

Abstract
T-helper 17 (Th17) cells play an important role in the pathogenesis of multiple sclerosis (MS), an autoimmune demyelinating disease that affects the CNS. In the present study, MicroRNA sequencing (miRNA-seq) was performed in mouse Th0 and Th17 cells to determine the critical miRNAs that are related to Th17 differentiation. We found that miR-30a was significantly downregulated during mouse Th17 differentiation. In addition, the level of miR-30a in CD4+ T cells from peripheral blood of MS patients and experimental autoimmune encephalomyelitis (EAE) animal models was also decreased and inversely correlated with the expression of interleukin 17a, the canonical cytokine of Th17 cells. Moreover, overexpression of miR-30a inhibited Th17 differentiation and prevented the full development of EAE, whereas interference of miR-30a promoted Th17 differentiation. Mechanism studies showed that miR-30a reduced IRF4 expression by specifically binding with the 3′-untranslated region. Through screening of 640 different Food and Drug Administration (FDA)-approved drugs, we found that disulfiram and diphenhydramine hydrochloride were effective candidates for inhibiting Th17 differentiation and ameliorating EAE development through upregulating miR-30a. To our knowledge, the present work is not only the first miRNA-seq study focusing on Th17 differentiation, but also the first chemical screening for FDA-approved drugs that inhibit Th17 differentiation through regulating miRNA expression.
SIGNIFICANCE STATEMENT The present work is the first miRNA sequencing (miRNA-seq) study focusing on T-helper 17 (Th17) differentiation. By miRNA deep sequencing, we found that miR-30a was downregulated during Th17 differentiation. miR-30a was also decreased in CD4+ T cells from multiple sclerosis patients and experimental autoimmune encephalomyelitis (EAE) mice. miR-30a reduced IRF4 expression by specific binding with the 3′-untranslated region and thus suppressed Th17 differentiation and prevented the full development of EAE. Interestingly, by performing a chemical screen with Food and Drug Administration-approved small-molecule drugs, we found that disulfiram and diphenhydramine upregulated miR-30a and suppressed Th17-associated autoimmune demyelination.
Keywords: EAE, FDA-approved drugs, miRNAs, multiple sclerosis, Th17 cells
Introduction
Multiple sclerosis (MS) is an autoimmune demyelinating disease that affects the CNS. Most patients with MS have an initial relapsing–remitting neurological course (relapsing-remitting MS, RRMS) that later develops into a secondary progressive course (secondary progressive MS, SPMS) characterized by gradual neurological deterioration (Rudick et al., 1997). Neuron injury and the failure of the maturation of oligodendrocyte progenitor cells (OPCs) in MS lesions are believed to be the critical drivers for the progression from RRMS to SPMS (Zamvil and Steinman, 2003; Franklin and Ffrench-Constant, 2008). However, current therapies for MS are largely ineffective in halting disease progression or are accompanied by severe side effects (Degenhardt et al., 2009; Methner and Zipp, 2013).
T-helper 17 (Th17) cells that produce interleukin-17 (IL-17) are important mediators of MS and experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Bettelli et al., 2006). IL-17-, IL-17 receptor-, or IL-23 (the key cytokine enhancing Th17 differentiation)-deficient mice are resistant to EAE (Chen et al., 2006; Komiyama et al., 2006). The detection of high levels of IL-17 in both plaques and CSF of patients with MS also implicated a role for Th17 cells in the pathogenesis of MS (Matusevicius et al., 1999; Yang et al., 2014). In general, IL-17 is a potent proinflammatory mediator that facilitates proinflammatory cytokine production (Matusevicius et al., 1999). In addition to these functions, the present studies also suggested that IL-17 exerted cytotoxic effects on OPCs (Paintlia et al., 2011). Moreover, an in vivo imaging study revealed that Th17 cells contacted neurons in EAE directly and induced neuronal dysfunction (Siffrin et al., 2010). These direct effects on neurons and OPCs indicate that Th17 cells may also contribute to the disease progression from RRMS to SPMS. Understanding the mechanisms in the differentiation of Th17 cells will be helpful in identifying new and promising targets for MS therapy that not only decrease inflammatory responses but also slow disease progression to the irreversible neurological disability.
Although the list of genes that regulate the differentiation of naive T cells toward the Th17 phenotype is ever growing, the knowledge of noncoding RNAs in Th17 polarization is still very limited and needs to be clarified. MicroRNAs (miRNAs), a class of noncoding RNAs (∼22 nt in length), modulate gene expression by base pairing with specific sequences in the 3′-untranslated region (UTR) of target mRNAs, causing degradation of mRNA or inhibition of translation (Ambros, 2004). Previous reports suggested the presence of some deregulated miRNAs in Th17 cells, but most of these reports were based on PCR analysis (Du et al., 2009; Escobar et al., 2014; Wang et al., 2014; Brusselle and Bracke, 2015; Murugaiyan et al., 2015). High-throughput screening with an miRNA array or the newly developed next-generation sequencing (Mardis, 2008) would be beneficial toward achieving a comprehensive transcriptional view of deregulated miRNAs during Th17 differentiation and may reveal novel deregulated miRNAs that are important for Th17 polarization.
Here, using miRNA deep sequencing, we found that miR-30a was downregulated during Th17 differentiation. miR-30a was also decreased in CD4+ T cells from MS patients and EAE mice. miR-30 reduced IRF4 expression by specific binding with the 3′-UTR region, thus suppressing Th17 differentiation and preventing the full development of EAE. By performing a chemical screen with Food and Drug Administration (FDA)-approved small-molecule drugs, we found that disulfiram and diphenhydramine upregulated miR-30a and suppressed IL-17-associated autoimmune demyelination.
Materials and Methods
Cell cultures.
Peripheral blood mononuclear cells (PBMCs) were isolated from human peripheral blood using lymphocyte separation medium (GE Healthcare). After labeling with APC-labeled anti-human CD4 antibody (Sungene Biotech), CD4+ T cells were sorted by FACS from PBMCs. The sorted T cells were cultured in RPMI 1640 containing 10% fetal bovine serum (Invitrogen).
Mouse CD4+ T cells were isolated by magnetic cell sorting from the spleens of female C57BL/6 mice at 6–8 weeks of age using the Mice CD4+ T cell Isolation Kit (Miltenyi Biotec). Naive CD4+CD62L+ helper T cells were isolated using the Mice CD4+CD62L+ T Isolation KitII(Miltenyi Biotec). Sorted mouse T cells were also cultured in RPMI 1640 containing 10% fetal bovine serum (Invitrogen).
In the drug screening, CD4+ T cells were sorted from mouse spleen and cultured in RPMI 1640. Cells were harvested after adding the drug for 24 h.
T-cell differentiation.
Cell culture plates were coated with anti-CD3 antibody (5–10 μg/ml; BD Biosciences) overnight at 4°C before T-cell planting. Sorted naive CD4+CD62L+ helper T cells were seeded in 24-well plates at a quantity of 106/well. Anti-CD28 (1 μg/ml; BD Biosciences), anti-IL-4 (5 μg/ml; BD Biosciences), and anti-IFN-γ (5 μg/ml; BD Biosciences) were added to the control group, which was defined as the Th0 cells. For Th17 induction, anti-CD28 (1 μg/ml; BD Biosciences), anti-IL-4 (5 μg/ml; BD Biosciences), anti-IFN-γ (5 μg/ml; BD Biosciences), anti-IL-2 (5 μg/ml; BD Biosciences), anti-IL-12 (5 μg/ml; BD Biosciences), TGF-β (2 ng/ml; R&D Systems), IL-6 (40 ng/ml; R&D Systems), IL-1β (10 ng/ml; R&D Systems), and IL-23 (40 ng/ml; R&D Systems) were added. We also conducted Th17 differentiation in traditional Th17, skewing conditions by adding IL-6 (20 ng/ml; R&D Systems), TGF-β (3 ng/ml; R&D Systems), anti-CD28 (1 μg/ml; BD Biosciences), anti-IL-4 (5 μg/ml; BD Biosciences), and anti-IFN-γ (5 μg/ml; BD Biosciences). For regulatory T-cell (Treg) induction, anti-CD28 (1 μg/ml; BD Biosciences), anti-IL-4 (5 μg/ml; BD Biosciences), anti-IFN-γ (5 μg/ml; BD Biosciences), anti-IL-2 (5 μg/ml; BD Biosciences), and TGF-β (2 ng/ml; R&D Systems) were added. For Th1 induction, anti-CD28 (1 μg/ml; BD Biosciences), anti-IL-4 (5 μg/ml; BD Biosciences), and IL-12 (2 ng/ml; Peprotech) were added. For Th2 induction, anti-CD28 (1 μg/ml; BD Biosciences), anti-IFN-γ (5 μg/ml; BD Biosciences), and IL-4 (40 ng/ml; R&D Systems) were added. All of the cells were cultured for 6–7 d before further analysis.
miRNA-seq.
Total RNA was extracted from Th0 or Th17 cells using a mirVana miRNA Isolation Kit (Ambion). RNA quality was evaluated with a BioAnalyzer 2100 system (Agilent Technologies). Small RNAs had linkers ligated to them and bar-coded cDNAs were prepared using a TruSeq Small RNA Sample Prep Kit (Illumina) following the manufacturer's instructions. Individual libraries were analyzed for the presence of linked cDNA at the appropriate size (140–150 bp) as determined by the BioAnalyzer. Subsequently, the amplified cDNA constructs were purified from agarose gel in preparation for sequencing analysis using the Illumina HiSeq 2500 platform according to the manufacturer's instructions at the Shanghai Biotechnology.
The small RNA sequence reads were preprocessed using the FASTXToolkit to exclude low-quality reads (ambiguous N, quality <10 nt, and length <18 nt) and 3′ adapter, 5′ adapter and poly(A) sequences. Further annotation analyses were performed using the commercial software CLC Genomic Workbench version 5.5. After all annotation steps, the sequencing libraries were used for size distribution and saturation analysis.
RNA isolation and quantitative PCR.
Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA was synthesized using a RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). Quantitative PCR (qPCR) was performed on a LightCycler 96 apparatus (Roche) using the SYBR Green Real-time PCR Master Mix (Toyobo) or the TaqMan Gene Expression Assay (sno202 and miR-30a; Life Technologies). Gene expression was normalized to a standard housekeeping gene using the ΔΔCT method. Primer pairs are listed below as follows: IL-17a 5′-TTTAACTCCCTTGGCGCAAAA-3′ (sense); 5′-CTTTCCCTCCGCATTGACAC-3′ (antisense); IFN-γ 5′-ATGAACGCTACACACTGCATC-3′ (sense); 5′-CCATCCTTTTGCCAGTTCCTC-3′ (antisense); IL-4 5′-CCCCAGCTAGTTGTCATCC-3′ (sense); 5′-CCTCGTTCAAAATGCCGAT-3′ (antisense); TGF-β 5′-CACTGATACGCCTGAGTG-3′ (sense); 5′-GTGAGCGCTGAATCGAAA-3′ (antisense); IRF4 5′-ATTGTTTAAAGGCAAGTTCCGAGA-3′ (sense); 5′-CTCGACCAATTCCTCAAAGTCA-3′ (antisense); RORγt 5′-TCATCAATGCCAACCGTCCT-3′ (sense); 5′-AGCCAGTTCCAAATTGTATTGC-3′ (antisense); GAPDH 5′-AAATGGTGAAGGTCGGTGTG-3′ (sense); and 5′-AGGTCAATGAAGGGGTCGTT-3′ (antisense).
Animal experiments.
The animal experiments in this study were performed in adherence with the National Institutes of Health's Guidelines on the Care and Use of Laboratory Animals and were approved by the Animal Experimentation Ethics Committee of the Second Military Medical University. For disease induction of myelin oligodendrocyte glycoprotein (MOG)-EAE, female C57BL/6 mice (at 6 weeks) were purchased from Slac and further fed for 2–4 weeks before the induction. MOG35–55 (M-E-V-G-W-Y-R-S-P-F-S-R-V-V-H-L-Y-R-N-G-K, 150 ng per mouse; GL Biochem) in incomplete Freund's adjuvant (Sigma-Aldrich) containing heat-killed Mycobacterium tuberculosis (5 mg/ml, H37Ra strain; Difco) was subcutaneously injected at 3 points in the back of mice to be immunized and that day was defined as day 0. On days 0 and 2, pertussis toxin (250 ng in 200 μl of PBS per mouse; Calbiochem) in PBS was administered intraperitoneally.
For disease induction of PLP-EAE, female SJL/J mice (at 4 weeks) were obtained from Vitalriver and were further fed for 4 weeks before the induction. PLP139–151(H-S-L-G-K-W-L-G-H-P-D-K-F; GL Biochem) were used instead of MOG35–55 and the other procedures were as described above.
Clinical manifestations generally occurred at days 10–13 and were blindly examined daily from then onwards. The clinical score was determined on a scale of 0–5 as follows: 0, no clinical signs; 1, paralyzed tail; 2, ataxia and/or paresis of hind limbs; 3, paraplegia; 4, paraplegia with forelimb weakness or paralysis; and 5, moribund state or death.
In the study of the effects of miR-30a in MOG-EAE, 1 × 107 viral particles overexpressing miR-30a were injected via the tail vein on day 2 after the immunization. In the study of the effects of disulfiram and diphenhydramine hydrochloride in MOG-EAE, an intraperitoneal injection of 10 mg/kg disulfiram or diphenhydramine was administered daily after immunization.
Patients.
After written informed consent was obtained, we collected the peripheral blood of 30 relapsing (10 males and 20 females) patients and 30 remitting patients (10 males and 20 females) suffering from RRMS and 28 sex- and age-matched healthy controls. The blood samples were obtained in Shanghai Changhai Hospital and Affiliated Hospital of the Bengbu Medical College. The diagnosis of MS matched published criteria (McDonald et al., 2001). Patients were hospitalized due to relapse and did not receive hormone or immunosuppressive treatment for at least 1 month before hospitalization. The EDSS (expanded disability status scale) score was set by a well trained neurologist with extensive clinical experience. Healthy controls were individuals participating in a physical examination.
FISH.
The LNA microRNA probe that was used for FISH was purchased from EXIQON. The TSA-Plus Fluorescein System was purchased from PerkinElmer. For detailed procedures, please refer to a previous report (de Planell-Saguer et al., 2010).
Western blot and ELISA.
Primary cell cultures were homogenized in NP40 buffer (Beyotime) that was supplemented with protease cocktail inhibitors (Roche). Cell lysates were subjected to Western blotting using anti-IRF4 (1:500; Santa Cruz Biotechnology) antibody and HRP-conjugated anti-GAPDH (Kangcheng). The protein bands were analyzed using Image Lab analysis (Bio-Rad).
The level of IL-17a in culture supernatant was quantified using an ELISA kit (Shanghai Jiayuan Biotech) according to the manufacturer's instructions. The sample concentrations were calculated using an equation generated from a standard curve.
Methyl tetrazolium (MTT) assay.
Mouse naive CD4+CD62L+ helper T cells were isolated by magnetic cell sorting from the spleens of female C57BL/6 mice at 6–8 weeks of age as described above and cultured in 96-well plate for 6–7 d after adding drugs of different concentrations as well as anti-CD3 (0.5 μg/ml; BD Biosciences) and anti-CD28 (1 μg/ml; BD Biosciences) antibodies. At the end of the incubation period, 20 μl of MTT solution (stock concentration, 5.0 mg/ml in PBS) was added to each well and incubated for 5 h. Thereafter, T cells were collected by centrifugation, resuspended in 200 μl of DMSO to dissolve formazan crystals, and the absorbance values were read at 550 nm. Cell viability was compared with the control adding drug solvent.
Luciferase.
HEK293T cells were maintained in DMEM containing 10% fetal bovine serum (Invitrogen) and cells of 30–40% confluence in 12-well plates were transfected with Lipofectamine 2000 (Invitrogen). A dual luciferase reporter system was used in this work and the binding between miR-30a and the 3′-UTR of IRF4 was shown by a decrease in the fluorescence intensity. The 3′-UTR of IRF4 was cloned after luciferase in pGL3-basic vector and the binding between miR-30a and the 3′-UTR of IRF4 linked to the mRNA of luciferase impeded the translation of luciferase. The luciferase reporter gene construct (500 ng per well), the pRL-SV40 Renilla luciferase construct (1 ng per well; for normalization), and AgomiR-30a (500 ng per well; GenePharma) were all cotransfected. Cell extracts were prepared 24 h after transfection and the luciferase activity was measured with the Dual-Luciferase Reporter Assay system (Promega).
Lentivirus transduction and electroporation.
The sequence of primary miR-30a was synthesized and ligated into the GV271 plasmid (GeneChem). The cloned sequence starts from GTTTACAGAATGTTGCCTGT and ends at TTTCTCTTTGATTTATTTTT. The titer of concentrated viral particles was 1 × 109 transducing units/ml.
GenePharma synthesized AgomiR-30a and AntagomiR-30a. Primary mouse CD4+ T cells were transfected using electroporation (program X-01; Amaxa) with AgomiR-30a, or AntagomiR-30a, or the “scrambled” control at 72 h after activation by adding anti-CD3 (0.5 μg/ml; BD Biosciences) and anti-CD28 (1 μg/ml; BD Biosciences) antibodies.
Flow cytometry analysis.
For the isolation of CD4+ T cells in the CNS from EAE and control, mice were killed and the cerebra and spinal cords were collected for homogenization and digestion. Then cells were suspended in 37% Percoll (GE Healthcare) and underlaid with 70% Percoll while overlaid with 30% Percoll for gradient centrifugation. The majority of the mononuclear cells were collected and stained with FITC-CD4 (BD Biosciences) antibody. CD4+ T cells were sorted on a Moflo XDP (Beckman Coulter) and assessed by qPCR.
For cell-type analysis, cells were collected and centrifuged. Surface staining was performed after resuspending the cells in FACS buffer with the corresponding antibodies. After surface staining, the cells were resuspended in fixation buffer (eBioscience) then permeabilization buffer (eBioscience) and intracellular cytokine staining was performed according to the manufacturer's protocol. APC-labeled anti-human CD4 antibody (Sungene) and FITC-labeled anti-mouse CD4 antibody (BD Biosciences) was used for surface staining. PE-labeled anti-mouse IL-17a antibody (Sungene) and APC-labeled anti-mouse IFN-γ antibody (Sungene) were used for intracellular staining. Cells were assessed or sorted on a Moflo XDP (Beckman Coulter).
H&E staining and Luxol fast blue (LFB) staining.
Lumbar spinal cords of mice were isolated and cut into cryosections (14 μm) or paraffin sections (5 μm). One of every 5 (cryosection) or 10 (paraffin section) successive sections was collected from each animal. The sections were stained with H&E solution or LFB and periodic acid Schiff. For H&E staining, the infiltrating inflammatory cells in one whole spinal cord slide were counted and all of the successive sections were calculated (total cell number − mean cell number in normal control) for one EAE mice. For LFB staining, both the area of the demyelinated white matter and the total white matter in one whole spinal cord slide were measured and all of the successive sections were calculated for one EAE mice, then the ratio of the demyelinated WM/ total WM was calculated. Independent readers blindly scored H&E or LFB sections and at least three animals in different treatment groups were included.
Statistical analysis.
A two-tailed Student's t test was applied for statistical comparison of two groups and a one-way ANOVA with a Tukey's or Dunnett's post hoc test was used for multiple groups. The EAE model was analyzed using the nonparametric Mann–Whitney U test. The data are presented as the mean ± SEM unless otherwise indicated and mean ± SEM generally represents biological replicates. p < 0.05 was considered statistically significant.
Results
miR-30a is downregulated in Th17 cells
We applied massively parallel signature sequencing to carry out an in-depth analysis of the miRNomes in activated, but undifferentiated CD4+ T cells (Th0) and Th17 cells. The abundance value of each known miRNA was normalized using ‘transcripts per million (TPM). Because minimum threshold amounts must be reached for miRNAs to repress their target mRNAs (Brown et al., 2007; Sarasin-Filipowicz et al., 2009) and the abundance of miRNAs in the entire miRNome of specific cell may be an important indicator for their functions (Hou et al., 2011), we only considered miRNAs with TPM >500 in Th17 or Th0 (accounting for 98% of the miRNome) and >1.5-fold alteration for further analysis. Using this strategy, 25 downregulated and 16 upregulated miRNAs were screened out (Fig. 1A). Furthermore, we selected 15 miRNAs (12 downregulated, three upregulated) that were highly conserved in both human and mouse (conservation value >0.99) (Kiezun et al., 2012) to retrieve their possible targets in the miRBASE database and five miRNAs possessed targets concerning Th17 differentiation. After excluding the miRNAs that have been reported previously to play a role in T-cell differentiation, miR-30a-5p was chosen for further study.
Figure 1.

Downregulation of miR-30a in Th17 cells and CD4+ T cells from EAE mice and MS patients. A, Screening of miRNAs that are differentially expressed in Th0 and Th17 cells. The volcano plots showing genes that are upregulated (red points) or downregulated (green points) for >1.5-fold (p < 0.05) after Th17 differentiation. B, qPCR analysis of miR-30a level in Th0, pathogenic, and traditional Th17 and Treg cells differentiated in vitro (n = 4 independent experiments; **p < 0.01, ***p < 0.005 vs Th0 cells). C, miR-30a level in mice CD4+ T cells stimulated with Th17 regulators IL-6 (40 ng/ml), TGF-β (20 ng/ml), or IL-23 (40 ng/ml) for 24 h (n = 4 independent experiments). D, qPCR analysis of miR-30a in CD4+ T cells during EAE development. Control: CFA without MOG, preclinic: day 10 and peak: day 16 after EAE induction (n = 5 mice per group). E, qPCR analysis of miR-30a in CD4+ T cells sorted from the CNS of PLP-EAE SJS/J mice at remitting phase by FACS (n = 3 mice per group). F, Linear regression analysis of miR-30a with IL-17a mRNA level in CD4+ T cells isolated from EAE mice. Each point represents one mouse (n = 9 mice). G, Linear regression analysis of miR-30a with TGF-β mRNA level in CD4+ T cells isolated from EAE mice (n = 9 mice). H, qPCR analysis of miR-30a in CD4+ T cells from relapsing or remitting MS patients (n = 30 per group) and their controls (n = 28). I, Linear regression analysis of miR-30a with IL-17a mRNA level in CD4+ T cells isolated from relapsing MS patients. Each point represents one patient (n = 30). J, FISH analysis of miR-30a (green) costained with anti-IL-17a antibody (red) in CD4+ T cells from patients with relapsing MS. White arrow indicates CD4+ T cell containing high miR-30a with low IL-17a; yellow arrows indicate the opposite. Scale bar, 10 μm. K, Fluorescence intensity of miR-30a in IL-17a+ and IL-17a− cells. L, Linear regression analysis of the fluorescence intensity of IL-17a with miR-30a (n = 15 representative cells; *p < 0.05, **p < 0.01, **p < 0.01 vs control). B–D, Spleen; H, one-way ANOVA with Dunnett's post hoc test; D, E, CNS; K, Student's t test. Data are shown as mean ± SEM.
We then induced naive CD4+ T cells to differentiate into Th17 and Treg cells in vitro and evaluated miR-30a levels using qPCR. The miR-30a level was found significantly downregulated in Th17 cells but upregulated in Treg cells compared with Th0 cells (Th17/Th0: 0.08 ± 0.01, p < 0.001; Treg/Th0: 1.44 ± 0.16, p = 0.015; one-way ANOVA with Dunnett's post hoc test; n = 4 independent experiments; Fig. 1B). To determine which cytokine is responsible for the regulation of miR-30a, we added IL-6, TGF-β, or IL-23, which are required in Th17 or Treg differentiation, to mice CD4+ T cells. The qPCR showed that the miR-30a level was significantly reduced after IL-6 treatment for 24 h, suggesting that the downregulation of miR-30a in Th17 cells may be caused by IL-6 (IL-6/control: 0.49 ± 0.10, p = 0.007; TGF-β/control: 1.43 ± 0.04, p = 0.02; IL-23/control: 0.73 ± 0.16, p = 0.154; one-way ANOVA with Dunnett's post hoc test; n = 4 independent experiments; Fig. 1C). Interestingly, we also found that the miR-30a level was increased after treatment with TGF-β, which can induce Treg differentiation (Fig. 1C).
miR-30a is downregulated in CD4+ T cells and Th17 cells from EAE mice and MS patients
Because the Th17 and Treg cells are critical subtypes of CD4+ T cells in the pathogenesis of EAE, we next explored the correlation between miR-30a level and EAE. We examined the miR-30a level in the spleen CD4+ T cells sorted using magnetic beads at the onset and acute phases of EAE. The results showed that miR-30a was downregulated at the onset phase and an even lower level was observed at acute phase (Pre/control: 0.44 ± 0.02, p < 0.001; Peak/control: 0.33 ± 0.02, p < 0.001; one-way ANOVA with Dunnett's post hoc test; n = 5 mice; Fig. 1D). In addition, we also sorted CD4+ T cells in the CNS by FACS at peak phase in EAE mice (day 16) and miR-30a was also found downregulated (0.39 ± 0.05, p < 0.001, unpaired t test, n = 5 mice; Fig. 1D). PLP-EAE induced in SJL/J mice showed a remitting–relapsing pattern of disease and was considered similar to RRMS in humans. We conducted the PLP-EAE induction and sorted CD4+ T cells in the CNS by FACS at remitting phase (day 19). miR-30a was also found to be downregulated (0.17 ± 0.01, p < 0.001, unpaired t test, n = 3 mice; Fig. 1E). These results indicated miR-30a might be involved in the pathogenesis of EAE. Moreover, because the in vitro experiments showed that the miR-30a level was changed during Th17 and Treg development, we also analyzed the in vivo relationship between miR-30a and Th17 or Treg cells, which specifically produce IL-17a and TGF-β, respectively. We isolated spleen CD4+ T cells from EAE mice (n = 9 mice) and analyzed the relationship between the levels of miR-30a and the mRNA of IL-17a or TGF-β using a linear regression analysis. The results showed a significant negative correlation of the miR-30a level with IL-17a (R2 = 0.7255, linear regression; n = 9 mice; Fig. 1F), but no significant correlation with TGF-β (R2 = 0.0014, linear regression; n = 9 mice; Fig. 1G). These results indicated that the miR-30a level was correlated with Th17, but not with Treg cells, in vivo.
Furthermore, we explored the miR-30a level in CD4+ T cells from MS patients (Table 1). We isolated PBMCs from 30 relapsing and 30 remitting patients with RRMS and 28 healthy donors using lymphocyte separation medium. Then, CD4+ T cells were sorted using FACS and the miR-30a level was evaluated using qPCR. The results showed that the miR-30a level in patients was significantly decreased (relapsing: 0.28 ± 0.02, p < 0.001; remitting: 0.72 ± 0.05, p = 0.012; control = 28, relapsing patients = 30, remitting patient = 30, one-way ANOVA with Dunnett's post hoc test; Fig. 1H). Moreover, the mRNA level of IL-17a showed a significant negative correlation with the miR-30a level at the relapsing phase of MS patients, which is consistent with the negative correlation between miR-30a level and IL-17a in EAE mice (R2 = 0.5933, linear regression; n = 30 relapsing patients; Fig. 1I). Furthermore, our FISH experiments using the CD4+ T cells from MS patients showed low expression of IL-17a in cells with a high expression of miR-30a (Fig. 1J), whereas the signal intensity of miR-30a was significantly lower in IL-17a+ cells (0.45 ± 0.03, p < 0.001, unpaired t test, n = 15 representative cells; Fig. 1K). The fluorescence intensity of miR-30a and IL-17a showed a negative correlation (R2 = 0.6316, linear regression; n = 15 representative cells; Fig. 1L). These results indicate that the decreased miR-30a level in MS patients and during the acute phase of EAE may be associated with increased Th17 cells.
Table 1.
Characteristics of patients and controls
| Control | Relapsing RRMS | Remitting RRMS | |
|---|---|---|---|
| Sample size | 28 | 30 | 30 |
| Age | 44.82 ± 7.43 | 44.10 ± 11.79 | 43.00 ± 11.38 |
| Sex | |||
| Female | 16 (57.14%) | 20 (66.67%) | 20 (66.67%) |
| Male | 12 (42.86%) | 10 (33.33%) | 10 (33.33%) |
| EDSS | NA | 4.32 ± 1.72 | 1.05 ± 0.53 |
Shown is relevant information about human subjects recruited for this study. Sample size is total number of subjects; age and EDSS are presented as mean ± SD; sex is total number (with percent of group in parentheses). Subjects were patients from the outpatient clinic of Changhai Hospital (Shanghai, China) or Affiliated Hospital of Bengbu Medical College (Anhui, China) with clinically defined RRMS or age- and sex-matched healthy volunteers from personnel of the institute. NA, Not applicable.
miR-30a inhibits Th17 differentiation in vitro
To investigate whether miR-30a can regulate the differentiation of Th17 cells directly, we transfected miR-30a mimics (AgomiR-30a) using electroporation into mouse spleen CD4+CD62+ T cells that were sorted using magnetic beads. Next, the cells were cultured for 6 d under Th17 differentiation conditions before testing. The results showed that the miR-30a level was significantly increased after AgomiR-30a transfection (327.39 ± 39.01, p = 0.004, unpaired t test, n = 4 independent experiments; Fig. 2A), whereas the mRNA levels of IL-17a and RORγt (key transcription factor in Th17 differentiation) were significantly reduced (IL-17a: 0.01 ± 0.01, p < 0.001; RORC: 0.08 ± 0.01, p < 0.001; IL-4: 1.15 ± 0.08, p = 0.107; unpaired t test, n = 5 independent experiments; Fig. 2B). Flow cytometry showed that IL-17a+ cells were significantly reduced after AgomiR-30a transfection [negative control (NC): 21 ± 1.20, 30a: 10.34 ± 0.59, p < 0.001, unpaired t test, n = 4 independent experiments; Fig. 2C,D]. ELISA also revealed lower IL-17a levels in the supernatant after the transfection with AgomiR-30a (NC: 4.11 ± 0.15, 30a: 2.79 ± 0.29, p = 0.007, unpaired t test, n = 4 independent experiments; Fig. 2E). In contrast, after transfection of the miR-30a inhibitors (AntagomiR-30a) by electroporation, the expression levels of IL-17a and RORγt were significantly increased (IL-17a: 4.32 ± 0.46, p = 0.002; RORC: 2.67 ± 0.30, p = 0.005; IL-4: 0.9 ± 0.13, p = 0.489; unpaired t test, n = 5 independent experiments; Fig. 2F). Flow cytometry showed increased IL-17a+ cells (NC: 20.32 ± 1.97, 30a: 35.59 ± 0.80, p = 0.002, unpaired t test, n = 4 independent experiments; Fig. 2G,H), and ELISA showed elevated IL-17a levels in the supernatant (NC: 4.11 ± 0.15, 30a: 5.79 ± 0.35, p = 0.005, unpaired t test, n = 4 independent experiments; Fig. 2I). These results suggest that miR-30a can negatively regulate the differentiation of Th17 cells.
Figure 2.
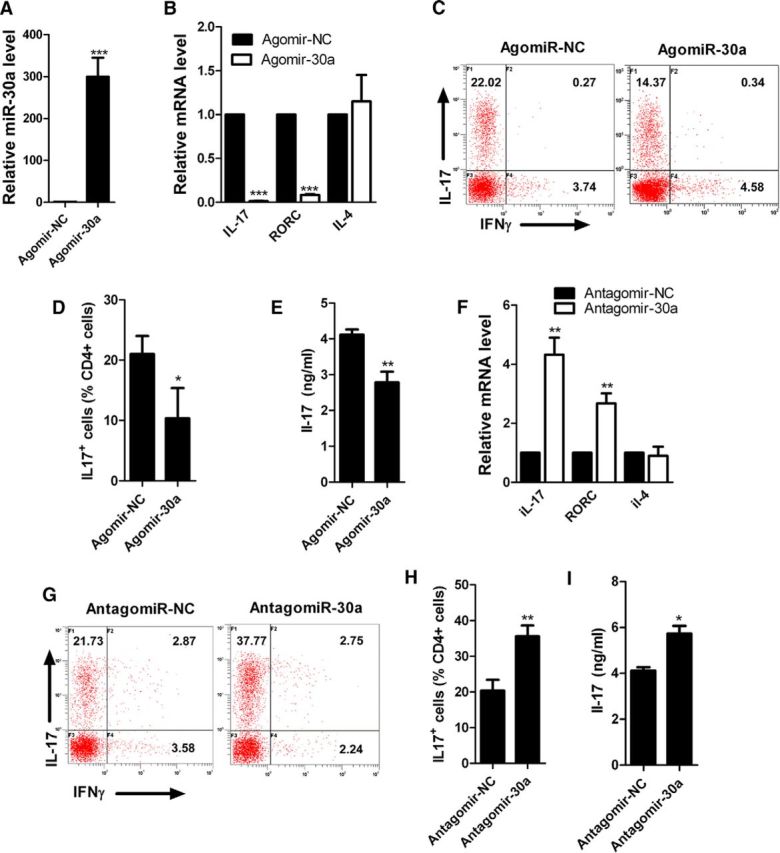
Inhibition of Th17 differentiation by miR-30a in vitro. A, qPCR analysis of miR-30a in CD4+CD62L+ T cells nucleotransfected with AgomiR-30a or its negative control (Agomir-NC) for 3 d in vitro (n = 4 independent experiments). B, qPCR analysis of IL-17a and RORγt in CD4+CD62L+ T cells nucleotransfected with AgomiR-30a/Agomir-NC and cultured for 6 d in Th17-polarizing conditions in vitro (n = 5 independent experiments). C, Intracellular staining of IL-17a of cells in B by FACS. Numbers in quadrant C indicate the percentage of IL-17a+IFN-γ− cells (top left) or IL-17a−IFN-γ+ cells (bottom right) (n = 4 independent experiments). D, Frequency of CD4+IL-17a+ (Th17) cells in C. E, ELISA analysis of IL-17a in culture supernatant of cells in B. F, qPCR analysis of IL-17a and RORγt in CD4+CD62L+ T cells transfected with AntagomiR-30a and its negative control (Antagomir-NC) and cultured for 6 d in Th17-polarizing conditions (n = 5 independent experiments). G, Intracellular staining of IL-17a of cells in F. H, Frequency of CD4+IL-17a+ (Th17) cells in G (n = 4 independent experiments). I, ELISA analysis of IL-17a in culture supernatant of cells in F (*p < 0.05, **p < 0.01, ***p < 0.001 vs control, Student's t test). Data are shown as mean ± SEM.
miR-30a prevents disease from developing fully and suppresses Th17 differentiation during EAE
Because Th17 cells are one of the most important participants in the pathogenesis of EAE and miR-30a can regulate Th17 differentiation negatively, to investigate whether miR-30a affects the progression of EAE, we intravenously injected lentivirus encoding a pre-miR-30a sequence (LV-miR-30a) into EAE mice at day 2 after immunization and evaluated the expression of miR-30a in vivo. The miR-30a level was significantly increased in the liver, spleen, lymph nodes, and brain after the injection of LV-miR-30a (liver: 6.36 ± 0.49, p < 0.001; spleen: 18.12 ± 1.25, p < 0.001; lymph nodes: 5.20 ± 0.65, p < 0.001; brain: 1.58 ± 0.20, p = 0.026; unpaired t test, n = 4 mice; Fig. 3A). We found that the mice injected with LV-miR-30a showed decreased EAE scores (p = 0.002, Mann–Whitney U test, n = 10 mice; Fig. 3B). Moreover, the spinal cords of EAE mice were taken for histological analysis. H&E staining showed decreased infiltration of inflammatory cells (NC: 823.33 ± 44.49, 30a: 373.33 ± 29.08, p < 0.001, unpaired t test, n = 4 mice; Fig. 3C,E) and LFB staining showed attenuated white matter demyelination after LV-miR-30a injection (NC: 23.33 ± 1.92, 30a: 4.53 ± 0.96, p < 0.001, unpaired t test, n = 4 mice; Fig. 3D,F).
Figure 3.
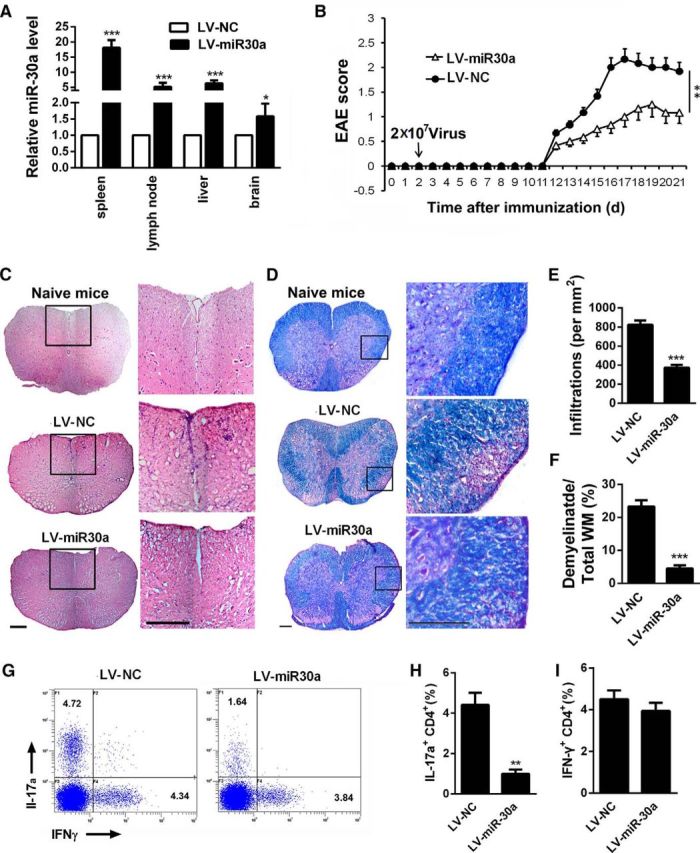
Preventing the full development of EAE by miR-30a. A, qPCR analysis of miR-30a in the liver, spleen, PLNs, and brain of mice injected with lentivirus encoding miR-30a (LV-miR-30a) or its control (LV-ctrl) at day 7 after lentivirus administration (n = 4 mice per group). B, Clinical scores of EAE in mice infected with lentivirus in A (n = 10 mice per group). EAE scores were analyzed using nonparametric Mann–Whitney U test. C, D, Representative spinal cord sections of H&E staining (C) and LFB staining (D) from EAE mice at day 21 after immunization. Scale bars, 70 μm. E, F, Quantification of spinal cord infiltration in white matter (WM) (E) and the percentage of demyelinated WM in total WM (F) in C and D (n = 3 mice per group). G, Intracellular staining of IL-17a and IFN-γ in DLN cells isolated from lentivirus-infected mice on day 14 after immunization. Numbers in quadrants indicated frequency of CD4+ cells. H, I, Frequency of CD4+IL-17a+ (Th17) cells (H) and CD4+IFN-γ+ (Th1) cells (I) in G (n = 3 mice per group; *p < 0.05, p < 0.01, ***p < 0.001 vs control; A, E, F, H, I, Student's t test; B, Mann–Whitney U test). Data are shown as mean ± SEM.
We then sought to determine whether miR-30a can influence Th17 differentiation in vivo. Therefore, we examined draining lymph nodes (DLNs, pooled peripheral lymph nodes from subiliac, proper axillary, and accessory axillary lymph nodes) from LV-miR-30a injected mice for the presence of IL-17a (Th17)- or IFN-γ (Th1)-producing CD4+ T cells during EAE. On day 14 after immunization with MOG35–55, LV-miR-30a-injected mice had significantly diminished amounts of Th17 cells in their DLNs compared with control mice. The amount of IFN-γ-producing Th1 CD4+ cells in the DLNs of LV-miR-30a injected mice were not significantly changed (NC: 4.41 ± 0.65, 30a: 0.99 ± 0.24, p = 0.006, unpaired t test, n = 3 mice; Fig. 3G,H; NC: 4.50 ± 0.47, 30a: 3.94 ± 0.42, p = 0.39, unpaired t test, n = 3 mice; Fig. 3G,I). These results demonstrated that miR-30a decreased Th17 differentiation and prevented the full development of pathological injuries during EAE, which indicates that increasing the miR-30a level in Th17 cells may have effects on the prevention of EAE and MS.
IRF4 is a functional target of miR-30a
To elucidate how miR-30a inhibits the differentiation of Th17 cells, we used Targetscan and miRBase to search for potential target genes; the 3′-UTR region of IRF4 was predicted to contain a complementary binding sequence for miR-30a (Fig. 4A). We then verified the binding using a luciferase reporter system. Cotransfecting 293T cells with miR-30a/IRF4 3′-UTR inhibited luciferase activity, whereas cotransfection of mutant miR-30a/IRF4 3′-UTR or miR-30a/mutant IRF4 3′-UTR failed to affect luciferase activity (WT 3′UTR + WT 30a: 0.43 ± 0.06, mutant 3′UTR + WT 30a: 0.95 ± 0.13, WT 3′UTR + mutant 30a: 0.98 ± 0.13; (WT + WT)/(mutant/mutant): p = 0.004, one-way ANOVA with Dunnett's post hoc test, n = 3 independent experiments; Fig. 4B). These results validated the specific binding between miR-30a and the 3′-UTR region of IRF4.
Figure 4.
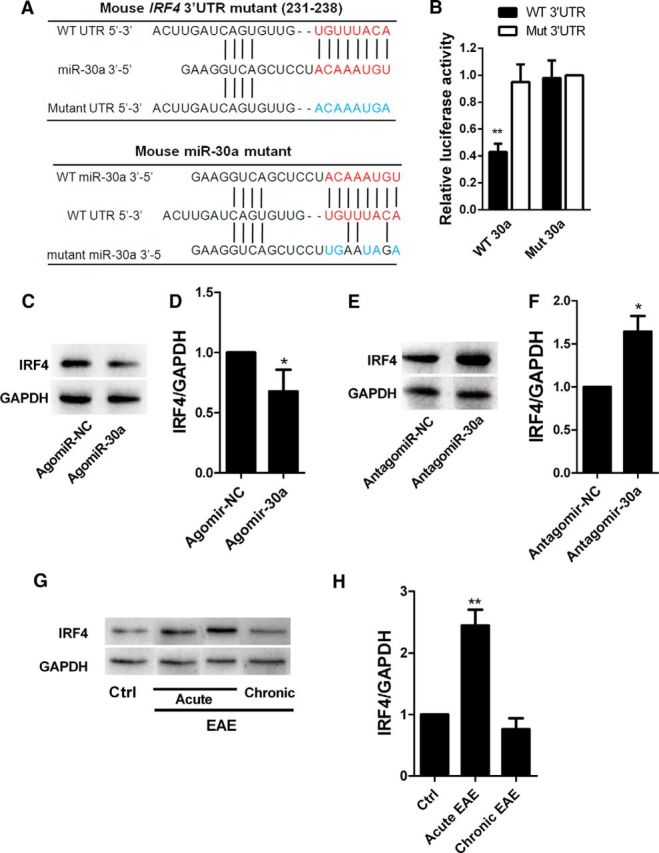
IRF4 is the functional target of miR-30a. A, Putative miR-30a-binding sites in IRF4 mRNA 3′-UTR and mutations in the IRF4 mRNA 3′-UTR (top) and miR-30a (bottom). B, Luciferase activity of reporter carrying the mutated (Mut3′-UTR) or wild-type (WT3′-UTR) IRF4 mRNA 3′-UTR cotransfected into HEK293T cells with wild-type miR-30a (WT 30a) or its mutant (mutant 30a) (n = 3 independent experiments). C, Western blot analysis of IRF4 in CD4+ T cells after transduction with AgomiR-30a or its control (Agomir-NC) for 6 d. D, Quantification of IRF4 densitometry normalized to GAPDH in C (n = 3 independent experiments). E, Western blot analysis of IRF4 in CD4+ T cells after transduction with AntagomiR-30a or its control (Antagomir-NC) for 6 d. F, Quantification of IRF4 densitometry normalized to GAPDH in E (n = 3 independent experiments). G, Western blot analysis of IRF4 in CD4+ T cells from EAE mice at different phases. H, Quantification of IRF4 densitometry normalized to GAPDH in G (n = 3 independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001 vs control; D, F, Student's t test; B, H, one-way ANOVA with Dunnett's post hoc test). Data are from at least three experiments and are shown as mean ± SEM.
Furthermore, we transfected mouse spleen CD4+ T cells with AgomiR-30a or AntagomiR-30a and detected the IRF4 expression level 4 d later. Western blots showed downregulated expression of IRF4 in CD4+ T cells overexpressing miR-30a (0.66 ± 0.08, p = 0.011, unpaired t test, n = 3 independent experiments; Fig. 4C,D), whereas interfering miR-30a had the opposite effect (1.68 ± 0.11, p = 0.027, unpaired t test, n = 3 independent experiments; Fig. 4E,F). These results demonstrated that miR-30a regulated the protein expression level of IRF4. In addition, we examined the expression level of IRF4 in CD4+ T cells in different phases of EAE. The results showed that IRF4 expression increased in the acute phase but decreased back to normal level in the chronic phase (acute: 2.44 ± 0.43, p = 0.008; chronic: 0.46 ± 0.67, p = 0.245; one-way ANOVA with Dunnett's post hoc test, n = 3 independent experiments; Fig. 4G,H), which was consistent with the expression pattern of miR-30a in CD4+ T cells from EAE mice. Because IRF4 is essential in Th17 differentiation and EAE development (Huber et al., 2008; Huber et al., 2013), our results indicated that the effect of miR-30a on Th17 differentiation and EAE development might occur through the regulation of IRF4 expression.
disulfiram and diphenhydramine hydrochloride upregulate miR-30a and suppress Th17 differentiation
As a negative regulator of Th17 differentiation, the miR-30a level should be negatively associated with the differentiation of Th17 cells. Therefore, we sought drugs that can inhibit Th17 differentiation by increasing the miR-30a level. We screened 640 FDA-approved small-molecule drug compounds based on this strategy and found 72 drugs that could increase the miR-30a level in CD4+ T cells significantly. We then excluded the compounds that may cause severe side effects, such as antineoplastic agents, anesthetic drugs, drugs resulting in significant mental changes, and drugs that significantly change blood pressure and the respiratory system. We also excluded compounds that already have been reported to reduce Th17 differentiation or promote EAE repair, including steroid hormones (Tischner and Reichardt, 2007; Gold and Voskuhl, 2009), artemisinin (Zhao et al., 2012) and sildenafil (Pifarré et al., 2014). We took the remaining 11 compounds for further use in a Th17 differentiation assay and found that diphenhydramine hydrochloride and disulfiram could inhibit Th17 differentiation significantly (Fig. 5A,B). We then investigated the concentration-dependent effects of the two drugs on Th17 differentiation and miR-30a level. The two drugs were added independently during Th17 differentiation at different concentrations and qPCR was performed 6 d later. The results showed that both drugs could decrease the IL-17a mRNA level significantly while increasing the miR-30a level in a concentration-dependent manner (IL-17a, diphenhydramine: 0.01, 98.38 ± 7.02, p = 0.965; 0.1, 61.53 ± 7.17, p = 0.001; 1, 24.37 ± 5.44, p < 0.001; 10, 10.44 ± 2.38, p < 0.001; 100, 6.3 ± 2.35, p < 0.001; 1000, 5.06 ± 1.82, p < 0.001; disulfiram: 0.01, 105.66 ± 6.89, p = 0.677; 0.1, 88.51 ± 3.98, p = 0.183; 1, 47.54 ± 3.37, p < 0.001; 10, 18.27 ± 3.97, p < 0.001; 100, 10.41 ± 0.79, p < 0.001; 1000, 8.15 ± 0.01, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 3; Fig. 5C; miR-30a, diphenhydramine: 0.01, 1.78 ± 0.09, p = 0.028; 0.1, 1.67 ± 0.02, p = 0.006; 1, 2.69 ± 0.08 p = 0.012; 10, 10.80 ± 0.79, p = 0.008; 100, 3.12 ± 0.23, p = 0.020; 1000, 1.87 ± 0.01, p < 0.001; disulfiram: 0.01, 2.50 ± 0.09, p = 0.022; 0.1, 2.48 ± 0.07, p = 0.003; 1, 3.95 ± 0.41, p = 0.050; 10, 4.31 ± 0.05, p = 0.001; 100, 10.72 ± 0.64, p = 0.007; 1000, 6.66 ± 0.20, p = 0.008; one-way ANOVA with Dunnett's post hoc test, n = 3 independent experiments; Fig. 5D). The toxicity of the two drugs to T cells were evaluated by MTT and both were proven nontoxic, whereas diphenhydramine even slightly promoted cell viability (diphenhydramine: 0.01, 1.32 ± 0.40, p = 0.990; 0.1, 1.25 ± 0.18, p = 0.844; 1, 1.30 ± 0.10, p = 0.338; 10, 1.42 ± 0.17, p = 0.480; 100, 1.38 ± 0.19, p = 0.632; 1000, 1.27 ± 0.17, p = 0.770; disulfiram: 0.01, 0, 97 ± 0.20, p = 1.000; 0.1, 0.88 ± 0.05, p = 0.540; 1, 0.86 ± 0.37, p = 0.725; 10, 0.92 ± 0.21, p = 1.000; 100, 1.07 ± 0.20, p = 1.000; 1000, 0.57 ± 0.22, p = 0.626; one-way ANOVA with Dunnett's post hoc test, n = 6 technical replicates; Fig. 5E). Flow cytometry showed that both drugs reduced the number of IL-17a+ cells significantly (Th0: 0.55 ± 0.13; Th17: 27.25 ± 2.63, p < 0.001; diphenhydramine: 1.09 ± 0.21, p < 0.001; disulfiram: 2.69 ± 0.46, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 6 independent experiments; Fig. 5F,G). ELISA showed that both drugs reduced the IL-17a concentration in the supernatant (Th0: 0.09 ± 0.08; Th17: 5.63 ± 0.67, p < 0.001; diphenhydramine: 0.85 ± 0.23, p < 0.001; disulfiram: 1.35 ± 0.31, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 6 independent experiments; Fig. 5H). These results suggest that disulfiram and diphenhydramine hydrochloride can inhibit Th17 differentiation. Furthermore, we found that interference of miR-30a could attenuate the inhibitory effects of the two drugs on Th17 differentiation (one-way ANOVA with Tukey's post hoc test, n = 3 independent experiments; Fig. 5I), indicating that disulfiram and diphenhydramine hydrochloride inhibit Th17 differentiation at least partially by upregulating miR-30a.
Figure 5.
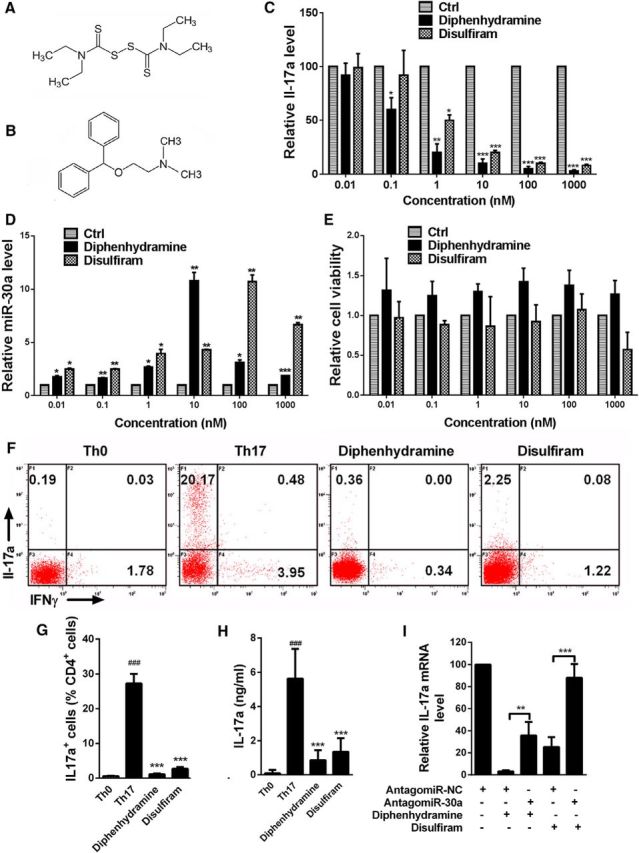
Diphenhydramine hydrochloride and disulfiram upregulate miR-30a and suppress Th17 differentiation. A, B, Molecular structure of diphenhydramine (A) and disulfiram (B). C, qPCR analysis of IL-17a in CD4+CD62L+ T cells with diphenhydramine hydrochloride or disulfiram added at different concentrations and cultured for 6 d in Th17-polarizing conditions (n = 3 independent experiments). D, qPCR analysis of miR-30a in CD4+CD62L+ T cells with diphenhydramine hydrochloride or disulfiram added at different concentrations and cultured for 6 d in Th17-polarizing conditions (n = 6 technical replicates). E, Cell viability detected by MTT assay of mouse naive CD4+CD62L+ Th cells after adding drugs of different concentrations as well as anti-CD3 (0.5 μg/ml; BD Biosciences) and anti-CD28 (1 μg/ml; BD Biosciences) antibody (n = 6 technical replicates). F, Representative intracellular staining of cytokine production in CD4+CD62L+ T cells with diphenhydramine hydrochloride or disulfiram added and cultured for 6 d in Th17-polarizing conditions. G, Ratio number of IL-17a+ cells in D (Th17 vs Th0, diphenhydramine and disulfiram vs Th17; n = 6 independent experiments). H, ELISA analysis of IL-17a in the supernatant of CD4+CD62L+ T cells with either of the two drugs or solvent added and cultured for 6 d in Th17-polarizing conditions (Th17 vs Th0, diphenhydramine and disulfiram vs Th17, n = 6 independent experiments). I, qPCR analysis of IL-17a of CD4+CD62L+ T cells cells transfected with AntagomiR-30a/NC in the presence of either diphenhydramine hydrochloride or disulfiram and cultured for 6 d in Th17-polarizing conditions (n = 3 independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001 vs control, one-way ANOVA with Dunnett's or Tukey's post hoc test). Data are from at least three experiments and are shown as mean ± SEM.
disulfiram and diphenhydramine hydrochloride prevent the full development of EAE
To investigate the role of disulfiram and diphenhydramine hydrochloride in MS, we administered the two drugs via intraperitoneal injection daily to EAE mice. The results showed that both drugs could significantly delay the onset and reduce the behavior score of the EAE mice (control/disulfiram: p = 0.005; control/diphenhydramine: p = 0.001; Mann–Whitney U test, n = 10 mice; Fig. 6A). H&E staining showed reduced inflammatory cell infiltration in the spinal cord after administration of either drug (control: 516.67 ± 32.75; disulfiram: 117.25 ± 61.91, p < 0.001; diphenhydramine: 74.75 ± 33.12, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 4 mice; Fig. 6B,D). Luxol fast blue staining showed attenuated white matter demyelination after drug administration (control: 20.62 ± 1.22; disulfiram: 4.49 ± 2.2, p < 0.001; diphenhydramine: 3.01 ± 0.89, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 4; Fig. 6C,E). These results suggest that disulfiram and diphenhydramine hydrochloride effectively prevent the full development of EAE.
Figure 6.

Disulfiram and diphenhydramine hydrochloride prevent the full development of EAE. A, EAE scores of mice administrated with disulfiram or diphenhydramine (10 mg/kg) by intraperitoneal injection daily after immunization. Data were analyzed using nonparametric Mann–Whitney U test (n = 10 mice per group). B, C, Representative spinal cord sections of H&E staining (B) and LFB staining (C) from EAE mice at day 15 after immunization. Arrows in B indicate inflammatory infiltration (n = 4 mice per group). Scale bars, 80 μm. D, E, Quantification of spinal cord infiltration in WM (D) and the percentage of demyelinated WM in total WM (E) in B and C. F, G, qPCR analysis of miR-30a (F) and IRF4 (G) in CD4+ T cells from mice administrated with disulfiram or diphenhydramine for 15 d (n = 3 mice; *p < 0.05, **p < 0.01, ***p < 0.001 vs control; A, Mann–Whitney U test; D–G, one-way ANOVA with Dunnett's post hoc test). Data are from at least three experiments and are shown as mean ± SEM.
Finally, the influence of disulfiram and diphenhydramine hydrochloride on miR-30a and IRF4 expression in vivo was investigated. The results showed that the two drugs upregulated miR-30a and downregulated IRF4 expression in CD4+ T cells isolated from EAE mice (disulfiram: 2.01 ± 0.10, p < 0.001; diphenhydramine: 3.63 ± 0.08, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 3 mice; Fig. 6F; disulfiram: 62.59 ± 6.43, p < 0.001; diphenhydramine: 30.29 ± 1.52, p < 0.001; one-way ANOVA with Dunnett's post hoc test, n = 3 mice; Fig. 6G). Considering the critical effects of IRF4 and miR-30a in EAE development, these results suggested that disulfiram and diphenhydramine hydrochloride might inhibit the full development of EAE by raising the miR-30a level and decreasing IRF4 expression.
Discussion
The miR-30 family includes five members (miR-30a– miR-30e) that share the same eight-nucleotide seed sequence and are highly conserved from zebrafish to humans. Recent studies have revealed that the miR-30 family plays important roles in pronephros development, hepatobiliary duct formation, and myogenesis in zebrafish (Agrawal et al., 2009; Hand et al., 2009); the miR-30 family also regulates cancer metastasis in humans and mice (Zhong et al., 2013; Zhang et al., 2016). However, the roles of the miR-30 family in neuroimmunological diseases have not been reported. The present study performed an in-depth RNA-sequencing analysis to identify the potentially important miRNAs that regulate Th17 differentiation. We found that miR-30a was decreased significantly in Th17 cells. Along with being downregulated in mouse Th17 cells, miR-30a also exhibited an abnormally low expression in the CD4+ T cells of MS patients. It is reported that miR-30a played an anti-inflammatory role in IL-17-mediated signaling transduction. These suppressive effects of miR-30a were mediated by targeting Act1, an important downstream signaling molecule in IL-17 signaling pathway (Wan et al., 2015), directly. However the role of miR-30a on the production of IL-17, especially the differentiation of Th17 cells, is still unclear (Wan et al., 2015). In the present study, miR-30a was found to regulate Th17 differentiation negatively and prevent EAE from being developed fully. Importantly, we found that disulfiram and diphenhydramine hydrochloride could effectively upregulate miR-30a expression and inhibit the differentiation of Th17 cells, thereby preventing the development of full-blown EAE and demonstrating their potential application in the prevention of MS.
To study how miR-30a regulates Th17 differentiation, we used target gene prediction software and found that miR-30a targeted IRF4, a member of the interferon regulatory factor family that is a key transcriptional factor in Th17 differentiation (Huber et al., 2008). IRF4 knock-out mice showed significant EAE resistance and this resistance was related to the differentiation failure of Th17 cells. IRF4 regulates the differentiation of Th17 cells by binding with the transcription factors RORγt, STAT3, and BATF (Schraml et al., 2009; Durant et al., 2010; Jadidi-Niaragh and Mirshafiey, 2011; Ciofani et al., 2012). In the present study, we found that miR-30a could bind with the 3′-UTR of IRF4 and thus inhibit its translation. In addition, AgomiR-30a and AntagomiR-30a studies demonstrated that miR-30a regulated the protein expression level of IRF4. Moreover, the expression of IRF4 in CD4+ T cells was significantly upregulated at the peak phase of EAE, whereas the expression of miR-30a was downregulated. These results indicated that the effects of miR-30a on Th17 differentiation might be mediated by IRF4. The discovery of the miR-30a/IRF4 pathway in Th17 differentiation enriches the understanding of the functional regulatory network of Th17 cells.
Currently, the goal of MS treatment is to prevent relapse, control the symptoms, and reduce the damage in the CNS. Because there is still no cure available, it is important to develop new safe and effective drugs for the treatment of MS (Barten et al., 2010; Salvetti et al., 2015). Given the strong association between excessive Th17 activity and MS, it has been suggested that targeting Th17 differentiation may be a promising therapeutic strategy (Becher and Segal, 2011; Jadidi-Niaragh and Mirshafiey, 2011; Zepp et al., 2011). In the present study, injection of lentivirus overexpressing miR-30a into the tail vein effectively suppressed Th17 differentiation and prevented disease from being fully developed during EAE. However, exogenous lentivirus injection has potential risks, such as random insertion of foreign genes into host genome, insertional mutagenesis by enhancer-mediated dysregulation of neighboring genes, and aberrant splicing (Rothe et al., 2013). Moreover, miRNA mimics or inhibitors are very difficult to transfect into unstimulated T cells, with the exception of using silicon nanowires (McManus et al., 2002; Yosef et al., 2013). Therefore, compared with exogenous transfection, activating or inhibiting endogenous expression of miRNAs using drugs is a more practical and effective approach. Effective compounds have been screened for their ability to suppress Th17 differentiation by targeting IL-17a or key transcription factors for Th17 differentiation such as STAT3 and RORγt (Huh et al., 2011; Ishiguro et al., 2011; Pallandre et al., 2015). However, there is currently no drug-screening study based on identifying miRNAs that regulate Th17 differentiation. In the present study, miR-30a was chosen as a marker for screening drugs that suppress Th17 differentiation. The screening library that we used was an FDA-approved small-molecule drug library because new functions in Th17 differentiation for existing drugs can be rapidly applied into clinical practice. We found that 72 drugs could increase the expression of miR-30a in CD4+ T cells significantly. Among these drugs, the steroid hormones sildenafil and artemisinin have been reported to suppress Th17 differentiation and EAE progress. Our study provides a new molecular mechanism for these drugs in the regulation of Th17 differentiation or EAE progression. Finally, we identified that disulfiram and diphenhydramine hydrochloride could upregulate miR-30a effectively and suppress Th17 differentiation. More importantly, intraperitoneal administration of either of these drugs could significantly delay the onset and reduce the behavior score of EAE in mice and prevent the full development of white matter demyelination. Diphenhydramine hydrochloride is a commonly used anti-allergy drug that inhibits histamine H1 receptor functions (Sharma and Hamelin, 2003). Disulfiram can inhibit the activity of aldehyde dehydrogenase, thereby blocking the oxidative metabolism of ethanol, which leads to the in vivo accumulation of acetaldehyde (Petrakis et al., 2006). Further study is needed to elucidate how disulfiram and diphenhydramine hydrochloride regulate the level of miR-30a.
Interestingly, miR-30a inhibited the expression of IL-4 during Th2 differentiation (data not shown). The role of miR-30a in Th2 cells may be mediated by the inhibition of IRF4, which promotes Th2 differentiation (Rengarajan et al., 2002; Tominaga et al., 2003). IL-4 is not affected by miR-30a during Th17 differentiation, possibly due to the low expression level of IL-4 in Th17 cells (Fig. 2). However, we found that disulfiram and diphenhydramine hydrochloride promoted the expression of IL-4 in Th2 cells (data not shown), which may be mediated by a mechanism other than miR-30a.
Our work identified miR-30a as an important participant in the neuroimmunological process of MS and EAE because it is downregulated in CD4+ T cells from the peripheral blood of MS patients, inhibiting Th17 differentiation and preventing the development of full-blown EAE. Further work will be done to elucidate whether miR-30a can be applied in the treatment of EAE and MS by administrating miR-30a after the clinical onset of EAE. The same goes for diphenhydramine hydrochloride and disulfiram.
In conclusion, our study identifies miR-30a as a novel MS-related hematological marker and demonstrates a novel drug-screening strategy for the intervention of MS and Th17 differentiation using miR-30a as a target. Moreover, our results provide a rationale for the potential clinical application of diphenhydramine hydrochloride and disulfiram in MS.
Footnotes
This work was supported by the National Natural Science Foundation of China (Key Program Grant 31130024; Grants 81371326, 31371068, and 31571066; and International Cooperation and Exchange Grant 81461138035).
The authors declare no competing financial interests.
References
- Agrawal R, Tran U, Wessely O. The miR-30 miRNA family regulates Xenopus pronephros development and targets the transcription factor Xlim1/Lhx1. Development. 2009;136:3927–3936. doi: 10.1242/dev.037432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Barten LJ, Allington DR, Procacci KA, Rivey MP. New approaches in the management of multiple sclerosis. Drug Des Devel Ther. 2010;4:343–366. doi: 10.2147/DDDT.S9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Segal BM. T(H)17 cytokines in autoimmune neuro-inflammation. Curr Opin Immunol. 2011;23:707–712. doi: 10.1016/j.coi.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, Baccarini A, Lazzari G, Galli C, Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- Brusselle GG, Bracke KR. MicroRNA miR-22 drives TH17 responses in emphysema. Nat Immunol. 2015;16:1109–1110. doi: 10.1038/ni.3295. [DOI] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, Littman DR. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt A, Ramagopalan SV, Scalfari A, Ebers GC. Clinical prognostic factors in multiple sclerosis: a natural history review. Nat Rev Neurol. 2009;5:672–682. doi: 10.1038/nrneurol.2009.178. [DOI] [PubMed] [Google Scholar]
- de Planell-Saguer M, Rodicio MC, Mourelatos Z. Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat Protoc. 2010;5:1061–1073. doi: 10.1038/nprot.2010.62. [DOI] [PubMed] [Google Scholar]
- Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, Li Z, Wu Z, Pei G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10:1252–1259. doi: 10.1038/ni.1798. [DOI] [PubMed] [Google Scholar]
- Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, O'Shea JJ. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, Bhairavabhotla RK, Northrup D, Zahr R, Burr P, Liu X, Zhao K, Sher A, Jankovic D, Zhu J, Muljo SA. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity. 2014;40:865–879. doi: 10.1016/j.immuni.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Prog Brain Res. 2009;175:239–251. doi: 10.1016/S0079-6123(09)17516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand NJ, Master ZR, Eauclaire SF, Weinblatt DE, Matthews RP, Friedman JR. The microRNA-30 family is required for vertebrate hepatobiliary development. Gastroenterology. 2009;136:1081–1090. doi: 10.1053/j.gastro.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, Zhang Q, Zhuang SM, Zheng L, Liang A, Tao W, Cao X. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–243. doi: 10.1016/j.ccr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Huber M, Brüstle A, Reinhard K, Guralnik A, Walter G, Mahiny A, von Löw E, Lohoff M. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci U S A. 2008;105:20846–20851. doi: 10.1073/pnas.0809077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, Guralnik A, Bollig N, Jeltsch K, Heinemann C, Wittmann E, Buch T, Prazeres da Costa O, Brüstle A, Brenner D, Mak TW, Mittrücker HW, Tackenberg B, Kamradt T, Lohoff M. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. 2013;123:247–260. doi: 10.1172/JCI63681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, Cuesta A, Santori FR, Lafaille JJ, Xu HE, Gin DY, Rastinejad F, Littman DR. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro A, Akiyama T, Adachi H, Inoue J, Nakamura Y. Therapeutic potential of anti-interleukin-17A aptamer: suppression of interleukin-17A signaling and attenuation of autoimmunity in two mouse models. Arthritis Rheum. 2011;63:455–466. doi: 10.1002/art.30108. [DOI] [PubMed] [Google Scholar]
- Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74:1–13. doi: 10.1111/j.1365-3083.2011.02536.x. [DOI] [PubMed] [Google Scholar]
- Kiezun A, Artzi S, Modai S, Volk N, Isakov O, Shomron N. miRviewer: a multispecies microRNA homologous viewer. BMC Res Notes. 2012;5:92. doi: 10.1186/1756-0500-5-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, McFarland HF, Paty DW, Polman CH, Reingold SC, Sandberg-Wollheim M, Sibley W, Thompson A, van den Noort S, Weinshenker BY, Wolinsky JS. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- McManus MT, Haines BB, Dillon CP, Whitehurst CE, van Parijs L, Chen J, Sharp PA. Small interfering RNA-mediated gene silencing in T lymphocytes. J Immunol. 2002;169:5754–5760. doi: 10.4049/jimmunol.169.10.5754. [DOI] [PubMed] [Google Scholar]
- Methner A, Zipp F. Multiple sclerosis in 2012: Novel therapeutic options and drug targets in MS. Nat Rev Neurol. 2013;9:72–73. doi: 10.1038/nrneurol.2012.277. [DOI] [PubMed] [Google Scholar]
- Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, Yosef N, Vaidya VS, Weiner HL. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. 2015;125:1069–1080. doi: 10.1172/JCI74347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116:508–521. doi: 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallandre JR, Borg C, Rognan D, Boibessot T, Luzet V, Yesylevskyy S, Ramseyer C, Pudlo M. Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors. Eur J Med Chem. 2015;103:163–174. doi: 10.1016/j.ejmech.2015.08.054. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Nich C, Ralevski E. Psychotic spectrum disorders and alcohol abuse: a review of pharmacotherapeutic strategies and a report on the effectiveness of naltrexone and disulfiram. Schizophr Bull. 2006;32:644–654. doi: 10.1093/schbul/sbl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifarré P, Gutierrez-Mecinas M, Prado J, Usero L, Roura-Mir C, Giralt M, Hidalgo J, García A. Phosphodiesterase 5 inhibition at disease onset prevents experimental autoimmune encephalomyelitis progression through immunoregulatory and neuroprotective actions. Exp Neurol. 2014;251:58–71. doi: 10.1016/j.expneurol.2013.10.021. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M, Modlich U, Schambach A. Biosafety challenges for use of lentiviral vectors in gene therapy. Curr Gene Ther. 2013;13:453–468. doi: 10.2174/15665232113136660006. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Cohen JA, Weinstock-Guttman B, Kinkel RP, Ransohoff RM. Management of multiple sclerosis. N Engl J Med. 1997;337:1604–1611. doi: 10.1056/NEJM199711273372207. [DOI] [PubMed] [Google Scholar]
- Salvetti M, Landsman D, Schwarz-Lam P, Comi G, Thompson AJ, Fox RJ. Progressive MS: from pathophysiology to drug discovery. Mult Scler. 2015;21:1376–1384. doi: 10.1177/1352458515603802. [DOI] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat Med. 2009;15:31–33. doi: 10.1038/nm.1902. [DOI] [PubMed] [Google Scholar]
- Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, Hatton RD, Stormo GD, Weaver CT, Russell JH, Murphy TL, Murphy KM. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hamelin BA. Classic histamine H1 receptor antagonists: a critical review of their metabolic and pharmacokinetic fate from a bird's eye view. Curr Drug Metab. 2003;4:105–129. doi: 10.2174/1389200033489523. [DOI] [PubMed] [Google Scholar]
- Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, Leuenberger T, Lehmann SM, Luenstedt S, Rinnenthal JL, Laube G, Luche H, Lehnardt S, Fehling HJ, Griesbeck O, Zipp F. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. 2010;33:424–436. doi: 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol. 2007;275:62–70. doi: 10.1016/j.mce.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Tominaga N, Ohkusu-Tsukada K, Udono H, Abe R, Matsuyama T, Yui K. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int Immunol. 2003;15:1–10. doi: 10.1093/intimm/dxg001. [DOI] [PubMed] [Google Scholar]
- Wan Q, Zhou Z, Ding S, He J. The miR-30a negatively regulates IL-17-mediated signal transduction by targeting Traf3ip2. J Interferon Cytokine Res. 2015;35:917–923. doi: 10.1089/jir.2014.0146. [DOI] [PubMed] [Google Scholar]
- Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. 2014;15:393–401. doi: 10.1038/ni.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. 2014;35:493–500. doi: 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamvil SS, Steinman L. Diverse targets for intervention during inflammatory and neurodegenerative phases of multiple sclerosis. Neuron. 2003;38:685–688. doi: 10.1016/S0896-6273(03)00326-X. [DOI] [PubMed] [Google Scholar]
- Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Yan S, Wang J, Deng F, Guo Y, Li Y, Fan M, Song Q, Liu H, Weng Y, Shi Q. miR-30a regulates the proliferation, migration, and invasion of human osteosarcoma by targeting Runx2. Tumour Biol. 2016;37:3479–3488. doi: 10.1007/s13277-015-4086-7. [DOI] [PubMed] [Google Scholar]
- Zhao YG, Wang Y, Guo Z, Gu AD, Dan HC, Baldwin AS, Hao W, Wan YY. Dihydroartemisinin ameliorates inflammatory disease by its reciprocal effects on Th and regulatory T cell function via modulating the mammalian target of rapamycin pathway. J Immunol. 2012;189:4417–4425. doi: 10.4049/jimmunol.1200919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Bian Z, Wu Z. miR-30a suppresses cell migration and invasion through downregulation of PIK3CD in colorectal carcinoma. Cell Physiol Biochem. 2013;31:209–218. doi: 10.1159/000343362. [DOI] [PubMed] [Google Scholar]