteins leads to cell death via pathways that are not fully overlapping.
Figure 3.
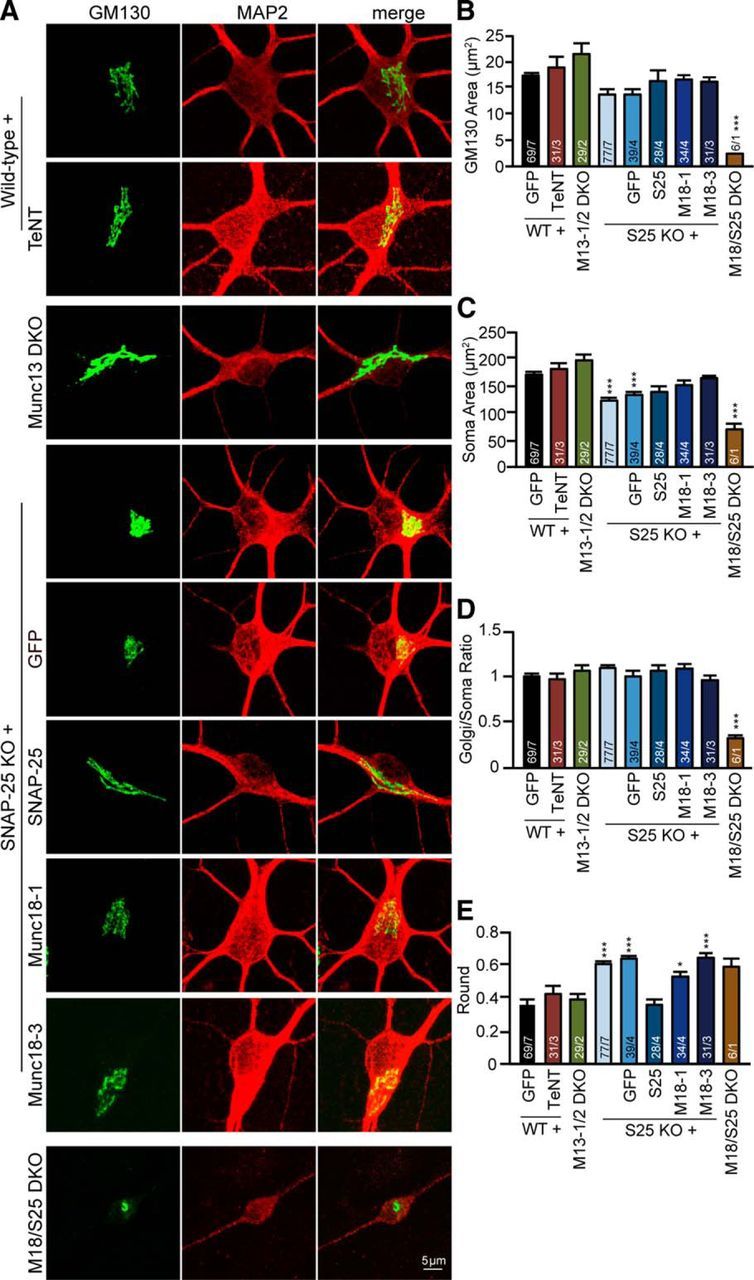
Depletion of SNAP-25, but not synaptobrevins or Munc13–1/2 DKO, leads to changes in cis-Golgi morphology. A, Cortical neuronal cultures from WT, WT treated with TeNT, Munc13–1/2 DKO (M13 DKO), SNAP-25 KO (S25 KO), SNAP-25 KO rescued with eGFP, SNAP-25, Munc18-1 or -3, and Munc18-1/SNAP-25 DKO (M18/S25 DKO) were fixed at DIV 3 and stained with a cis-Golgi marker (GM130) and a dendritic marker (MAP2). B, cis-Golgi (GM130) morphology was analyzed quantitatively using ImageJ software; WT: 17.2 ± 0.75 μm2; WT with TeNT: 19.0 ± 1.93 μm2; Munc13–1/2 DKO: 21.6 ± 1.68 μm2; SNAP-25 KO: 14.0 ± 0.65 μm2; SNAP-25 KO with GFP: 13.7 ± 0.99 μm2; SNAP-25 KO with SNAP-25: 16.2 ± 1.89 μm2; SNAP-25 KO with Munc18-1 (M18-1): 16.5 ± 1.07 μm2; SNAP-25 KO with Munc18-3 (M18-3): 16.1 ± 0.87 μm2; Munc18-1/SNAP-25 DKO: 2.4 ± 0.27 μm2. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). C, Soma (MAP2) area was measure quantitatively using ImageJ software; WT: 170.7 ± 6.16 μm2; WT with TeNT: 183.1 ± 10.94 μm2; Munc13–1/2 DKO: 197.0 ± 10.23 μm2; SNAP-25 KO: 125.5 ± 4.70 μm2; SNAP-25 KO with GFP: 132.8 ± 7.50 μm2; SNAP-25 KO with SNAP25: 139.6 ± 10.97 μm2; SNAP-25 KO with Munc18-1: 149.8 ± 7.50 μm2; SNAP-25 KO with Munc18-3: 162.0 ± 7.74 μm2; Munc18-1/SNAP-25 DKO: 73.4 ± 5.61 μm2. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). D, cis-Golgi area corrected for the soma area; WT: 1.0 ± 0.03; WT with TeNT: 1.0 ± 0.07; Munc13–1/2 DKO: 1.1 ± 0.06; SNAP-25 KO: 1.1 ± 0.04; SNAP-25 KO with GFP: 1.0 ± 0.05; SNAP-25 KO with SNAP-25: 1.1 ± 0.06; SNAP-25 KO with Munc18-1: 1.1 ± 0.06; SNAP-25 KO with Munc18-3: 1.0 ± 0.04; Munc18-1/SNAP-25 DKO: 0.3 ± 0.03. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). E, cis-Golgi shape was measured quantitatively using ImageJ software; WT: 0.4 ± 0.03; WT with TeNT: 0.4 ± 0.04; Munc13–1/2 DKO: 0.4 ± 0.04; SNAP-25 KO: 0.6 ± 0.02; SNAP-25 KO with GFP: 0.6 ± 0.03; SNAP-25 KO with SNAP-25: 0.4 ± 0.04; SNAP-25 KO with Munc18-1: 0.5 ± 0.04; SNAP-25 KO with Munc18-3: 0.6 ± 0.04; Munc18-1/SNAP-25 DKO: 0.6 ± 0.05. *p < 0.05 (one-way ANOVA followed by the post hoc Bonferroni's test). ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). Data are mean ± SEM.
Munc18-3 expression rescues survival and Golgi abnormalities, but not synaptic transmission and syntaxin-1 targeting defects in Munc18-1 KO neurons
Overexpression of a SNAP-25 isoform, SNAP-23, rescues cell death of SNAP-25 KO neurons (Delgado-Martínez et al., 2007). To investigate whether expression of a non-neuronal Munc18 isoform (Munc18-3) rescues survival of Munc18-1 KO neurons, Munc18-3 was overexpressed by lentiviral infection in Munc18-1 KO neurons and fixed at DIV 3, 7, 9, and 14 for immunocytochemistry. At DIV 3, Munc18-1 KO neurons rescued with Munc18-1 or Munc18-3 showed 100% and 90.9% viable cells, respectively, compared with 68.3% of Munc18-1 KO neurons expressing control virus (GFP only; Fig. 4A,B). At DIV 7 and 9, few Munc18-1 KO neurons remained, as shown in Figure 1 (4.7% and 5.8%, surviving cells, respectively), whereas in Munc18-1 KO neurons rescued with Munc18-1, but also with non-neuronal Munc18-3, no reduction in cell count was observed (Fig. 4A,B). Only after 2 weeks in culture, Munc18-1 KO neurons rescued with Munc18-3 started to show cell loss (Fig. 4A,B). The number of synapses and the length of dendrites analyzed at DIV 14 showed no significant differences between neurons rescued with Munc18-1 or -3 (Fig. 4C–E). These data show that a non-neuronal Munc18 isoform rescues the viability and neuronal morphology of Munc18-1 KO neurons until DIV 14.
Figure 4.
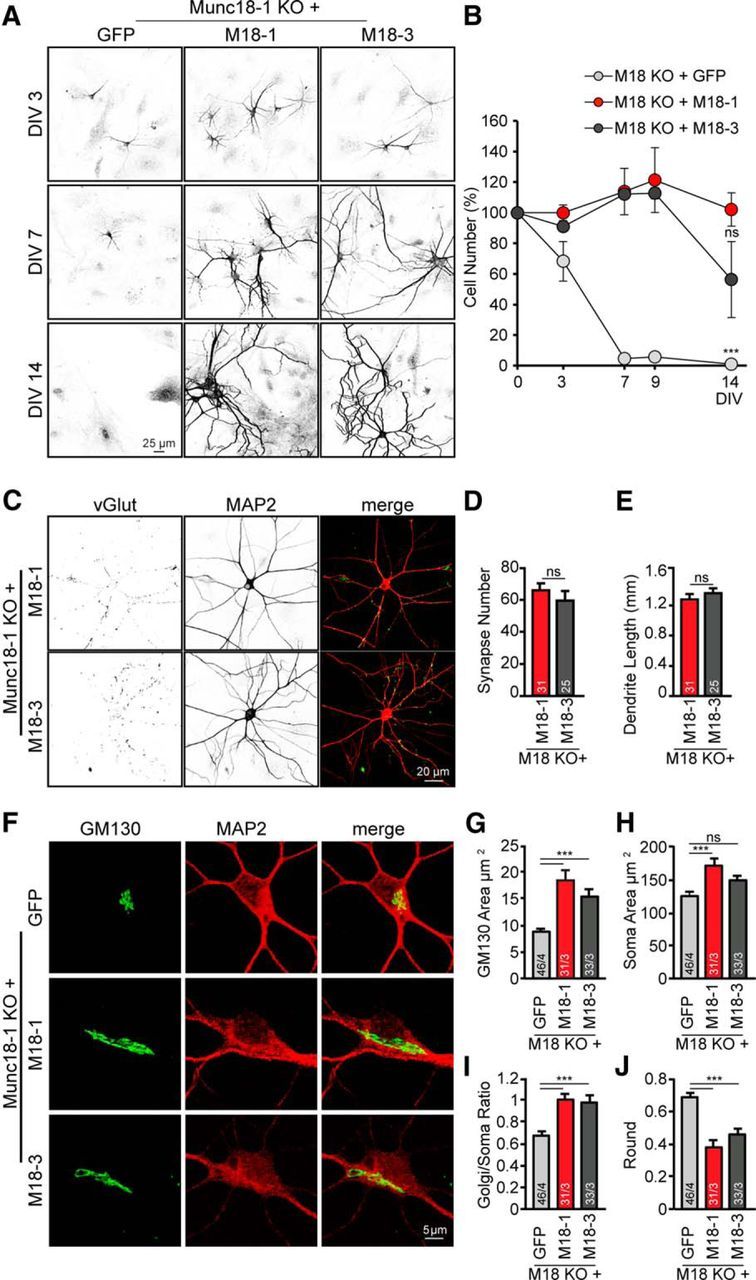
Munc18-3 expression rescues cell death, neuronal and cis-Golgi morphology in Munc18-1 KO neurons. A, Cortical neuronal cultures from Munc18-1 KO were infected by lentiviral particles with eGFP (control), Munc18-1 (M18–1), or Munc18-3 (M18–3), fixed at different time points (DIV 3, 7, 9, and 14), and stained with a dendritic marker (MAP2). B, Quantification of number of neurons per 97 fields of view; percentage of Munc18-1 KO with Munc18-1 cells: DIV 3, 100 ± 5.16%; DIV 7, 113.8 ± 15.22%; DIV 9, 121.3 ± 21.18%; DIV 14, 102.07 ± 10.97%; Munc18-1 KO with Munc18-3: DIV 3, 90.95 ± 1.21%; DIV 7, 112.17 ± 1.82%; DIV 9, 112.8 ± 3.64%; DIV 14, 56.4 ± 24.86%; Munc18-1 KO with GFP: DIV 3, 72.2% ± 9.93; DIV 7, 4.7 ± 1.52%; DIV 9, 5.8 ± 1.94%; DIV 14, 0.9 ± 0.58%; not significant, p > 0.05. ***p < 0.001 (two-way ANOVA followed by the post hoc Bonferroni's test). C, Cortical neuronal cultures from Munc18-1 KO were infected by lentiviral particles with Munc18-1 or -3, fixed at DIV 14, and stained for vGlut and MAP2. D, Total synapse number was comparable between Munc18-1 and Munc18-3 rescued neurons; synapses Munc18-1: 66.0 ± 4.4; Munc18-3: 59.6 ± 6.1; not significant, p > 0.05 (Student's t test). E, No difference was found in total dendrite length between Munc18-1 and Munc18-3 rescued neurons; dendrite length Munc18-1: 1279.6 ± 73.3 μm; Munc18-3: 1364.4 ± 64.7 μm; not significant, p > 0.05 (Student's t test). F, Cortical neuronal cultures from Munc18-1 KO were infected by lentiviral particles with eGFP (control), Munc18-1 or -3, fixed at DIV 3, and stained with a cis-Golgi marker (GM130) and a dendritic marker (MAP2). G, No changes in cis-Golgi (GM130) area in Munc18-1 and -3 rescued neurons; Munc18-1 KO with GFP: 8.9 ± 0.59 μm2; Munc18-1 KO with Munc18-1: 18.5 ± 1.74 μm2; Munc18-1 KO with Munc18-3: 15.5 ± 1.13 μm2; not significant, p > 0.05. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). H, Soma (MAP2) area was analyzed quantitatively using ImageJ software; Munc18-1 KO with GFP: 125.3 ± 5.98 μm2; Munc18-1 KO with Munc18-1: 171.7 ± 11.47 μm2; Munc18-1 KO with Munc18-3: 148.8 ± 6.62 μm2; not significant, p > 0.05. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). I, cis-Golgi area corrected for soma area showed an increase in the area for both Munc18-1 and -3 groups; Munc18-1 KO with GFP: 0.7 ± 0.04; Munc18-1 KO with Munc18-1: 1.0 ± 0.05; Munc18-1 KO with Munc18-3: 1.0 ± 0.06; not significant, p > 0.05. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). J, cis-Golgi shape showed a more elongated morphology in the Munc18-1 and -3 groups compared with control. Munc18-1 KO with GFP: 0.7 ± 0.02; Munc18-1 KO with Munc18-1: 0.4 ± 0.04; Munc18-1 KO with Munc18-3: 0.5 ± 0.04; not significant, p > 0.05. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). Data are mean ± SEM.
Staining for GM130 revealed that Munc18-3 expression in Munc18-1 KO neurons also restored cis-Golgi abnormalities (Fig. 4F–J). As before, cis-Golgi morphology was quantified as a total Golgi area (Fig. 4G), as fraction of the soma (Fig. 4G–I) and using the “round” parameter (Fig. 4J). These analyses showed no differences in cis-Golgi morphology between Munc18-1 and -3 expression in Munc18-1 KO neurons and significant differences to Munc18-1 KO neurons infected with GFP only (Fig. 4F–J).
Golgi integrity and dynamics are regulated by interactions between the Golgi and the cytoskeleton and actin depolymerization leads to a condensed Golgi (Lázaro-Diéguez et al., 2006) similar to the Golgi abnormalities observed in Munc18-1 KO neurons. Therefore, we analyzed the main cytoskeletal components in neurons, tubulin and actin, in Munc18-1 KO and control neurons. Cortical cultures were fixed at DIV 3 and immunohistochemistry was performed for tubulin, phalloidin (a high affinity F-actin probe), neurofilament, and MAP2. No difference in phalloidin intensity was observed between mutant and control neurons (1811.6 ± 106.88 A.U. and 1798.2 ± 153.67 A.U., WT and Munc18-1 KO, respectively; not significant, p > 0.05, Student's t test; Fig. 5A–C). Furthermore, cortical cultures were infected with ManII-GFP a cis-Golgi marker and calponin homology domain of utrophin (UtrCH-Cherry), a probe to visualize F-actin and imaged live. No difference was observed between WT and Munc18-1 KO neurons in UtrCH intensity at the cis-Golgi (2575.4 ± 117.11 A.U. and 2457.0 ± 160.61 A.U., WT and Munc18-1 KO, respectively; not significant, p > 0.05, Student's t test; Fig. 5D–F) at the area around the Golgi, quantified in a ring around the ManII-GFP area ((Intensity ROI 2 × Mean ROI 2) − (Intensity ROI 1 × Mean ROI 1)/(Area ROI 2 − Area ROI 1) 1442.3 ± 127.54 A.U. and 1606.0 ± 177.04 A.U., WT and Munc18-1 KO, respectively; not significant, p > 0.05, Student's t test; Figure 5D–F). Colocalization between UtrCH-Cherry and ManII-GFP also showed no differences (0.7 ± 0.03 and 0.6 ± 0.04 Pearson's correlation, WT and Munc18-1 KO, respectively; not significant, p > 0.05, Student's t test; Fig. 5D–G). Hence, both the tubulin and actin cytoskeleton had developed normally in Munc18-1 KO neurons, also specifically around the Golgi; therefore, we conclude that the early Golgi abnormalities cannot be explained by cytoskeletal defects.
Figure 5.
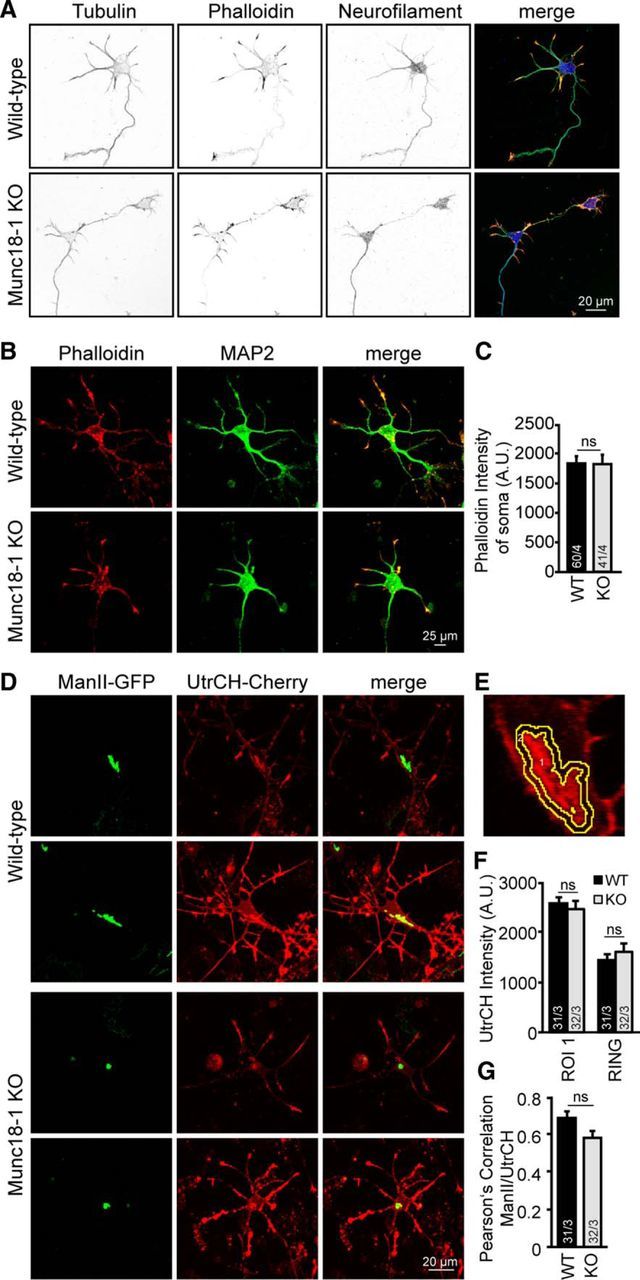
Munc18-1 KO neurons show no cytoskeleton organization defects. A, Cortical neuronal cultures of WT and Munc18-1 KO were fixed at DIV 3 and stained with neurofilament and tubulin antibodies and phalloidin (a F-actin probe). Merge image: Green represents tubulin; red represents phalloidin; blue represents neurofilament. B, Cortical neuronal cultures of WT and Munc18-1 KO were fixed at DIV 3 and stained with MAP2 and phalloidin. C, Quantification of somatic phalloidin intensity was performed using ImageJ software. No differences were observed between mutant and control neurons (WT: 1811.6 ± 106.88 A.U.; Munc18-1 KO, 1798.2 ± 153.67 A.U.; not significant, p > 0.05, Student's t test). D, WT and Munc18-1 KO cortical neurons expressing UtrCH-Cherry (a live probe to visualize F-actin), and ManII-GFP (a live cis-Golgi marker) were image at DIV 3. E, Example of ROI placement. F, Quantification of UtrCH intensity was performed at the cis-Golgi and the area around labeled as ring ((Intensity ROI 2 × Mean ROI 2) − (Intensity ROI 1 × Mean ROI 1)/(Area ROI 2 − Area ROI 1) using ImageJ software; ROI 1 WT: 2575.4 ± 117.11 A.U.; ROI 1 Munc18-1 KO: 2457.0 ± 160.61 A.U.; not significant, p > 0.05, Student's t test). Ring WT: 1442.3 ± 127.54 A.U.; Ring Munc18-1 KO: 1606.0 ± 177.04 A.U.; not significant, p > 0.05, (Student's t test). G, No differences in actin and Golgi colocalization in Munc18-1 KO neurons; WT: 0.69 ± 0.03; Munc18-1 KO: 0.58 ± 0.04; measured quantitatively using ImageJ software; not significant, p > 0.05 (Student's t test). Data are mean ± SEM.
Munc18-3 rescued synapse formation in Munc18-1 KO neurons (Fig. 4D); we therefore studied synaptic transmission in these cells using whole-cell voltage-clamp experiments (Wierda et al., 2007). The amplitude and decay time of spontaneous release events (mEPSC) were comparable between neurons rescued with either Munc18-1, -2, or -3. However, Munc18-2- and -3-expressing neurons showed virtually no spontaneous release: the frequency was ∼4% and 2% of the frequency observed in Munc18-1-expressing neurons (Fig. 6A,B). In addition, action potential driven evoked EPSC amplitudes were reduced in Munc18-2-expressing neurons and virtually absent in Munc18-3-expressing neurons, to 22% and 8% of Munc18-1 amplitude, respectively, whereas evoked EPSC decay time was normal (Fig. 6C,D). The decrease in miniature frequency and evoked EPSC amplitude can be caused by either a decrease in the RRP vesicles or a decrease in vesicular release probability (Pves). We probed the RRP by application of hyperosmotic sucrose solution (500 mm) and found a large reduction in RRP size in both Munc18-2- and -3-expressing neurons, to 3% and 1.5% of Munc18-1 RRP size, respectively (Fig. 6E,F). Evoked EPSC amplitude and RRP size were proportionally reduced in Munc18-2- and -3-expressing neurons. Therefore, the initial release probability (evoked EPSC charge/initial sucrose charge) (Rosenmund and Stevens, 1996) of the few vesicles that were released upon stimulation appeared to be similar between Munc18-1- and Munc18-2- and -3-expressing neurons (Fig. 6E,F). Hence, whereas neuronal viability is rescued by expressing Munc18-2 and -3, synaptic transmission is not.
Figure 6.
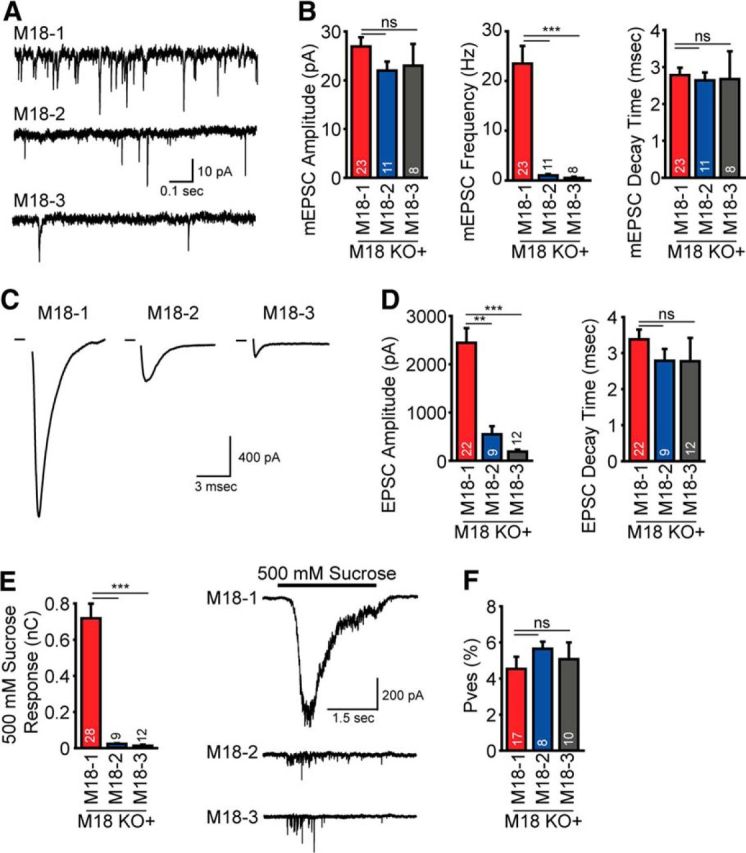
Munc18-2/-3 expression does not rescue synaptic transmission in Munc18-1 KO neurons. A, Example traces of spontaneous release in neurons rescued with Munc18-1 (M18–1), Munc18-2 (M18–2), or Munc18-3 (M18–3). B, Spontaneous frequency is impaired in neurons expressing Munc18-2 or -3, whereas spontaneous amplitude and decay time are normal (amplitude: Munc18-1, 26.9 ± 1.9 pA; Munc18-2, 22.0 ± 1.8 pA; Munc18-3, 23.0 ± 4.5 pA; p > 0.05, frequency: Munc18-1, 23.5 ± 3.5 Hz; Munc18-2, 1.0 ± 0.2 Hz; Munc18-3, 0.5 ± 0.3 Hz; p < 0.0001, decay time: Munc18-1, 2.8 ± 0.2 ms; Munc18-2, 2.6 ± 0.2 ms; Munc18-3, 2.7 ± 0.7 ms; p > 0.05). C, Examples of evoked EPSCs in neurons expressing Munc18-1, -2, and -3. D, EPSC amplitude is significantly reduced in neurons expressing non-neuronal isoforms of Munc18, whereas EPSC decay time is normal (EPSC amplitude: Munc18-1, 2441.5 ± 304.8 pA; Munc18-2, 548.8 ± 167.9 pA; Munc18-3, 188.3 ± 39.7 pA; p > 0.05, EPSC decay time: Munc18-1, 3.4 ± 0.3 ms; Munc18-2, 2.8 ± 0.3 ms; Munc18-3, 2.8 ± 0.6 ms; p > 0.05). E, RRP size, probed by hyperosmotic sucrose application, is severely decreased in neurons expressing Munc18-2 or -3 (Munc18-1, 0.72 ± 0.08 nC; Munc18-2, 0.02 ± 0.003 nC; Munc18-3, 0.01 ± 0.005 nC; p < 0.01). F, Release probability of fusion competent vesicles is normal in neurons expressing Munc18-2 and -3 (Munc18-1, 4.5 ± 0.7%; Munc18-2, 5.6 ± 0.4%; Munc18-3, 5.1 ± 0.9%; p < 0.05). Data are mean ± SEM.
It has been reported before that targeting of syntaxin-1 to the plasma membrane is impaired in the absence of Munc18-1 (Rowe et al., 2001; Gulyás-Kovács et al., 2007; Arunachalam et al., 2008; de Wit et al., 2009). As such defects might contribute to cis-Golgi abnormalities observed in Munc18-1 KO neurons, costaining of syntaxin-1 and GM130 was performed. At DIV 3, Munc18-1 KO neurons showed a significant increase in colocalization of syntaxin-1 and GM130 compared with WT neurons (0.08 and 0.45 Pearson's correlation, WT and Munc18-1 KO, respectively; p < 0.001, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7A,B). SNAP-25 KO neurons, although having Golgi abnormalities, showed no significant differences in syntaxin-1 colocalization with cis-Golgi compared with WT neurons (0.14 Pearson's correlation; not significant, p > 0.05, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7A,B). At DIV 14, WT neurons and Munc18-1 KO neurons rescued with Munc18-1 or -2 showed a negative Pearson's correlation between syntaxin-1 and GM130 (−0.358, −0.359, and −0.255 Pearson's correlation, WT, Munc18-1 KO rescued with Munc18-1 and -2, respectively; not significant, p > 0.05, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7C,D). Despite the fact that cis-Golgi morphology is normal in Munc18-1 KO neurons expressing Munc18-3, analysis of colocalization between syntaxin-1 and GM130 showed a positive Pearson's correlation of 0.17, significantly different from WT and Munc18-1 KO expressing Munc18-1 or -2 (p < 0.001, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7C,D). The synaptic syntaxin-1 levels were also analyzed by costaining Munc18-1 KO neurons expressing Munc18-1, -2, or -3 at DIV 14 with syntaxin-1 and synaptobrevin-2/VAMP2. No differences in synaptobrevin-2/VAMP2 intensity were observed (163.1 ± 7.0 A.U., 150.3 ± 5.8 A.U., and 169.0 ± 6.1 A.U. Munc18-1 KO rescued with Munc18-1, -2, and -3, respectively; not significant, p > 0.05, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7E,F). A significant decrease in the synaptic syntaxin-1 staining was observed in Munc18-1 KO neurons expressing Munc18-2 or -3 compared with Munc18-1 (ratio syntaxin-1/synaptobrevin-2, 1.12 ± 0.04, 0.91 ± 0.07, and 0.87 ± 0.05 Munc18-1 KO expressing Munc18-1, -2, and -3, respectively (p < 0.05, one-way ANOVA followed by the post hoc Bonferroni's test; Fig. 7G). Together, these data show that, whereas viability, neuronal morphology, and Golgi abnormalities are rescued by expressing Munc18-3 and Munc18-2, syntaxin-1 targeting is not rescued by Munc18-3 while Munc18-2 produces an intermediate phenotype, suggesting that syntaxin-1 targeting defects are not causal to cell death observed upon Munc18-1 loss.
Figure 7.

Syntaxin-1 targeting is hampered in Munc18-1 KO neurons and in Munc18-1 KO neurons expressing Munc18-3. A, Cortical neuronal cultures fixed at DIV 3 were stained for syntaxin-1, a cis-Golgi marker (GM130), and a dendritic marker (MAP2). B, Munc18-1 (M18-1) KO show a 0.45 ± 0.03 Pearson's correlation coefficient, between GM130 and syntaxin-1 (WT: 0.08 ± 0.02; SNAP-25 (S25) KO: 0.14 ± 0.02), analyzed quantitatively using ImageJ software. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). C, Cortical neuronal cultures from WT neurons and Munc18-1 KO were rescued with Munc18-1, -2, and -3, fixed at DIV 14 and stained for syntaxin-1, a cis-Golgi marker (GM130) and a dendritic marker (MAP2). D, At DIV 14, Munc18-3 did not rescue syntaxin-1 localization (Pearson's correlation coefficient, WT: −0.36 ± 0.01; Munc18-1 KO with Munc18-1 (M18-1): −0.36 ± 0.02; Munc18-1 KO with Munc18-2 (M18-2): −0.26 ± 0.03; Munc18-1 KO with Munc18-3 (M18-3): 0.17 ± 0.03), measured quantitatively using ImageJ software. ***p < 0.001 (one-way ANOVA followed by the post hoc Bonferroni's test). E, Syntaxin-1 and synaptobrevin-2/VAMP2 staining in Munc18-1, -2, and -3 expressing neurons. F, No difference was found in synaptobrevin-2/VAMP2 expression levels (Munc18-1: 163.1 ± 7.0 A.U.; Munc18-2: 150.3 ± 5.8 A.U.; Munc18-3: 169.0 ± 6.1 A.U.). not significant, p > 0.05, (one-way ANOVA followed by the post hoc Bonferroni's test). G, Syntaxin-1 levels were not completely restored in Munc18-2 or -3 compared with Munc18-1-expressing neurons (Syntaxin/VAMP: Munc18-1, 1.12 ± 0.04; Munc18-2, 0.91 ± 0.07; Munc18-3, 0.87 ± 0.05). *p < 0.05 (one-way ANOVA followed by the post hoc Bonferroni's test). Data are mean ± SEM.
Munc18-1 KO DRG neurons survive in culture without compensatory Munc18-2 or -3 expression
In contrast to Munc18-1 KO neurons in the CNS and most notably in the spinal cord, the sensory neurons of the DRG of Munc18-1 KO mice do not show signs of degeneration until birth (Verhage et al., 2000; Heeroma et al., 2003). To investigate whether Munc18-1 KO DRG neurons are also distinct from other neurons with respect to the cellular abnormalities observed in this study, DRG neurons were isolated from E14 animals, cultured as whole explants, fixed at different time points (DIV 1, 6, 13, and 21) and stained using anti-neurofilament antibody. We found that Munc18-1 KO DRG neurons survived in culture as long as WT DRG neurons, until DIV 21 (Fig. 8A). For comparison, we also analyzed cortical neurons obtained at the same age as the DRGs (E14 instead of E18 as before; Fig. 1). In agreement with the data obtained from E18 cultures, the E14 Munc18-1 KO CNS neurons also died, albeit slightly slower: at DIV 5, ∼50% of Munc18-1 KO neurons were still viable, followed by rapid deterioration leaving <8% of viable cells at DIV 7 (Fig. 8B,C). Hence, Munc18-1 KO CNS neurons cultured at an earlier developmental stage (E14) also deteriorate within a week, but in cultured DRG neurons Munc18-1 is not crucial for survival.
Figure 8.

Munc18-1 KO DRG neurons survive in culture without compensatory expression of Munc18-2 or -3. A, DRG cultures form E14 animals of WT and Munc18-1 (M18–1) KO were fixed at different time points (DIV 1, 6, 13, and 21) and stained with neurofilament antibody; no degeneration was observed in Munc18-1 KO DRG neurons. B, Cortical neuronal cultures from E14 animals, WT and Munc18-1 KO, were fixed at different time points and stained for a dendritic marker (MAP2), at DIV 7. Munc18-1 KO neurons were few and underdeveloped. C, Quantification of number of neurons per 97 fields of view showed a decrease in percentage of surviving Munc18-1 KO cells, but not WT cells. D, mRNA levels of Munc18-1, -2, and -3 were quantified in DRG neurons from E14 animals, no changes were observed; Munc18-1 WT: 1.0 ± 0.19 A.U.; KO: 0.0 ± 0.00 A.U.; Munc18-2 (M18-2) WT: 1.0 ± 0.12 A.U.; KO: 1.0 ± 0.06 A.U.; Munc18-3 (M18-3) WT: 1.0 ± 0.07 A.U.; KO: 0.84 ± 0.13 A.U.; not significant, p > 0.05 (Mann–Whitney U test). *p < 0.05 (Mann–Whitney U test). Data are mean ± SEM. E, mRNA levels of Munc18-1, -2, and -3 were quantified in the cerebral cortex from E18 animals; no changes were observed; Munc18-1 WT: 1.0 ± 0.00 A.U.; KO: 0.0 ± 0.00 A.U.; Munc18-2 WT: 1.0 ± 0.00 A.U.; KO 1.1 ± 0.04 A.U.; Munc18-3 WT: 1.0 ± 0.00 A.U.; KO: 1.05 ± 0.21 A.U.; not significant, p > 0.05 (Mann–Whitney U test). *p < 0.05 (Mann–Whitney U test). Data are mean ± SEM.
Survival in DRG neurons might be explained by (compensatory) expression of other Munc18 isoforms that does not occur in other neurons. Therefore, mRNA levels of Munc18-1, -2, and -3 were quantified, relative to a mEEF1A control (Mouse Eukaryotic Elongation Factor 1A), in cerebral cortex and DRG neurons of WT and Munc18-1 KO animals. No differences in the relative levels of the three Munc18 isoforms (normalized to mEEF1A) were observed between cerebral cortex and DRG neurons (Fig. 8D,E). Thus, the distinctive viability of Munc18-1 KO DRG neurons cannot be explained by expression of Munc18 isoforms and/or by compensatory changes in their expression profile in the absence of Munc18-1.
Munc18-1 KO DRG neurons have a similar syntaxin-1 targeting phenotype as CNS neurons
Munc18-1 KO DRG neurons were further analyzed for similar cellular changes as in CNS neurons using immunohistochemistry for a cis-Golgi marker, syntaxin-1 and neurofilament. The cis-Golgi size was significantly smaller in Munc18-1 KO DRG neurons (Fig. 9A,B), as well as the soma size (Fig. 9C), similar to Munc18-1 KO CNS neurons (Fig. 2A–E). When normalized to soma area, the decrease in Munc18-1 KO DRG Golgi was still significant (Fig. 9D). However, the Golgi shape (roundness) was not different (Fig. 9E). Syntaxin-1 mislocalization was apparent in Munc18-1 KO DRG neurons, as a positive Pearson's correlation was found between syntaxin-1 and GM130 in Munc18-1 KO DRG neurons, significantly different from WT DRG neurons (−0.12 and 0.20, WT and Munc18-1 KO, respectively (p < 0.001, Mann–Whitney U test; Fig. 9F). Thus, syntaxin-1 targeting defects and reduction in cis-Golgi size, but not shape, were also evident in DRG neurons, although these neurons do not die until DIV 21.
Figure 9.
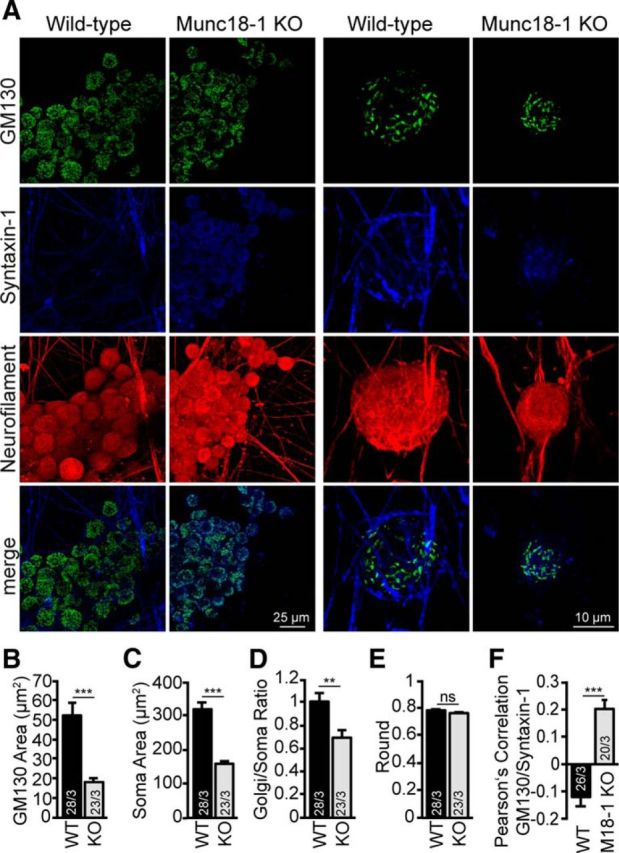
Munc18-1 KO DRG neurons are underdeveloped and show a smaller cis-Golgi and syntaxin-1 target defects. A, DRG cultures from E14 animals, WT and Munc18-1 (M18–1) KO, were fixed at DIV 21 and stained for syntaxin-1, cis-Golgi (GM130), and neurofilament. B, Munc18-1 KO DRG neurons show a smaller cis-Golgi area WT: 52.0 ± 6.20 μm2; Munc18-1 KO: 18.2 ± 2.35 μm2. ***p < 0.001 (Mann–Whitney U test). C, Munc18-1 KO DRG neurons show a smaller soma area WT: 318.9 ± 20.24 μm2; Munc18-1 KO: 159.1 ± 9.26 μm2. ***p < 0.001 (Mann–Whitney U test). D, cis-Golgi area corrected for soma area shows that Munc18-1 KO cells have a reduced cis-Golgi, WT: 1.0 ± 0.08; Munc18-1 KO: 0.7 ± 0.07. **p < 0.01 (Mann–Whitney U test). E, cis-Golgi shape shows no difference in cis-Golgi morphology; WT: 0.8 ± 0.02; Munc18-1 KO: 0.8 ± 0.02; not significant, p > 0.05 (Mann–Whitney U test). F, Munc18-1 KO DRG neurons show a Pearson's correlation coefficient between syntaxin-1 and GM130 of 0.20 ± 0.03 compared with Pearson's correlation coefficient in WT DRG neurons, −0.12 ± 0.03. ***p < 0.001 (Mann–Whitney U test). Data are mean ± SEM.
Discussion
In this study, we showed that, under the same conditions, t-SNAREs and Munc18-1 are crucial for neuronal survival, but v-SNAREs are not. A condensed cis-Golgi was observed as an early feature of neuronal cell death. Defects in synaptic transmission and, in the case of cell death in Munc18-1 KO, syntaxin-1 targeting defects were excluded as causal to cell death.
Similar roles for t-SNAREs and Munc18-1 but not v-SNAREs in neuronal viability
We observed massive cell loss in the absence of presynaptic proteins Munc18-1, and t-SNAREs (SNAP-25 or syntaxins) in cultured neurons (Fig. 1A,B), but not upon loss of all major neuronal v-SNAREs (treating TI-VAMP KO neurons with tetanus toxin; Fig. 1D,E). These data are consistent with previous studies showing that loss of t-SNAREs and Munc18-1 leads to neuronal cell death (Verhage et al., 2000; Washbourne et al., 2002; Heeroma et al., 2004; Berliocchi et al., 2005; Delgado-Martínez et al., 2007; Peng et al., 2013; Kofuji et al., 2014; Vardar et al., 2016) and that synaptobrevin-2/VAMP2 KO neurons and WT neurons treated with TeNT do not degenerate (Schiavo et al., 1992a, b; Schoch et al., 2001; Peng et al., 2013). Hence, t-SNAREs and the SM-protein Munc18-1, but not v-SNAREs, have a crucial function in neuronal viability. Although t-SNAREs and Munc18-1 appear to have similar roles in neuronal viability, their impact is not identical. Under the same conditions created in this study, cell loss proceeded at a different pace in the absence of the different proteins. Syntaxin proteolysis led to the fastest cell loss, within 3 DIV, as shown previously (Berliocchi et al., 2005; Peng et al., 2013; Vardar et al., 2016), whereas in SNAP-25 KO cultures, a fraction of neurons survived beyond the first week, as shown previously (Washbourne et al., 2002; Delgado-Martínez et al., 2007) (Fig. 1A,B). We have now shown that Munc18-1/SNAP-25 DKO leads to cell death earlier than the two single KOs (Fig. 1A,B). This indicates that the survival pathway(s) supported by t-SNAREs and Munc18-1 depends on these proteins to a different extent during different phases of neuronal development. It seems plausible that functionally related proteins operate in the same pathway(s) until a given developmental phase and that the timing of expression of such proteins determines the exact time point where survival becomes dependent on t-SNAREs and Munc18-1.
Loss of synaptic transmission is not causally related to cell loss
In all conditions studied here, in vitro cell loss in the absence of t-SNAREs or Munc18-1 occurred well before synapse formation. This confirms previous findings on Munc18-1 KO (Heeroma et al., 2004), SNAP-25 KO (Delgado-Martínez et al., 2007), and syntaxin KO (Vardar et al., 2016) neurons and indicates that the role of these proteins in neuronal survival is unrelated to their synaptic function. Furthermore, in contrast to cell loss observed in neurons exposed to BoNT/C, neurons exposed to BoNT/A, which selectively cleaves SNAP-25 (Blasi et al., 1993b), show no signs of cell death, whereas synaptic transmission is blocked and mEPSCs are abolished (Peng et al., 2013). The current study provides a different kind of evidence for this conclusion: Munc18-2 and -3 expression in Munc18-1 KO neurons did not rescue synaptic transmission defects (Fig. 6), as expected for Munc18-3 because it does not bind syntaxin-1 in its closed conformation (Pevsner et al., 1994; Tellam et al., 1995; Tellam et al., 1997; Hu et al., 2007; Hu et al., 2011) but does rescue viability. Hence, loss of synaptic transmission is not causally related to the loss of neuronal viability. In addition to their established role in synaptic transmission, t-SNAREs and Munc18-1 have a separate, earlier role in neuronal viability.
Syntaxin-1 targeting defects are not causal to cell loss in Munc18-1 KO neurons
Neurons lacking Munc18-1 showed syntaxin-1 mislocalization at the Golgi (Figs. 7; 9), suggesting that, without Munc18-1, syntaxin-1 export from the Golgi apparatus to the PM and synapses is impaired. This is consistent with previous studies in non-neuronal cells where syntaxin-1 overexpression alone produces Golgi abnormalities (Rowe et al., 1999; Khvotchev and Südhof, 2004), but if transfected together with Munc18-1, localizes to the PM and no Golgi abnormalities were observed (Rowe et al., 1999, 2001; Khvotchev and Südhof, 2004; Arunachalam et al., 2008). A similar mislocalization of syntaxin was observed in the current study upon expression of Munc18-3 in Munc18-1 KO neurons. However, in these neurons, the cis-Golgi abnormalities and cell death were rescued (Fig. 7C,D). Similarly, in Munc18-1 KO DRG neurons, syntaxin-1 targeting was also impaired, but no cell death was observed (Figs. 8; 9). Together, these data indicate that syntaxin-1 targeting defects are not causal to the Golgi abnormalities or cell death in Munc18-1 KO neurons.
Trophic factor secretion defects cannot explain cell loss upon t-SNARE/Munc18 depletion
It has been proposed that Munc18-1 and SNAP-25, in addition to synaptic transmission, are involved in trophic factor release, from neurons or glia, and this may provide an explanation for the cell loss in the absence of t-SNAREs or Munc18-1 (Bronk et al., 2007; Delgado-Martínez et al., 2007; Kofuji et al., 2014). Consistent with this idea, SNAP-25 neurons survived at least 3 weeks when cultured at very high density (Bronk et al., 2007) and syntaxin-1B KO neurons survived on WT, but not KO glial feeders (Kofuji et al., 2014). In the current study, we used selective BoNT/C (light chain) expression in neurons, not glia, whereas previous studies used the holotoxin or syntaxin-1 inactivation, which do not discriminate between neurons and glia. With selective BoNT/C expression in neurons only, cell loss is still massive (Fig. 1) (Vardar et al., 2016). Furthermore, upon sparse Munc18-1 inactivation only in Purkinje cells of mouse cerebellum, loss of these neurons was still massive, despite the presence of surrounding neurons and glia providing trophic support (Heeroma et al., 2004). Together, these considerations suggest that inhibition of trophic factor release is insufficient to explain the cell loss upon depletion of at least syntaxin and Munc18-1. Instead, early Golgi abnormalities and the fact that viability can be rescued by expression of non-synaptic isoforms (see below) suggest that cell-intrinsic trafficking defects are the main cause of neurodegeneration upon t-SNARE or Munc18-1 depletion.
Condensed cis-Golgi morphology is an early hallmark of neurodegeneration
Although defects in synaptic transmission and syntaxin-1 targeting can be excluded as causal for degeneration, and lack of trophic factor secretion appears to be an insufficient explanation, the identity of the degenerative mechanism(s) remains unclear. Previous studies have reported neurite fragmentation followed by cytochrome C release from mitochondria and apoptosis after BoNT/C holotoxin application (Berliocchi et al., 2005; Peng et al., 2013). The current study shows that depletion of Munc18-1, SNAP-25, or both produced cis-Golgi abnormalities at an early development stage (Figs. 2; 3), whereas no differences in ER, lysosomes, recycling endosomes, or mitochondria were observed (Fig. 2K–M). Conversely, in neurons that do not degenerate (e.g., Munc13–1/2 and synaptobrevin/VAMP loss), no Golgi defects were observed. Although the cis-Golgi abnormalities were not identical upon depletion of Munc18-1, SNAP-25, or both, together they are a selective, early feature of neurodegeneration in all these models. This feature was distinct from the fragmented cis-Golgi observed in apoptosis (Fig. 2) (Philpott et al., 1996; Sesso et al., 1999; Lowe et al., 2004), suggesting that this phenomenon is upstream of the apoptosis observed in BoNT/C treated neurons (Berliocchi et al., 2005; Peng et al., 2013). Interestingly, Golgi defects are also an early preclinical feature that precedes neurodegenerative loss of cell bodies and axons in Alzheimer's disease (Stieber et al., 1996; Joshi et al., 2014), amyotrophic lateral sclerosis (Mourelatos et al., 1990; Gonatas et al., 1992), and Parkinson's disease (Fujita et al., 2006). Thus, our data suggest Golgi defects as an early, shared hallmark preceding cell death upon depletion of t-SNAREs or Munc18-1.
Rescue of viability by nonsynaptic isoforms of t-SNAREs/Munc18 suggests generic membrane trafficking defects
Expression of Munc18-2 and -3 in Munc18-1 KO neurons rescued neuronal viability (Figs. 4; 7). Similarly, expression of SNAP-23 rescues viability of SNAP-25 KO neurons (Delgado-Martínez et al., 2007) and syntaxin-2, -3, and -4 rescue viability of syntaxin-1-depleted neurons (Peng et al., 2013). Hence, rescue of viability by noncognate isoforms is a shared characteristic among the presynaptic proteins that cause neuronal loss upon their depletion. It seems plausible that these isoforms compensate in generic membrane trafficking pathways in neurons, required for neuronal maintenance and are more tolerant to differences among isoforms (Munc18-1 or -2/-3; SNAP-23 or -25, syntaxin-1 or -2/-4) than in the case of synaptic transmission. Selective defects in cis-Golgi were observed in at least two of the cases (Munc18-1 and SNAP-25 depletion), whereas in the last case (syntaxin depletion), cell loss was too rapid to detect Golgi defects reliably. This places these generic membrane trafficking pathways most upstream in the regulated secretory pathway, probably ER to cis-Golgi or a retrograde pathway to the Golgi.
Footnotes
This work was supported by the Netherlands Organization for Scientific Research ZonMw Pionier/VICI 900-01-001 and by the European Union ERC Advanced Grant 322966 and HEALTH-F2-2009-242167 SynSys to M.V. We thank Robbert Zalm for expert help with virus production and cloning; Desiree Schut for preparing glia feeders and culturing neurons; Rob Zwart for performing the qPCR experiments; Ingrid Saarloos for performing Western blot experiments; and Joost Hoetjes, Joke Wortel, Christiaan van der Meer, and Frank den Oudsten for breeding and genotyping mutant mice.
The authors declare no competing financial interests.
References
- Arunachalam L, Han L, Tassew NG, He Y, Wang L, Xie L, Fujita Y, Kwan E, Davletov B, Monnier PP, Gaisano HY, Sugita S (2008) Munc18-1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol Biol Cell 19:722–734. 10.1091/mbc.E07-0662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliocchi L, Fava E, Leist M, Horvat V, Dinsdale D, Read D, Nicotera P (2005) Botulinum neurotoxin C initiates two different programs for neurite degeneration and neuronal apoptosis. J Cell Biol 168:607–618. 10.1083/jcb.200406126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Yamasaki S, Binz T, Niemann H, Jahn R (1993a) Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J 12:4821–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Südhof TC, Niemann H, Jahn R (1993b) Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365:160–163. 10.1038/365160a0 [DOI] [PubMed] [Google Scholar]
- Bronk P, Deák F, Wilson MC, Liu X, Südhof TC, Kavalali ET (2007) Differential effects of SNAP-25 deletion on Ca2+-dependent and Ca2+-independent neurotransmission. J Neurophysiol 98:794–806. 10.1152/jn.00226.2007 [DOI] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, Bement WM (2007) Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton 64:822–832. 10.1002/cm.20226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Lüthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99:179–188. 10.1016/S0092-8674(00)81649-9 [DOI] [PubMed] [Google Scholar]
- Danglot L, Zylbersztejn K, Petkovic M, Gauberti M, Meziane H, Combe R, Champy MF, Birling MC, Pavlovic G, Bizot JC, Trovero F, Della Ragione F, Proux-Gillardeaux V, Sorg T, Vivien D, D'Esposito M, Galli T (2012) Absence of TI-VAMP/Vamp7 leads to increased anxiety in mice. J Neurosci 32:1962–1968. 10.1523/JNEUROSCI.4436-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO (1987) A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci 78:151–164. 10.1016/0022-510X(87)90057-8 [DOI] [PubMed] [Google Scholar]
- Dekkers MP, Nikoletopoulou V, Barde YA (2013) Cell biology in neuroscience: death of developing neurons: new insights and implications for connectivity. J Cell Biol 203:385–393. 10.1083/jcb.201306136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Martínez I, Nehring RB, Sørensen JB (2007) Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci 27:9380–9391. 10.1523/JNEUROSCI.5092-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M (2009) Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell 138:935–946. 10.1016/j.cell.2009.07.027 [DOI] [PubMed] [Google Scholar]
- Farina M, van de Bospoort R, He E, Persoon CM, van Weering JR, Broeke JH, Verhage M, Toonen RF (2015) CAPS-1 promotes fusion competence of stationary dense-core vesicles in presynaptic terminals of mammalian neurons. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran P, Lawrence GW, Shone CC, Foster KA, Dolly JO (1996) Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry 35:2630–2636. 10.1021/bi9519009 [DOI] [PubMed] [Google Scholar]
- Fujita Y, Ohama E, Takatama M, Al-Sarraj S, Okamoto K (2006) Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson's disease. Acta Neuropathol 112:261–265. 10.1007/s00401-006-0114-4 [DOI] [PubMed] [Google Scholar]
- Galli T, Zahraoui A, Vaidyanathan VV, Raposo G, Tian JM, Karin M, Niemann H, Louvard D (1998) A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol Biol Cell 9:1437–1448. 10.1091/mbc.9.6.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC (2008) Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321:1507–1510. 10.1126/science.1163174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Mourelatos Z, Chen Y, Gonatas JO, Appel SH, Hays AP, Hickey WF, Hauw JJ (1992) Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am J Pathol 140:731–737. [PMC free article] [PubMed] [Google Scholar]
- Guégan C, Przedborski S (2003) Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest 111:153–161. 10.1172/JCI200317610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás-Kovács A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sørensen JB (2007) Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci 27:8676–8686. 10.1523/JNEUROSCI.0658-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeroma JH, Plomp JJ, Roubos EW, Verhage M (2003) Development of the mouse neuromuscular junction in the absence of regulated secretion. Neuroscience 120:733–744. 10.1016/S0306-4522(03)00258-6 [DOI] [PubMed] [Google Scholar]
- Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, Brussaard A, Matus A, Verhage M (2004) Trophic support delays but does not prevent cell-intrinsic degeneration of neurons deficient for Munc18-1. Eur J Neurosci 20:623–634. 10.1111/j.1460-9568.2004.03503.x [DOI] [PubMed] [Google Scholar]
- Hu SH, Latham CF, Gee CL, James DE, Martin JL (2007) Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc Natl Acad Sci U S A 104:8773–8778. 10.1073/pnas.0701124104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL (2011) Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc Natl Acad Sci U S A 108:1040–1045. 10.1073/pnas.0914906108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW (2002) Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J Biol Chem 277:16278–16284. 10.1074/jbc.M111141200 [DOI] [PubMed] [Google Scholar]
- Joshi G, Chi Y, Huang Z, Wang Y (2014) Abeta-induced Golgi fragmentation in Alzheimer's disease enhances Abeta production. Proc Natl Acad Sci U S A 111:E1230–E1239. 10.1073/pnas.1320192111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvotchev M, Südhof TC (2004) Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J Biol Chem 279:47101–47108. 10.1074/jbc.M408474200 [DOI] [PubMed] [Google Scholar]
- Kofuji T, Fujiwara T, Sanada M, Mishima T, Akagawa K (2014) HPC-1/syntaxin 1A and syntaxin 1B play distinct roles in neuronal survival. J Neurochem 130:514–525. 10.1111/jnc.12722 [DOI] [PubMed] [Google Scholar]
- Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW (1995) Staurosporine-induced neuronal apoptosis. Exp Neurol 135:153–159. 10.1006/exnr.1995.1074 [DOI] [PubMed] [Google Scholar]
- Kurokawa Y. (1987) [Binding and degenerative effect of Clostridium botulinum type A, C and E toxins on cultured neurons] Hokkaido Iqaku Zasshi 62:381–391. [PubMed] [Google Scholar]
- Lázaro-Diéguez F, Jiménez N, Barth H, Koster AJ, Renau-Piqueras J, Llopis JL, Burger KN, Egea G (2006) Actin filaments are involved in the maintenance of Golgi cisternae morphology and intra-Golgi pH. Cell Motil Cytoskeleton 63:778–791. 10.1002/cm.20161 [DOI] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Südhof TC, Niemann H, Jahn R (1992) Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun 189:1017–1023. 10.1016/0006-291X(92)92305-H [DOI] [PubMed] [Google Scholar]
- Link E, McMahon H, Fischer von Mollard G, Yamasaki S, Niemann H, Südhof TC, Jahn R (1993) Cleavage of cellubrevin by tetanus toxin does not affect fusion of early endosomes. J Biol Chem 268:18423–18426. [PubMed] [Google Scholar]
- Lowe M, Lane JD, Woodman PG, Allan VJ (2004) Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J Cell Sci 117:1139–1150. 10.1242/jcs.00950 [DOI] [PubMed] [Google Scholar]
- McEwen JM, Kaplan JM (2008) UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol Biol Cell 19:3836–3846. 10.1091/mbc.E08-02-0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O'Toole E, Ferguson S, Cremona O, De Camilli P (2011) Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72:587–601. 10.1016/j.neuron.2011.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison R, Kinoshita Y, Johnson M, Ghatan SJ (2000) Neuronal survival and cell death signaling pathways (Databa MC, ed). Austin, TX: Landes Bioscience. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Adler H, Hirano A, Donnenfeld H, Gonatas JO, Gonatas NK (1990) Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc Natl Acad Sci U S A 87:4393–4395. 10.1073/pnas.87.11.4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC (1996) The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A 93:5472–5477. 10.1073/pnas.93.11.5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osen-Sand A, Staple JK, Naldi E, Schiavo G, Rossetto O, Petitpierre S, Malgaroli A, Montecucco C, Catsicas S (1996) Common and distinct fusion proteins in axonal growth and transmitter release. J Comp Neurol 367:222–234. [DOI] [PubMed] [Google Scholar]
- Peng L, Liu H, Ruan H, Tepp WH, Stoothoff WH, Brown RH, Johnson EA, Yao WD, Zhang SC, Dong M (2013) Cytotoxicity of botulinum neurotoxins reveals a direct role of syntaxin 1 and SNAP-25 in neuron survival. Nat Commun 4:1472. 10.1038/ncomms2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, Scheller RH (1994) n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci U S A 91:1445–1449. 10.1073/pnas.91.4.1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott KL, McCarthy MJ, Becker D, Gatchalian C, Rubin LL (1996) Morphological and biochemical changes in neurons: apoptosis versus mitosis. Eur J Neurosci 8:1906–1915. 10.1111/j.1460-9568.1996.tb01334.x [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/S0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P (1999) Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci 112:1865–1877. [DOI] [PubMed] [Google Scholar]
- Rowe J, Calegari F, Taverna E, Longhi R, Rosa P (2001) Syntaxin 1A is delivered to the apical and basolateral domains of epithelial cells: the role of munc-18 proteins. J Cell Sci 114:3323–3332. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Poulain B, Rossetto O, Benfenati F, Tauc L, Montecucco C (1992a) Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J 11:3577–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C (1992b) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359:832–835. 10.1038/359832a0 [DOI] [PubMed] [Google Scholar]
- Schiavo G, Shone CC, Bennett MK, Scheller RH, Montecucco C (1995) Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J Biol Chem 270:10566–10570. 10.1074/jbc.270.18.10566 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. 10.1126/science.1064335 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. (2002) Alzheimer's disease is a synaptic failure. Science 298:789–791. 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- Sesso A, Fujiwara DT, Jaeger M, Jaeger R, Li TC, Monteiro MM, Correa H, Ferreira MA, Schumacher RI, Belisário J, Kachar B, Chen EJ (1999) Structural elements common to mitosis and apoptosis. Tissue Cell 31:357–371. 10.1054/tice.1999.0042 [DOI] [PubMed] [Google Scholar]
- Stieber A, Mourelatos Z, Gonatas NK (1996) In Alzheimer's disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. J Pathol 148:415–426. [PMC free article] [PubMed] [Google Scholar]
- Tellam JT, McIntosh S, James DE (1995) Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. J Biol Chem 270:5857–5863. 10.1074/jbc.270.11.5857 [DOI] [PubMed] [Google Scholar]
- Tellam JT, Macaulay SL, McIntosh S, Hewish DR, Ward CW, James DE (1997) Characterization of Munc-18c and syntaxin-4 in 3T3–L1 adipocytes: putative role in insulin-dependent movement of GLUT-4. J Biol Chem 272:6179–6186. 10.1074/jbc.272.10.6179 [DOI] [PubMed] [Google Scholar]
- Toonen RF, de Vries KJ, Zalm R, Südhof TC, Verhage M (2005) Munc18-1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. J Neurochem 93:1393–1400. 10.1111/j.1471-4159.2005.03128.x [DOI] [PubMed] [Google Scholar]
- Vaidyanathan VV, Yoshino K, Jahnz M, Dörries C, Bade S, Nauenburg S, Niemann H, Binz T (1999) Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J Neurochem 72:327–337. 10.1046/j.1471-4159.1999.0720327.x [DOI] [PubMed] [Google Scholar]
- van de Bospoort R, Farina M, Schmitz SK, de Jong A, de Wit H, Verhage M, Toonen RF (2012) Munc13 controls the location and efficiency of dense-core vesicle release in neurons. J Cell Biol 199:883–891. 10.1083/jcb.201208024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardar G, Chang S, Arancillo M, Wu YJ, Trimbuch T, Rosenmund C (2016) Distinct functions of syntaxin-1 in neuronal maintenance, synaptic vesicle docking, and fusion in mouse neurons. J Neurosci 36:7911–7924. 10.1523/JNEUROSCI.1314-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A 99:9037–9042. 10.1073/pnas.122623799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venderova K, Park DS (2012) Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med 2:piia009365. 10.1101/cshperspect.a009365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Villeneuve J, Scarpa M, Ortega-Bellido M, Malhotra V (2013) MEK1 inactivates Myt1 to regulate Golgi membrane fragmentation and mitotic entry in mammalian cells. EMBO J 32:72–85. 10.1038/emboj.2012.329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M (2001) Munc18-1 promotes large dense-core vesicle docking. Neuron 31:581–591. 10.1016/S0896-6273(01)00391-9 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. 10.1038/nn783 [DOI] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M (2007) Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54:275–290. 10.1016/j.neuron.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Williamson LC, Neale EA (1998) Syntaxin and 25-kDa synaptosomal-associated protein: differential effects of botulinum neurotoxins C1 and A on neuronal survival. J Neurosci Res 52:569–583. [DOI] [PubMed] [Google Scholar]
- Zhao LC, Yang B, Wang R, Lipton SA, Zhang D (2010) Type C botulinum toxin causes degeneration of motoneurons in vivo. Neuroreport 21:14–18. 10.1097/WNR.0b013e328330dcca [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, Südhof TC (2013) Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32:159–171. 10.1038/emboj.2012.307 [DOI] [PMC free article] [PubMed] [Google Scholar]