

Abstract
The striatum is a large brain nucleus with an important role in the control of movement and emotions. Medium spiny neurons (MSNs) are striatal output neurons forming prominent descending axon tracts that target different brain nuclei. However, how MSN axon tracts in the forebrain develop remains poorly understood. Here, we implicate the Wnt binding receptor Frizzled3 in several uncharacterized aspects of MSN pathway formation [i.e., anterior–posterior guidance of MSN axons in the striatum and their subsequent growth into the globus pallidus (GP), an important (intermediate) target]. In Frizzled3 knock-out mice, MSN axons fail to extend along the anterior–posterior axis of the striatum, and many do not reach the GP. Wnt5a acts as an attractant for MSN axons in vitro, is expressed in a posterior high, anterior low gradient in the striatum, and Wnt5a knock-out mice phenocopy striatal anterior–posterior defects observed in Frizzled3 mutants. This suggests that Wnt5a controls anterior–posterior guidance of MSN axons through Frizzled3. Axons that reach the GP in Frizzled3 knock-out mice fail to enter this structure. Surprisingly, entry of MSN axons into the GP non–cell-autonomously requires Frizzled3, and our data suggest that GP entry may be contingent on the correct positioning of “corridor” guidepost cells for thalamocortical axons by Frizzled3. Together, these data dissect MSN pathway development and reveal (non)cell-autonomous roles for Frizzled3 in MSN axon guidance. Further, they are the first to identify a gene that provides anterior–posterior axon guidance in a large brain nucleus and link Frizzled3 to corridor cell development.
SIGNIFICANCE STATEMENT Striatal axon pathways mediate complex physiological functions and are an important therapeutic target, underscoring the need to define how these connections are established. Remarkably, the molecular programs regulating striatal pathway development remain poorly characterized. Here, we determine the embryonic ontogeny of the two main striatal pathways (striatonigral and striatopallidal) and identify novel (non)cell-autonomous roles for the axon guidance receptor Frizzled3 in uncharacterized aspects of striatal pathway formation (i.e., anterior–posterior axon guidance in the striatum and axon entry into the globus pallidus). Further, our results link Frizzled3 to corridor guidepost cell development and suggest that an abnormal distribution of these cells has unexpected, widespread effects on the development of different axon tracts (i.e., striatal and thalamocortical axons).
Keywords: axon guidance, corridor cell, development, Frizzled3, striatum
Introduction
The formation of longitudinal axon tracts in the CNS is complex and requires a myriad of molecular signals and specialized cell types, such as intermediate target and guidepost cells. Progress has been made in defining the mechanisms that regulate the wiring of long ascending and descending pathways in the brainstem and spinal cord (for review, see Mastick et al., 2010; Stoeckli, 2006; Van den Heuvel and Pasterkamp, 2008; Zou, 2012). However, how longitudinal axon tracts that originate in the forebrain are established is less well understood. Furthermore, our understanding of how intermediate targets and guidepost cells develop into choicepoints and permissive corridors for growing axons is rather rudimentary (Dickson and Zou, 2010; Garel and López-Bendito, 2014).
Among the most prominent descending longitudinal fiber systems in the forebrain are axon projections from medium spiny neurons (MSNs) in the striatum. The striatum is part of the basal ganglia and controls the output of this structure through two distinct axon tracts: the direct and indirect pathways. These pathways arise from morphologically similar but biochemically and functionally distinct subpopulations of MSNs that are intermingled within the striatum. The direct pathway originates from striatonigral MSNs that, in mice, project axons to the entopeduncular nucleus and substantia nigra (SN). In contrast, axons from striatopallidal MSNs form the indirect pathway and innervate the globus pallidus (GP) and indirectly influence the SN via the subthalamic nucleus (Gerfen and Surmeier, 2011). The striatonigral (direct) and striatopallidal (indirect) pathways provide antagonistic but balanced outputs that control psychomotor function (Graybiel, 2005). Changes in striatal connectivity cause behavioral deficits and underlie disorders, such as Huntington's disease (Crittenden and Graybiel, 2011; Gerfen and Surmeier, 2011; Russo and Nestler, 2013), underscoring the need to understand how these connections are established and maintained.
Surprisingly little is known about the extracellular cues that regulate the development of MSN axon projections. The transmembrane protein OL-protocadherin (OL-pc or Pcdh10) cell-autonomously mediates MSN axon outgrowth, whereas interactions between the chemorepellent semaphorin3E (Sema3E) and its receptor plexinD1 regulate striatonigral axon pathfinding (Chauvet et al., 2007; Uemura et al., 2007). The transcriptional regulators Islet1 (Isl1) and COUP-TF-interacting protein 2 (Ctip2) control plexinD1 expression in embryonic MSNs, whereas Isl1 also regulates sema3E expression along the trajectory of striatonigral axons (Arlotta et al., 2008; Ehrman et al., 2013; Lu et al., 2014). Isl1−/− mice show altered striatonigral pathfinding in part phenocopying defects observed in sema3E−/− mice (Ehrman et al., 2013). Knock-out mice for the transcription factor early B-cell factor 1 (Ebf1) also show reduced striatonigral pathway development, but the extrinsic effectors involved are unknown (Lobo et al., 2006, 2008). Despite this recent progress in our understanding of the intrinsic regulation of MSN axon pathway development, the extrinsic cues involved remain largely unknown. For example, molecules controlling anterior–posterior (AP) guidance and GP navigation have yet to be determined.
Here, we determine the ontogeny of the mouse striatal efferent pathways using BAC transgenic reporter mice and identify a requirement for the Wnt binding receptor Frizzled3 (Fzd3) (Zou, 2012; Tissir and Goffinet, 2013) in striatal pathway formation. We find that, within the striatum, Fzd3, and its ligand Wnt5a are required in vivo for extension of MSN axons along the AP axis. In addition, Fzd3 non–cell-autonomously regulates the entry of MSN axons into an important (intermediate) target, the GP, and the positioning of “corridor” cells. Corridor cells are a population of guidepost cells for thalamocortical axons located in between the GP and the medial ganglionic eminence (MGE). The mechanisms that regulate corridor cell development remain incompletely understood (Bielle and Garel, 2013). Our data indicate that corridor cells repel MSN axons and that their mislocalization in the GP of Fzd3−/− mice may render the GP nonpermissive for MSN axon growth. Conclusively, our data reveal previously uncharacterized aspects of striatal pathway development and identify cell autonomous and non–cell-autonomous roles for Fzd3 in MSN axon guidance. Further, they implicate Fzd3 in the poorly defined mechanisms underlying corridor cell development.
Materials and Methods
Animals and tissue treatment.
All animal use and care were in accordance with local regulations and institutional guidelines. Mice of either sex were used and maintained in a 12 h light-dark cycle and housed 1–4 per cage with food and water ad libitum. C57BL/6 mice were obtained from Charles River. Frizzled3 knock-out mice were kindly provided by Jeremy Nathans (Johns Hopkins University School of Medicine, Baltimore) (Wang et al., 2002), Pitx3-Cre mice by Marten Smidt (University of Amsterdam) (Smidt et al., 2012), Islet1-Cre mice by Sonia Garel (Ecole Normale Supérieure, Paris, France) (Srinivas et al., 2001); with permission from Thomas Jessell, Ryk knock-out mice by Steven Stacker (Ludwig Institute for Cancer Research, Melbourne, Australia) (Halford et al., 2000), Gbx2-CreERT2 mice by James Li (University of Connecticut Health Center, Farmington, CT) (Chen et al., 2009), and Nkx2-1-Cre mice by Oscar Marin (MRC Center for Developmental Neurobiology, London) (Xu et al., 2008); with permission from Stewart Anderson. Wnt5a knock-out mice (Yamaguchi et al., 1999) and ROSA26-DTA mice (Ivanova et al., 2005) were obtained from The Jackson Laboratory, and Drd2-EGFP, Chrm4-EGFP, and Drd2-Cre (ER44) BAC transgenic mice from Mutant Mouse Regional Resource Center (www.gensat.org) (Gong et al., 2003, 2007). Frizzled3fl/fl mice were as described previously (Chai et al., 2014). The morning of detection of the vaginal plug was defined as embryonic (E) day 0.5, and the day of birth as postnatal (P) day 0. For in situ hybridization experiments, embryonic brains were directly frozen, and 20 μm sections were cut on a cryostat. For immunohistochemistry, embryonic brains were collected in PBS and fixed by immersion for 0.5–8 h in 4% PFA in PBS at 4°C. Postnatal mice were transcardially perfused with ice-cold saline followed by 4% PFA and postfixed for 2 h. Samples intended for immunohistochemistry on cryosections were washed in PBS and cryoprotected in 30% sucrose. Brains were frozen and stored at −80°C. Cryostat sections were cut at 16–25 μm. To generate vibratome sections, fixed brains were embedded in 3% low melting point agarose in PBS and sectioned coronally at 150 μm using a vibratome (Leica).
Enzymatic dissociation and FACS of striatal neurons.
E15.5 or E17.5 embryos from hemizygous D2-EGFP or M4-EFGP mice were collected in ice-cold L-15 (Invitrogen) followed by dissection of the striatum in L-15 with 10% FCS. Striatal tissue was dissociated using the Papain Dissociation System (Worthington Biochem), as previously described (Lobo et al., 2006). Cells were resuspended in L-15 containing 25 μg/ml DnaseI (Sigma) and filtered through a 70 μm mesh. Cells were treated with propidium iodide (20 μg/ml, Invitrogen) to label dead cells and sorted in a Cytopeia Infux cell sorter (528/38 filter for EGFP fluorescence). Wild-type cells were used to calibrate the FITCH and propidium iodide signals. Approximately 70,000–100,000 striatal cells were obtained per embryo from each FACS run. EGFP+ cells were directly collected in RLT lysis buffer (QIAGEN).
RNA extraction and quantitative PCR.
Total RNA extraction was performed from FACS purified striatal neurons using the RNeasy Micro Kit (QIAGEN) according to the manufacturer's protocol. RNA quality and relative concentration were determined using a spectrophotometer and Bioanalyzer Nanochip (Agilent). Each experimental sample consisted of pooled RNA derived from at least two hemizygous D2-EGFP or M4-EGFP embryos. cDNA synthesis, cRNA double amplification, and quality control and fragmentation were performed using an automated system (Caliper Life Sciences) and starting with 70 ng total RNA from each sample, as described previously (Chakrabarty et al., 2012). The qPCR was performed in a 10 μl reaction solution using Roche SYBR Green I (LC-FastStart DNA MasterPlus) and 0.5 μm of each primer. PCRs were performed in a 7900HT Fast Real-Time PCR System (Applied Biosystems) (95°C for 3 min, followed by 45 cycles of denaturation at 95°C, annealing at 55°C, and extension at 72°C for 30 s each). Primer sequences are as follows: Fzd1-10 and Gapdh (Shah et al., 2009), Drd2 (Bice et al., 2008), and Hprt1 (QuantiTec primer Assay; QIAGEN). To detect Chmr4, the following primers were used: 5′-AGCCGAGCATTAAGAAACCTCCAC-3′ (forward); and 5′-TCATTGGAAGTGTCCTTGTCAGCC-3′ (reverse). Samples were run in triplicate, and a common threshold signal was chosen manually in the linear amplification range of all samples by inspecting the log-transformed fluorescence signals plotted against cycle number using SDS software (Applied Biosystems). For each gene, relative expression was calculated using the ddCt method (Livak and Schmittgen, 2001) correcting for the amplification efficiency of each primer pair and using Gapdh and Hprt1 as reference genes.
Explant, dissociated neuron, and hemislice cultures.
Three-dimensional collagen matrix assays were performed as described previously (Schmidt et al., 2012). In brief, E13.5-E14.5 wild-type or Fzd3−/− and littermate control embryos were collected in ice-cold L-15 and the striatum, GP, or corridor cells were carefully dissected in ice-cold L-15 containing 2% FCS. Small explants were then cut from the dissected tissue at 200–350 μm and plated in custom-made rat tail collagen. Striatal explants were placed 300–500 μm from either (1) aggregates of HEK293 cells transiently transfected with EGFP, Wnt5a, or Wnt5b constructs using Lipofectamine 2000; or (2) GP or corridor cell explants in 4-well tissue culture dishes (Nunc). Explants were cultured in Neurobasal with B27 supplement (Invitrogen), HEPES, β-mercaptoethanol, and antibiotics, and incubated in a humidified incubator at 37°C and 5% CO2 for 2–3 d. Then, cultures were fixed in 4% PFA for 1 h at room temperature and immunostained with anti-β-tubulin (Covance) and anti-Nkx2-1 antibodies (Biopat), as previously described (Schmidt et al., 2012). Staining was visualized using epifluorescent illumination on a Zeiss Aksioskop A1 or by confocal laser scanning microscopy (Olympus FV1000). Nkx2-1 staining was used to monitor the success of GP dissection and only cocultures containing the entire GP were analyzed. Quantifications were performed on explants deriving from at least three independent experiments. To calculate proximal/distal (P/D) ratios in cocultures, the total area covered by striatal axons in the proximal and distal quadrants of the cultures was determined and used to calculate P/D ratios per explant (Schmidt et al., 2012). For quantification of corridor-striatal explant cocultures, the length of the 40 longest axons extending into the quadrants proximal and distal to the corridor explant was measured with ImageJ software. Once the mean P/D ratio of each explant was determined, a guidance index was calculated as described previously (Bielle et al., 2011b). Data were statistically analyzed by unpaired two-tailed Mann–Whitney test, one-way ANOVA (α = 5%) or Kruskal–Wallis test and expressed as mean ± SEM.
To generate dissociated striatal neuron cultures, the striatum was microdissected from E15.5 D2-EGFP and M4-EGFP embryos. Tissue from several different embryos was pooled and incubated in trypsin in DMEM/F-12 for 20 min at 37°C. Trypsin activity was inhibited through addition of 20% FCS in DMEM/F12 and tissue was triturated with a fire-polished glass pipette. Then, cells were pelleted by mild centrifugation and resuspended in Neurobasal medium supplemented with B27, antibiotics, and glutamine. A single-cell suspension was generated by forcing the cells through a 70 μm filter, after which cells were plated on poly-d-lysine- and laminin-coated glass coverslips in a 12-well plate for 3 d in Neurobasal medium. Cultures were fixed in 4% PFA for 10 min at room temperature and coimmunostained with rabbit anti-Fzd3 (1:500, a gift from Jeremy Nathans) and chicken anti-EGFP (1:500, Abcam) antibodies. Images were captured on a Zeiss Aksioskop A1 or by confocal laser scanning microscopy (Olympus FV1000). The length of axons emerging from EGFP-labeled MSNs was determined in three randomly selected areas in each culture using OpenLab (Improvision) software. Data from three independent experiments were statistically analyzed by unpaired two-tailed Student's t test and expressed as mean ± SEM.
For transplantation experiments, striatal explants (200 μm) were microdissected from E14.5 Fzd3−/− embryos or littermate controls, and a small DiI crystal (Invitrogen) was placed in each explant, as described previously (Uemura et al., 2007). Striatal explants containing DiI crystals were then placed into the rostral part of the striatum in E14.5 wild-type or Fzd3−/− hemislices. Hemislices consist of half brains cultured on membrane inserts (Corning) with the cortical surface facing up (Pasterkamp et al., 2003; Schmidt et al., 2014). Transplant cultures were grown in Neurobasal medium supplemented with B27, antibiotics, and glutamine and maintained for 3 d in culture. Then cultures were fixed in PBS containing 4% PFA and 4% sucrose for 1 h at room temperature, washed in PBS, incubated in blocking solution (1% PBS, 1% FCS, 1 mg/ml digitonin) for 1 h at room temperature, and incubated in primary antibodies overnight 4°C. Cultures were washed extensively in PBS, incubated with AlexaFluor secondary antibodies for 3 h at room temperature, rinsed in PBS, and embedded in Fluosave (Calbiochem) or 95% glycerol. To quantify the transplantation experiments, the presence of DiI-labeled axons from striatal explants was assessed using Photoshop software. The GP was visualized by Nkx2-1 immunostaining, and fluorescence intensity of striatal axons was measured in three bins corresponding to regions rostral to the GP, in the GP, and caudal to the GP followed by background correction. Data from at least three independent experiments were statistically analyzed by Kruskal–Wallis test, and data were presented as mean ± SEM.
In situ hybridization and immunohistochemistry.
Nonradioactive in situ hybridization was performed as described previously (Pasterkamp et al., 2007). Digoxigenin-labeled Fzd3, Wnt5a, Sema3A, Sema3F, ephrinA5, and Netrin-1 in situ probes were as described previously (Kolk et al., 2009; Fenstermaker et al., 2010; Schmidt et al., 2014). For Sema3E probes, the following primers were used to generate probe templates: 5′-CCGTACTGTGCCTGGGATGGC-3′ (forward), 5′-GGGGTTGGCATACTTCCACTT-3′ (reverse); and for Ebf1 5′-GCTCACTTTGAGAAGCAGCCG-3′ (forward), 5′-CGTACCTTCCGAGGGGTCAAG-3′ (reverse). Probe hybridization was performed overnight, and sense probes were included as specificity controls. Sections subjected to the entire in situ hybridization procedure, but with no or sense probe added, did not exhibit specific hybridization signals.
Immunohistochemistry on cryostat or vibratome sections was as described previously (Kolk et al., 2009). In brief, sections were incubated in blocking solution (1% PBS, 1% BSA, 0.1% Triton X-100) for 30 min at room temperature followed by incubation in primary antibodies in blocking buffer overnight at 4°C. The next day, sections were rinsed several times in PBS and incubated with appropriate AlexaFluor secondary antibodies for 1 h at room temperature. Sections were counterstained with DAPI (Sigma), washed extensively in PBS, and embedded in Prolong Gold Antifade reagent (Invitrogen). Staining was visualized using epifluorescent illumination on a Zeiss Aksioskop A1 or by confocal laser scanning microscopy (Olympus FV1000).
The following primary antibodies were used: rabbit anti-EGFP (1:500, Invitrogen, A11122), chicken anti-EGFP (1:500, Abcam AB13970), rabbit anti-DARPP32 (1:500, Santa Cruz Biotechnology SC-11365), rabbit anti-Nkx2-1 (1:2000, Biopat), rabbit anti-Frizzled3 (1:500; a gift from Jeremy Nathans), rabbit anti-tyrosine hydroxylase (TH) (1:1000; Pel-Freeze), rat anti-OL-protocadherin (1:2000, Millipore, clone 5G10), mouse anti-βIII-tubulin (1:3000, Sigma, T8660), mouse anti-Islet1 (1:100; Developmental Studies Hybridoma Bank, 394D5), mouse anti-neurofilament (1:50; Developmental Studies Hybridoma Bank, 2H3), rat anti-Ctip2 (1:500, Abcam, ab18465), and rabbit anti-Foxp1 (1:250; Abcam, ab16645). Secondary antibodies were AlexaFluor-488, AlexaFluor-555, or AlexaFluor-594 conjugated (1:500, Sigma).
To assess and quantify MSN axon orientation defects, sagittal sections from E17.5-E18.5 mutant embryos (n = 3) and littermate controls (n = 3) were immunostained with anti-DARPP32 antibodies and counterstained with DAPI. Images were captured from these sections at identical medial to lateral locations in the middle portion of the striatum using a Zeiss Axioskop A1 microscope. Then, using Axiovision software (Zeiss), angles were determined between baseline (a line dividing the striatum in a dorsal and ventral half, represents 0°) and the trajectory of the initial axon segment just proximal to the DARPP32-positive cell body. Fifty to 100 DARPP32-positive neurons were measured per embryo, and angles were grouped and calculated as percentage of total (Fenstermaker et al., 2010). For assessing DARPP32 axon density at the GP, two to five embryos were analyzed at E17.5-E18.5 and two or three well-spaced (80 μm) sections at the same AP level were imaged and assessed. A 250 μm rectangle was placed 50 μm from the dorsal surface of the Nkx2-1-positive GP and DARPP32 axon density was assessed in the rectangle using ImageJ software (Kolk et al., 2009). In each embryo, signals were normalized to DARPP32 staining in the cortex, which was similar in wild-type and mutant mice. Data were averaged per embryo, and data from several individual animals were pooled. Finally, data from mutant mice were normalized to control and statistically analyzed by unpaired two-tailed Student's t test.
Quantification and statistics methods.
Statistical analyses were performed using IBM SPSS Statistics by Student's t test, one-way ANOVA, or Kruskal–Wallis. All data in this manuscript are derived from at least three independently performed experiments. All data were expressed as mean ± SEM, and significance was defined as p < 0.05.
Results
Development of striatopallidal and striatonigral axon projections
MSNs account for the vast majority of all striatal neurons (90%–95%) and are the output projection neurons of the striatum. The remaining 5%–10% of striatal neurons are interneurons with local connections within the striatum. The two classes of MSNs that occupy the striatum, striatopallidal and striatonigral neurons, are morphologically indistinguishable and intermingled in the striatum. As a result of these anatomical features, relatively little is known about the spatiotemporal development of striatopallidal and striatonigral MSNs and their axon projections. Here we exploited BAC transgenic mouse lines in which striatopallidal and striatonigral MSNs are specifically and independently labeled with GFP (Gong et al., 2003). In Dopamine receptor D2 (Drd2)-EGFP (D2-EGFP) mice, striatopallidal MSNs and their axons are labeled, whereas in Cholinergic receptor, muscarinic 4 (Chrm4)-EGFP (M4-EGFP) mice striatonigral MSNs and axons are labeled. These mouse lines have been used previously to examine functional and molecular properties of postnatal and adult striatopallidal and striatonigral neurons (Lobo, 2009), but a comprehensive analysis at embryonic and early postnatal stages is lacking. The bulk of MSNs is generated from E13 onwards (Passante et al., 2008), and we therefore started our analysis at E13.5. At E13.5, EGFP+ cells were observed in the striatum of both D2-EGFP and M4-EGFP embryos (Fig. 1A,B). In D2-EGFP mice, these cells were largely confined to the striatum, whereas cellular EGFP expression in M4-EGFP mice was broader with labeling in surrounding structures, such as the ganglionic eminences. In both lines, EGFP+ striatal axons were readily discernible and extended posteriorly to the GP at E13.5 (Fig. 1A–C). In addition, the first striatonigral axons already contacted their target, the SN, at this stage (Fig. 1C). Comparison of D2-EGFP and M4-EGFP mouse embryos showed that the development of the striatopallidal pathway appeared delayed compared with that of the striatonigral pathway. For example, at E15.5 and E17.5, a prominent striatonigral bundle was present in M4-EGFP mice, and numerous EGFP+ axons projected into midbrain. In contrast, despite the presence of a large number of strongly labeled EGFP+ neurons in the striatum, a relatively small subset of EGFP+ axons extended into the GP at these stages in D2-EGFP mice (Fig. 1D–H). However, by P7, patterns of innervation resembled those detected in the mature brain in both lines. The GP was heavily innervated in D2-EGFP mice, and EGFP+ axons traversed the GP in tight axon fascicles in M4-EGFP mice to further fasciculate into a compact axon bundle at more caudal levels and innervate the SN (Fig. 1I,J). Together, these results reveal previously unexplored aspects of striatonigral and striatopallidal axon pathway development and identify D2-EGFP and M4-EGFP mice as valuable tools for the analysis of striatal axon pathfinding and target innervation.
Figure 1.
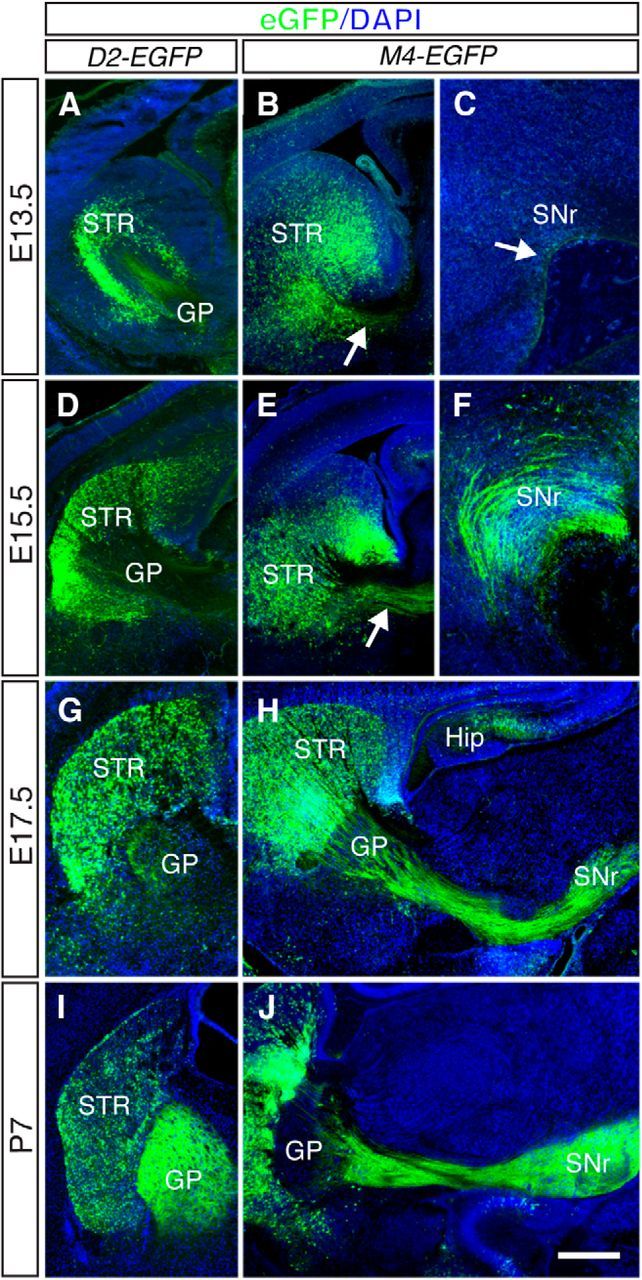
Development of striatopallidal and striatonigral axon pathways. Immunohistochemistry for EGFP (green) in coronal sections from Dopamine receptor D2 (D2)-EGFP mice (A, D, G, I) and in sagittal sections from Cholinergic receptor, muscarinic 4 (M4)-EGFP mice (B, C, E, F, H, J) at the indicated developmental stages. B, C, E, F, H, J, Rostral is to the left. Sections are counterstained with DAPI (blue). In D2-EGFP and M4-EGFP mice, the striatopallidal (indirect) and striatonigral (direct) pathways are labeled, respectively. Arrows indicate the striatonigral pathway. At least n = 3 embryos per time point per genotype were analyzed. Hip, Hippocampus; SNr, substantia nigra, pars reticulata; STR, striatum. Scale bars: A, B, D, E, G, I, 100 μm; C, F, 40 μm; H, J, 200 μm.
Molecular profiling of Frizzleds in striatopallidal and striatonigral neurons
The D2-EGFP and M4-EGFP mouse lines not only provide a tool for visualizing striatal pathways (Fig. 1) but also enable the purification of MSNs by FACS (Lobo et al., 2006). We exploited this property to search for molecular cues involved in striatal pathway development. Pure populations of striatopallidal and striatonigral MSNs were obtained from E15.5 and E17.5 D2-EGFP and M4-EGFP mice by FACS (Fig. 2A). At these developmental stages, striatal axons are actively navigating toward and into their target structures (Fig. 1). Analysis of the expression of Drd2 and Chmr4, markers for striatopallidal and striatonigral MSNs, respectively, confirmed the specificity of the purification procedure (Fig. 2B,C). Next, we performed quantitative PCR (qPCR) to detect expression of members of different axon guidance ligand and receptor families (Kolodkin and Pasterkamp, 2013) (data not shown). Among the most strongly expressed candidates was the Wnt binding and planar cell polarity receptor Frizzled3 (Fzd3), which was detected at equal levels in E15.5 and E17.5 striatopallidal and striatonigral MSNs (Fig. 2D). Fzd3 has previously been implicated in the development of longitudinal axon bundles that, analogous to striatal projections, extend along the AP axis of the CNS (Zou, 2012). In addition, Fzd3 is enriched in the embryonic striatum, and the striatum of Fzd3−/− embryos displays increased cell death from E18 onwards, hinting at reduced target innervation by striatal projections (Wang et al., 2002). Together, these observations identify Fzd3 as a strong candidate for controlling striatal axon development. To further examine the role of Fzd3 in MSNs, we performed immunohistochemistry for Fzd3 in dissociated neuron cultures from E15.5 D2-EGFP and M4-EGFP mice. Fzd3 was expressed in the somata, axons, and growth cones of most EGFP+ striatopallidal and striatonigral MSNs (Fig. 2E–J). Thus, Fzd3 is expressed in MSNs and their axons during the period of axonal growth and pathfinding.
Figure 2.
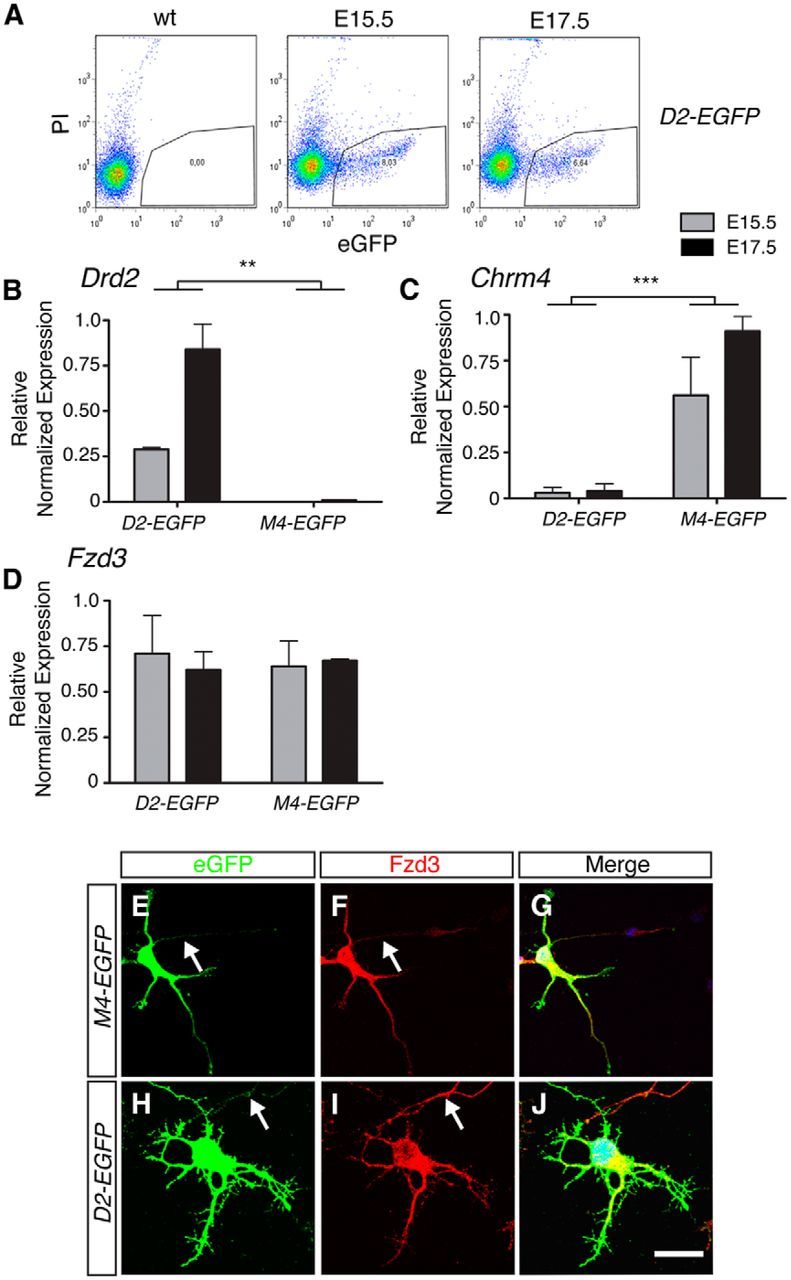
Molecular profiling of Frizzled expression in striatopallidal and striatonigral MSNs. A, Scatter plots showing the distribution of EGFP-positive MSNs purified from E15.5 and E17.5 D2-EGFP embryos compared with reference tissue. EGFP-positive neurons (outlined area) were selected by FACS. PI, Propidium iodide; wt, wild-type. B–D, Quantitative PCR was used to determine relative expression of Dopamine receptor D2 (Drd2; marks striatopallidal MSNs), Cholinergic receptor, muscarinic 4 (Chrm4; marks striatonigral MSNs), and Frizzled3 in D2-EGFP+ or M4-EGFP+ cells obtained by FACS at E15.5 or E17.5. Drd2 is expressed in D2-EGFP+ but not in M4-EGFP+ cells, whereas Chrm4 is expressed in M4-EGFP+ but not D2-EGFP+ cells, confirming the specificity of the FACS procedure. Of 10 Frizzleds tested, only Fzd2 (data not shown) and Fzd3 showed detectable expression in E15.5 and E17.5 MSNs. n = 3 independent experiments. **p < 0.01 (two-way ANOVA). ***p < 0.001 (two-way ANOVA). E–J, Coimmunostaining for EGFP and Frizzled3 in dissociated striatal neuron cultures generated from E15.5 M4-EGFP or D2-EGFP mice analyzed at 3 DIV. Arrows indicate axons. M4-EGFP n = 3 litters, D2-EGFP n = 3 litters. Scale bars: E–J, 40 μm.
Frizzled3 is required for growth of striatal axons toward and into the GP
To examine whether Fzd3 is required for striatal pathway development in vivo, we performed immunohistochemistry for dopamine- and cAMP-regulated phosphoprotein (DARPP32) in E17.5 wild-type and Fzd3−/− embryos. DARPP32 is expressed in ∼95% of MSNs but not by other cell types in the striatum (Anderson and Reiner, 1991). Although expression of DARPP32 was decreased in the striatum of Fzd3−/− embryos (Fig. 3A–E), analysis of the expression of various other genes known to be expressed in MSNs (OL-pc, Ebf1, Ctip2, forkhead box protein p1 (Foxp1), Isl1, Netrin-1) did not reveal a marked loss of MSNs (Figs. 4, Fig. 10). This is in line with previous work showing that the proliferation, distribution, and number of striatal neurons is similar in wild-type and Fzd3 mutant mice until E18 (Wang et al., 2002). Analysis of DARPP32+ axons revealed that in wild-type mice MSN axons projected caudally in the striatum toward the GP, entered the GP forming large bundles that eventually targeted the midbrain. In contrast, in Fzd3−/− embryos striatal axons accumulated at the GP and did not enter this nucleus nor project caudally toward the SN (Fig. 3A–I). In mice, striatopallidal axons target the GP, whereas striatonigral axons traverse the GP to innervate the entopeduncular nucleus and the SN (Gerfen and Surmeier, 2011). Because DARPP32 labels both axon types, we next generated Fzd3−/−;D2-EGFP and Fzd3−/−;M4-EGFP mice to assess whether or not both striatopallidal and striatonigral pathways are affected. MSN axons accumulated in very close proximity to the GP in Fzd3−/−;D2-EGFP and Fzd3−/−;M4-EGFP mice but never entered this structure, confirming the DARPP32 data and showing that both striatonigral and striatopallidal axons are affected (Fig. 3J–M).
Figure 3.
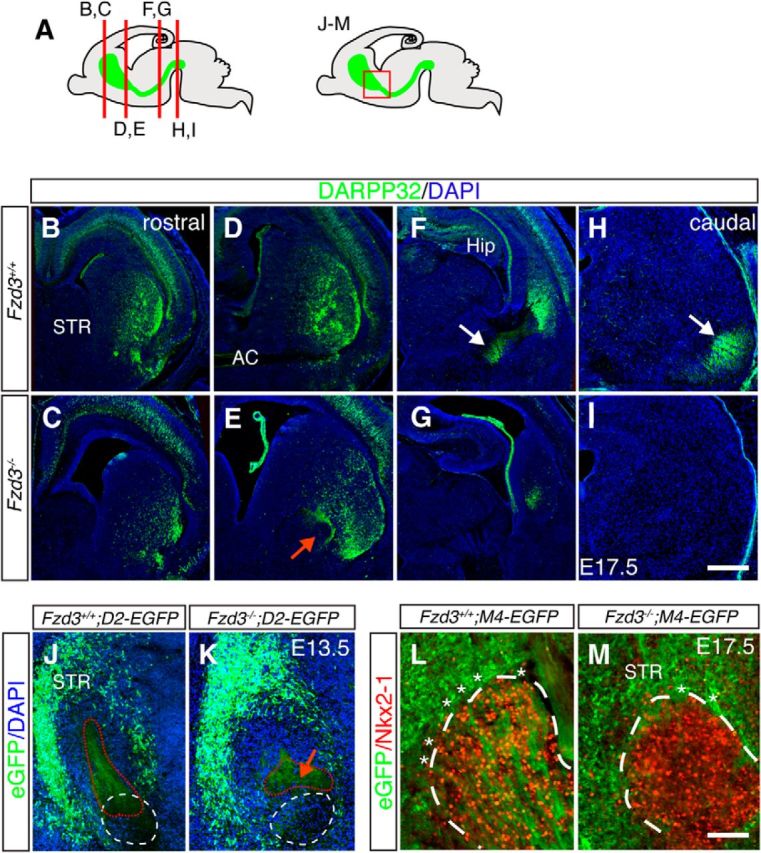
Frizzled3−/− mice show defects in striatal pathway development. A, Schematics of sagittal sections of the embryonic brain showing the striatum (STR) and its axon projections in green. Red lines and box indicate the origin of B–M. B–I, Immunohistochemistry for DARPP32 (green) in coronal sections from E17.5 Fzd3+/+ and Fzd3−/− mice obtained from different rostrocaudal levels. Sections are counterstained with DAPI (blue). White arrows indicate the striatonigral bundle. Red arrow indicates accumulation of striatal axons at the level of the GP. AC, Anterior commissure; Hip, hippocampus; STR, striatum. J, K, Immunohistochemistry for EGFP in coronal sections from E13.5 Fzd3+/+;D2-EGFP and Fzd3−/−;D2-EGFP embryos. DAPI in blue. Red arrow indicates accumulation of striatopallidal axons. White dotted lines indicate GP. Red dotted line indicates striatal axons. L, M, Immunohistochemistry for EGFP and Nkx2-1 (red) in coronal sections from E17.5 Fzd3+/+;M4-EGFP and Fzd3−/−;M4-EGFP embryos. White dotted lines indicate GP. Asterisks indicate axon bundles at the GP. Number of mice analyzed: B–I, E17.5 Fzd3+/+, n = 10; E17.5 Fzd3−/−, n = 12; J, K, E13.5 Fzd3+/+;D2-EGFP or Fzd3−/−;D2-EGFP, n = 3; L, M, E17.5 Fzd3+/+;M4-EGFP or Fzd3−/−;M4-EGFP, n = 5. Scale bars: B–I, 190 μm; J–M, 95 μm.
Figure 4.
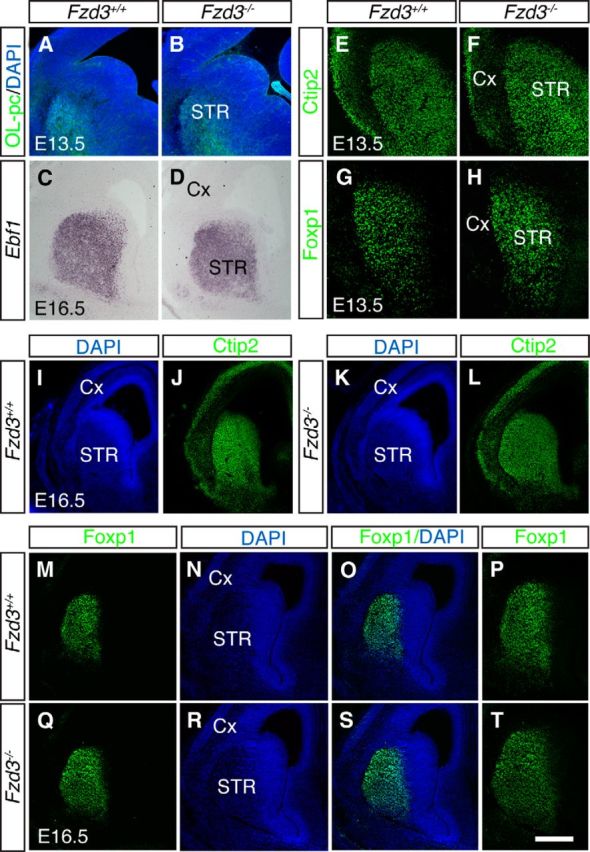
MSN differentiation is intact in Frizzled3−/− mice. Immunocytochemistry for OL-protocadherin (OL-pc) (A, B), COUP-TF-interacting protein 2 (Ctip2) (E, F, J, L), or forkhead box protein p1 (Foxp1) (G, H, M, P, Q, T) on coronal sections of E13.5 or E16.5 Fzd3+/+ or Fzd3−/− embryos. Several sections are counterstained with DAPI. C, D, In situ hybridization for early B-cell factor 1 (Ebf1) on coronal sections of E16.5 Fzd3+/+ or Fzd3−/− embryos. Fzd3−/− mice have a slightly enlarged brain but show no obvious defect in the expression or distribution of various striatal markers (OL-pc, Ctip2, Foxp1, Ebf1). Cx, Cortex; STR, striatum. Scale bars: A, B, 400 μm; C, D, 400 μm; E–H, 550 μm; I–L, 650 μm; M–O, Q–S, 500 μm; P, T, 250 μm.
Figure 10.
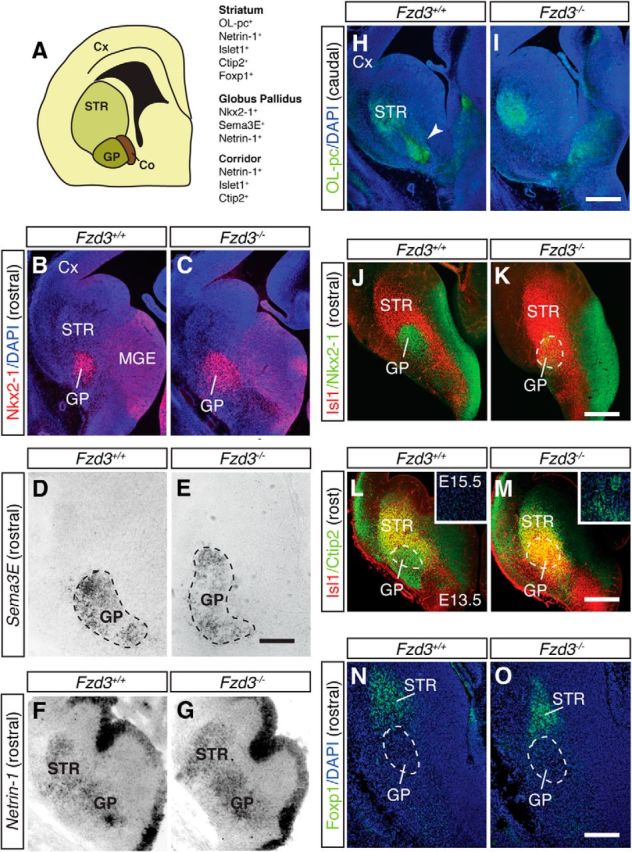
Mislocalization of corridor cells in the GP of Frizzled3−/− mice. A, Schematic of a coronal section indicating the cortex (Cx), striatum (STR), GP, and corridor cells (Co). Listed at the right are proteins expressed by the indicated structures. B, C, Immunohistochemistry for Nkx2-1 (red) in coronal sections from E13.5 embryos of the rostral part of the GP. Sections are counterstained with DAPI (blue). The GP covers an expanded area in Fzd3−/− embryos. D–G, In situ hybridization for Semaphorin 3E (D, E) and Netrin-1 (F, G) in coronal sections from E13.5 embryos. Sema3E and Netrin-1 expression is intact in the GP of Fzd3−/− embryos but covers an expanded region. H, I, Immunohistochemistry for OL-protocadherin (OL-pc; green) in coronal sections from E13.5 embryos of the caudal part of the GP. Arrowhead indicates striatal axons in GP. J–O, Double immunostaining for Islet1 (Isl1; red) and Nkx2-1 (green) (J, K) and for Isl1 (red) and Ctip2 (green) (L, M), and immunohistochemistry for Foxp1 (green; DAPI in blue) (N, O), in coronal sections from E13.5 Fzd3+/+ and Fzd3−/− embryos. L, M, Insets, Ctip2 immunostaining (green) in the GP at E15.5 (blue; DAPI staining). Dotted lines indicate rostral GP. The GP in Fzd3−/− embryos is invaded by ectopic Isl1+;Ctip2+;Foxp1− corridor cells. Number of mice analyzed: E13.5 Fzd3+/+, n = 6; E15.5 Fzd3+/+, n = 3; E13.5 Fzd3−/−, n = 5; E15.5 Fzd3−/−, n = 3. Scale bars: B, C, H–M, 400 μm; D, E, 85 μm; F, G, 155 μm; N, O, 145 μm.
In addition to a failure to grow into the GP, we noted that fewer EGFP+ axon bundles appeared to arrive at the rostral border of GP in E17.5 Fzd3−/−;D2-EGFP and Fzd3−/−;M4-EGFP mice, compared with wild-type controls (Fig. 3L,M). Quantification of axonal EGFP signals at the GP at E17.5 confirmed this observation (relative axonal intensity at GP: Fzd3+/+;D2-EGFP 100 ± 3.3, Fzd3−/−;D2-EGFP 81.1 ± 2.8; Fzd3+/+;M4-EGFP 100 ± 3.9, Fzd3−/−;M4-EGFP 76.0 ± 4.1; n = 3 mice per genotype; p < 0.01). Thus, our results indicate that, in Fzd3−/− mice, (1) fewer MSN axons project from the striatum to the GP, and (2) no MSN axons enter the GP.
To further characterize these MSN axon defects, we genetically ablated Fzd3 from (1) striatonigral or striatopallidal MSNs by crossing conditional Fzd3fl/fl mice (Chai et al., 2014) with Isl1-Cre (Srinivas et al., 2001) or D2-Cre (Gong et al., 2007) mice, respectively, or (2) GP neurons by crossing Fzd3fl/fl mice with Nkx2-1-Cre mice (Xu et al., 2008). Isl1-Cre;ROSA26-EYFP and D2-Cre;ROSA26-EYFP mice revealed robust recombination in the early embryonic striatum but not in the GP (Fig. 5A,B,E). MSN axons entered and crossed the GP in Isl1-Cre;Fzd3fl/fl and D2-Cre;Fzd3fl/fl mice (Fig. 5C,D,F,G). However, the number of axons arriving at the GP was reduced in Isl1-Cre;Fzd3fl/fl and D2-Cre;Fzd3fl/fl mice compared with littermate controls (relative axonal intensity at GP: Isl1-Cre;Fzd3+/+, 100 ± 5.1; Isl1-Cre;Fzd3fl/fl, 78.2 ± 3.8; D2-Cre;Fzd3+/+, 100 ± 4.5; D2-Cre;Fzd3fl/fl, 81.8 ± 3.2; n = 3 per genotype; p < 0.01). Nkx2-1-Cre;ROSA26-EYFP mice revealed robust recombination in the GP and MGE but not in the early embryonic striatum, except for a small number of interneurons (Fig. 5H). MSN axons entered and crossed the GP in Nkx2-1-Cre;Fzd3fl/fl mice (Fig. 5I,J), and the number of axons arriving at the GP was similar in Nkx2-1-Cre;Fzd3+/+ and Nkx2-1-Cre;Fzd3fl/fl mice (relative axonal intensity at GP: Nkx2-1-Cre;Fzd3+/+, 100 ± 5.1; Nkx2-1-Cre;Fzd3fl/fl, 99.8 ± 4.7; n = 3 per genotype; p = 0.8351). Together, these data support a cell-autonomous role for Fzd3 in MSNs during the growth of MSN axons toward the GP and a non–cell-autonomous role in their subsequent entry into the GP.
Figure 5.
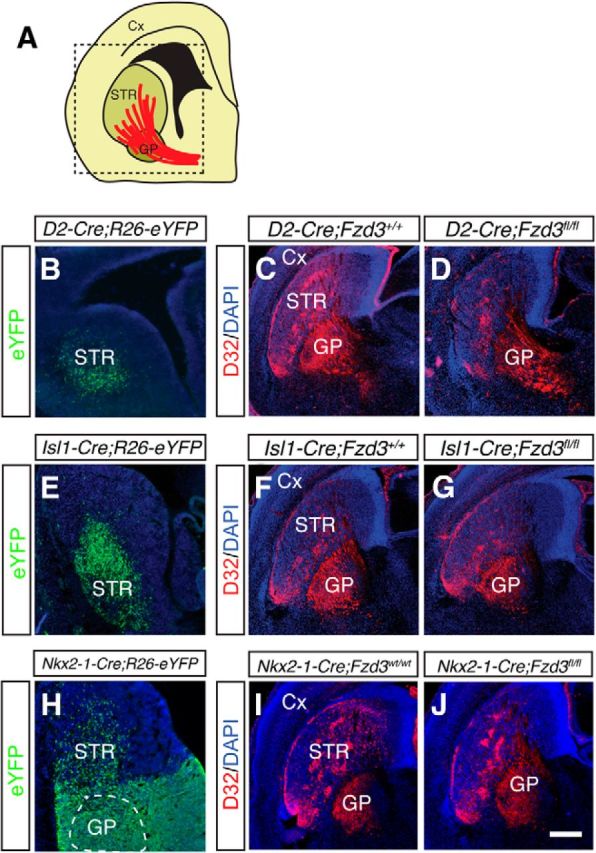
Conditional ablation of Frizzled3 in the striatum and GP. A, Schematic of a coronal section indicating the location of B–J. Cx, Cortex; STR, striatum. B, E, H, Immunohistochemistry for EYFP on E13.5 coronal sections in D2-Cre;Rosa26-EYFP, Isl1-Cre;Rosa26-EYFP, and Nkx2-1-Cre;Rosa26-EYFP mice. D2-Cre;Rosa26-EYFP and Isl1-Cre;Rosa26-EYFP mice show specific labeling of the STR (B, E). Nkx2-1-Cre;Rosa26-EYFP mice show labeling of the GP (dotted line as determined by Nkx2-1 immunolabeling; data not shown). EYFP+ cells in the STR are interneurons. C, D, F, G, I, J, Immunohistochemistry for DARPP32 (red) in coronal sections from E18.5 mice. Sections are counterstained with DAPI (blue). Specific ablation of Fzd3 in MSNs (in D2-Cre;Fzd3fl/fl or Isl1-Cre;Fzd3fl/fl mice) reduces MSN axon growth toward GP but does not affect entry of axons into the GP. Ablation of Fzd3 in the GP (in Nkx2-1-Cre;Fzd3fl/fl mice) does not affect striatal axon growth toward or into the GP. n = 3 mice analyzed for each genotype. Scale bars: B, E, 375 μm; H, 400 μm; C, D, F, G, 600 μm; I, J, 900 μm.
Frizzled3 dictates the AP guidance of striatal axons
A decrease in the number of MSN axons arriving at the rostral border of the GP in Fzd3−/−, Isl1-Cre;Fzd3fl/fl, or D2-Cre;Fzd3fl/fl mice could simply reflect a reduction in MSN axon outgrowth. However, analysis of axon outgrowth in dissociated MSN cultures derived from E15.5 Fzd3−/−;D2-EGFP or Fzd3−/−;M4-EGFP mice, and littermate controls did not reveal a difference in axon outgrowth between wild-type and mutant cultures (data not shown). Frizzleds have been shown to regulate AP guidance of axons in the brainstem and spinal cord. Therefore, another explanation for the decrease in the number of striatal axons, reaching the GP is a defect in axonal AP guidance in the striatum. To test this model, we analyzed the orientation of MSN cell bodies and axons in the striatum of wild-type and Fzd3−/−, Isl1-Cre;Fzd3fl/fl, or D2-Cre;Fzd3fl/fl embryos. In wild-type embryos, many DARPP32+ MSN cell bodies were oriented toward the GP with their axons running posteriorly in a specific medial–lateral plane within the striatum. MSN cell bodies at the same anatomical location in (conditional) knock-out mice were oriented more randomly, and their axons were misrouted along the lateral–medial and rostral–caudal axes (i.e., no longer confined to their normal posterior direction) (Fig. 6A–C). The only known ligands for Fzd3 are Wnts, and gradients of Wnt proteins can guide longitudinal axon tracts along the AP axis in the brainstem and spinal cord in a Fzd3-dependent manner (Zou, 2012). Whether similar mechanisms function in the forebrain is unknown. Several Wnts are expressed in the embryonic striatum, but only Wnt5a and Wnt5b display an anterior low, posterior high expression pattern consistent with a role in AP guidance (Fig. 6D,E) (Fenstermaker et al., 2010). Therefore, we determined whether or not Wnt5a and Wnt5b can direct the growth of striatal axons using a previously established collagen matrix assay (Schmidt et al., 2012). When confronted with cell aggregates releasing Wnt5a or Wnt5b, axons emanating from striatal explants displayed biased growth toward the cells (Fig. 6F). Quantification of P/D ratios in these cultures confirmed the axon attractive effect of Wnt5a and Wnt5b (con, P/D ratio, 1.05 ± 0.05; n = 14; Wnt5a, P/D ratio, 1.45 ± 0.11; n = 10; Wnt5b, P/D ratio, 1.38 ± 0.09; n = 14, p < 0.001, Kruskal–Wallis test). Previous work identifies Fzd3 as an axonal receptor for instructive Wnt5a gradients during axon pathfinding (Fenstermaker et al., 2010). To determine whether, similar to Fzd3, Wnt5a is required in vivo for striatal axon guidance, Wnt5a−/− mice (Yamaguchi et al., 1999) were analyzed. DARPP32+ axons entered the GP in Wnt5a−/− mice and did reach the SN. However, innervation of the SN was dramatically reduced in the absence of Wnt5a, and the number of axons that arrived at the GP was decreased (Fig. 6G–J). Although DARPP32 expression in the striatum was reduced, quantification showed that a smaller subset of axons arrived at the GP in Wnt5a−/− mice compared with wild-type mice (relative axonal intensity at GP normalized to neuronal DARPP32 expression: Wnt5a+/+, 100 ± 3.3; Wnt5a−/−, 71.2 ± 3.9; n = 3 mice per genotype; p < 0.01). Similar to Fzd3−/− mice, many DARPP32+ axons in Wnt5a−/− mice were no longer confined to their normal posterior trajectory in the striatum (Fig. 6K). Analysis of mice deficient for Ryk (Halford et al., 2000), another Wnt5a receptor, did not reveal obvious defects (Fig. 6K). These results, together with Wnt expression analysis and data from collagen matrix assays, suggest that Fzd3 and Wnt5a cooperate to attract MSN axons into their normal posterior direction in the striatum toward the GP.
Figure 6.
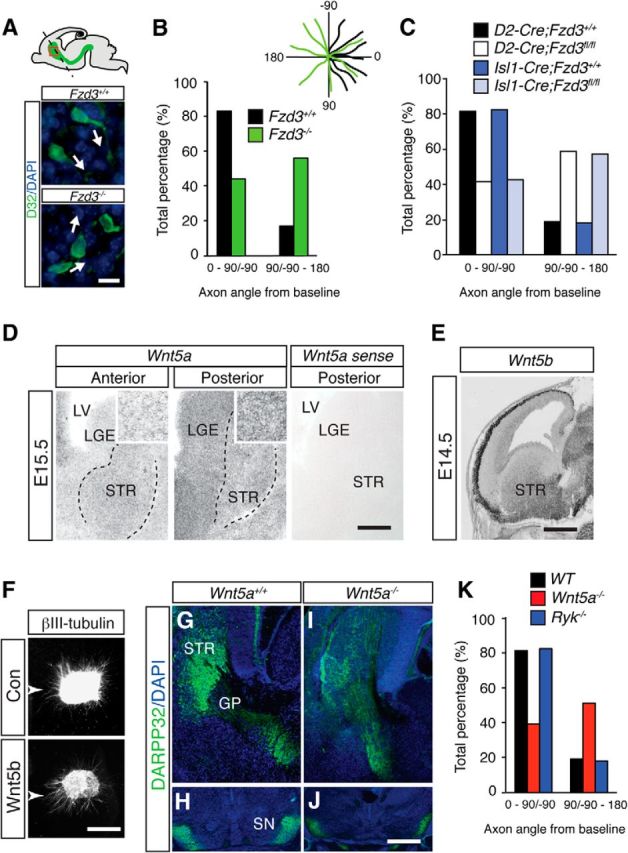
Frizzled3 and Wnt5a regulate AP guidance of striatal axons. A, Top, The embryonic brain with the striatum and its projections in green. Red box represents position of bottom panels. Dotted line indicates baseline for quantification in B, C, and K. Bottom, Immunohistochemistry for DARPP32 (green) in sagittal sections from E17.5 Fzd3+/+ and Fzd3−/− mice obtained from similar medial–lateral levels. In Fzd3+/+ mice, most MSN cell bodies and axons are oriented posteriorly (white arrows), whereas in Fzd3−/− mice MSNs project their axons more randomly (white arrows). MSN axons project in an abnormal lateral–medial direction and are therefore for the most part not visible. B, Quantification of the angles between baseline (0°, black dotted line in A) and the trajectory of the axon segment just proximal to the cell body. Inset, Axon orientation plots for a few randomly selected axons. Number of mice analyzed: E17.5 Fzd3+/+, n = 4; E17.5 Fzd3−/−, n = 4. C, Quantification of MSN axon orientation in E18.5 D2-Cre;Fzd3fl/fl or Isl1-Cre;Fzd3fl/fl mice and littermate controls as in B. n = 3 mice for the different groups. D, In situ hybridization for Wnt5a on coronal sections from E15.5 mouse brain. Images containing the anterior and posterior striatum (STR) are shown. Dotted lines outline the striatum. Insets, Higher magnification of ISH signals in the striatum. Wnt5a expression displays an anterior low, posterior high gradient in the striatum and surrounding regions (cortex, LGE). Sections incubated with Wnt5a sense probes do not exhibit specific labeling. LV, Lateral ventricle. E, In situ hybridization for Wnt5b on a sagittal section from E14.5 mouse brain. Image obtained from www.genepaint.org (EH3735) (Visel et al., 2004). Wnt5b is expressed in an anterior low, posterior high gradient in the striatum (STR). F, Collagen matrix cultures of E14.5 striatal explants cultured adjacent to HEK293 cells (arrowhead) secreting either control protein, Wnt5a (data not shown) or Wnt5b. Con, n = 14 explants; Wnt5a, n = 10; Wnt5b, n = 14. G–J, Immunohistochemistry for DARPP32 (green) in coronal sections from E17.5 Wnt5a+/+ and Wnt5a−/− mice obtained at the level of the STR and GP (G, I) or the SN (H, J). Sections are counterstained with DAPI (blue). K, Quantification of the angles as in B. Number of mice analyzed: E17.5 Wnt5a+/+, n = 3; E17.5 Wnt5a−/−, n = 3; E17.5 Ryk−/−, n = 3. Scale bars: A, 20 μm; D, 90 μm; D, insets, 30 μm; E, 1 mm; F, 250 μm; G–J, 100 μm.
Non–cell-autonomous role for Frizzled3 during entry of MSN axons into the GP
MSN axons that do reach the GP in Fzd3−/− mice fail to enter this nucleus. The observation that selective ablation of Fzd3 from MSNs in Isl1-Cre;Fzd3fl/fl or D2-Cre;Fzd3fl/fl embryos does not cause MSN axon stalling at the GP hints at a non–cell-autonomous role for Fzd3 in GP entry. To further confirm this model, we performed in vitro transplantation experiments. Striatal explants were dissected from wild-type or Fzd3−/− embryos and grafted into the striatum of wild-type and mutant hemislice cultures adjacent to the GP, at the onset of striatal axon pathfinding (Fig. 7A). Although this setup does not allow analysis of striatal AP guidance given the close proximity of the striatal explants to the GP, it can be used to monitor axon growth into the GP. As the majority of MSNs are generated from E13 onwards (Passante et al., 2008), explants and hemislices were dissected at E14.5 and cultured for 3 d in vitro (DIV). Immunohistochemistry for DARPP32 in DIV3 hemislices revealed prominent striatal projections emanating from the striatum toward synaptic targets, such as the SN, as observed in vivo, confirming the physiological relevance of the culture system (Fig. 7B). Explants were labeled with the lipophilic dye DiI and the GP, in the hemislices, was visualized using whole-mount immunohistochemistry for Nkx2-1. At DIV3, wild-type grafts send out DiI-labeled axons into the endogenous striatum and the GP. In addition, a subset of DiI-labeled axons already extended beyond the GP toward the midbrain (Fig. 7C–F,K). Intriguingly, axons derived from Fzd3−/− grafts extended into and beyond the GP in wild-type hemislices (Fig. 7G,H,K). The irregular trajectories of individual DiI-labeled wild-type and knock-out axons, which are clearly distinct from endogenous axon tracts, suggest that axons emanating from the grafts do not simply follow endogenous axons but rather engage in active pathfinding. In striking contrast to the ability of wild-type and Fzd3−/− striatal axons to navigate the GP in wild-type hemislices, axonal projections from wild-type grafts stalled at the border of the GP in hemislices derived from Fzd3−/− mice, resembling the defects observed in Fzd3−/− mice in vivo (Fig. 7I–K). These data together with the intact growth of MSN axons into the GP in Isl1-Cre;Fzd3fl/fl or D2-Cre;Fzd3fl/fl embryos (Fig. 5) support a non–cell-autonomous role for Fzd3 during the entry of MSN axons into the GP.
Figure 7.
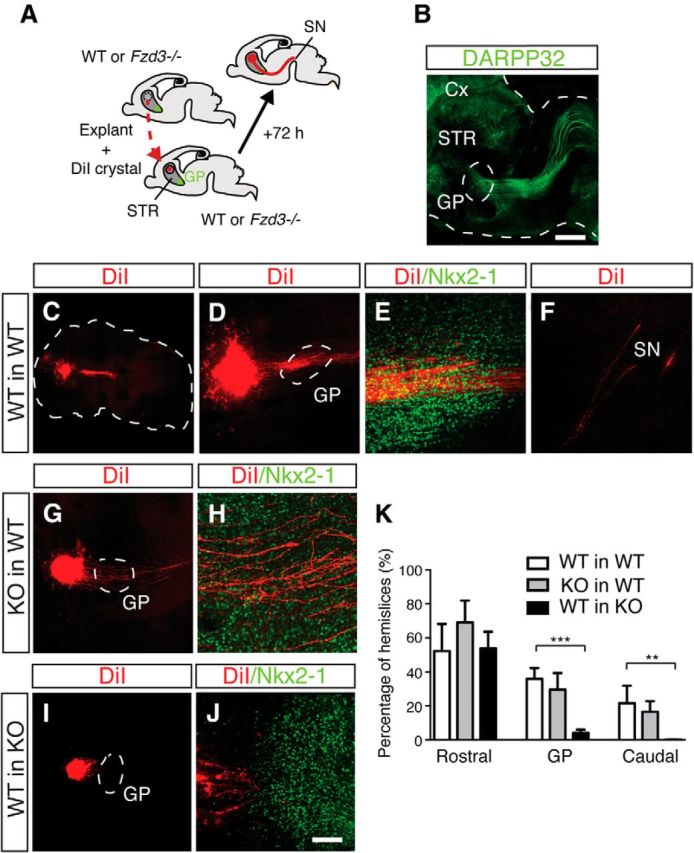
Frizzled3 non–cell-autonomously regulates the entry of striatal axons into the GP. A, Schematic illustrating the transplantation of Fzd3+/+ (WT) or Fzd3−/− explants into WT or Fzd3−/− hemislices followed by striatal pathway analysis using DiI labeling. STR, Striatum. B, E14.5 hemislice cultured for 3 DIV and subjected to whole-mount immunostaining for DARPP32 (green). Cx, Cortex. Dotted lines indicate boundaries of hemislice and GP. C–J, Examples of WT and Fzd3−/− explants transplanted into hemislices and analyzed at DIV3. DiI labeling in red and immunostaining for Nkx2-1, to label the GP, in green. C, Dotted line outlines hemislice. D, G, I, Dotted line outlines the GP. DiI-labeled striatal axons from WT explants fail to enter the GP in KO hemislices (I, J). K, Quantification of the number of hemislices showing DiI-labeled projections from transplanted explants rostral to the GP, in the GP, or caudal to the GP. **p < 0.01 (Kruskal–Wallis test). ***p < 0.001 (Kruskal–Wallis test). Data represent mean ± SEM. Scale bars: B, 100 μm; C, 2.2 mm; D, G, I, 75 μm; E, F, H, J, 20 μm.
Axon entry into the GP is independent from reciprocal axon-axon interactions
One mechanism through which Fzd3 could non–cell-autonomously regulate striatal pathfinding at the GP is by controlling the development of other axon tracts on which striatal axons depend for guidance. For example, interactions between reciprocal axon projections regulate pathfinding in the embryonic subpallium and midbrain (Deck et al., 2013; Schmidt et al., 2014). During early embryonic stages, striatal axons at the GP grow in close proximity to dopaminergic and thalamocortical axons en route to the striatum and cortex, respectively (Fig. 8A–D) (Uemura et al., 2007). Other projections, such as corticothalamic and cortifugal axons, arrive at the GP after striatal axons have entered this nucleus (Uemura et al., 2007; Leyva-Díaz and López-Bendito, 2013). In Fzd3−/− mice, dopaminergic and thalamocortical pathways are disrupted and fail to enter the subpallium (Wang et al., 2002; Fenstermaker et al., 2010). To evaluate a model in which striatal axons use reciprocal dopaminergic or thalamocortical projections to traverse the GP, we performed in vivo genetic ablation studies. DTA mice, which conditionally express subunit A of the diphtheria toxin, were crossed with Pitx3-Cre or Gastrulation Brain Homeobox 2 (Gbx2)-CreERT2 mice to ablate the midbrain dopamine system and principal nuclei of the thalamus, respectively (Chen et al., 2009; Smidt et al., 2012). E17.5 Pitx3-Cre;DTA embryos displayed a loss of midbrain dopamine neurons and their axon projections in the midbrain and forebrain, including those in the medial forebrain bundle, as visualized by immunohistochemistry for TH (Fig. 8E,G). However, DARPP32+ MSN axons were present and projected into and beyond the GP in Pitx3-Cre;DTA mice (Fig. 8F,H). Similarly, in Gbx2-CreERT2;DTA mice, in which thalamocortical axons from principal thalamic nuclei were ablated, navigation of the GP by DARPP32+ MSN axons was comparable with control (Fig. 8I–L). Thus, MSN axons do not rely on reciprocal dopaminergic and thalamalocortical axons for navigation into and beyond the GP.
Figure 8.

Striatal axons do not require dopaminergic or thalamocortical projections to enter the GP. A–D, Immunohistochemistry for EGFP (green) and TH (red) in coronal sections of E13.5 or E15.5 M4-EGFP embryos. Arrowhead indicates dopaminergic axons. Dotted line indicates GP. Striatal and dopaminergic axons grow in close proximity during early development. Cx, Cortex; ic, internal capsule; STR, striatum; Thal, thalamus. E–H, Immunohistochemistry for TH (E, G) or DARPP32 (F, H) in sagittal cryosections of E17.5 WT;DTA or Pitx3-Cre;DTA mice. Sections are counterstained with DAPI (blue). Arrowheads indicate medial forebrain (E) and striatonigral (F, H) bundles. Hip, Hippocampus; SN, substantia nigra. I–L, Immunohistochemistry for neurofilament (NF; I, K) or DARPP32 (J, L) in coronal vibratome sections of E17.5 WT;DTA or Gbx2-CreERT2;DTA mice. Sections are counterstained with DAPI (blue). I, Arrowhead indicates thalamocortical pathway. Whereas dopaminergic and thalamocortical pathways are lost in Pitx3-Cre;DTA and Gbx2-CreERT2;DTA mice, respectively, DARPP32-positive striatal projections are intact. Number of mice analyzed: E13.5 and E15.5 M4-EGFP, n = 2; E17.5 WT;DTA or Pitx3-Cre;DTA, n = 4; E17.5 WT;DTA or Gbx2-CreERT2;DTA mice, n = 3. Scale bars: A, B, 100 μm; E–H, 200 μm; I, K, 560 μm; J, L, 250 μm.
The permissive effect of the GP on striatal axons is dependent on Frizzled3
An alternative explanation for the accumulation of axons at the GP in Fzd3−/− mice is that Fzd3 deficiency affects the GP and thereby striatal axon pathfinding at this nucleus. Complex target-mediated molecular mechanisms regulate the entry of axons at other (intermediate) targets, such as the spinal cord midline (Derijck et al., 2010; Dickson and Zou, 2010; Chédotal, 2011; Izzi and Charron, 2011). Therefore, we first examined whether the GP releases instructive cues for striatal axons. Striatal and GP explants were cocultured in collagen matrices (Schmidt et al., 2014). Axons emanating from wild-type striatal explants showed preferred growth toward wild-type GP explants, indicating that this nucleus secretes attractive or permissive cues for striatal axons (Fig. 9A,E). Interestingly, Fzd3−/− striatal explants also displayed biased growth toward wild-type GP explants. In contrast, quantification showed that wild-type and Fzd3−/− striatal explants do not display biased growth toward Fzd3−/− GP (Fig. 9B–E). Thus, Fzd3 deficiency causes loss of GP-derived permissive signals but leaves the ability of striatal axons to sense these signals in wild-type GP tissue intact. It is unlikely that Fzd3 directly promotes the expression of permissive cues in GP cells, or represses inhibitory ones, because in situ hybridization for Fzd3 showed Fzd3 expression in the embryonic striatum, but no labeling in the mid-embryonic GP (Fig. 9F–H). In addition, we found no difference in the ability of striatal axons to enter the GP in Nkx2-1-Cre;Fzd3fl/fl mice, in which Fzd3 is genetically ablated from most GP neurons (Fig. 5). Together, these data argue against a direct role for Fzd3 in the regulation of molecular cues produced by GP neurons.
Figure 9.
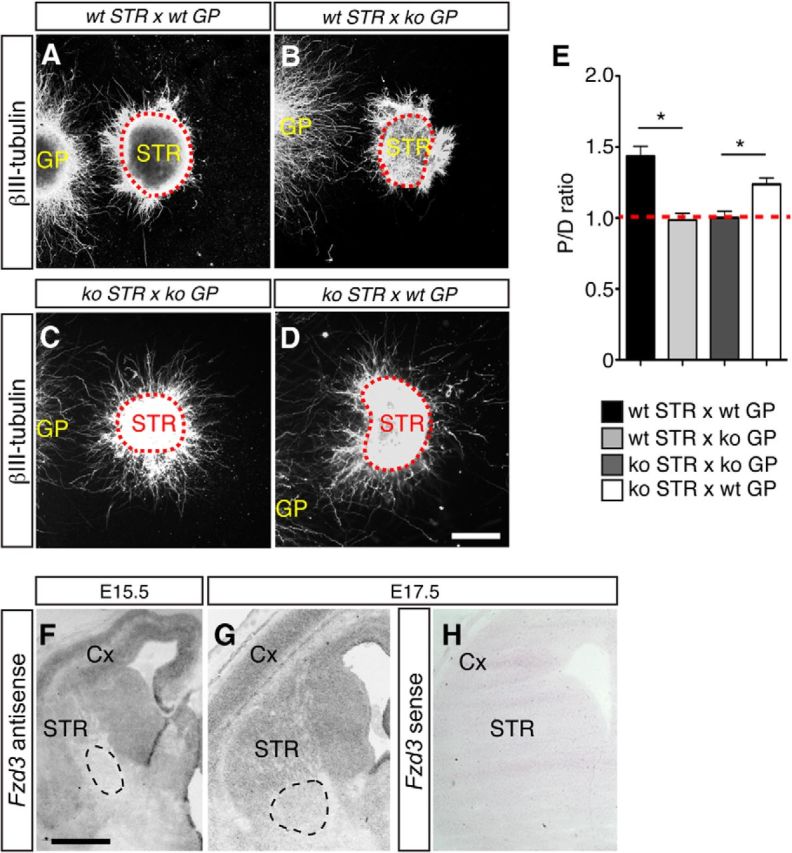
GP-mediated chemoattraction of striatal axons is lost in Frizzled3−/− mice. A–D, Collagen matrix cocultures of E14.5 Fzd3+/+ (WT) or Fzd3−/− (KO) striatal (STR) and GP explants maintained for 3 DIV and immunostained for βIII-tubulin. GP explants are on the left and dotted line indicates STR explants. E, Quantification of the P/D ratios of striatal explants in collagen matrix cultures as shown in A–D. P/D > 1.0 indicates attraction. wt STR versus wt GP, n = 18; wt STR versus ko GP, n = 29; ko STR versus ko GP, n = 11; ko STR versus wt GP, n = 28. *p < 0.05 (Kruskal–Wallis test). Red dotted line indicates P/D = 1.0. F–H, In situ hybridization for Fzd3 in coronal sections of E15.5 and E17.5 wild-type mice. Dotted line indicates GP, which does not show specific Fzd3 signal. No specific staining is detected following incubation with Fzd3 sense probes (H). Cx, Cortex. Scale bars: A–D, 180 μm; F, 580 μm; G, H, 650 μm.
As an alternative model, we examined whether Fzd3 controls the cellular make-up of the GP and thereby indirectly the permissive effect of this nucleus. Immunohistochemistry was performed for different molecular markers known to be expressed in and around the GP (Fig. 10A). Immunostaining for Nkx2-1 revealed that Nkx2-1+ GP neurons occupied a larger area in Fzd3−/− mice, but that their number was unchanged (E17.5 Fzd3+/+ 481.7 ± 1.2 Nkx2-1+ cells, Fzd3−/− 480.7 ± 1.3; n = 3 per genotype; p = 0.607) (Fig. 10B,C). A similar result was obtained by using in situ hybridization for two other GP markers, Sema3E and Netrin-1. Sema3E, and Netrin-1 expression was detected but occupied an expanded area (Fig. 10D–G). Expression of OL-pc, a marker for MSN neurons and axons, was unaffected in the striatum of Fzd3−/− embryos, but as expected OL-pc+ striatal axons did not project into the GP (Fig. 10H,I). Intriguingly, double immunolabeling for Nkx2-1 and Islet1 (Isl1) revealed an invasion of Isl1+ cells into the GP of Fzd3−/− mice (Fig. 10J,K). Isl1 is expressed in striatonigral MSNs and in “corridor” cells, a population of guidepost cells for thalamocortical axons located in between the GP and MGE (López-Bendito et al., 2006). Double labeling for Isl1 and Ctip2 confirmed the presence of ectopic Isl1+ cells in the Fzd3−/− GP (Fig. 10L,M). At early embryonic stages, Ctip2 labels the GP, striatum, and corridor cells, but at later stages (E15.5 onwards) becomes restricted to the striatum and corridor cells. Interestingly, at E15.5 ectopic Ctip2+ cells were observed in the E15.5 GP of Fzd3−/− but not control mice (Fig. 10L,M, inset). To determine whether the ectopic Isl1+;Ctip2+ cells in the Fzd3−/− GP represent MSNs or corridor cells, we performed immunostaining for Foxp1, which is expressed in MSNs but not in corridor cells. Immunolabeling revealed no Foxp1+cells in the GP of Fzd3−/− mice. This suggests that corridor cells are misplaced into the GP in the Fzd3−/− mice (Fig. 10N,O). Corridor cells express repulsive axon guidance proteins, such as Slit1, ephrinA5, and class 3 semaphorins (Sema3s), that guide thalamocortical axons in the subpallium (Garel and López-Bendito, 2014). The ectopic expression of these cues in the GP, due to mislocalization of corridor cells, could counteract the normally attractive effect of the GP on MSN axons. To test this model, we determined whether repulsive guidance molecules are ectopically expressed in the Fzd3−/− GP and assessed whether corridor cells affect MSN axon growth. In situ hybridization revealed that cues, such as ephrinA5, Sema3A, and Sema3F, are absent from wild-type GP but expressed in Fzd3−/− GP (Fig. 11A–D; data not shown). In addition, cocultures of corridor and striatal explants showed that striatal axons are repelled by corridor cells (Fig. 11E,F). Together, our results are consistent with a model in which mislocalization of corridor cells into the GP due to Fzd3 deficiency causes loss of the permissive effect of the GP on MSN axons due to the ectopic expression of axon repulsive cues.
Figure 11.
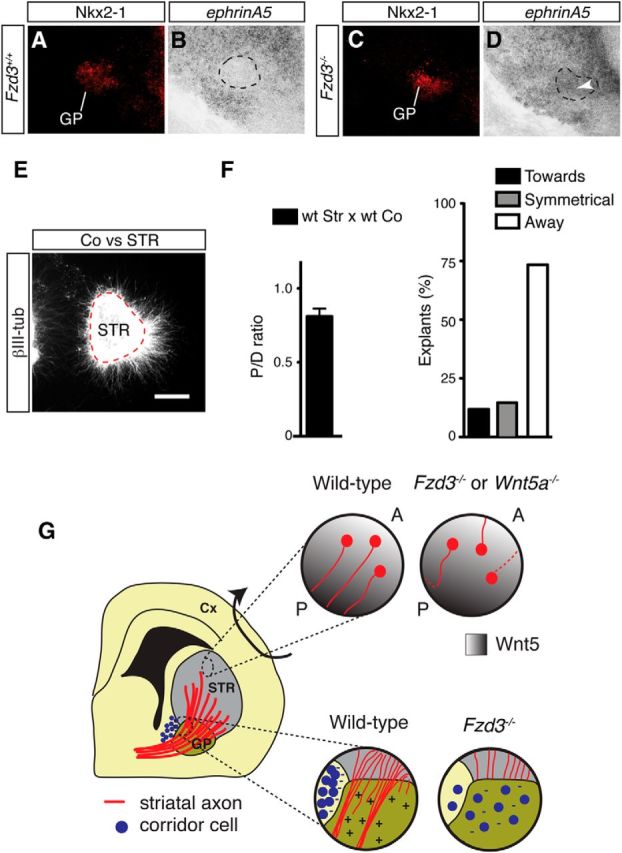
Corridor cells induce striatal axon repulsion. A–D, Double staining combining immunohistochemistry for Nkx2-1 (A, C) and in situ hybridization for ephrinA5 (B, D) in coronal sections from E13.5 embryos. EphrinA5-positive cells are present in the GP of Fzd3−/− but not wild-type embryos (indicated by arrowhead). Number of mice analyzed: E13.5 Fzd3+/+, n = 3; E13.5 Fzd3−/−, n = 3. E, Collagen matrix cocultures of E14.5 striatal (STR) and corridor cell (Co) explants maintained for 2 DIV and immunostained for βIII-tubulin. Coexplants are at the left. Dotted line indicates STR explant. F, Left graph, Quantification of the P/D ratios of STR explants in cocultures as in E. P/D < 1.0 indicates repulsion. n = 34 cocultures. Right graph, Quantification of the number of STR explants that show symmetrical axon growth or growth toward or away from coexplants. Scale bars: A–D, 220 μm; E, 160 μm. G, Schematic representation of a coronal section of the embryonic mouse brain showing the striatum (STR) and GP. In wild-type embryos, MSNs in the striatum send their axons posteriorly. These axons project into (striatopallidal pathway) or through (striatonigral pathway) the GP. An anterior (A) low, posterior (P) high Wnt5a/Wnt5b gradient is present in the striatum, and MSN axons are attracted by Wnt5s in vitro. Fzd3−/−, and Wnt5a−/− mice display aberrant caudal, lateral, and medial MSN projections. These data support a model in which Wnt5s guide Fzd3-postive MSN axons along the AP axis of the STR. In Fzd3−/− mice, all striatal axons accumulate at the GP and do not enter this structure. Corridor cells express axon repellents for MSN axons and are mislocalized in the GP of Fzd3−/− mice. The normally attractive effect of the GP on MSN axons is lost in the absence of Fzd3. This suggests that patterning of corridor cells by Fzd3 is required to create permissive corridors for MSN axon growth. Cx, Cortex.
Discussion
Striatal axon pathways mediate complex physiological functions and are a known therapeutic target, underscoring the need to define how these connections are established. MSN axon projections are remarkably complex and their development is contingent on the completion of several highly stereotypic events, such as AP extension and intermediate target navigation. The molecular programs regulating these disparate developmental processes remain mostly unknown. Here, we determine the embryonic ontogeny of the two main striatal pathways (striatonigral and striatopallidal) and identify a role for Fzd3 in uncharacterized aspects of striatal pathway formation (i.e., AP guidance in the striatum and axon entry into the GP). Further, our results link Fzd3 to corridor cell development.
Frizzled3 and Wnt5a control AP guidance of striatal axons
Frizzleds can pattern axonal connections along the AP axis different species (Zou, 2012; Avilés et al., 2013; Ackley, 2014). In mice, these molecules provide directional guidance for ascending and descending axons along the AP axis of the spinal cord and brainstem (Lyuksyutova et al., 2003; Wolf et al., 2008; Fenstermaker et al., 2010; Shafer et al., 2011; Hua et al., 2014). Until now, it was unknown whether AP guidance of longitudinal axon tracts originating in the forebrain also requires Frizzleds. Here, we show that indeed Fzd3 is required for AP guidance of striatal axons in the forebrain in vivo. In Fzd3−/− mice, MSN axons are misrouted along the lateral–medial and rostral–caudal axes of the striatum (i.e., no longer confined to their normal posterior trajectory). As a result, fewer MSN axons arrive at the rostral border of the GP (Fig. 11G). In the mouse brainstem and spinal cord, Fzd3-mediated AP guidance is controlled by complex Wnt protein gradients that bind Fzd3 to repel or attract axons into the appropriate direction (Lyuksyutova et al., 2003; Fenstermaker et al., 2010). Our data show expression of two Wnt family members, Wnt5a and Wnt5b, in an anterior low, posterior high pattern in the striatum and reveal chemoattractive effects of both Wnts on striatal axons in vitro. In addition, Wnt5a−/− mice display defects in AP guidance of MSN axons similar to those observed in Fzd3−/− mice. These observations together with previous work identifying Fzd3 as an axonal receptor for instructive Wnt5a gradients during axon pathfinding (Fenstermaker et al., 2010) are consistent with a model in which Fzd3 and Wnt5a cooperate to attract MSN axons posteriorly in the developing striatum (Fig. 11G). More generally, these data are, to our knowledge, the first to identify a cue, Fzd3, that dictates AP axon guidance within a large brain nucleus, the striatum.
Entry of striatal axons into the GP non–cell-autonomously requires Frizzled3
In the mouse, the GP is a final target for striatopallidal axons and an intermediate target for striatonigral axons (Loopuijt and van der Kooy, 1985). We show that, in Fzd3−/− mice, MSN axons are present and the outgrowth of Fzd3−/− MSN axons in vitro is intact. However, both striatopallidal and striatonigral axons stall at the rostal border of the GP failing to enter this structure (Fig. 11G). Our data show that the GP releases permissive cues for striatal axons and provide support for a model in which Fzd3 deficiency induces loss of this permissive nature of the GP, thereby causing axon stalling at this nucleus. First, in in vitro transplant studies, Fzd3−/− MSN axons successfully navigate the wild-type GP, but wild-type axons stall at the Fzd3−/− GP, as observed in vivo. Second, genetic ablation of Fzd3 in striatopallidal and striatonigral MSNs in D2-Cre;Fzd3fl/fl and Isl1-Cre;Fzd3fl/fl mice, respectively, does not prevent MSN axons from entering the GP. Third, axons derived from wild-type and Fzd3−/− striatal explants grow toward wild-type but not knock-out GP explants in vitro. Finally, genetic ablation of axonal populations that traverse the GP at early embryonic stages (i.e., dopaminergic and thalamocortical axons) and that may serve as a scaffold for MSN axons through reciprocal axon–axon interactions, does not influence the entry of MSN axons into the GP.
How does Fzd3 control the permissive nature of the GP? A plausible explanation would be that Fzd3 deficiency causes decreased expression of axon attractants or enhanced expression of axon inhibitory cues in GP cells. However, the lack of Fzd3 expression in the embryonic GP and the normal trajectory of striatal axons in Nkx2-1-Cre;Fzd3fl/fl mice, in which Fzd3 is inactivated in a major subset of GP cells (Nóbrega-Pereira et al., 2010), argue against direct regulation of guidance cue expression by Fzd3 in GP cells. Indeed, expression of several guidance cues, such as Netrin-1, Sema3E, and Sfrp4, was unchanged in the GP of Fzd3−/− embryos. Further, striatal axons enter the GP in knock-out mice lacking Netrin-1, Sema3E, or Sfrp4 (Chauvet et al., 2007; Ehrman et al., 2013; F.M. and R.J.P., unpublished observations).
We report that cells displaying a molecular signature of corridor cells (Isl1+ and Ctip2+, but Foxp1−) (López-Bendito et al., 2006; Bielle and Garel, 2013) are detected in the GP of Fzd3−/− but not wild-type embryos. These data are in line with previous work showing thalamocortical axon pathfinding defects and an abnormal distribution of cells expressing Ebf1, a marker for MSNs and corridor cells, in Dlx5/6-Cre;Fzd3fl/fl mice (Hua et al., 2014; Qu et al., 2014). Corridor cells are guidepost cells for thalamocortical axons and are located in between the GP and MGE (López-Bendito et al., 2006). Corridor cells originate in the lateral ganglionic eminence (LGE) and express molecular cues to sort and guide thalamocortical axons (López-Bendito et al., 2006; Bielle et al., 2011b; Garel and López-Bendito, 2014; Leyva-Díaz et al., 2014). These cues include several known axon repellents, including Slit1, Sema3A, and ephrinA5. It is therefore tempting to speculate that axon repellents expressed by mislocalized corridor cells in the Fzd3−/− GP disrupt molecular mechanisms that normally would facilitate MSN axon target entry into the GP (Fig. 11G). Our observations are consistent with this model. First, ectopic corridor cells in the GP of Fzd3−/− mice express axon repellents, such as ephrinA5 and Sema3s. Second, MSN axons are repelled by molecular cues released by corridor cell explants in vitro. Therefore, a possible explanation for our observations is that Fzd3 deficiency causes a mislocalization of corridor cells expressing axon repellents in the GP rendering this structure nonpermissive for MSN axon extension.
Interestingly, recent studies analyzing Fzd3−/− mice report motor axon stalling at an important intermediate target in the peripheral nervous system, the plexus region, and a failure of sympathetic axons to innervate peripheral targets, such as the heart and kidneys (Armstrong et al., 2011; Hua et al., 2013). These phenotypes have been attributed to defects in axon-intrinsic mechanisms. These results coupled with our own findings in the CNS indicate that Fzd3 can regulate (intermediate) target innervation through cell-autonomous and non–cell-autonomous mechanisms (i.e., by controlling axon growth and guidance or by regulating the cellular composition of intermediate targets, respectively).
A role for Frizzled3 in corridor cell positioning
Corridor cells migrate tangentially from the LGE to the MGE where they form an instructive corridor for thalamocortical axons. The molecules that control corridor cell migration or that confine these cells to their final position are largely unknown (Bielle and Garel, 2013). Here, we show that Fzd3 is required for restraining corridor cells to their normal position between the GP and MGE. This effect of Fzd3 is most likely non–cell-autonomous as analysis of Isl1-Cre;Fzd3fl/fl mice, in which Fzd3 is ablated in striatonigral MSNs (Ehrman et al., 2013; Lu et al., 2014) and corridor cells (López-Bendito et al., 2006), does not reveal defects in striatal axon pathfinding nor in the location of corridor cells (Fig. 5F,G; data not shown). Fzd3 is known to regulate neuronal migration (Wada et al., 2006; Vivancos et al., 2009; Fenstermaker et al., 2010; Qu et al., 2010; Hua et al., 2013), and one model to explain our observations is therefore that Fzd3 deficiency triggers the aberrant migration of corridor cells from the LGE into the GP. Given the important role of Slit2 in corridor cell migration (Bielle et al., 2011a) and expansion of slit2 expression following fzd3a knockdown in zebrafish (Hofmeister et al., 2012), it is tempting to speculate that Fzd3 regulates Slit2 expression along the presumptive trajectory of corridor cells and thereby controls their distribution. Future studies will address this and other possible mechanisms.
Importantly, our data suggest that the proper positioning of corridor cells may not only be crucial for the development of thalamocortical axons but also for the formation of permissive territories for other axonal populations, such as MSN axons. In addition, the contribution of Fzd3 to guidepost cell development is most likely not restricted to corridor cells. For example, Fzd3−/− mice show defects in the distribution of putative calretinin+ guidepost cells in the midbrain (Hua et al., 2014) and knockdown of fzd3a in zebrafish embryos perturbs the development of a population of glial guidepost cells at the midline of the forebrain (Hofmeister et al., 2012). Several different populations of guidepost cells have been defined in the brain (Bielle and Garel, 2013), and it will be interesting to determine whether, and if so how, Fzd3 contributes to the development of these cells.
In conclusion, our study provides tools and a conceptual framework for further dissection of striatal circuit assembly. Further, our data identify cell autonomous and non–cell-autonomous requirements for Fzd3 in several fundamental and previously uncharacterized aspects of striatal pathway formation in vivo. More generally, our findings identify Fzd3 as the first cue that controls AP guidance in a large brain nucleus and unveil a novel role for this transmembrane protein in regulating (intermediate) target entry of CNS axons. Further, they implicate Fzd3 in the positioning of corridor cells and indicate that an abnormal distribution of these cells may have unexpected, widespread effects on the development of different axon tracts.
Footnotes
This work was supported by The Netherlands Organization for Health Research and Development (ZonMW-VIDI and ZonMW-TOP), Stichting ParkinsonFonds, and the European Union (mdDANeurodev, FP7/2007–2011, Grant 222999) to R.J.P., Actions de Recherches Concerteés (ARC-10/15-026) and Fondation médicale Reine Elisabeth to F.T., and the Spanish MINECO BFU2012-34298 to G.L.-B. (who is an EMBO Young Investigator). This study was performed in part within the framework of Dutch Top Institute Pharma Project T5-207 to R.J.P. We thank Yimin Zou for critically reading this manuscript; Ger Arkestein for technical assistance during FACS; the Mutant Mouse Regional Resource Center facility at University of California–Davis; Jeremy Nathans, Marten Smidt, Oscar Marin, Stewart Anderson, Sonia Garel, and Thomas Jessell for providing mice; Jeremy Nathans for providing rabbit anti-Fzd3 antibody; and Eljo van Battum for help with the figures.
The authors declare no competing financial interests.
References
- Ackley BD. Wnt-signaling and planar cell polarity genes regulate axon guidance along the anteroposterior axis in C. elegans. Dev Neurobiol. 2014;74:781–796. doi: 10.1002/dneu.22146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KD, Reiner A. Immunohistochemical localization of DARPP-32 in striatal projection neurons and striatal interneurons: implications for the localization of D1-like dopamine receptors on different types of striatal neurons. Brain Res. 1991;568:235–243. doi: 10.1016/0006-8993(91)91403-N. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Jabaudon D, Yoshida Y, Macklis JD. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J Neurosci. 2008;28:622–632. doi: 10.1523/JNEUROSCI.2986-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong A, Ryu YK, Chieco D, Kuruvilla R. Frizzled3 is required for neurogenesis and target innervation during sympathetic nervous system development. J Neurosci. 2011;31:2371–2381. doi: 10.1523/JNEUROSCI.4243-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilés EC, Wilson NH, Stoeckli ET. Sonic hedgehog and Wnt: antagonists in morphogenesis but collaborators in axon guidance. Front Cell Neurosci. 2013;7:86. doi: 10.3389/fncel.2013.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bice PJ, Liang T, Zhang L, Strother WN, Carr LG. Drd2 expression in the high alcohol-preferring and low alcohol-preferring mice. Mamm Genome. 2008;19:69–76. doi: 10.1007/s00335-007-9089-2. [DOI] [PubMed] [Google Scholar]
- Bielle F, Garel S. Neuronal migration of guidepost cells. In: L R Rubenstein J, Rakic P, editors. Cellular migration and formation of neuronal connections. Amsterdam: Elsevier; 2013. pp. 457–479. [Google Scholar]
- Bielle F, Marcos-Mondejar P, Keita M, Mailhes C, Verney C, Nguyen Ba-Charvet K, Tessier-Lavigne M, Lopez-Bendito G, Garel S. Slit2 activity in the migration of guidepost neurons shapes thalamic projections during development and evolution. Neuron. 2011a;69:1085–1098. doi: 10.1016/j.neuron.2011.02.026. [DOI] [PubMed] [Google Scholar]
- Bielle F, Marcos-Mondéjar P, Leyva-Díaz E, Lokmane L, Mire E, Mailhes C, Keita M, García N, Tessier-Lavigne M, Garel S, López-Bendito G. Emergent growth cone responses to combinations of Slit1 and Netrin 1 in thalamocortical axon topography. Curr Biol. 2011b;21:1748–1755. doi: 10.1016/j.cub.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Chai G, Zhou L, Manto M, Helmbacher F, Clotman F, Goffinet AM, Tissir F. Celsr3 is required in motor neurons to steer their axons in the hindlimb. Nat Neurosci. 2014;17:1171–1179. doi: 10.1038/nn.3784. [DOI] [PubMed] [Google Scholar]
- Chakrabarty K, Von Oerthel L, Hellemons A, Clotman F, Espana A, Groot Koerkamp M, Holstege FC, Pasterkamp RJ, Smidt MP. Genome wide expression profiling of the mesodiencephalic region identifies novel factors involved in early and late dopaminergic development. Biol Open. 2012;1:693–704. doi: 10.1242/bio.20121230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet S, Cohen S, Yoshida Y, Fekrane L, Livet J, Gayet O, Segu L, Buhot MC, Jessell TM, Henderson CE, Mann F. Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron. 2007;56:807–822. doi: 10.1016/j.neuron.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédotal A. Further tales of the midline. Curr Opin Neurobiol. 2011;21:68–75. doi: 10.1016/j.conb.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Chen L, Guo Q, Li JY. Transcription factor Gbx2 acts cell-non-autonomously to regulate the formation of lineage-restriction boundaries of the thalamus. Development. 2009;136:1317–1326. doi: 10.1242/dev.030510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Graybiel AM. Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat. 2011;5:59. doi: 10.3389/fnana.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deck M, Lokmane L, Chauvet S, Mailhes C, Keita M, Niquille M, Yoshida M, Yoshida Y, Lebrand C, Mann F, Grove EA, Garel S. Pathfinding of corticothalamic axons relies on a rendezvous with thalamic projections. Neuron. 2013;77:472–484. doi: 10.1016/j.neuron.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijck AA, Van Erp S, Pasterkamp RJ. Semaphorin signaling: molecular switches at the midline. Trends Cell Biol. 2010;20:568–576. doi: 10.1016/j.tcb.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Dickson BJ, Zou Y. Navigating intermediate targets: the nervous system midline. Cold Spring Harb Perspect Biol. 2010;2:a002055. doi: 10.1101/cshperspect.a002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrman LA, Mu X, Waclaw RR, Yoshida Y, Vorhees CV, Klein WH, Campbell K. The LIM homeobox gene Isl1 is required for the correct development of the striatonigral pathway in the mouse. Proc Natl Acad Sci U S A. 2013;110:E4026–E4035. doi: 10.1073/pnas.1308275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenstermaker AG, Prasad AA, Bechara A, Adolfs Y, Tissir F, Goffinet A, Zou Y, Pasterkamp RJ. Wnt/planar cell polarity signaling controls the anterior–posterior organization of monoaminergic axons in the brainstem. J Neurosci. 2010;30:16053–16064. doi: 10.1523/JNEUROSCI.4508-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garel S, López-Bendito G. Inputs from the thalamocortical system on axon pathfinding mechanisms. Curr Opin Neurobiol. 2014;27C:143–150. doi: 10.1016/j.conb.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia: learning new tricks and loving it. Curr Opin Neurobiol. 2005;15:638–644. doi: 10.1016/j.conb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Halford MM, Armes J, Buchert M, Meskenaite V, Grail D, Hibbs ML, Wilks AF, Farlie PG, Newgreen DF, Hovens CM, Stacker SA. Ryk-deficient mice exhibit craniofacial defects associated with perturbed Eph receptor crosstalk. Nat Genet. 2000;25:414–418. doi: 10.1038/78099. [DOI] [PubMed] [Google Scholar]
- Hofmeister W, Devine CA, Rothnagel JA, Key B. Frizzled-3a and slit2 genetically interact to modulate midline axon crossing in the telencephalon. Mech Dev. 2012;129:109–124. doi: 10.1016/j.mod.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Hua ZL, Smallwood PM, Nathans J. Frizzled3 controls axonal development in distinct populations of cranial and spinal motor neurons. Elife. 2013;2:e01482. doi: 10.7554/eLife.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua ZL, Jeon S, Caterina MJ, Nathans J. Frizzled3 is required for the development of multiple axon tracts in the mouse central nervous system. Proc Natl Acad Sci U S A. 2014;111:E3005–E3014. doi: 10.1073/pnas.1406399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis. 2005;43:129–135. doi: 10.1002/gene.20162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi L, Charron F. Midline axon guidance and human genetic disorders. Clin Genet. 2011;80:226–234. doi: 10.1111/j.1399-0004.2011.01735.x. [DOI] [PubMed] [Google Scholar]
- Kolk SM, Gunput RA, Tran TS, van den Heuvel DM, Prasad AA, Hellemons AJ, Adolfs Y, Ginty DD, Kolodkin AL, Burbach JP, Smidt MP, Pasterkamp RJ. Semaphorin 3F is a bifunctional guidance cue for dopaminergic axons and controls their fasciculation, channeling, rostral growth, and intracortical targeting. J Neurosci. 2009;29:12542–12557. doi: 10.1523/JNEUROSCI.2521-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Pasterkamp RJ. SnapShot: axon guidance II. Cell. 2013;153:722.e1. doi: 10.1016/j.cell.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Leyva-Díaz E, López-Bendito G. In and out from the cortex: development of major forebrain connections. Neuroscience. 2013;254:26–44. doi: 10.1016/j.neuroscience.2013.08.070. [DOI] [PubMed] [Google Scholar]
- Leyva-Díaz E, del Toro D, Menal MJ, Cambray S, Susín R, Tessier-Lavigne M, Klein R, Egea J, López-Bendito G. FLRT3 is a Robo1-interacting protein that determines Netrin-1 attraction in developing axons. Curr Biol. 2014;24:494–508. doi: 10.1016/j.cub.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lobo MK. Molecular profiling of striatonigral and striatopallidal medium spiny neurons past, present, and future. Int Rev Neurobiol. 2009;89:1–35. doi: 10.1016/S0074-7742(09)89001-6. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Yeh C, Yang XW. Pivotal role of early B-cell factor 1 in development of striatonigral medium spiny neurons in the matrix compartment. J Neurosci Res. 2008;86:2134–2146. doi: 10.1002/jnr.21666. [DOI] [PubMed] [Google Scholar]
- Loopuijt LD, van der Kooy D. Organization of the striatum: collateralization of its efferent axons. Brain Res. 1985;348:86–99. doi: 10.1016/0006-8993(85)90363-4. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Cautinat A, Sánchez JA, Bielle F, Flames N, Garratt AN, Talmage DA, Role LW, Charnay P, Marín O, Garel S. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KM, Evans SM, Hirano S, Liu FC. Dual role for Islet-1 in promoting striatonigral and repressing striatopallidal genetic programs to specify striatonigral cell identity. Proc Natl Acad Sci U S A. 2014;111:E168–E177. doi: 10.1073/pnas.1319138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyuksyutova AI, Lu CC, Milanesio N, King LA, Guo N, Wang Y, Nathans J, Tessier-Lavigne M, Zou Y. Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science. 2003;302:1984–1988. doi: 10.1126/science.1089610. [DOI] [PubMed] [Google Scholar]
- Mastick GS, Farmer WT, Altick AL, Nural HF, Dugan JP, Kidd T, Charron F. Longitudinal axons are guided by Slit/Robo signals from the floor plate. Cell Adh Migr. 2010;4:337–341. doi: 10.4161/cam.4.3.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Gelman D, Bartolini G, Pla R, Pierani A, Marín O. Origin and molecular specification of globus pallidus neurons. J Neurosci. 2010;30:2824–2834. doi: 10.1523/JNEUROSCI.4023-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passante L, Gaspard N, Degraeve M, Frisén J, Kullander K, De Maertelaer V, Vanderhaeghen P. Temporal regulation of ephrin/Eph signalling is required for the spatial patterning of the mammalian striatum. Development. 2008;135:3281–3290. doi: 10.1242/dev.024778. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424:398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Kolk SM, Hellemons AJ, Kolodkin AL. Expression patterns of semaphorin7A and plexinC1 during rat neural development suggest roles in axon guidance and neuronal migration. BMC Dev Biol. 2007;7:98. doi: 10.1186/1471-213X-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Glasco DM, Zhou L, Sawant A, Ravni A, Fritzsch B, Damrau C, Murdoch JN, Evans S, Pfaff SL, Formstone C, Goffinet AM, Chandrasekhar A, Tissir F. Atypical cadherins Celsr1–3 differentially regulate migration of facial branchiomotor neurons in mice. J Neurosci. 2010;30:9392–9401. doi: 10.1523/JNEUROSCI.0124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Huang Y, Feng J, Alvarez-Bolado G, Grove EA, Yang Y, Tissir F, Zhou L, Goffinet AM. Genetic evidence that Celsr3 and Celsr2, together with Fzd3, regulate forebrain wiring in a Vangl-independent manner. Proc Natl Acad Sci U S A. 2014;111:E2996–E3004. doi: 10.1073/pnas.1402105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ERE, Morello F, Pasterkamp RJ. Dissection and culture of mouse dopaminergic and striatal explants in three-dimensional collagen matrix assays. J Vis Exp. 2012;61:pii3691. doi: 10.3791/3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ER, Brignani S, Adolfs Y, Lemstra S, Demmers J, Vidaki M, Donahoo AL, Lilleväli K, Vasar E, Richards LJ, Karagogeos D, Kolk SM, Pasterkamp RJ. Subdomain-mediated axon-axon signaling and chemoattraction cooperate to regulate afferent innervation of the lateral habenula. Neuron. 2014;83:372–387. doi: 10.1016/j.neuron.2014.05.036. [DOI] [PubMed] [Google Scholar]
- Shafer B, Onishi K, Lo C, Colakoglu G, Zou Y. Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev Cell. 2011;20:177–191. doi: 10.1016/j.devcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SM, Kang YJ, Christensen BL, Feng AS, Kollmar R. Expression of Wnt receptors in adult spiral ganglion neurons: frizzled 9 localization at growth cones of regenerating neurites. Neuroscience. 2009;164:478–487. doi: 10.1016/j.neuroscience.2009.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, von Oerthel L, Hoekstra EJ, Schellevis RD, Hoekman MF. Spatial and temporal lineage analysis of a Pitx3-driven Cre-recombinase knock-in mouse model. PLoS One. 2012;7:e42641. doi: 10.1371/journal.pone.0042641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckli ET. Longitudinal axon guidance. Curr Opin Neurobiol. 2006;16:35–39. doi: 10.1016/j.conb.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Shaping the nervous system: role of the core planar cell polarity genes. Nat Rev Neurosci. 2013;14:525–535. doi: 10.1038/nrn3525. [DOI] [PubMed] [Google Scholar]
- Uemura M, Nakao S, Suzuki ST, Takeichi M, Hirano S. OL-Protocadherin is essential for growth of striatal axons and thalamocortical projections. Nat Neurosci. 2007;10:1151–1159. doi: 10.1038/nn1960. [DOI] [PubMed] [Google Scholar]
- Van den Heuvel DM, Pasterkamp RJ. Getting connected in the dopamine system. Prog Neurobiol. 2008;85:75–93. doi: 10.1016/j.pneurobio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivancos V, Chen P, Spassky N, Qian D, Dabdoub A, Kelley M, Studer M, Guthrie S. Wnt activity guides facial branchiomotor neuron migration, and involves the PCP pathway and JNK and ROCK kinases. Neural Dev. 2009;4:7. doi: 10.1186/1749-8104-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Tanaka H, Nakayama S, Iwasaki M, Okamoto H. Frizzled3a and Celsr2 function in the neuroepithelium to regulate migration of facial motor neurons in the developing zebrafish hindbrain. Development. 2006;133:4749–4759. doi: 10.1242/dev.02665. [DOI] [PubMed] [Google Scholar]
- Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J. Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J Neurosci. 2002;22:8563–8573. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AM, Lyuksyutova AI, Fenstermaker AG, Shafer B, Lo CG, Zou Y. Phosphatidylinositol-3-kinase-atypical protein kinase C signaling is required for Wnt attraction and anterior-posterior axon guidance. J Neurosci. 2008;28:3456–3467. doi: 10.1523/JNEUROSCI.0029-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- Zou Y. Does planar cell polarity signaling steer growth cones? Curr Top Dev Biol. 2012;101:141–160. doi: 10.1016/B978-0-12-394592-1.00009-0. [DOI] [PubMed] [Google Scholar]