

Abstract
Denervation of the neuromuscular junction (NMJ) precedes the loss of motor neurons (MNs) in amyotrophic lateral sclerosis (ALS). ALS is characterized by a motor unit (MU)-dependent vulnerability where MNs with fast-fatigable (FF) characteristics are lost first, followed by fast fatigue-resistant (FR) and slow (S) MNs. However, changes in NMJ properties as a function of MU types remain debated. We hypothesized that NMJ synaptic functions would be altered precociously in an MU-specific manner, before structural alterations of the NMJ. Synaptic transmission and morphological changes of NMJs have been explored in two nerve–muscle preparations of male SOD1G37R mice and their wild-type (WT) littermates: the soleus (S and FR MU); and the extensor digitorum longus (FF MU). S, FR, and FF NMJs of WT mice showed distinct synaptic properties from which we build an MU synaptic profile (MUSP) that reports MU-dependent NMJ synaptic properties. At postnatal day 180 (P180), FF and S NMJs of SOD1 already showed, respectively, lower and higher quantal content compared with WT mice, before signs of MN death and before NMJ morphological alterations. Changes persisted in both muscles until preonset (P380), while denervation was frequent in the mutant mouse. MN death was evident at this stage. Additional changes occurred at clinical disease onset (P450) for S and FR MU. As a whole, our results reveal a reversed MUSP in SOD1 mutants and highlight MU-specific synaptic changes occurring in a precise temporal sequence. Importantly, changes in synaptic properties appear to be good predictors of vulnerability to neurodegeneration.
SIGNIFICANCE STATEMENT The inadequate excitability of motor neurons and their output, the neuromuscular junctions (NMJs), has been considered a key factor in the detrimental outcome of the motor function in amyotrophic lateral sclerosis. However, a conundrum persists at the NMJ whereby persistent but incoherent opposite neurotransmission changes have been reported to take place. This article untangles this conundrum by systematically analyzing the changes in synaptic properties over the course of the disease progression as a function of the motor unit type. This temporal analysis reveals that early synaptic alterations evolve with disease progression but precede NMJ neurodegeneration. These data provide a novel framework of analysis and comparison of synaptic transmission alterations in neurodegenerative disorders.
Keywords: ALS, motor unit, neuromuscular junction, quantal analysis, synaptic transmission, transmitter release
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal late-onset neurodegenerative disease characterized by the progressive loss of upper and lower motor neurons (MNs). Neuromuscular junction (NMJ) degeneration is an early event in disease pathogenesis, and NMJ morphological and physiological alterations have been reported in several genetic mouse models (Fischer et al., 2004; Narai et al., 2009; Moloney et al., 2014; Arbour et al., 2015). Denervation and loss of synaptic proteins (e.g., SV2) were observed in SOD1G93A mice, while early stalling of synaptic vesicles in the nerve terminal has been preferentially observed in fast-twitch fibers (Pun et al., 2006). These changes occur before the onset of symptoms and the loss of ventral root axons in the spinal cord (Frey et al., 2000; Fischer et al., 2004; Narai et al., 2009). This loss affects primarily fast-fatigable (FF) motor units (MUs), as they show the greatest vulnerability, followed by fast fatigue-resistant (FR) and slow (S) MUs, the latter being the most resistant that persist until end-stage (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007, 2008). Large MUs in fast-twitch muscles are then partially replaced by compensatory reinnervation by axons from small (S and FR) MUs (Schaefer et al., 2005; Hegedus et al., 2008).
Hence, the development of this MN disease appears to be characterized by profound synaptic alterations at the NMJ. However, this led to a number of discrepancies, generating a conundrum in the literature. For instance, Souayah et al. (2012) reported a reduced probability of release at NMJs of the diaphragm muscle in the presymptomatic SOD1G93A mice, while, using the same neuromuscular preparation at the same stage, Rocha et al. (2013) reported that a distinct group of mutant NMJs presented a higher quantal content. After disease onset, however, a decreased quantal content was subsequently observed in some NMJs. In addition, a diminished synaptic fidelity as well as decreased spontaneous activity was reported in spinal MNs of the mutant TDP-43 zebrafish larvae (Armstrong and Drapeau, 2013a). Also, either a gain or a loss of fused in sarcoma (FUS) function impaired neurotransmission by reducing quantal content and synaptic current amplitude in Drosophila (Shahidullah et al., 2013; Machamer et al., 2014) or zebrafish (Armstrong and Drapeau, 2013b). The difficulty in reconciling these observations could be due in part to the developmental stage, the animal model itself, or even to the specific mutation. However, an important aspect that was not considered in these studies is the type of MU to which the NMJs belong.
Consistent with the selective vulnerability of specific groups of MUs and the temporal pattern of denervation observed in various models of ALS, we postulate that synaptic activity is altered differentially depending on the MU type and also evolves differently during the disease process. We used SOD1G37R mice, a late-onset, slow, and progressive model of ALS (Wong et al., 1995), to record neurotransmission from three different MU types of two different muscles: the fast-twitch extensor digitorum longus (EDL; FF MU) and the slow-twitch Soleus (SOL; S and FR MU). We show that MUs at wild-type (WT) NMJs are characterized by a distinctive synaptic profile that we define as a MU synaptic profile (MUSP). We further show that opposite synaptic changes are seen in FF and S MUs of SOD1G37R mice before motor neuron degeneration in the spinal cord. NMJs of mutant FF MUs present a persistent reduced synaptic strength, while those of S MUs first show a substantial increase in synaptic evoked release that disappears after clinical onset of the disease. FR MUs show a reduced quantal content only after disease onset. These characteristics generated a distinctive MUSP that reflected the various stages of disease progression. These synaptic alterations preceded NMJ structural changes, underlying the preferential vulnerability of NMJs from FF MUs. This work highlights the importance of considering MU types in the study of NMJ functions in ALS, given their differential susceptibility to the disease and the specific intrinsic properties that confer their unique response to disease progression.
Materials and Methods
Animals
Mice overexpressing the human mutated SOD1G37R transgene were obtained from The Jackson Laboratory and bred at our animal facilities on a C57BL/6 background. These mice, referred to as line 29, expressed twofold to fivefold the level of mutant proteins in the brain, spinal cord, and brainstem and developed a slow motor neuron syndrome that resembles the human pathology characterized by a progressive weakness and a late onset of hindlimb paralysis originally occurring ∼12–13 months of age (Wong et al., 1995). However, this mouse strain currently exhibits an extended lifetime up to >500 d of age, confirmed by The Jackson Laboratory (Fig. 1A, time course of the disease of the SOD1G37R mouse in our facilities). Three specific time periods were investigated in the present experiment in the SOD1G37R mice and their WT littermates: the asymptomatic stage, postanatal day 170 (P170) to P190 (P180), the presymptomatic stage, P370 to P400 (P380), and the symptomatic stage, P420 to 460 (P450). Only males were used in this study.
Figure 1.
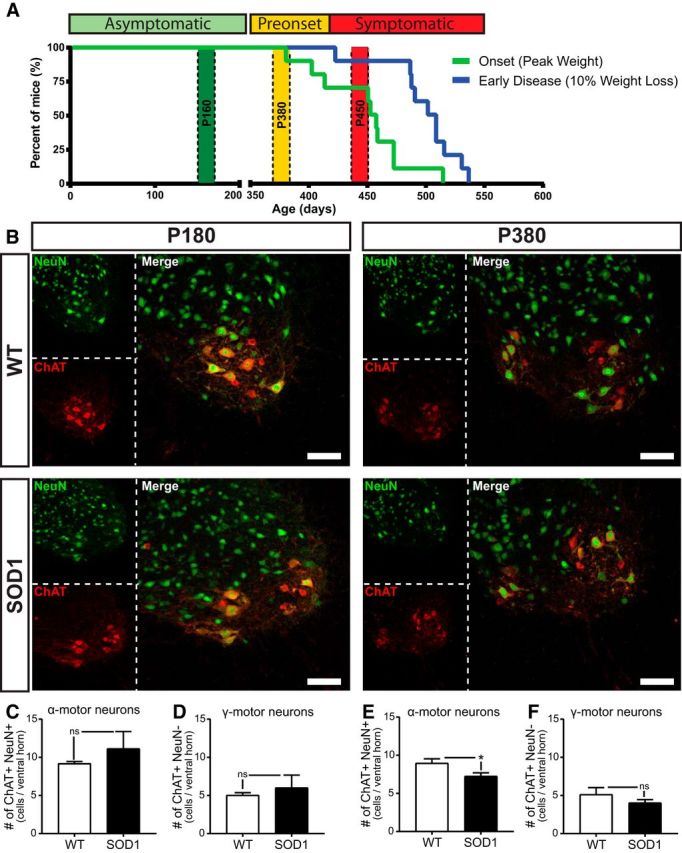
Disease progression and motor neuron death in the SOD1G37R mouse model. A, Percentage of mice reaching disease onset (pale green line) and early disease (blue line). Based on symptoms and clinical nomenclature, the asymptomatic period (green zone) was defined from birth until P350, the presymptomatic (preonset) stage (yellow zone) from P350 until P400 approximately, and the symptomatic stage (red zone) started at ∼P400 lasting until the end-stage at P525 to P600. We investigated three time periods corresponding to the three stages represented in the figure: P180, P380, and P450. WT, N = 10; SOD1, N = 9. B, Confocal images of immunohistological labeling of motor neurons in the ventral horn of a lombar spinal cord section: neuronal-specific nuclear protein (NeuN; green) and choline acetyltransferase (ChAT; red) were immunolabeled on spinal cord cross sections of WT mice (top panels) and SOD1 mice (bottom panels) at P180 (left) and P380 (right). Neurons in the ventral horn were identified as α-MNs when brightly labeled for both NeuN and ChAT and were identified as γ- MNs when labeled with ChAT but weakly or not labeled for NeuN. C–F, Histograms showing the mean ± SEM of the number of α-MNs (C) and γ-MNs (D) at P180 and α-MNs (E) and γ-MNs (F) at P380. Note the selective reduction of α-MNs in the mutant mouse at P380, without any loss of γ-MNs. Scale bars, 100 μm. *p < 0.05.
Mice genotyping was performed at the time of the weaning by PCR amplification of the human SOD1 transgene using a small tail sample. All experiments were performed in accordance with the guidelines of the Canadian Council of Animal Care and the Comité de déontologie animale of Université de Montréal.
Nerve–muscle preparations
Nerve–muscle preparations of the SOL and EDL were dissected in oxygenated Ringer's solution (in mm), as follows: 110 NaCl, 5 KCl, 1 MgCl2, 25 NaHCO3, 2 CaCl2, 11 glucose, 0.3 glutamic acid, 0.4 glutamine, 5 BES (N,N-Bis(2-hydroxyethyl)-2-aminoethanesulfonic acid sodium salt), 0.036 choline chloride, and 4.34 × 10−7 cocarboxylase. After dissection, nerve muscle preparations were pinned in a Sylgard-coated recording chamber constantly perfused with oxygenated Ringer's solution (95% O2, 5% CO2). The pH (7.4) and temperature (28 ± 2°C) were continuously regulated.
Electrophysiological recordings of synaptic transmission
Suprathreshold stimulation (2–5×) of the tibial nerve (SOL) or the deep peroneal nerve (EDL) was performed using a suction electrode filled with Ringer's solution. Muscle contractions were prevented by continuous perfusion of the preparations with a modified Ringer's solution prepared with low Ca2+ (1 mm) and high Mg2+ concentrations (6.4 mm), or by incubating the preparations with μ-conotoxin GIIIB (3–4 μm; Peptides International) in normal Ringer's physiological solution in a closed bath for 20 min before the experiments. Recordings were considered successful when the initial membrane potential (Em) of a muscle fiber was between −65 and −80 mV, an important indicator of muscle quality. In addition, a recording was considered valid and included for further analysis only if the Em variation was <5 mV during the experiment.
Importantly, soon after disease onset (defined by the start of weight loss), the nerve–muscle preparations of the EDL were too fragile to sustain the experimental procedures, and spontaneous contractions would inevitably occur. This further confirmed the vulnerability of these muscles in the disease process. In addition, extensive denervation in the EDL greatly reduced the availability of surface NMJs, further reducing the success rate and preventing us from obtaining successful recordings after disease onset. The nerve–muscle preparation quality of the SOL could be maintained for a longer time window, allowing us to perform recordings at the beginning of the symptomatic period.
Intracellular recordings of spontaneous and nerve-evoked synaptic responses were performed using a sharp glass microelectrode (1.0 mm; WPI) filled with 3 mm KCl (35–70 MΩ) as previously described (Darabid et al., 2013; Arbour et al., 2015). Briefly, spontaneous activity was monitored, first, where miniature end plate potentials (mEPPs) were recorded for a period of 5–10 min without any motor nerve stimulation. mEPP frequency was obtained by dividing the number of mEPPs occurring in a period of at least 5 min (minimum of 100 mEPPs). EPPs were then evoked by paired (10–15 ms interval) motor nerve stimulations at 0.2 Hz for a period of at least 10 min (120 repetitions). Paired-pulse facilitation (PPF) was calculated as the mean amplitude of the second EPP divided by the mean amplitude of the first EPP (10 min period). Quantal content was calculated according to the study by del Castillo and Katz (1954): m is the mean amplitude of the first EPP (including failures)/mean amplitude of the mEPP. At least 100 mEPPs were included in the calculation of m. Quantal content and PPF are presynaptic markers of the synaptic strength of the recorded NMJs.
Fiber type identification and classification were performed on previously recorded NMJs in the SOL muscle. Preparations were incubated right after the physiological experiment for 10 min with α-bungarotoxin (BTX; 1.3–2.0 μg/ml; Alexa Fluor 594 conjugate, Thermo Fisher Scientific), an irreversible marker of nicotinic acetylcholine receptors (nAChRs) at the NMJ. This labeling revealed the unique pretzel-like pattern of each NMJ, allowing us to retrieve each recorded NMJ. Additionally, after the completion of the experiment and to further ensure an accurate post hoc NMJ identification, surrounding fibers were gently impaled with a sharp microelectrode in a characteristic way to make the NMJ even more salient and eventually use it as a map. The number of fibers separating the recorded NMJ from the main nerve entry was also counted, which served as a supplemental reference. Images on live muscles were then acquired on an Olympus BX-51W1 Upright Microscope with a BrightLine Pinkel filter set (single-band exciter FF01–560/25, Semrock).
Immunohistochemistry and fiber type identification
The SOL muscle contains mainly two fiber types in mice: S fibers expressing the myosin heavy chain I (MHCI); and FR fibers expressing the MHCIIa. Triple staining was performed on SOL muscle after each physiological experiment to identify the two MHCs and the nAChRs (see Fig. 5B). Muscles were fixed with 4% formaldehyde (Mecalab) diluted in PBS solution (in mm: 137 NaCl, 2.7 KCl, 10 Na2HPO4, and 2 KH2PO4) over 10 min and rinsed three times with PBS. Membrane permeabilization was then performed in cold methanol for 6 min (−20°C). The following steps of the immunostaining were performed at room temperature. After a rinsing period (three times with PBS), muscles were incubated in a PBS-Triton X (0.01%) solution containing 10% normal donkey serum (NDS; Jackson ImmunoResearch) for 20 min to limit nonspecific antibody labeling. Muscles were then incubated for 2 h with a mouse anti-MHC type I IgG2B and a mouse anti-MHC type IIa IgG1 [BAD5c (1:100) and SC-71c (1:200), Developmental Studies Hybridoma Bank]. Between each immunolabeling step, muscles were subsequently washed three times (5 min each) in PBS-Triton 0.01%. SOL preparations were incubated for 1 h at room temperature consecutively in an Alexa Fluor 647-goat α-mouse IgG2b (1:500; Jackson ImmunoResearch) and then with the Alexa Fluor 488-goat α-mouse IgG1 (1:500; Jackson ImmunoResearch). Finally, a 30 min staining with an Alexa Fluor 594-conjugated-α-BTX (1.33–2.0 μg/ml; Thermo Fisher Scientific) was performed to label postsynaptic receptors. Muscles were then mounted on a glass slide in Prolong Gold antifade (Thermo Fisher Scientific). Images were subsequently acquired using a Zeiss LSM 510 Confocal Microscope.
Figure 5.

Unaltered spontaneous activity at P180 in S, FR, and FF NMJs. A, Typical traces of mEPP for S (left), FR (center), and FF (right) NMJs of WT and SOD1G37R mice. B, Confocal false color images showing fiber type identification and labeling of the SOL muscle: S MHC (MHC 1, purple), FR MHC (MHC IIa, green), and postsynaptic nAChRs (α-BuTX, red). Note the alternance of the two MU types at the surface of the SOL muscle. C, D, Histograms showing the mean ± SEM of mEPP frequency (C) and mEPP amplitude (D) for S (in green), FR (in blue), and FF NMJs (in black). Note the gradual increase in mEPP frequency as a function of the MU type, which is similar for WT and SOD1G37R NMJs. However, the relative mEPP amplitude of SOD1 NMJs did not follow the pattern observed in WT littermates as the mEPP amplitude of FR MU was larger in SOD1 than WT mice. **p < 0.01; ***p < 0.001. Scale bar in B, 10 μm.
Immunolabeling on cross sections of the EDL was performed as previously described (Gouspillou et al., 2014). Briefly, EDL muscles were mounted in cold optimal cutting temperature compound (OCT; Tissue-Tek) and frozen in isopentane cooled to −80°C in liquid nitrogen. Transverse sections (10 μm) were made and incubated in blocking solution (10% NDS in PBS). Sections were incubated with either mouse IgG1 anti-MHC type IIa (1:200; SC-71), mouse IgG2b anti-MHC type I (1:100; BA-D5), and mouse IgM anti-MHC type IIb (1:200; BF-F3), or mouse IgM anti-MHC type IIx (1:10; 6H1) and mouse IgG1 anti-MHC all but IIx (1:200; BF-35) for 1 h at room temperature (all from Developmental Studies Hybridoma Bank). Sections were then rinsed in PBS three times and incubated with the following secondary antibodies: goat anti-mouse IgG1 Alexa Fluor 488; goat anti-mouse IgM Alexa Fluor 405; and goat anti-mouse IgG2b Alexa Fluor 647 (all 1:500; Jackson ImmunoResearch). Sections were then mounted in ProLong Gold Antifade Reagent (Invitrogen).
MU identification was not performed after each experiment in the EDL muscle since we found that the specific zone on the muscle surface where NMJs were recorded was composed only of MHC type IIb-expressing fibers (see Fig. 8H, example at preonset stage). In contrast, immunolabeling was necessary after each physiological experiment to identify the FR NMJ (MHC type IIa) and the S NMJ (MHC type I) of the SOL muscle (see Fig. 5B), as they are intermingled and present in similar proportions in WT and SOD1G37R mice at the surface of the SOL muscle (Arbour et al., 2015).
Figure 8.

Persistent and additional synaptic changes at P380 in the EDL. A, Examples of mEPPs in control (WT) and SOD1G37R mice. B, C, Histograms showing the mean ± SEM of mEPP frequency (B) and amplitude (C) in FF NMJs. D, Traces of EPPs from WT and SOD1 FF NMJs. E–G, Histograms showing the mean ± SEM of EPP amplitude (E), quantal content (F), and paired-pulse facilitation ratio (G). H, Transverse cross sections of an EDL muscle (WT, top; SOD1, bottom) showing fiber type labeling of three MHCs. Left panels, MHC IIa (in green) and MHC IIb (in red); right panels: MHCs all but MHCIIx (in green) and MHCIIx (by extension, in red). Note that the type of MHC at the surface recorded in our experiments (arrow) did not change during the course of the disease despite the atrophy of the muscle (bottom panels). Scale bar, 100 μm. *p < 0.05; ***p < 0.001.
Immunohistochemistry of NMJs and lumbar spinal cord sections
Tissue preparation.
Mice were anesthetized with a lethal dose of ketamine (375–450 mg/kg) and xylazine (25–30 mg/kg) and then were perfused through the heart with cold PBS 1× for 5 min, followed by 4% formaldehyde diluted in PBS 1× (4% PFA) for ∼15 min. SOL and EDL muscles were then dissected and postfixed for 10 min in 4% PFA at room temperature. The whole mouse was then further fixed overnight in 4% PFA at 4°C, and lumbar spinal cords were dissected, postfixed in 4% PFA for 2 h at room temperature, and cryoprotected in a 30% sucrose PBS 1× solution for 72 h at 4°C. Lumbar spinal cords were then embedded in OCT and snap frozen in cooled isopentane (−40 to −50°C).
Spinal cord cross sections and motor neuron counting.
Lumbar spinal cord sections were prepared as previously described (Parone et al., 2013; Pickles et al., 2013). Floating 30 μm-thick spinal cord cryosections were washed twice (5 min each) in PBS 1× then incubated in a blocking solution (10% NDS, 0.3% Triton X-100 in PBS) for 1 h at room temperature. Sections were incubated overnight with primary antibodies against choline acetyltransferase (ChAT; 1:100; goat, catalog #AB144P, Millipore) and neuronal-specific nuclear protein (NeuN; 1:300; mouse IgG1, catalog #MAB377, Millipore) in blocking solution at room temperature. Slices were then rinsed three times with PBS 1× (5 min each) and incubated with the secondary antibody Alexa Fluor 594-donkey anti-goat (1:500; Jackson ImmunoResearch) in blocking solution for 1 h at room temperature. Slices were then rinsed three times with PBS 1× and incubated with Alexa Fluor 488-goat anti-mouse IgG1 secondary antibody for 1 h. Finally, sections were rinsed three times with PBS 1× (5 min each) and mounted on Superfrost Plus Gold Slides (Thermo Fisher Scientific) with Prolong Gold Antifade Reagent (Thermo Fisher Scientific).
Image acquisition was performed on an Olympus FV1000 Confocal Microscope with a 20× water-immersion objective (numerical aperture 0.95). Motor neurons in both ventral horns were counted from 15 to 20 sections per animal, at least 90 μm apart. ChAT- and NeuN-positive cells in the ventral horn were counted as α-motor neurons, whereas ChAT-positive and NeuN-negative cells were counted as γ-motor neurons as previously described (Lalancette-Hebert et al., 2016). Results are expressed as the average number of cells counted per ventral horn in each animal.
NMJ identification and labeling.
NMJ immunolabeling to label the three synaptic compartments of the NMJ was performed as previously described (Valdez et al., 2012; Darabid et al., 2013; Arbour et al., 2015). EDL and SOL muscles were permeabilized in 100% cold methanol at −20°C for 6 min. To prevent nonspecific labeling, preparations were incubated for 1 h at room temperature in a blocking solution composed of 10% NDS and 1% of Triton X-100 diluted in PBS 1×.
For SOL muscles, type I muscle fibers were first labeled by incubating muscles with an antibody against MHC type I (1:100; mouse IgG2b, catalog #BA-D5c, Developmental Studies Hybridoma Bank) diluted in blocking solution for 24 h at 4°C. SOL and EDL muscles were incubated with a rabbit anti-S100β antibody (1:250; Agilant-Dako) in blocking solution for 24 h at 4°C to label Schwann cells. Next, axons [1:2000; chicken anti-neurofilament M (NF-M), Rockland Immunochemicals] and nerve terminals [1:2000; mouse IgG1 anti-synaptic vesicular protein 2 (SV2), Developmental Studies Hybridoma Bank] were labeled in blocking solution for 24 h. SOL muscles were then incubated with the secondary antibody Alexa Fluor 488-goat anti-mouse IgG2b (1:500; Jackson ImmunoResearch) in blocking solution for 2 h at room temperature. SOL and EDL muscles were then incubated simultaneously with Alexa Fluor 594-conjugated-α-BTX (1.33–2.0 μg/ml; Thermo Fisher Scientific) and the following three secondary antibodies diluted in blocking solution for 2 h at room temperature: goat anti-mouse IgG1 Alexa Fluor 405 (SOL) or Alexa Fluor 488 (EDL); donkey anti-chicken Alexa Fluor 405 (SOL) or Alexa Fluor 488 (EDL); and Alexa Fluor 647-donkey anti-rabbit (all 1:500; Jackson ImmunoResearch). Finally, whole muscle preparations were mounted in Prolong Gold Antifade Reagent (Thermo Fisher Scientific). Muscles were washed six times (10 min each) in PBS-Triton 0.01% following each antibody incubation. Some EDL muscles were dissected from unfixed mice in normal Ringer's solution and fixed in 4% PFA at room temperature for 10 min. Immunolabeling of the three synaptic components was performed as previously described for these muscles (Darabid et al., 2013; Arbour et al., 2015). Observations and image acquisition were performed on a Zeiss LSM 510 or a Zeiss LSM 880 Confocal Microscope. No image manipulations were performed after acquisition, except for linear contrast adjustments for figure presentation.
Statistical analysis
Results are represented as the mean ± SEM where the number of animals is identified as N and the number of NMJs is represented by n. Unpaired t tests were used in most cases where two different groups were compared. In addition, two-way ANOVA with post hoc Bonferroni multiple comparison test was used to compare the values obtained for the three MUs of SOD1 and WT mice. The confidence level used was 95% (α = 0.05).
Data from the NMJ morphological analysis (number of events over a number of observations, expressed as a percentage of events) follow a logistic distribution rather than a Gaussian distribution. This implies that variance is not constant and will vary depending on the value of the mean (i.e., variance is smaller toward the extreme values of 0% and 100%; Crawley, 2007). Classical statistical tests (such as t test or two-way ANOVA) assume that variance is constant and therefore are not appropriate for this analysis. Thus, a generalized linear model (GLM) using a logistic distribution (binomial error structure, logit link function) was created to test the effect of the MU type and the genotype of animals on the measured morphological criteria (see Fig. 2). The MU type was defined as paired within each subject (“within-subject factor”). All pairwise comparisons were performed post hoc using Sidak's test. Only post hoc p values are reported in the text. All analyses were made with SPSS version 24.0.0.0 software (IBM). The number of animals used (N; number of replicates) and the number of NMJs (n; number of observations) are indicated in the text.
Figure 2.

Definition and examples of the criteria used for NMJ morphological analysis. The state of the three components (left column) of the NMJ, presynaptic (green), postsynaptic (red), and glia (blue), was analyzed using seven criteria. Immunohistochemical analysis of the three components was performed based on the criteria defined in Table 1 of Arbour et al. (2015). Three criteria were defined to analyze the presynaptic component: denervation, nerve sprouting, and polyinnervation. Postsynaptic receptor organization was assessed using the following two criteria: faint clustered nAChRs or ectopic receptors. Finally, the structure and integrity of PSCs were evaluated using the following two criteria: incomplete glial coverage and glial sprouting. Confocal false color images of the three components of the NMJ obtained from the mutant mouse illustrating each criterion (right column), as follows: presynaptic terminal (synaptic vesicles and the neurofilament, SV2 + NF-M, in green); end plate acetylcholine receptors (postsynaptic, α-BTX, in red), and perisynaptic Schwann cells (Glia, S100β, in blue). Scale bars, 10 μm.
Results
Considering the progressive differential susceptibility of fast MU in ALS (Frey et al., 2000; Atkin et al., 2005; Pun et al., 2006; Hegedus et al., 2007, 2008), we postulated that previous inconsistencies seen in synaptic transmission at the NMJ in various ALS models may be explained by the differential effect of the mutation on the different MU types. In addition, we hypothesized that synaptic alterations presented by the various NMJ types (S-FR-FF) might be transient and evolve differently as disease progresses. Hence, using the SOD1G37R mouse model, we tested whether spontaneous and nerve-evoked activity were altered in the slow-twitch SOL and the fast-twitch EDL, in an MU-specific manner and at different time points during the course of the disease. We investigated three defined time periods to dissect the temporal effect of SOD1 mutation on neuromuscular transmission and NMJ structure: the asymptomatic stage (P180), the presymptomatic (preonset) stage (P380), and disease onset (P450; Fig. 1, for details).
α-Motor neurons numbers are preserved at P180 but not at P380 in the SOD1G37R mouse
We first characterized the time of onset of motor neuron loss in the slow and progressive SOD1G37R model with respect to disease progression (Fig. 1A). We used the general neuronal nuclei marker NeuN and the cholinergic neuron marker ChAT to specifically identify MNs in lumbar spinal cords cross sections of mutant and control mice. Neurons in the ventral horn of the spinal cord positive for both markers were counted as α-MN, while those weakly-labeled or unlabeled for NeuN were considered γ-MNs. As expected, analysis revealed no change in α-MN or γ-MN numbers at P180, 9 months before disease onset (Fig. 1B–D; WT α-MN: 9.19 ± 0.26, N = 3; SOD1 α-MN: 11.14 ± 2.23, N = 3; WT γ-MN: 5.05 ± 0.32, N = 3; SOD1 γ-MN: 6.02 ± 1.65, N = 3). At P380, however, 1 or 2 months before the symptomatic stage, a significant loss of α-MN was found in the ventral horn of SOD1 spinal cord, while no change in γ-MN number was observed (Fig. 1B,E, F; WT α-MN: 8.98 ± 0.56, N = 4; SOD1 α-MN: 7.26 ± 0.43, N = 5; WT γ-MN: 5.13 ± 0.90, N = 4; SOD1 γ-MN: 4.04 ± 0.42, N = 5). This indicates that α-MN degeneration was already ongoing at this stage.
At preonset stage: NMJ morphology is altered in the EDL and SOL muscles
At P180, several months before the start of overt motor symptoms, there was no evidence of morphological alterations of the presynaptic, postsynaptic, and glial components in the WT and SOD1G37R NMJs (Fig. 2, details on the seven criteria analyzed), in either the SOL or the EDL (data not shown).
However, since several morphological alterations have been reported in the SOD1G93A mouse before symptom onset, especially in FF MUs (Frey et al., 2000; Pun et al., 2006; Valdez et al., 2012), we expected substantial denervation and postsynaptic changes in the fast-twitch EDL that could impair synaptic transmission during the preonset stage. The three components of the NMJ were then labeled at P380 on EDL and SOL whole-mounted muscle preparations, and each NMJ was analyzed according to the seven criteria previously described (Fig. 2; Arbour et al., 2015).
Denervation and partial innervation were already noticeable and frequent in the SOD1G37R in all fiber types compared with WT mice (Fig. 3A–C; SOD1 S, 18.89 ± 5.95%, n = 81, N = 4; SOD1 FR, 12.67 ± 4.52%, n = 109, N = 4; SOD1 FF, 38.27 ± 6.26%, n = 153, N = 6; WT S, 0.74 ± 0.74%, n = 94, N = 4; WT FR, 2.18 ± 2.18%, n = 69, N = 4; WT FF, 6.68 ± 1.50%, n = 125, N = 6; p = 0.007, p = 0.025, and p < 0.001, respectively, GLM). In comparison, only one NMJ was found completely denervated in WT muscles at this age and only a few were partially denervated (Fig. 3A–C). Denervation was, however, clearly higher in the EDL than in the SOL muscle, with FF MUs presenting significantly more partially or completely denervated end plates than FR MUs (Fig. 3A–C; GLM, p = 0.0307). In all mutant MU types, postsynaptic end plate areas showed more ectopic receptors, a diffuse α-BTX labeling (Fig. 2, examples), and a clustered appearance in SOD1 mice compared with WT mice (Fig. 3D; WT S, 26.66 ± 8.69%; WT FR, 36.90 ± 5.39%; WT FF, 18.28 ± 2.25%; SOD1 S, 77.00 ± 3.20%; SOD1 FR, 67.48 ± 8.75%; SOD1 FF, 38.88 ± 3.71%; p > 0.001, p = 0.006, and p = 0.001, respectively, GLM). However, positive NMJ repair signs, including nerve sprouting, polyinnervation, and perisynaptic Schwann cell (PSC) process extensions were observed more frequently in both mutant S and FR synapses compared with FF synapses (Fig. 3E; SOD1 S, 40.32 ± 7.42%; SOD1 FR, 35.79 ± 8.02%; SOD1 FF, 12.67 ± 3.56%; p < 0.001 and p = 0.006, respectively, GLM), suggesting that some regenerative processes likely occur in the SOD1 SOL muscle at this stage. This is consistent with the high plasticity capacity of these synapses compared with FF synapses (Frey et al., 2000). Interestingly, in the WT mice as well, FR MUs presented greater signs of NMJ reinnervation than FF MUs (Fig. 3E; WT S, 19.82 ± 7.29%; WT FR, 33.79 ± 6.66%; WT FF, 5.09 ± 2.47; p < 0.001, GLM), further confirming the increased adaptive capacity of the SOL muscle in healthy and pathological conditions.
Figure 3.
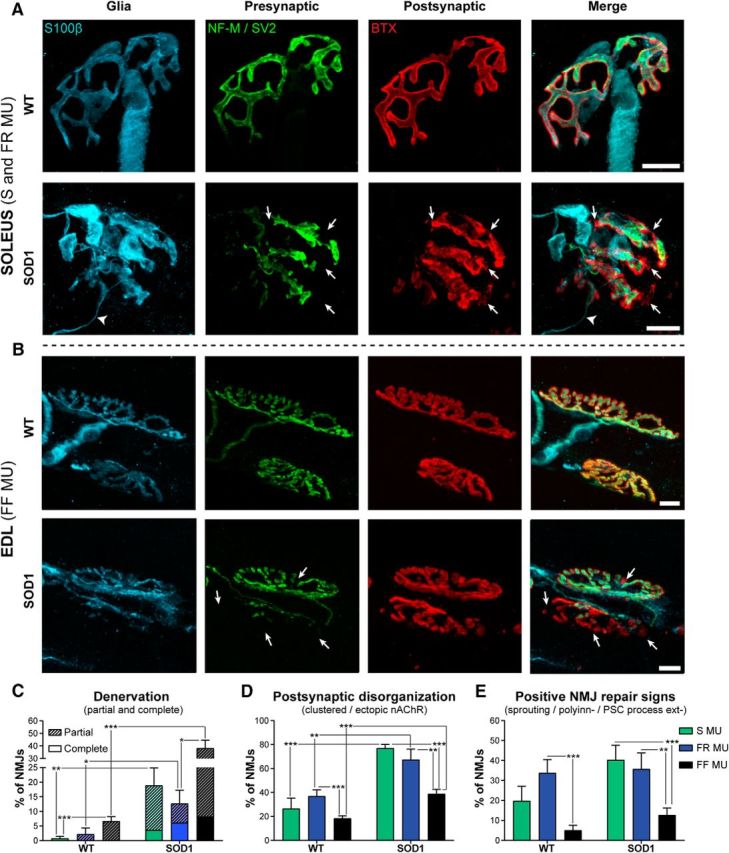
Morphological alterations at P380 of FF, FR, and S NMJs in EDL and SOL muscles. A, B, Confocal images of immunohistochemical labeling of the three components of the NMJ: the perisynaptic Schwann cells (Glia, S100β, in blue), the presynaptic terminal (SV2 + NF-M, in green), and AChRs (postsynaptic, α-BTX, in red) in the SOL (A) and the EDL (B) of WT (top panel) and SOD1G37R mice (bottom panel). Note the presence of partially innervated NMJs in the SOL (A, bottom) and EDL (B, bottom) of SOD1 mice (arrows) and a glial process extension in the SOD1 SOL (A, arrowheads). C–E, Histograms showing the mean ± SEM of the percentage of NMJs with denervation, including partially innervated and completely denervated NMJs (C); postsynaptic end plate disorganization, including faint, clustered, or ectopic nAChRs (D); and positive NMJ repair signs, namely nerve sprouting, polyinnervation and PSC bridges (E). Note that all MU types already present significant denervation and postsynaptic disorganization at the presymptomatic stage in the mutant mouse compared with the control. *p < 0.05; **p < 0.01; ***p < 0.001, GLM. Scale bars, 10 μm.
At disease onset: selective morphological vulnerability of NMJs from FF MUs
We next analyzed morphological NMJ properties at disease onset (P450) and performed similar immunostainings as described above (Fig. 3). Consistent with their well known relative vulnerability in the SOD1G93A model (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007, 2008; Kryściak et al., 2014), denervation and partial innervation were frequently observed at FF NMJs, significantly higher than in FR and S NMJs. Indeed, only 12.94 ± 5.61% of S NMJs and 12.62 ± 5.20% of FR NMJs were denervated and partially innervated as opposed to 49.24 ± 3.62% of FF NMJs (Fig. 4A–C; SOD1 S, n = 64, N = 4; SOD1 FR, n = 97, N = 4; SOD1 FF, n = 153, N = 4, p < 0.001 and p < 0.001, GLM). By comparison, denervation was observed on 5.73 ± 1.73% of S WT NMJs, 3.65 ± 1.40% of FR NMJs, and 3.62 ± 1.58% of FF NMJs, only the latter being significantly different from the mutant (see Fig. 4A–C; WT S, n = 107, N = 5; WT FR, n = 174, N = 5; WT FF, n = 249, N = 5; p > 0.05, p > 0.05, and p < 0.01, GLM).
Figure 4.
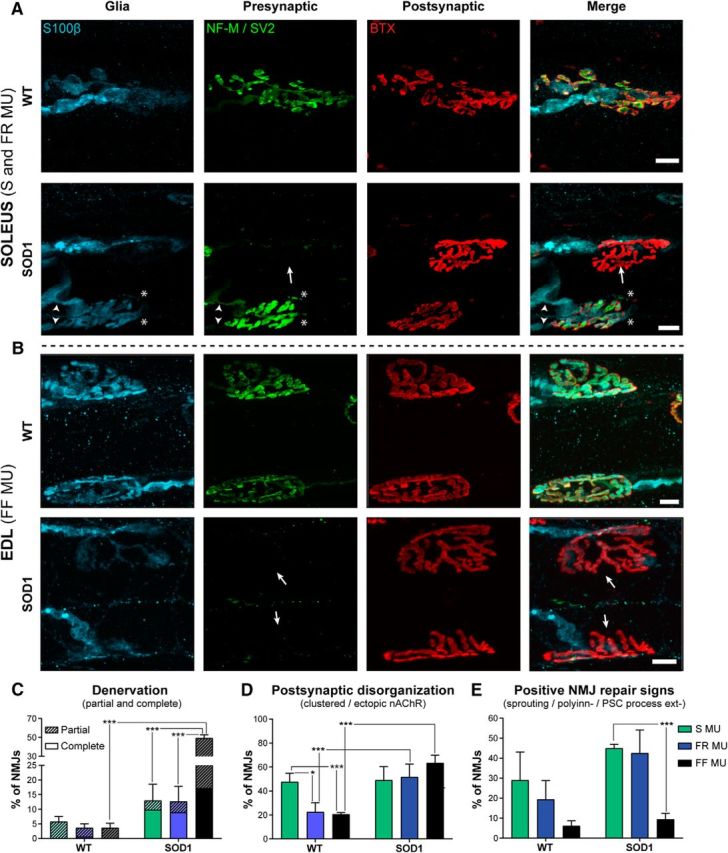
Differential NMJ morphological alterations at clinical disease onset (P450). A, B, Confocal false color images of the three synaptic compartments of the NMJ: glia (S100β, blue), presynaptic nerve terminal (NF-M and SV2, green), and postsynaptic end plate (nAChRs; α-bungarotoxin, red) for WT and SOD1 NMJs. Note the presence of denervated NMJs in the SOL and the EDL (A, bottom) and EDL (B, bottom) of SOD1 mice (arrows), as well as polyinnervation and nerve terminal sprouting in the SOD1 SOL (A, arrowheads and asterisks, respectively). Immunohistochemical analysis was performed as in Figure 3. C–E, Histograms showing the mean ± SEM of the percentage of NMJs with partial or complete denervation (C), with faint, clustered, or ectopic nAChRs (D), or with nerve sprouting, polyinnervation, or PSC process extensions (E). Note the major deterioration of NMJs of the EDL compared with the ones of the SOL. *p < 0.05; ***p < 0.001, GLM.
While postsynaptic end plates of NMJs from S MUs in WT mice were more disorganized than FR and FF synapses (Fig. 4D; WT S, 47.63 ± 7.16%; WT FR, 22.58 ± 7.66%; WT FF, 20.55 ± 1.39; p = 0.013 and p < 0.001, GLM), only mutant FR and FF MUs showed more disorganized postsynaptic end plates (increased ectopic receptors, diffuse α-BTX labeling, and clustered nAChRs) than WT mice (Fig. 4D; SOD1 FR, 51.71 ± 10.74%; SOD1 FF, 63.45 ± 6.38%; p = 0.005 and p < 0.001, respectively, GLM).
In addition, mutant NMJs from S MUs had significantly more frequent positive signs of reinnervation, including nerve sprouting, polyinnervation, and PSCs process extensions, than the FF NMJs (Fig. 4E; SOD1 S, 44.98 ± 1.99%; SOD1 FR 42.52 ± 11.57%; SOD1 FF, 9.38 ± 3.08%; p < 0.001, GLM). Consistent with the positive influence of these parameters on the reinnervation of NMJs (Son et al., 1996), we observed numerous NMJs that were relatively spared in SOL muscles of SOD1 mice, showing a well preserved presynaptic component and a complete PSC coverage. This result is consistent with the relative resistance of this muscle to denervation in ALS (Valdez et al., 2012). Furthermore, our data confirm that the specific vulnerability of FF MUs reported in other SOD1 models of the disease is also present in the SOD1G37R model.
At asymptomatic stage: spontaneous activity is unaltered in all MU types
Following the characterization of the state of innervation, we examined the NMJ properties and synaptic changes throughout disease progression as a function of the MU type.
Frequency and amplitude of spontaneous synaptic events were first analyzed at P180 in the SOL and EDL muscles of WT and SOD1G37R mice. Figure 5 shows that the properties of spontaneous activity are MU dependent (F(2,73) = 86.86, p < 0.0001, two-way ANOVA). In WT mice, NMJs of slow MUs showed the lowest mEPP frequency with 0.50 ± 0.03 Hz (Fig. 5A, C; WT S, n = 9, N = 6), followed by the FR NMJs at 1.16 ± 0.22 Hz (Fig. 5A, C; WT FR, n = 11, N = 6), and finally the FF NMJs at 5.51 ± 0.56 Hz (Fig. 5A, C; WT FF, n = 15, N = 7). Importantly, the same pattern was observed in the SOD1 mice, without any difference compared with control NMJs (Fig. 5A, C; SOD1 S: 0.57 ± 0.06 Hz, n = 13 N = 7; SOD1 FR: 1.46 ± 0.23 Hz, n = 14, N = 6; SOD1 FF: 4.66 ± 0.46 Hz, n = 17, N = 6; p > 0.05, Bonferroni post hoc test). This is consistent with a previous report of unaltered spontaneous transmitter release at a presymptomatic stage in the SOD1G93A mouse diaphragm (Rocha et al., 2013).
We next investigated the amplitude of the spontaneous events, knowing that this variable is usually inversely correlated with the fiber diameter (Sieck and Prakash, 1997). Accordingly, we predicted that mEPP amplitude should be the smallest in FF MUs and the largest in S MUs in WT mice. Alteration of this criterion could highlight postsynaptic defects in the mutant mice. Our results show that mEPP amplitude varied as a function of the MU type (F(2,69) = 58.72, p < 0.0001, two-way ANOVA). In the WT mice, FF MUs had the smallest mEPP amplitude while FR MUs and S MUs had the greatest mEPP amplitude (Fig. 5D; WT S, 0.35 ± 0.02 mV, n = 10, N = 6; WT FR, 0.31 ± 0.01 mV, n = 12, N = 7; WT FF, 0.24 ± 0.01 mV, n = 15, N = 7; p < 0.001, Bonferroni post hoc test). Mutant NMJs followed approximately the same pattern, but, in contrast, mEPP amplitude of mutant FR NMJs was significantly higher than in control mice (Fig. 5D; SOD1 S, 0.37 ± 0.02 mV, n = 13, N = 7; SOD1 FR, 0.36 ± 0.02 mV, n = 12, N = 6; SOD1 FF, 0.24 ± 0.01 mV, n = 14, N = 6; p < 0.01, Bonferroni post hoc test). Unlike previously reported for the SOD1G93A diaphragm (Rocha et al., 2013), giant mEPPs (>1 mV) were not predominant in any MU (<1 event/5 min recording period) in both WT and SOD1 mice.
At asymptomatic stage: evoked activity is high in S MUs and low in FF MUs
We next tested nerve-evoked synaptic transmission in an MU-specific manner at P180 in the SOD1G37R mouse model. We performed intracellular electrophysiological recordings using a PPF protocol (two stimuli, 0.1 ms duration at 10 ms interval) elicited at 0.2 Hz, in a low Ca2+/high Mg2+ Ringer's solution. These properties were then studied in normal Ca2+ concentrations.
The amplitude of EPPs evoked by stimulation of the tibial nerve (SOL) and the deep peroneal nerve (EDL) was first measured using the first EPP evoked by the PPF protocol. Overall, we found a significant interaction effect (F(2,73) = 8.836, p = 0.0004, two-way ANOVA). For instance, EPP amplitude in the WT SOL was not significantly different in S, FR, or FF MUs (Fig. 6A,B; WT S, 0.74 ± 0.08 mV, n = 10, N = 6; WT FR, 0.97 ± 0.07 mV, n = 13, N = 7; WT FF, 0.95 ± 0.08 mV, n = 16, N = 6; p > 0.05, Bonferroni post hoc test). In contrast, mutant mice showed a completely reversed pattern, with EPP amplitude significantly larger at S NMJs than at FF NMJs (Fig. 6A,B; SOD1 S, 1.21 ± 0.13 mV, n = 11, N = 7; SOD1 FF, 0.98 ± 0.12 mV, n = 16, N = 6; p < 0.001, Bonferroni post hoc test). In addition, significant differences emerged between WT and SOD1 mice where EPP amplitude was 64% larger in mutant S NMJs than in WT S NMJs (Fig. 6A,B; p < 0.001, Bonferroni post hoc test), while it was 36% smaller in the FF NMJs of SOD1 mice (Fig. 6A,B; p < 0.01, Bonferroni post hoc test). However, no difference in EPP amplitude was detected at FR NMJs from SOD1 mice compared with WT mice (Fig. 6A,B; SOD1 FR, 0.61 ± 0.05 mV, n = 14, N = 7; p > 0.05, Bonferroni post hoc test).
Figure 6.
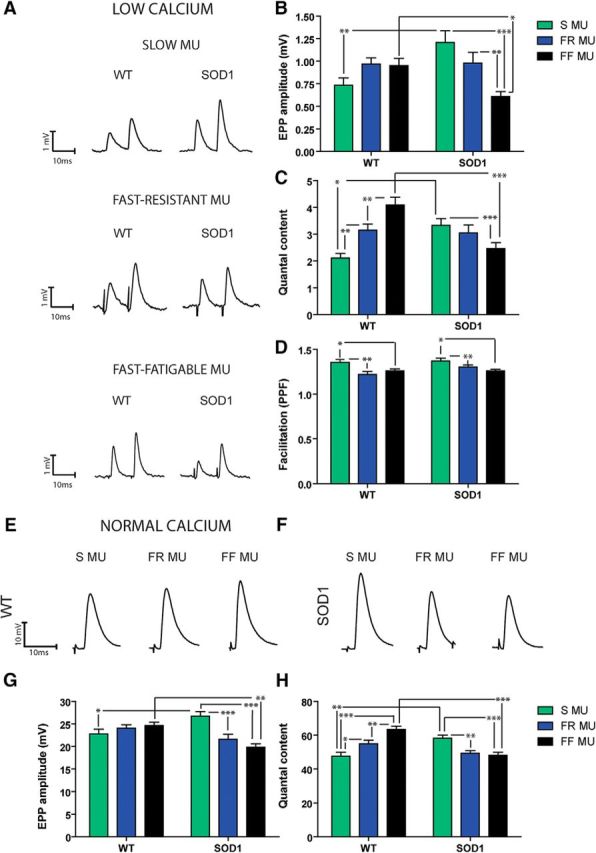
EPP amplitude and quantal content are increased at P180 in S NMJs but decreased in FF NMJs. A, Typical traces of EPPs evoked by a paired-pulse protocol (0.2 Hz, 10 ms interval) in a low Ca2+/high Mg2+ solution for S (top), FR (middle), and FF NMJs (bottom) of WT and SOD1G37R mice. B–D, Histograms of the mean ± SEM of EPP amplitude (B), quantal content (C), and paired-pulsed facilitation ratio (D) of the S (green), FR (blue), and FF NMJs (black) from WT and SOD1G37R mice. Note that SOD1 NMJs showed a reverse MU pattern. E, F, Example traces of EPPs in physiological Ca2+ concentration with μ-conotoxin GIIIB to block muscle contractions, for WT (E) and SOD1 (F) mice. G, H, Histograms of the mean ± SEM of EPP amplitude (G) and quantal content (H) performed in normal extracellular Ca2+ concentration. Note the similarities between data obtained in low vs normal Ca2+ conditions, confirming the striking synaptic differences observed in the mutant mice. *p < 0.05; **p < 0.01; ***p < 0.001.
We next determined the quantal content as a function of the MU type in WT and SOD1 animals. Based on the well known differences in MU functions and synaptic properties (Reid et al., 1999; Kanning et al., 2010), we predicted a gradient of quantal content in WT mice, from S to FF NMJs. Indeed, S NMJs had the smallest quantal content with 2.11 ± 0.17 (Fig. 6C; p < 0.01, Bonferroni post hoc test), while FF NMJs presented the largest one with 4.10 ± 0.29 (Fig. 6C; WT S, n = 10, N = 6, WT FF, n = 16, N = 7; p < 0.001, Bonferroni post hoc test). Furthermore, FR NMJs showed an intermediate value that was significantly different from the others (Fig. 6C; WT FR, 3.15 ± 0.23, n = 14, N = 7; p < 0.05 and p < 0.001, respectively, Bonferroni post hoc test).
Interestingly, this MU dependency was altered at SOD1 NMJs. Indeed, well before onset of motor symptoms, opposite synaptic changes were already prominent in the S fibers of the SOL and the FF fibers of the EDL, reversing the normal physiological properties observed at WT NMJs. Interestingly, the presence of the mutation influenced the MU types differently, as shown by a significant interaction effect (F(2,68) = 15.01, p < 0.0001, two-way ANOVA). Indeed, S MUs of the SOD1G37R SOL presented a 57% increase in the mean quantal content compared with WT (Fig. 6C; SOD1 S, 3.33 ± 0.26, n = 13, N = 6; p < 0.01, Bonferroni post hoc test) while it was reduced by 66% in the FF NMJs of the mutant mice (Fig. 6C; SOD1 FF, 2.46 ± 0.23, n = 16, N = 7; p < 0.001, Bonferroni post hoc test). These results demonstrate the presence of early and striking synaptic alterations in mutant mice, which contradicts the established roles and functions of the various MU types (Kanning et al., 2010).
Last, we analyzed the PPF as an indicator of presynaptic release probability and synaptic efficacy. Consistent with the idea that the initial probability of release of a NMJ is normally inversely proportional to the PPF (Mallart and Martin, 1968), we expected, based on our quantal analysis results, to observe MU-specific variation of this variable in WT mice. Because of the quantal content in SOD1 mice described above, we further hypothesized that the PPF ratio should be reduced at SOD1 S NMJs and increased at FF NMJs in SOD1 mice compared with WT mice.
Once again, MU type had the greatest influence on these properties, regardless of the genotype (F(2,75) = 8.847, p = 0.004, two-way ANOVA). Indeed, the PPF ratio was significantly greater at S and FR NMJs than at FF NMJs in WT (Fig. 6D; WT S, 1.35 ± 0.03, n = 10, N = 6, WT FR, 1.22 ± 0.03, n = 12, N = 6, WT FF, 1.26 ± 0.02, n = 16, N = 6; p < 0.05, Bonferroni post hoc test) and in SOD1 mice (Fig. 6D; SOD1 S, 1.37 ± 0.02, n = 13, N = 6, SOD1 FR, 1.30 ± 0.02, n = 15, N = 7, SOD1 FF, 1.26 ± 0.02, n = 16, N = 6; p < 0.05, Bonferroni post hoc test). Surprisingly, however, the PPF ratio was unchanged in SOD1 compared with WT mice in any MU type (Fig. 6D; p > 0.05, Bonferroni post hoc test). This is inconsistent with the typical physiology of the NMJ, whereby a large PPF is associated with a low quantal content. However, these results are in accordance with the MU-specific differences in PPF reported in WT mice.
In the last sets of experiments, we tested that the various alterations in the synaptic properties described above are also present in regular Ca2+ conditions and occur in normal synaptic physiological conditions (Fig. 6E–H). We performed electrophysiological recordings in normal Ca2+ Ringer's solution, using μ-conotoxin GIIIB (3–4 μm) to selectively block Na+ channel in muscle fibers and prevent muscle contractions. Properties of nerve-evoked synaptic activity were again clearly altered as a function of the MU type (F(2,86) = 4.055, p = 0.0207, two-way ANOVA) and the interaction with the genotype (F(2,86) = 12.03, p < 0.0001, two-way ANOVA) for EPP amplitude. In addition, there was a clear influence of the genotype (F(1,75) = 4.543, p = 0.0363, two-way ANOVA) and an interaction of the genotype with MU type for the quantal content (F(2,75) = 21.91, p < 0.0001, two-way ANOVA). Indeed, consistent with our previous findings in low Ca2+/high Mg2+, quantal content in the WT was smaller at S NMJs compared with FR and FF NMJs, with FF NMJs showing the largest one (Fig. 6H; WT S, 47.61 ± 2.32, n = 11, N = 5; WT FR; 54.98 ± 2.06, n = 14, N = 5; WT FF, 63.44 ± 1.83, n = 15, N = 5; SOD1 S, 58.24 ± 1.88, n = 14, N = 5; SOD1 FR, 49.20 ± 1.71, n = 10, N = 5; SOD1 FF, 48.04 ± 1.84, n = 19, N = 5; p < 0.05, p < 0.001, and p < 0.01, respectively, Bonferroni post hoc test). Importantly, the NMJs of SOD1 mice presented the opposite MU-dependent quantal content distribution whereby the mutant S MUs had an elevated mean EPP amplitude and quantal content compared with WT mice (Fig. 6E–H; EPP amplitude: SOD1 S, 26.71 ± 1.03, n = 14, N = 5; SOD1 FR, 21.61 ± 1.07, n = 12, N = 5; SOD1 FF, 19.86 ± 0.74, n = 20, N = 5; WT S, 22.80 ± 1.06, n = 11, N = 5; WT FR; 24.10 ± 0.72, n = 16, N = 5; WT FF, 24.63 ± 0.77, n = 15, N = 5; p < 0.05 and p < 0.001, respectively, Bonferroni post hoc test). In addition, EPP amplitude and quantal content were significantly smaller in the FF NMJs of mutant mice compared with controls (Fig. 6E–H; p < 0.01 and p < 0.001, Bonferroni post hoc test). Also, similar to changes observed in low Ca2+/high Mg2+ conditions, mEPP amplitude was altered as a function of the MU type (F(2,72) = 12.92, p < 0.0001, two-way ANOVA), with FF NMJs having a smaller mEPP amplitude compared with S NMJs in both control and mutant mice and FR NMJs having a median value (data not shown; p < 0.01 and p < 0.05, Bonferroni post hoc test). These experiments further confirm the profound synaptic alterations that are present in physiological conditions in the SOD1G37R mouse. These occur before the onset of symptoms, NMJ denervation, and motor neuron loss in the ventral horn of the spinal cord. Moreover, the great similarity of synaptic signatures in the different Ca2+ conditions further highlights the importance of the MU-dependent gradient of synaptic properties.
Building a motor unit synaptic profile
To better describe and evaluate synaptic variations as a function of MU type and subsequently to facilitate comparison with mutant synaptic deficits, we developed a motor unit synaptic profile (MUSP) based on the characteristics of WT spontaneous and evoked neurotransmission at P180 (Fig. 7). The comparison of the spontaneous and evoked synaptic properties allowed us to generate a profile for the properties of each MU type based on four variables that represent synaptic strength, namely, mEPP frequency and amplitude, quantal content, and PPF. This reflects the normal profile that one should expect at adult mouse NMJs based on the MU types. This synaptic profile reveals that the various MU types present a relative continuum of properties.
Figure 7.

Motor unit synaptic profile (MUSP) based on synaptic transmission of WT mice. Schematic representation of relative variations in synaptic properties as a function of the MU type (S, yellow; FR, green; FF, purple/orange) based on our experimental data obtained in low and normal Ca2+ conditions. The following four parameters were used to determine the profile: mEPP amplitude and frequency, quantal content and PPF. Note the relative gradation of spontaneous and nerve-evoked properties following the MU types, which allowed us to generate the MUSP.
This distinct MU synaptic signature then allows one to subsequently identify and classify MUs based on measures of their synaptic strength. This represents a tool to study changes in synaptic transmission at the NMJ in ALS, not only as a comparison between SOD1 and WT NMJs but also as a function of the intrinsic gradient of synaptic properties across MU types. Hence, we expanded the characterization of the SOD1 NMJ synaptic properties to further build the MUSP for SOD1 mice over the course of disease progression.
At presymptomatic stage: alterations of synaptic properties persist in the EDL muscle
We then performed electrophysiological recordings on the presymptomatic EDL (P380) of SOD1G37R mice and age-matched controls to examine whether the properties observed at the asymptomatic stage persisted while denervation and NMJ morphological alterations were present. As shown in Figure 8, spontaneous neurotransmission was now significantly changed in the EDL, as opposed to P180. Indeed, mEPP frequency was considerably decreased in SOD1 mice (Fig. 8A,B; WT, 6.33 ± 0.35 Hz, n = 15, N = 6; SOD1, 3.36 ± 0.80 Hz, n = 12, N = 6; p = 0.0082, unpaired t test), while mEPP amplitude was increased (Fig. 8C; WT, 0.21 ± 0.01 mV, n = 13, N = 6; SOD1, 0.25 ± 0.02 mV, n = 12, N = 5; p = 0.05, respectively, unpaired t test). On the other hand, similar to the data obtained at P180, EPP amplitude at FF NMJs was 31% smaller in SOD1 mice than in WT mice (Fig. 8D,F; WT, 0.85 ± 0.05 mV, n = 19, N = 7; SOD, 0.59 ± 0.05, n = 14, N = 6; p = 0.0005, unpaired t test). Additionally, quantal content was still considerably reduced at this age (Fig. 8F; WT, 3.97 ± 0.21, n = 15, N = 7; SOD1, 2.49 ± 0.33, n = 13, N = 6; p = 0.0007, unpaired t test), and no significant difference in PPF was observed between WT and SOD1 mice (Fig. 8G; WT, 1.25 ± 0.02, n = 19, N = 8; SOD, 1.31 ± 0.03, n = 14, N = 6; p > 0.05, unpaired t test). To confirm that only NMJs from FF MUs were recorded, immunolabeling for the MHCIIb and all-but-MHCIIX was performed to identify the two fiber types on transverse cross sections of the EDL muscle. As shown in Figure 8H, type IIB fibers (FF MUs) were still predominantly present at the surface of the EDL muscle, despite the presence of clear muscle atrophy. This atrophy was also accompanied by a reduction in muscle weight (data not shown).
After disease onset: spontaneous activity in the SOL remains unaltered
We next tested whether the alterations of synaptic transmission in the SOL muscle persisted after disease onset, since vulnerability to neurodegeneration, specifically for FR MUs, has been observed in this period in SOD1 mice (Frey et al., 2000; Hegedus et al., 2008). The properties and quality of the SOL preparations still allowed us to perform the recording experiments even after the start of the weight loss because of its late degeneration in ALS (Hegedus et al., 2007; Valdez et al., 2012).
As expected, significant differences were observed in mEPP frequencies in S MUs and FR MUs, which were significantly higher at FR NMJs than at S NMJs, for WT and SOD1 mice (Fig. 9A–C; WT FR, 1.14 ± 0.12 Hz, n=14, N = 8; SOD1 FR, 1.23 ± 0.22 Hz, n = 8, N = 5; WT S, 0.55 ± 0.01 Hz, n = 12, N = 8; SOD1 S, 0.60 ± 0.04 Hz, n = 11, N = 6; p < 0.05 and p < 0.01, respectively, Bonferroni post hoc test). In addition, there was again a clear effect of the MU type on mEPP amplitude (F(2,69) = 58.72, p < 0.0001, two-way ANOVA), where it was larger at S NMJs than at FR NMJs in WT mice (Fig. 9D; WT S, 0.38 ± 0.02 mV, n = 15, N = 6; WT FR, 0.33 ± 0.02 mV, n = 14, N = 6; p = 0.001, Bonferroni post hoc test). However, there was no difference between WT and SOD1 mice (SOD1 S, 0.39 ± 0.02 mV, n = 11, N = 6; SOD1 FR, 0.32 ± 0.01 mV, n = 9, N = 5; p > 0.05). Consistent with the data obtained at P180, spontaneous activity was unchanged in the mutant, where mEPP frequency of the WT NMJs was not different from that of the SOD1 NMJs (Fig. 9C; p > 0.05, Bonferroni post hoc test).
Figure 9.

At disease onset, quantal content is no longer enhanced in S MUs and is decreased in FR MUs. A, B, Typical traces of spontaneous events in the S (A) and FR (B) NMJs from WT (top) and SOD1G37R (bottom) mice. C, D, Histograms showing the mean ± SEM of mEPP frequency (C) and amplitude (D) for S (green) and FR (blue) MUs. E, F, Traces of EPPs in the S (E) and FR (F) MUs in WT (left) and mutant (right) mice. G–I, Histograms showing the mean ± SEM of EPP amplitude (G), quantal content (H), and paired-pulse facilitation (I), for S (green) and FR (blue) MUs. Note that the quantal content of the SOD1 S MU is no longer larger than the WT MU, while it is now significantly smaller in the FR MU. *p < 0.05; **p < 0.01.
After disease onset: nerve-evoked activity evolved differently in S and FR MUs
Unlike the long-lasting changes found in the FF MUs of the EDL muscle, none of the MU-dependent nerve-evoked synaptic alterations previously reported in the SOL muscle persisted at P450 in SOD1 mice. Indeed, EPP amplitude was no longer significantly different between WT and SOD1 mice in S MUs (Fig. 9E,G; WT, 0.85 ± 0.05 mV, n = 15, N = 8, SOD1, 1.01 ± 0.09 mV, n = 12, N = 7; p > 0.05, Bonferroni post hoc test). Moreover, the EPP amplitude at FR NMJs of SOD1 mice was now lower compared with WT FR NMJs (Fig. 9F–G; WT, 0.85 ± 0.06 mV, n = 17, N = 9; SOD1, 0.68 ± 0.06 mV, n = 9, N = 6; p < 0.01, Bonferroni post hoc test).
At this age, WT mice still presented a specific pattern of synaptic properties in accordance with the MUSP developed at P180 (Fig. 7). However, several prominent synaptic changes were observed in SOD1 mice: quantal content of FR NMJs, which was normal during the presymptomatic stage, was now considerably reduced (by 27%) in SOD1 FR NMJs (Fig. 9H; WT, 2.89 ± 0.19, n = 14, N = 8; SOD1, 2.12 ± 0.19, n = 12, N = 7; p < 0.05, Bonferroni post hoc test), while at the same time the increase in neurotransmission reported earlier in mutant S MUs was no longer observed (Fig. 9H; WT, 2.29 ± 0.15, n = 15, N = 8; SOD, 2.61 ± 0.18, n = 9, N = 6; p > 0.05, Bonferroni post hoc test). Finally, again at this age, paired-pulse facilitation was only significantly influenced by the MU type (F(1,48) = 12.73, p = 0.0008, two-way ANOVA), while it remained unchanged at SOD1 NMJs compared with WT NMJs (F(1,48) = 3.533, p = 0.0663, two-way ANOVA). In both cases, S NMJs presented a higher PPF ratio than FR NMJs (Fig. 9I; WT S, 1.39 ± 0.03, n = 15, N = 8; SOD1 S, 1.32 ± 0.02, n = 12, N = 7; WT FR, 1.27 ± 0.02, n = 17, N = 8; SOD1 FR, 1.22 ± 0.03, n = 8, N = 6; p < 0.05, Bonferroni post hoc test), following the synaptic property identified earlier in the MUSP.
Reversal of the MUSP at SOD1 NMJs
Our comprehensive analysis of MU-dependent deficits in NMJ synaptic transmission and the corresponding timeline alterations of the MUSP in SOD1 mice are summarized in Figure 10. We observed that NMJs of FF MUs, which should show the strongest synaptic output according to their intrinsic properties (Kanning et al., 2010), showed a reduced quantal content at P180, when NMJs presented a normal morphology. This reduced quantal content persisted at P380, when denervation became frequent in the EDL muscle. In addition, in complete reverse fashion, the quantal content of the S MUs was large at P180, but eventually decreased at disease onset (P450). The reduction of quantal content in FR NMJs at P450, before the appearance of pronounced morphological alterations, indicates that changes in neurotransmission are early indicators of NMJ malfunction. Hence, the decreased evoked transmission precedes the extensive NMJ denervation observed in the pathology and appears predictive of subsequent NMJ denervation and axonal degeneration.
Figure 10.
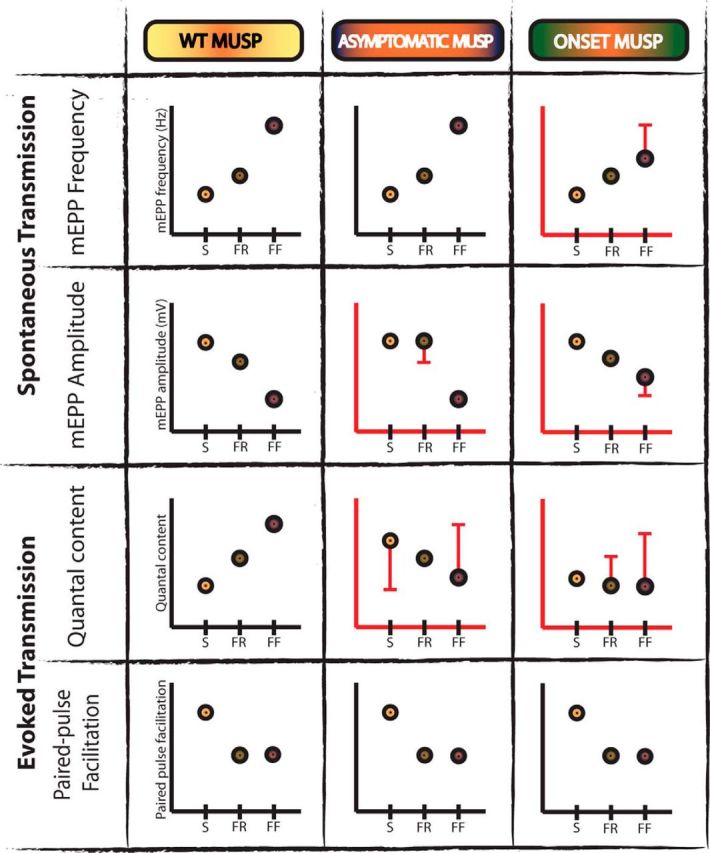
Motor unit synaptic profile (MUSP) alterations over the course of the disease in SOD1 mice. Schematic representation of changes in synaptic properties of NMJs as a function of MU type (S, FR, and FF) and disease stage for the mutant mice. WT synaptic profile of the four parameters (mEPP amplitude and frequency, quantal content, and PPF) derived from Figure 5 is shown again for ease of comparison (left column). To simplify the figure, data from the EDL (P380) and SOL (P450) muscles are represented in the onset category (right column), while data at P180 is represented in the asymptomatic category (middle column). Synaptic alterations between WT and SOD1 NMJs are represented with red bars, highlighting the statistical difference with the value of the WT MUSP. Note the relative evolution of the alterations of quantal content, mEPP frequency, and mEPP amplitude throughout the disease at SOD1 NMJs. Only PPF remained unaltered at all stages.
Together, our results reveal prominent opposite MU-specific synaptic alterations in FF and S NMJs of SOD1 mice, reversing the pattern of synaptic properties normally seen in the MUSP of WT mice (Fig. 7). These early persistent synaptic changes will subsequently evolve differently after the onset of symptoms in the SOL muscle. In particular, the quantal content is the hallmark of the ALS-dependent changes in NMJ synaptic functions with a particular signature at a given stage of the disease.
Discussion
Our study provides a unique characterization of synaptic transmission recorded in three MU types over time in mutant SOD1G37R mice and their WT littermates. A MUSP was developed based on WT neurotransmission that showed a distinct signature of S, FR, and FF NMJs. This strategy revealed several synaptic alterations in SOD1 mice that provided not only a MUSP signature for the SOD1 mutant, but also a precise and unique MUSP for each stage of the disease. Furthermore, synaptic alterations were observed before MN loss, axonal degeneration, and NMJ structural changes. Reduction of nerve-evoked transmission appears to be predictive of the NMJ denervation in FF and FR NMJs.
MU-dependent NMJ morphological alterations during disease progression
To our knowledge, this is the first extensive MU-specific analysis of NMJ properties in a model of ALS. Our characterization of NMJs allowed us to specifically track the morphological alterations in all MUs during disease progression and to confirm key characteristics of the animal model. The fact that denervation was more frequent in the EDL than in the SOL muscle at P380 and P450 supports an early selective vulnerability of the FF MU also observed in other ALS models. However, somewhat surprisingly, partially or completely denervated NMJs were more frequent in all MU types of SOD1 mice compared with the control, but only at P380. Our data are consistent with a recent study in SOD1G93A mice that showed that innervation of SOL NMJs was initially as affected as other muscles but showed a slower rate of decline (Valdez et al., 2012). In addition, S and FR MUs showed more positive signs of nerve repair (increased polyinnervation, nerve sprouting, and PSC process extensions). Their presence during disease progression is consistent with the relative resistance of the SOL muscle in ALS. The importance of these processes to NMJ reinnervation (Son et al., 1996) may contribute to the better preserved NMJ structure in the S and FR MUs than in FF MUs at the symptomatic stage. Of course, the presence of NMJ repair signs does not warrant NMJ protection since NMJs of the SOL muscle eventually become denervated at end-stage (Valdez et al., 2012).
As a whole, our work supports the findings on the widely used model SOD1G93A (Frey et al., 2000; Atkin et al., 2005; Pun et al., 2006; Hegedus et al., 2007, 2008; Kryściak et al., 2014) as it confirms the selective vulnerability of the FF MUs and the pattern of spinal MN loss. Interestingly, the comprehensive characterization provided by our work may facilitate the use of an SOD1G37R mouse model that more closely mimics disease progression in patients.
The MUSP reflects MU properties
Our work allowed the development of a novel tool to characterize NMJ properties over time and in a MU-dependent manner. The MU-specific synaptic properties of the WT NMJs used to build the MUSP are in perfect accordance with known differences between slow and fast NMJs (Reid et al., 1999; Atkin et al., 2005), the MN type (Leroy et al., 2014), and the muscle contractile properties. Indeed, the S NMJs had the smallest evoked and spontaneous activity, consistent with their high resistance to fatigue (Hennig and Lømo, 1985) and the slow contractile properties of type I fibers (Kryściak et al., 2016). In contrast, the FF MUs that develop strength rapidly, but only transiently (Hennig and Lømo, 1985), presented the highest mEPP frequency and nerve-evoked synaptic transmission.
However, SOD1 NMJs do not follow this profile, demonstrating striking alterations of quantal content early in the pathogenesis. Importantly, these salient differences in the quantal content were also observed in normal extracellular Ca2+ conditions, suggesting that these properties are expressed during normal synaptic activity in SOD1 mice and that they occur in the course of ALS. This further confirms the differential MU-specific synaptic phenotype as a biomarker of the disease process.
Early and progressive reduction of neurotransmission in FF NMJs highlights their relative vulnerability
The reduced neurotransmitter release in FF SOD1 NMJs is in accordance with a reduced quantal transmission reported in fast twitch-like muscle in mutTARDBP and mutFUS zebrafish larvae (Armstrong and Drapeau, 2013a,b) and the Drosophila model of FUS (Shahidullah et al., 2013; Machamer et al., 2014). Similar results were observed at NMJs from ALS patients (Maselli et al., 1993). These observations suggest that reduced neurotransmission at the NMJ is a prominent feature of the various forms of ALS. This is consistent with the reported stalling of synaptic vesicles in the nerve terminal observed in fast NMJs before denervation (Pun et al., 2006) and the impaired axonal transport of acetylcholine transferase in SOD1G93A MNs (Tateno et al., 2009) that could reduce neurotransmitter synthesis at the NMJ. In addition, the selective increase in endoplasmic reticulum stress in a subset of vulnerable MNs observed in the SOD1G93A mouse in vivo (Saxena et al., 2009) could be involved in early synaptic alterations. However, our observation of changes in mEPP amplitude is indicative of possible postsynaptic effects, including fragmentation and disorganization of the end plate and changes in muscle fiber diameter (Sieck and Prakash, 1997). Hence, changes in the NMJ properties of FR and FF motor units likely imply changes in both presynaptic and postsynaptic elements that evolve over the course of the disease process.
The reduction in neurotransmitter release first appeared in the mutant EDL muscle at P180, without NMJ morphological changes and before MN degeneration. Moreover, FR MUs also showed similar reductions in evoked neurotransmission after disease onset, before the appearance of major NMJ structural changes in the SOL muscles. Hence, functional synaptic alterations are early indicators of NMJ malfunction that precede NMJ denervation.
Increased nerve-evoked transmission observed in S NMJs only
Our study also reveals abnormal elevated synaptic activity in NMJs of S MUs observed at P180 that persisted until P380 (Arbour et al., 2015). Increased synaptic transmission has been reported at unidentified NMJs of the diaphragm in presymptomatic SOD1G93A mice (Rocha et al., 2013) and is consistent with early intrinsic MN hyperexcitability that is thought to contribute to neurodegeneration in ALS. However, contradictory results have been reported such that, while hyperexcitability of SOD1G93A MNs in culture was reported (Pieri et al., 2003; Kuo et al., 2004, 2005), others reported either an unchanged excitability (Pambo-Pambo et al., 2009; Quinlan et al., 2011) or even a decreased excitability in mutant SOD1 MNs (Bories et al., 2007). Interestingly, a recent elegant in vivo study reported that adult MNs are not hyperexcitable (Delestree et al., 2014) and that fast MNs have an unchanged or reduced excitability compared with WT mice while the more resistant S MNs show a transient intrinsic hyperexcitability (Leroy et al., 2014). These results strongly suggest that increased excitability does not play a crucial role in neurodegeneration in ALS but rather appears neuroprotective of the MNs during the presymptomatic stage of the disease and perhaps is related to the relative resilience of S MUs (Saxena et al., 2013).
Synaptic candidates underlying the MU-dependent changes in ALS
A number of synaptic mechanisms may underlie the MU-dependent differential NMJ changes. A first possible mechanism relates to adenosine, which decreases (via A1 receptors) or increases (via A2A receptors) presynaptic ACh release at the NMJ (Sebastião and Ribeiro, 2009a,b). The facilitating effect of adenosine is enhanced at NMJs of SOD1G93A mice at the presymptomatic stage (Nascimento et al., 2014). Although the role of A1 receptors remains ill-defined at the NMJ in ALS, their involvement could explain the reduction of spontaneous activity at P380 in the EDL muscle.
Another possible mechanism is a toxic gain of function of the SOD1 enzyme, affecting neurotransmission in ALS. Indeed, an abnormal persistent Na+ current has been observed in cultured mutant SOD1 MNs (Pieri et al., 2003; Kuo et al., 2004, 2005) and in humans (Vucic and Kiernan, 2006). Ca2+-dependent mechanisms may also be involved as indicated by the increase of NMJ neurotransmission by ALS-derived antibodies (Appel et al., 1995; Engelhardt et al., 1995), by Ca2+ dysregulation in the MNs (Siklós et al., 1998), and by the perturbations of mitochondrial Ca2+ homeostasis in SOD1 mice (Jaiswal and Keller, 2009; Tradewell et al., 2011; Jaiswal, 2014). Any change of presynaptic Ca2+ handling will alter transmitter release and lead to differential outcomes according to the MN type (Kanning et al., 2010).
Another possibility relates to glial cell functions at the NMJ. A recent study showed an increased Ca2+-dependent activity of PSCs, glial cells at NMJs of S and FR MUs (Arbour et al., 2015). Interestingly, Ca2+-dependent mechanisms in PSCs regulate transmitter release and synaptic plasticity (Todd et al., 2010). Hence, the alteration of PSC Ca2+-dependent activity could alter neurotransmission and impact NMJ functions. In addition, because of their role in NMJ maintenance and repair (Son and Thompson, 1995; Son et al., 1996; Georgiou et al., 1999), alterations in PSCs early in the disease process may contribute to disease pathogenesis and further exacerbate the effect of SOD1 mutation on synaptic transmission (Arbour et al., 2017).
In conclusion, this extensive NMJ morphological and synaptic characterization, which evolves during disease progression, highlights the importance of the MU type in ALS and appears to be a strong and early biomarker of the disease. It underlies a specific gradient of synaptic properties (MUSP) that is completely altered in SOD1 mice. Understanding the specific implications of the selective long-term NMJ physiological changes in ALS will help to develop novel forms of functional biomarkers and to identify novel therapeutic targets in ALS.
Footnotes
This work was supported by grants to R.R. from the Canadian Institutes for Health research (MOP-14137 and MOP-111070) and The Robert Packard Research Center for ALS, and an infrastructure grant from Fonds Recherche Quebec-Santé to the Groupe de Recherche sur le Système Nerveux Central. E.T. holds a Canada Vanier Doctoral Award, and É.M. holds an ALS Canada Doctoral Research Award. We thank Drs. Christine Vande Velde and Danielle Arbour for their constant support and for exciting and stimulating discussions. We also thank them and Dr. Heather Durham for reading the manuscript. In addition, we thank Joanne Vallée and Julie Pépin for technical support.
The authors declare no competing financial interests.
References
- Appel SH, Smith RG, Alexianu M, Siklos L, Engelhardt J, Colom LV, Stefani E (1995) Increased intracellular calcium triggered by immune mechanisms in amyotrophic lateral sclerosis. Clin Neurosci 3:368–374. [PubMed] [Google Scholar]
- Arbour D, Tremblay E, Martineau É, Julien JP, Robitaille R (2015) Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35:688–706. 10.1523/JNEUROSCI.1379-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour D, Vande Velde C, Robitaille R (2017) New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol 595:647–661. 10.1113/JP270213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GA, Drapeau P (2013a) Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J Neurosci 33:1741–1752. 10.1523/JNEUROSCI.4003-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GA, Drapeau P (2013b) Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet 22:4282–4292. 10.1093/hmg/ddt278 [DOI] [PubMed] [Google Scholar]
- Atkin JD, Scott RL, West JM, Lopes E, Quah AK, Cheema SS (2005) Properties of slow- and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul Disord 15:377–388. 10.1016/j.nmd.2005.02.005 [DOI] [PubMed] [Google Scholar]
- Bories C, Amendola J, Lamotte d'Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:451–459. 10.1111/j.1460-9568.2007.05306.x [DOI] [PubMed] [Google Scholar]
- Crawley MJ. (2007) The R book. Chichester, UK, Hoboken, NJ: Wiley; 10.1002/9780470515075 [DOI] [Google Scholar]
- Darabid H, Arbour D, Robitaille R (2013) Glial cells decipher synaptic competition at the mammalian neuromuscular junction. J Neurosci 33:1297–1313. 10.1523/JNEUROSCI.2935-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol 124:560–573. 10.1113/jphysiol.1956.sp005610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D (2014) Adult spinal motoneurons are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592:1687–1703. 10.1113/jphysiol.2013.265843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JI, Siklós L, Kömüves L, Smith RG, Appel SH (1995) Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse 20:185–199. 10.1002/syn.890200302 [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240. 10.1016/j.expneurol.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20:2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou J, Robitaille R, Charlton MP (1999) Muscarinic control of cytoskeleton in perisynaptic glia. J Neurosci 19:3836–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Norris B, Barbat-Artigas S, Aubertin-Leheudre M, Morais JA, Burelle Y, Taivassalo T, Hepple RT (2014) The relationship between muscle fiber type-specific PGC-1alpha content and mitochondrial content varies between rodent models and humans. PLoS One 9:e103044. 10.1371/journal.pone.0103044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28:154–164. 10.1016/j.nbd.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586:3337–3351. 10.1113/jphysiol.2007.149286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig R, Lømo T (1985) Firing patterns of motor units in normal rats. Nature 314:164–166. 10.1038/314164a0 [DOI] [PubMed] [Google Scholar]
- Jaiswal MK. (2014) Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol Cell Ther 2:26. 10.1186/2052-8426-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal MK, Keller BU (2009) Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol 75:478–489. 10.1124/mol.108.050831 [DOI] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE (2010) Motor neuron diversity in development and disease. Annu Rev Neurosci 33:409–440. 10.1146/annurev.neuro.051508.135722 [DOI] [PubMed] [Google Scholar]
- Kryściak K, Grieb P, Celichowski J (2014) Changes in motor unit properties in SOD1 (G93A) rats. Muscle Nerve 50:577–586. 10.1002/mus.24196 [DOI] [PubMed] [Google Scholar]
- Kryściak K, Celichowski J, Drzymała-Celichowska H, Gardiner PF, Krutki P (2016) Force regulation and electrical properties of motor units in overloaded muscle. Muscle Nerve 53:96–106. 10.1002/mus.24690 [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bär PR, Anelli R, Heckman CJ, Kroese AB (2004) Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91:571–575. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R, Heckman CJ (2005) Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol 563:843–854. 10.1113/jphysiol.2004.074138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Sharma A, Lyashchenko AK, Shneider NA (2016) Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc Natl Acad Sci U S A 113:E8316–E8325. 10.1073/pnas.1605210113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Lamotte d'Incamps B, Imhoff-Manuel RD, Zytnicki D (2014) Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3:pii:e04046. 10.7554/elife.04046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer JB, Collins SE, Lloyd TE (2014) The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum Mol Genet 23:3810–3822. 10.1093/hmg/ddu094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallart A, Martin AR (1968) The relation between quantum content and facilitation at the neuromuscular junction of the frog. J Physiol 196:593–604. 10.1113/jphysiol.1968.sp008525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maselli RA, Wollman RL, Leung C, Distad B, Palombi S, Richman DP, Salazar-Grueso EF, Roos RP (1993) Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve 16:1193–1203. 10.1002/mus.880161109 [DOI] [PubMed] [Google Scholar]
- Moloney EB, de Winter F, Verhaagen J (2014) ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci 8:252. 10.3389/fnins.2014.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narai H, Manabe Y, Nagai M, Nagano I, Ohta Y, Murakami T, Takehisa Y, Kamiya T, Abe K (2009) Early detachment of neuromuscular junction proteins in ALS mice with SODG93A mutation. Neurol Int 1:e16. 10.4081/ni.2009.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento F, Pousinha PA, Correia AM, Gomes R, Sebastião AM, Ribeiro JA (2014) Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre-symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS One 9:e104081. 10.1371/journal.pone.0104081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pambo-Pambo A, Durand J, Gueritaud JP (2009) Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J Neurophysiol 102:3627–3642. 10.1152/jn.00482.2009 [DOI] [PubMed] [Google Scholar]
- Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33:4657–4671. 10.1523/JNEUROSCI.1119-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Destroismaisons L, Peyrard SL, Cadot S, Rouleau GA, Brown RH Jr, Julien JP, Arbour N, Vande Velde C (2013) Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet 22:3947–3959. 10.1093/hmg/ddt249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C (2003) Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 351:153–156. 10.1016/j.neulet.2003.07.010 [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419. 10.1038/nn1653 [DOI] [PubMed] [Google Scholar]
- Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ (2011) Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol 589:2245–2260. 10.1113/jphysiol.2010.200659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B, Slater CR, Bewick GS (1999) Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J Neurosci 19:2511–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA (2013) Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8:e73846. 10.1371/journal.pone.0073846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12:627–636. 10.1038/nn.2297 [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. 10.1016/j.neuron.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW (2005) A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol 490:209–219. 10.1002/cne.20620 [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA (2009a) Adenosine receptors and the central nervous system. Handb Exp Pharmacol 471–534. 10.1007/978-3-540-89615-9_16 [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA (2009b) Tuning and fine-tuning of synapses with adenosine. Curr Neuropharmacol 7:180–194. 10.2174/157015909789152128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Le Marchand SJ, Fei H, Zhang J, Pandey UB, Dalva MB, Pasinelli P, Levitan IB (2013) Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J Neurosci 33:19590–19598. 10.1523/JNEUROSCI.3396-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck GC, Prakash YS (1997) Morphological adaptations of neuromuscular junctions depend on fiber type. Can J Appl Physiol 22:197–230. 10.1139/h97-014 [DOI] [PubMed] [Google Scholar]
- Siklós L, Engelhardt JI, Alexianu ME, Gurney ME, Siddique T, Appel SH (1998) Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J Neuropathol Exp Neurol 57:571–587. 10.1097/00005072-199806000-00005 [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995) Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 14:133–141. 10.1016/0896-6273(95)90247-3 [DOI] [PubMed] [Google Scholar]
- Son YJ, Trachtenberg JT, Thompson WJ (1996) Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci 19:280–285. 10.1016/S0166-2236(96)10032-1 [DOI] [PubMed] [Google Scholar]
- Souayah N, Coakley KM, Chen R, Ende N, McArdle JJ (2012) Defective neuromuscular transmission in the SOD1 G93A transgenic mouse improves after administration of human umbilical cord blood cells. Stem Cell Rev 8:224–228. 10.1007/s12015-011-9281-3 [DOI] [PubMed] [Google Scholar]
- Tateno M, Kato S, Sakurai T, Nukina N, Takahashi R, Araki T (2009) Mutant SOD1 impairs axonal transport of choline acetyltransferase and acetylcholine release by sequestering KAP3. Hum Mol Genet 18:942–955. 10.1093/hmg/ddn422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30:11870–11882. 10.1523/JNEUROSCI.3165-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell ML, Cooper LA, Minotti S, Durham HD (2011) Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol Dis 42:265–275. 10.1016/j.nbd.2011.01.016 [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. 10.1371/journal.pone.0034640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC (2006) Axonal excitability properties in amyotrophic lateral sclerosis. Clin Neurophysiol 117:1458–1466. 10.1016/j.clinph.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. 10.1016/0896-6273(95)90259-7 [DOI] [PubMed] [Google Scholar]