

Abstract
Purpose of review
Recurrent loss of function mutations within genes of the cohesin complex have been identified in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). STAG2 is the most commonly mutated cohesin member in AML as well as solid tumors. STAG2 is recurrently, mutated in Ewing’s Sarcoma, bladder cancer, and glioblastoma, and is one of only ten genes known to be recurrently mutated in over four distinct tissue types of human cancer
Recent findings
The cohesin complex, a multiprotein ring, is canonically known to align and stabilize replicated chromosomes prior to cell division. Although initially thought to lead to unequal chromosomal separation in dividing cells, data in myeloid malignancies show this is not observed in cohesin mutant MDS/AML, either in large patient cohorts or mouse models. Mounting evidence supports a potential alternate mechanism whereby drivers of cell-type specific gene expression and hematopoietic development are impaired through alteration in three-dimensional nuclear organization and gene structure.
Summary
Understanding the functional consequences of cohesin mutations in regulating lineage-specific and signal-dependent defects and in myeloid transformation will identify novel pathophysiologic mechanisms of disease and inform the development of novel therapeutic targets.
Keywords: cohesin, leukemia, myeloid, transcription
INTRODUCTION
Recurrent somatic mutations in the genes that constitute the cohesin complex, have been identified in various cancers [1,2▪▪]. Cohesin is a tripartite ring composed of three structural proteins, SMC1A, SMC3, and RAD21, bound to STAG1 or STAG2 and is canonically known to align and stabilize sister chromatids during metaphase [3,4]. In isolation, cohesin complex mutations are not sufficient for malignant transformation either in mouse models [5,6] or whenever present as germline alleles [7]. Cohesin genes are mutated in the germline setting in the pediatric diseases Cornelia de Lange syndrome and Roberts syndrome [8–10]. These craniofacial and cognitive disorders result in an abbreviated lifespan; thus, the full oncogenic potential is unknown, nonetheless, these patients do not have an observed predisposition to cancer.
Prior studies have characterized the role of the cohesin complex in transcriptional activation [11,12], specifically in DNA-loop formation between enhancers and promoters within topologically associated domains (TADs) bounded by CCCTC-binding factor (CTCF) [12,13,14▪▪]. The key role of cohesin in facilitating the essential runx1 gene-expression signatures required for normal hematopoiesis in zebra-fish was strongly shown using an RNAi screen. Knockdown of rad21 resulted in loss of runx1 target gene expression including gata1 [15]. The function of cohesin in transcriptional regulation is tissue specific. In murine embryonic stem cells, the essential Yamanaka factor, Oct4 is lost upon shRNA expression against any member of the cohesin complex [11]. Thus, it is likely that somatic cohesin mutations mediate transcriptional dysregulation [16,17] through altered DNA-loop formation [11,18] and certainly cohesin mutations have biologic rationale as candidate disease alleles in human cancer.
INCIDENCE AND CLINICAL SIGNIFICANCE IN MYELOID MALIGNANCIES
A tremendous fund of knowledge for the entire field of cancer genetics has come from the Cancer Genome Atlas (TCGA) initiative. In addition to finding many known disease alleles for AML, several lesser known genes were identified and this included recurrent somatic mutations in the cohesin complex [19]. Similar large AML sequencing efforts identified a spectrum of cohesin component mutations throughout the ring complex and its regulators [19,20,21▪▪,25,26]. Mutations were found to have no obvious hotspots and were mostly frame-shift (17%) or nonsense (70%) mutations suggesting that the result was molecular loss of function [22–24]. Mutational frequency ranged from 12 to 20% of AML and myelodysplastic syndrome (MDS) and was most prevalent in high-risk MDS and secondary AML [24]. One additional finding from these reports was that several human leukemic cell lines had low expression of cohesin despite having no identified mutation [26] and that a distinct subset of cohesin wild type patients had markedly low cohesin expression [24]. Similar STAG2 low-expressers have also been described in gastric, colorectal, and prostate cancers [27]. Moreover, AML patients with a mutation in a given cohesin gene, had reduced expression of the other cohesin components - suggesting that known and unknown feedback regulatory elements are likely at play in maintaining cohesin stoichiometry.
Clonal analysis by several groups has shown that cohesin mutations are early events [22,24] and discrimination by variant allele frequency suggests that mutations are in the dominant clone [23,24]. Histologically, cohesin-mutant AML arises from a very immature myeloid-committed stem cell and is almost exclusively French-American-British (FAB) classification M1 [19,25] suggesting that cohesin mutations exert their effect mainly in the naive stem cell niche. However, as cohesin mutations have also been described at lower frequencies in chronic myeloid leukemia (CML), chronic myelomonocytic leukemia (CMML), and myeloproliferative neoplasms (MPN), modifiers to the cell of origin for cohesin-mutant leukemia must exist [22,24]. Recently cohesin mutations have been identified as a rare contributor to the entity clonal hematopoiesis [28▪,29–31]. Given that cohesin has been well described to be a key transcriptional coactivator over the last two decades, it is reasonable to conclude that cohesin mutations in isolation may in fact have a negative impact on functional hematopoiesis but are clinically transparent as a small single mutant clone. However, this may represent an ancestral clone susceptible to a ‘second-hit’ resulting in leukemic transformation.
As to the clinical and prognostic significance of cohesin mutations, retrospective clinical outcomes by several groups found that, although not independently prognostic, cohesin mutations trend towards a worse prognosis [20,23,24]. Moreover, recent genomic AML classification identified that STAG2 helps to define a chromatin-spliceosome AML group [21▪▪] whose prognosis is similar to secondary AML regardless of whether an antecedent MDS was present. Taken together, this suggests that STAG2 either defines a specific genetic context for the acquisition of other specific disease alleles, or that STAG2 represents a transformative event in a primed premalignant clone. Biologic precedence for such a phenomenon was demonstrated in single cell sequencing efforts that identified an SMC1A mutation in the preleukemic clone of a patient with FLT3-ITD mutant AML [32]. The SMC1A mutation carried over to the malignant clone in the transformed AML blasts.
PATHOPHYSIOLOGY: THE CASE AGAINST ANEUPLOIDY IN HEMATOPOIETIC TUMORS
It was originally reported that STAG2 mutations in solid tumors led to chromosomal instability in glioblastoma, bladder, and colon cancers [33,34]. Moreover, in the Trisomy 21-specific Down syndrome-associated acute megakaryoblastic leukemia, it was found that over 50% of such cases harbored cohesin mutations [26]. Taken together with the solid tumor data and the high frequency of chromosomal abnormalities in myeloid disease, there was a linear thought process to assume that impaired cohesin sister chromatid alignment and missegregation was the pathophysiologic consequence. In stark contrast, however, recurrent somatic cohesin mutations found in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are not associated with aneuploidy, and in fact found a statistically significant inverse correlation with aneuploidy for chromosome 5 or 7 [24]. Rather, there is evidence that the mechanism of tumorigenesis in cohesin mutant cancers is through defects in cell-type specific gene expression programs by loss in fidelity of chromatin loops. In hematopoiesis, the looping interaction of promoters and enhancers act in concert with key tissue-specific transcription factors including RUNX1 and C/EBPA. In support of this, mutations in these genes were found to significantly co-occur in one large study with STAG2-mutant AmL [21▪▪]. Other reports identify significant co-occurrence with NPM1 [23,35], RAS [36], as well as splicing mutations and epigenetic chromatin modifiers such as SRSF2 [21▪▪], BCOR, and ASXL1 [24].
To better understand the pathophysiologic role of cohesin mutations, model systems using viral transduction of cohesin mutations, shRNA knock-down, and transgenic mouse models were used to evaluate hematopoiesis [5,6,37]. Despite differences in the model systems, these studies consistently found that cohesin mutations and/or loss of function result in in-vivo and in-vitro expansion of hematopoietic stem cells (HSC) either in mice or in colony-forming assays. Transcriptional programming was skewed to an HSC signature and changes in chromatin accessibility were observed, potentially through regulation of the HoxA cluster [38], though the transcriptional effects of cohesin loss-of-function seem to be profoundly widespread. Important model-specific findings were also instructive. In the model using viral transduction of human HSC and progenitor cells, overexpression of mutant RAD21 in CD34+ HSC resulted in defective myelopoiesis and erythropoiesis, however, there was no affect of the mutant transgene in committed populations, demonstrating that effects of cohesin loss-of-function are developmental state-specific. This work suggests that once a cell passes a certain degree of commitment, essential transcriptional programs become hardwired and cohesin-independent in mediating further functional maturation. In the immature and stem-like state, the plasticity of the cell requires cohesin for key fate-decision thresholds.
In the transgenic mouse model, genetic deletion of obligate cohesin member Smc3 was lethal and resulted in bone marrow aplasia with premature sister chromatid separation on metaphase karyograms [6]. By comparison, in the heterozygous state, cooperation with Flt3-ITD resulted in a lethal and transplantable AML phenotype with a normal karyotype by metaphase cytogenetics and confirmed by low-coverage whole-genome sequencing. Of note, this same genetic background was found to promote clonal outgrowth in a CRISPR/Cas9 screen [39]. Hence, although low expression of cohesin is sufficient for cell survival, complete cohesin loss is incompatible with cellular viability, consistent with the sister chromatid alignment function requiring less cohesin than intranuclear architecture, demonstrating that the effects of cohesinopathies are dose-dependent.
Although the full mechanism remains incompletely understood, the effect of cohesin loss, with respect to transcriptionally active regions of euchromatin, is depicted (Fig. 1). Multiple model systems have shown an expansion of HSC gene signatures and these genes remain expressed in cohesin mutant HSC either because of redundant regulatory element structure or from an increased affinity to binding the residual cohesin. Complete loss of cohesin is lethal, likely impairing key genes for cell survival as well as leading to chromosomal catastrophe if cell division is attempted. A select number of genes - many of which are lineage-defining transcription factors-are highly unstable and are the most down-regulated with cohesin loss. These genes tend to have less complex local regulatory element profiles and leads to an overall more relaxed chromatin conformation. Such as the example with Smc3 and Flt3-ITD, the permissive chromatin structure cooperates with and amplifies FLT3 signaling through STAT5, resulting in clonal polarization and leukemic transformation.
FIGURE 1.
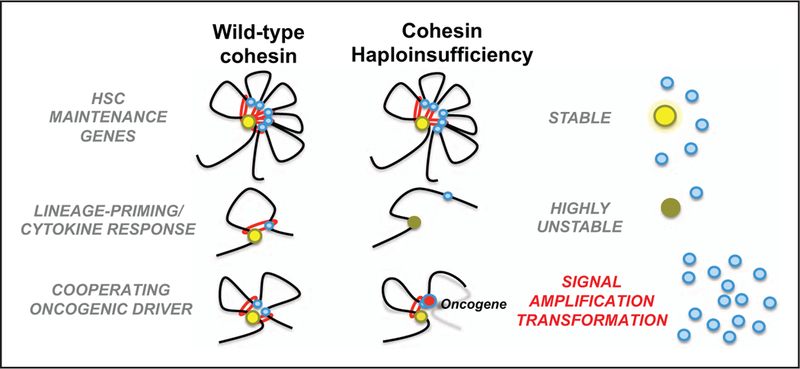
Proposed mechanism for tumor-suppressor function of cohesin in cancer. Complex chromatin structure at stem-maintenance genes either have increased affinity for remaining cohesin or have redundant structural stability and maintain expression. Genes essential for lineage priming, differentiation, and environmental response are highly sensitive and expression is impaired. Abnormal HSC maturation likely contributes to a myelodysplasia phenotype and with cooperating mutations leading to an overall relaxed chromatin state, which can amplify the signal of an oncogene-addicted clone resulting in clonal expansion and transformation.
THERAPEUTIC POTENTIAL
Data from retrospective clinical reports as well as translational mechanistic model systems have attempted to determine if specific cohesin-mutant dependencies might be exploited for therapeutic insight. Several studies are suggestive, but inconclusive that cohesin-mutant AML may have a better response to hypomethylating agents. It is also difficult to determine if this reflects a real cohesin-specific response or an MDS-specific response, given the enrichment of cohesin mutations in MDS and secondary AML. Data from the model systems have demonstrated that mutations in cohesin complex members contribute to transformation through reduced, but not absent cohesin function. As such, there is great therapeutic potential in that further impairment of cohesin function may result in synthetic lethality.
At least six targets already have chemical agents under investigation that may have cohesin-specific effects, with inhibitors of Aurora kinase, and PLK1 already in clinical trials for tumor site-specific indications, although no specific association with cohesin mutational status has been reported. Moreover, other key proteins that facilitate looping function such as BRD4 and the DNA damage repair function such as Poly ADP ribose protein (PARP) may also be unique sensitivities of cohesin mutant tumors. Cohesin has a role in maintaining the stability and integrity of the replication fork. Thus, it has been hypothesized that a stalled replication fork might be a terminal event whenever in concert with PARP inhibition. Two groups have shown in the SMCl-ortholog, him-1 deficient yeast [40], and in STAG2 mutant human glioblastoma cell lines [41] that PARP inhibition has a potential therapeutic window. It is evident that Smc3 acetylation is a regulatory component of replication fork speed [42], yet whether all cohesin mutations similarly affect fork stalling is not yet established.
Recently two groups have leveraged RNAi or CRISPR/Cas9 screen technology to determine if there are unique and specific targets in cell line systems with knockdown of STAG2. In both reports, a synthetic lethal interaction was found with codepletion of STAG1 [43,44▪▪]. This highlights a potential therapeutic target in STAG1, though unclear if this would be unique to STAG2 mutations alone or generalizable to all cohesin mutations.
STAG2 VERSUS THE FIELD
It is also important to note that caution and hesitancy should be taken whenever generalizing results for individual cohesin gene mutations to the entire cohesin family. Although all cohesin subunits are mutated in MDS/AML, this is not true of solid tumors where STAG2 is the sole recurrent mutation. There are likely unique characteristics of STAG2 function that remain incompletely understood. STAG2 is an X-linked gene (as is SMC1A), which may explain its enrichment in male predominant tumors such as bladder cancer and Ewing’s sarcoma, however, there is no sex disparity in STAG2 mutant myeloid malignancies. Interestingly, the paralog of STAG2, STAG1 has tissue-specific expression and is expressed in hematopoietic tumors, which may explain why there is perhaps equivalency with female STAG2 mutations on an expressed X-chromosome may equivalently be tolerated whenever compared with the male hemizygous mutation.
Several reports have suggested that STAG2 and STAG1 differ in their geographic location on chromosomes with the former being bound to centromeric regions and the latter resident at telomeric regions [45,46]. It must be stressed that these findings are specifically referring to morphologic differences observed in condensed chromosomes during replication and distinctly reflect the role of cohesin in sister chromatid cohesion. The distinct roles of STAG1 and STAG2 during interphase with regard to its essential role as orchestrator of transcriptional programming are not yet known. Given the viability of STAG2 mutant cells in clonal hematopoiesis, a redundant role, perhaps through STAG1 seems probable.
CONCLUSION
The effects of cohesin loss of function are anything but ‘simple’ as they are dependent on dosage, developmental state, and tissue type/cellular context. Cohesin has functions beyond canonical sister chromatid and its pathophysiologic function in human cancers is likely related to its role in orchestrating transcriptional regulatory networks. In model systems, cohesin loss of function maintains self-renewal/stem programs and impairs lineage priming/differentiation in HSC, but has little effect in committed cells. The broad impact of cohesin alterations on transcription is likely dependent on stoichiometry of the cohesin complex and its interaction with other epigenetic modifiers. Cohesin mutations may be an ideal genetic context for synthetic lethal dependencies, given the absolute requirement in HSC and multiple therapeutic opportunities.
KEY POINTS.
Models of hematopoietic cohesin dose-specific loss uniformly show stem cell expansion and impaired differentiation owing to a permissive and open-chromatin state, whereas complete cohesin loss is incompatible with cellular viability.
No affect on aneuploidy of chromosomal copy number has been observed in cohesin mutant patient data nor model systems of myeloid malignancies.
Cohesin alterations in myeloid malignancies are loss-of-function mutations, mutually exclusive, and most commonly in STAG2, which is also identified in solid tumors and clonal hematopoiesis.
The X-linked cohesin gene STAG2 has unique features compared with other cohesin complex members that are not completely understood including its exclusivity in solid tumors and presence of a redundant paralog in Stagl.
Acknowledgments
Financial support and sponsorship
A.V. was supported by a Damon Runyon Cancer Research Foundation Postdoctoral Fellowship Award (117-15).
Footnotes
Conflicts of interest
R.L. is a member of the Supervisory Board and its Science and Technology Committee QIAGEN, Scientific Advisory Board ofLoxo Pharmaceuticals, and is a Consultant for Novartis.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martincorena I, Raine K, Gerstung M, et al. Universal patterns of selection in cancer and somatic tissues. Cell 2017; 171:1029–1041.e21.■■ STAG2 mutations are identified in more than four distinct tumor histologies.
- 3.Haering CH, Lowe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell 2002; 9:773–788. [DOI] [PubMed] [Google Scholar]
- 4.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet 2009; 43:525–558. [DOI] [PubMed] [Google Scholar]
- 5.Mullenders J, Aranda-Orgilles B, Lhoumaud P, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med 2015; 212:1833–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viny AD, Ott CJ, Spitzer B, et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med 2015; 212:1819–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet 2009; 76:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deardorff MA, Bando M, Nakato R, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012; 489:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(R); 1993. [PubMed] [Google Scholar]
- 10.Xu B, Lu S, Gerton JL. Roberts syndrome: a deficit in acetylated cohesin leads to nucleolar dysfunction. Rare Dis 2014; 2:e27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010; 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wendt KS,Yoshida K,Itoh T, et al. Cohesin mediatestranscriptional insulation by CCCTC-binding factor. Nature 2008; 451:796–801. [DOI] [PubMed] [Google Scholar]
- 13.Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell 2013; 152:1285–1297. [DOI] [PubMed] [Google Scholar]
- 14.Rao SSP, Huang SC, St Hilaire BG, et al. Cohesin loss eliminates all loop domains. Cell 2017; 171:305–320.■■ Cohesin degradation results in loss of cis interactions within the boundaries of topologically associated domains.
- 15.Horsfield JA, Anagnostou SH, Hu JK, et al. Cohesin-dependent regulation of Runx genes. Development 2007; 134:2639–2649. [DOI] [PubMed] [Google Scholar]
- 16.Degner SC, Verma-Gaur J, Wong TP, et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture ofthe Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci USA 2011; 108: 9566–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaaf CA, Kwak H, Koenig A, et al. Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet 2013; 9:e1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaaf CA, Misulovin Z, Gause M, et al. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet 2013; 9:e1003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014; 28: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209–2221.■■ STAG2 mutations define AML patients in a chromatin-modified/spliceosome group with overall poor prognosis, although STAG2 mutations are not independently prognostic.
- 22.Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013; 45: 1232–1237. [DOI] [PubMed] [Google Scholar]
- 23.Thol F, Bollin R, Gehlhaar M, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood 2014; 123: 914–920. [DOI] [PubMed] [Google Scholar]
- 24.Thota S, Viny AD, Makishima H, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014; 124:1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478:64–69. [DOI] [PubMed] [Google Scholar]
- 27.Kim MS, Kim SS, Je EM, et al. Mutational and expressional analyses of STAG2 gene in solid cancers. Neoplasma 2012; 59:524–529. [DOI] [PubMed] [Google Scholar]
- 28.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with nonhematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017; 21:374–382.e4.■ STAG2 mutations are rare, but present in clonal hematopoiesis.
- 29.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep 2015; 10:1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 2012; 4:149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barber TD, McManus K, Yuen KW, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci USA 2008; 105:3443–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon DA, Kim T, Diaz-Martinez LA, et al. Mutational inactivation ofSTAG2 causes aneuploidy in human cancer. Science 2011; 333:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel JL, Schumacher JA, Frizzell K, et al. Coexisting and cooperating mutations in NPMI-mutated acute myeloid leukemia. Leuk Res 2017; 56: 7–12. [DOI] [PubMed] [Google Scholar]
- 36.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 2012; 120:e83–e92. [DOI] [PubMed] [Google Scholar]
- 37.Mazumdar C, Shen Y, Xavy S, et al. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 2015; 17:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher JB, Peterson J, Reimer M, et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 2017; 31:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tothova Z, Krill-Burger JM, Popova KD, et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell 2017; 21:547–555.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLellan JL, O’Neil NJ, Barrett I, et al. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet 2012; 8:e1002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey ML, O’Neil NJ, van Pel DM, et al. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther 2014; 13:724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terret ME, Sherwood R, Rahman S, et al. Cohesin acetylation speeds the replication fork. Nature 2009; 462:231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benedetti L, Cereda M, Monteverde L, et al. Synthetic lethal interaction between the tumour suppressor STAG2 and its paralog STAG1. Oncotarget 2017; 8:37619–37632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Lelij P, Lieb S, Jude J, et al. Synthetic lethality between the cohesion subunits STAG1 and STAG2 in diverse cancer contexts. Elife 2017; 6:pii: e26980.■ STAG1 represents a potential sythetic lethal target in STAG2-deficient cells.
- 45.Canudas S, Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol 2009; 187:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daniloski Z, Smith S. Loss of tumor suppressor STAG2 promotes telomere recombination and extends the replicative lifespan of normal human cells. Cancer Res 2017; 77:5530–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]