

ABSTRACT
Overall, NSCLC has a poor 5-year survival and new therapeutic approaches are urgently needed. ERBB-addicted NSCLC that have become resistant to ERBB inhibitors are often refractory to additional therapeutic interventions. The sphingosine-1-phosphate receptor modulator fingolimod (FTY720), approved for the treatment of multiple sclerosis, synergized with the NSCLC therapeutic pemetrexed to kill NSCLC and ovarian cancer cells. This occurred in lung cancer cells expressing mutated K-RAS, mutated ERBB1, or in NSCLC cells resistant to afatinib (an ERBB1/2/4 inhibitor). This drug combination appeared to use overlapping and distinct mechanisms of killing in different cell lines. Activation of AMP-dependent kinase (AMPK) and reduced expression and inactivation of mTOR were associated with increased autophagosome and autolysosome formation. Downregulation of Beclin1 considerably reduced formation of autophagosomes and protected the cells from drug combination-induced killing without significantly altering autolysosome formation. Autophagy protein 5 (ATG5) knock down afforded greater protection against the combination of pemetrexed with fingolimod. Treatment of cells with the mTOR inhibitor everolimus markedly enhanced the lethality of pemetrexed plus fingolimod combination. Our data suggest that the combination of fingolimod with the established NSCLC/ovarian cancer drug pemetrexed should be explored as a new therapy.
Keywords: autophagy, HDAC, sphingolipid
Introduction
Mutated active forms of ERBB1 and ERBB2, and ERBB2 over-expression, are very often biomarkers of an aggressive tumor.1 However, these biomarkers also (usually) predict for tumor cell addiction to their oncogenic signals and small molecule kinase inhibitors as well as inhibitory antibodies have been developed to block ERBB1/ERBB2 signaling.2,3 These approaches have been successful in the clinic.4,5 Cancer cells, however, are biologically, in the short-term, and genetically in the mid- to long-term, meta-stable. Thus, any evolutionary pressure placed on a tumor cell results in that cell attempting to evolve away from that pressure.
NSCLC is a devastating disease that is often diagnosed at a stage where effective therapies exhibit only partial efficacy.6,7 A frequently observed oncogenic driver mutation in NSCLC is the mutation of K-RAS that modulates its GTPase activity.8,9 Mutant K-RAS signals downstream into multiple downstream pathways including ERK1/2, AKT, JNK and NFkB.10–12 Because of the plasticity of cell signaling within tumor cells, inhibition of any single pathway downstream of mutant K-RAS only results in a partial growth retardation, which ultimately evolves the tumor cells to rely for their growth and viability on other pathways. We have recently examined the biology of H1975 NSCLC tumor cells, a line addicted to signaling by mutated active ERBB1.13–15 We found that resistance to the FDA approved suicide ERBB1/2/4 inhibitor afatinib of in vivo generated H1975 NSCLC tumors, that are addicted to signaling by mutated active ERBB1 is mediated by SRC/ERBB3.13–15 Ample evidence supports the notion that inhibition of one or even two signaling pathways downstream of mutated active plasma membrane signaling molecules such as ERBB1 or K-RAS will be insufficient to cause a prolonged durable anti-tumor response in patients.
Pemetrexed is an approved therapeutic for NSCLC, and is compendium listed for ovarian cancer, and NSCLC patients who have failed ERBB inhibitor therapy are often treated with this drug. Numerous preclinical studies have demonstrated that the sphingosine-1-phosphate (S1P) receptor modulator fingolimod (FTY720), the first orally available drug for treatment of multiple sclerosis, has potent anti-cancer actions.16 In colitis-associated cancer, fingolimod interfered with S1P receptor 1 feed-forward signaling to the pro-growth and pro-survival NFkB and STAT3 pathways.17,18 Inhibition of these pathways leads to tumor growth suppression. Similarly, in breast cancer, fingolimod not only reduced tumor growth, it also reduced lung metastasis.19 Because fingolimod can also enhance the efficacy of several anti-cancer drugs including sorafenib and regorafenib; in the present study, we examined enhancement of the killing effect of pemetrexed by fingolimod in a genetically diverse pool of NSCLC cells; determined their mechanisms of action; and effects of modulators of autophagy.20
Results
In agreement with previous studies, we found that low concentrations of pemetrexed or fingolimod induced cell death to a similar extent in a genetically diverse set of NSCLC cell lines (Figure 1A).20,21 Pemetrexed and fingolimod interacted in an additive to greater than additive fashion in short-term cell death assay (Figure 1A). Moreover, the combination of pemetrexed and fingolimod not only enhanced killing of wild type H1975 tumor clones but also similarly in the afatinib-resistant H1975 tumor clones indicating that afatinib resistance, including reduced PTEN expression, does not stop this drug combination from killing (Figure 1B).13,14 The drug combination also killed multiple established and PDX isolates of ovarian cancer (Figure 1C).
Figure 1.
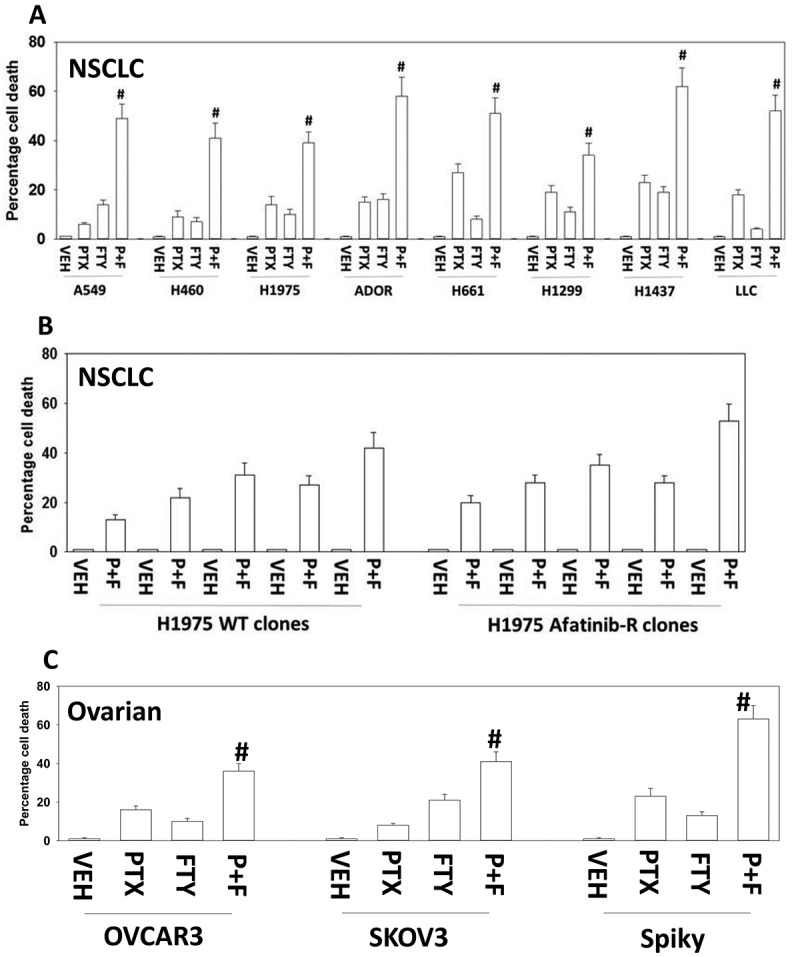
Pemetrexed and fingolimod interact to kill NSCLC cells. A. and B. Established human NSCLC lines (A549, H460, H1975, H661, H1299, H1437), a PDX NSCLC isolate ADOR, mouse Lewis Lung Carcinoma cells or tumor-derived H1975 (5 parental clones; 5 afatinib-resistant clones) were treated with vehicle control, pemetrexed (PTX, 500 nM), fingolimod (FTY, 200 nM) or the drugs in combination for 24h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). # p < 0.05 greater value than pemetrexed treatment alone. C. Established human ovarian cancer lines (OVCAR3, SKOV3) and a PDX isolate Spiky were treated with vehicle control, pemetrexed (PTX, 500 nM), fingolimod (FTY, 200 nM) or the drugs in combination for 24h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). # p < 0.05 greater value than pemetrexed treatment alone.
We next performed a series of agnostic screening studies to examine growth factor signaling pathways and DNA damage response pathways. Treatment of NSCLC cells with the combination of [pemetrexed + fingolimod] reduced the expression of the autophagy-regulatory kinase mTOR and in parallel reduced the phosphorylation of mTOR on S2448 and S2481, that are important for interactions with mTORC1 and mTORC2, respectively (Figure 2A). The phosphorylation of ULK1 S757, an mTORC1 substrate, was reduced and this was associated with ULK1 activation as shown by increased ATG S318 phosphorylation, also indicative for the potential of autophagosome formation. Further support for ULK1 activation was provided by the observation that drug exposure increased AMPKα T172 phosphorylation, and phosphorylation of the AMPK substrate, ULK1 S317 (Figure 2A) [20, 22 and references therein]. Similar protein phosphorylation data were obtained in a PDX ovarian cancer isolate (Figure S1A).
Figure 2.
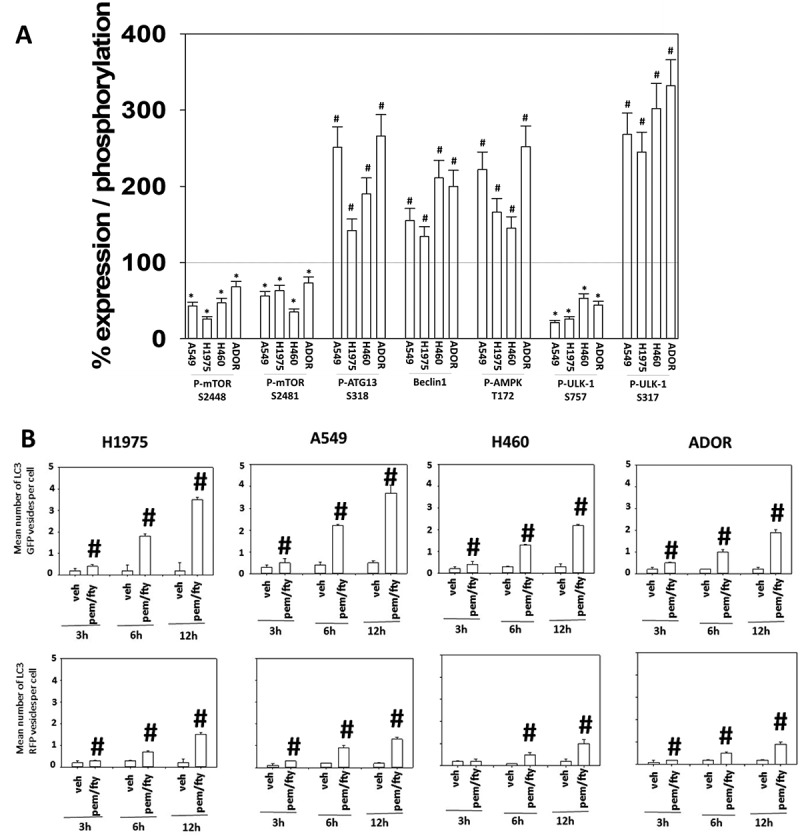
[Pemetrexed + fingolimod] treatment inactivates mTOR, activates the AMPK and ULK1 and simultaneously induces autophagosome and autolysosome formation. A. NSCLC cells were treated with vehicle control or with [pemetrexed (500 nM) + fingolimod (200 nM)] for 6h. After six h, cells were fixed in place and immunostaining performed to detect the expression and phosphorylation of the indicated proteins. Staining densities of phospho-/total-protein levels are determined in 40 cells in triplicate using software integral in the Hermes WiScan microscope (n = 120 cells ± SEM). # p < 0.05 greater than vehicle control value; * p < 0.05 less than vehicle control value. B. NSCLC cells were transfected with a plasmid to express LC3-GFP-RFP. Twenty-four h after transfection, cells were treated with vehicle control or with [pemetrexed (500 nM) + fingolimod (200 nM)] for 3h, 6h or 12h. At each time point the mean number of intense staining GFP+ and RFP+ vesicles in the cells was determined (at least 40 cells per condition were counted) (n = 120 cells ± SEM). # p < 0.05 greater than vehicle control value.
Pemetrexed can activate AMPK via two mechanisms; by DNA damage, and by increasing the levels of the allosteric activator ZMP [20, 22 and references therein]. It has also been recently shown that fingolimod/FTY720 can cause the internalization of the glucose transporter GLUT-1, which will lower glucose levels inside the cell, cause an increase in AMP levels, and allosterically activate AMPK.22 Consistent with previous reports treatment of H460 cells with fingolimod/FTY720 and, to a greater extent, the drug combination, reduced cell surface levels of GLUT-1 by almost 50% (data not shown). Taken together these data suggest that AMPK activation may be mediated via both ZMP and AMP.
Because the levels of Beclin1, essential for autophagosome formation, were also enhanced after exposure to the drug combination (see Figure 2A), we next investigated the impact of the drug combination on autophagosome and autolysosome formation using cells transiently transfected to express an LC3-GFP-RFP construct. Drug-treated cells simultaneously increased the levels of GFP+ staining autophagosomes and RFP+ autolysosomes (Figure 2B). Similar autophagosome and autolysosome formation data were obtained in ovarian cancer cells (Figure S1B). This finding is in contrast to other drug combinations we have developed using pemetrexed, e.g. [pemetrexed + sorafenib], where autophagosome formation preceded any observation of elevated autolysosome levels.20,21
Based on the data in Figure 2, and our prior publications, we predicted that knock down of the autophagy regulatory proteins ULK-1, Beclin1 or ATG5 would reduce drug combination toxicity.15 Knock down of either ULK-1, Beclin1 or ATG5 reduced the lethality of [pemetrexed + fingolimod], however in both H460 and A549 cells, knock down of ATG5 was more efficacious than knock down of Beclin1 (Figure 3A and B). Knock down of ATG5 or Beclin1 protected Spiky ovarian cancer cells from [pemetrexed + fingolimod] lethality (Figure S2). Prior studies from our laboratory using pemetrexed have demonstrated that the drug can play a role in facilitating endoplasmic reticulum stress signaling, which is mechanistically causal in enhancing the expression of Beclin1 [20, 22 and references therein]. In A549 cells, but not H460 cells, knock down of PERK or eIF2α did not reduce [pemetrexed + fingolimod] lethality. This observation correlated with data in Figure 2A which demonstrated: (a) that the basal expression of Beclin1 in A549 cells was high; (b) that the drug combination weakly induced Beclin1 in H460 cells that correlated with a very weak protective effect of Beclin1 knock down in this cell line.
Figure 3.
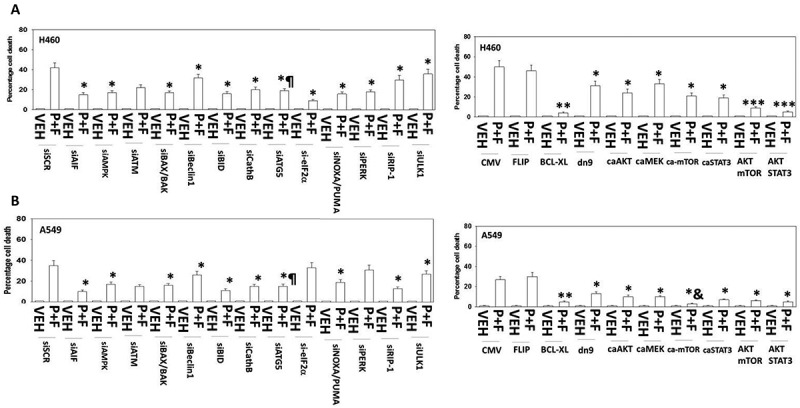
[Pemetrexed + fingolimod] lethality requires ATG5-dependent mitochondrial dysfunction leading to apoptotic and necroptotic death processes. A. and B. H460 and A549 NSCLC cells were transfected with the following plasmids to express: CMV/empty vector; c-FLIP-s; BCL-XL; dominant negative caspase 9; activated AKT; activated MEK1; activated mTOR; activated STAT3. Other H460 and A549 cells were transfected with the following siRNA molecules to knock down the expression of: control scramble, siSCR; apoptosis inducing factor, AIF; AMPKα subunit; ATM; BAX; BAK; Beclin1; BID; cathepsin B; ATG5; eIF2α; NOXA; PUMA; PERK; RIP-1; ULK1. Twenty-four h after transfection cells were treated with vehicle control or with [pemetrexed (500 nM) + fingolimod (200 nM)] for 24h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). * p < 0.05 lower value than the corresponding value in CMV/siSCR transfected cells; ** p < 0.01 lower value than the corresponding value in CMV/siSCR transfected cells; *** p < 0.05 less than corresponding value in activated AKT transfected cells; & p < 0.05 less death than corresponding value in H460 cells; ¶ p < 0.05 less than corresponding value in siBeclin1 transfected and in ULK1 transfected cells.
Consistent with activation of AMPK and inhibition of mTOR in both cell lines, downregulation of the ATM-AMPK signaling module reduced lethality induced by the combination of pemetrexed and FTY720 (Figure 3A and B). Moreover down-regulation of the knock down of toxic BH3 domain proteins including BAX, BAK, PUMA, NOXA and BID. BID can be cleaved and activated by caspases 8 and 10 that are downstream of death receptors, but also by lysosomal proteases such as cathepsins. Knock down of cathepsin B expression protected cells from [pemetrexed + fingolimod]. Signaling by RIP-1 has been linked to necroptotic signaling and knock down of RIP-1 reduced [pemetrexed + fingolimod] killing.23
We next examined contributions of survival signaling pathways. In both cell lines, expression of activated forms of AKT, MEK1, mTOR or STAT3 suppressed [pemetrexed + fingolimod] lethality (Figure 3A and B). In H460 cells activated AKT cooperated with activated mTOR or with activated STAT3 to further suppress cell killing. Over-expression of the mitochondrial protective protein BCL-XL prevented drug combination lethality in both lines, whereas expression of dominant negative caspase 9 that acts downstream of the mitochondrion was protective, though significantly less effective. Inhibition of death receptor signaling via over-expression of the caspase 8/10 inhibitor c-FLIP-s did not protect the cells, which argues autophagy-dependent cathepsin B signaling must play a key role in mediating mitochondrial dysfunction.
The data in Figure 3 indicate that ULK-1, Beclin1 and ATG5 played an essential role in killing by [pemetrexed + fingolimod]. In addition, we demonstrated in Figure 2 that autophagosome and autolysosome formation occurred simultaneously after drug exposure and that expression of activated mTOR was significantly more protective in A549 cells than in H460 cells (Figure 3). This correlated with a stronger drugs-induced expression of Beclin1 in A549 cells. Taken together, these findings suggest that promoting further autophagosome formation by use of an mTOR inhibitor may increase lethality of the drug combination. Treatment of NSCLC cells with the FDA approved mTOR inhibitor everolimus enhanced basal and drug combination induced levels of autophagosomes and autolysosomes (Figure 4), an effect that was reduced by knock down of Beclin1. Although knock down of Beclin1 initially also reduced autolysosome formation, however its effect was lost after 12 h. This data suggests that the drug combination stimulates formation of autolysosome vesicle (stained with RFP-LC3) not only by enhancing autophagic flux, but also via a pathway that directly activates lysosomal digestive processes.
Figure 4.
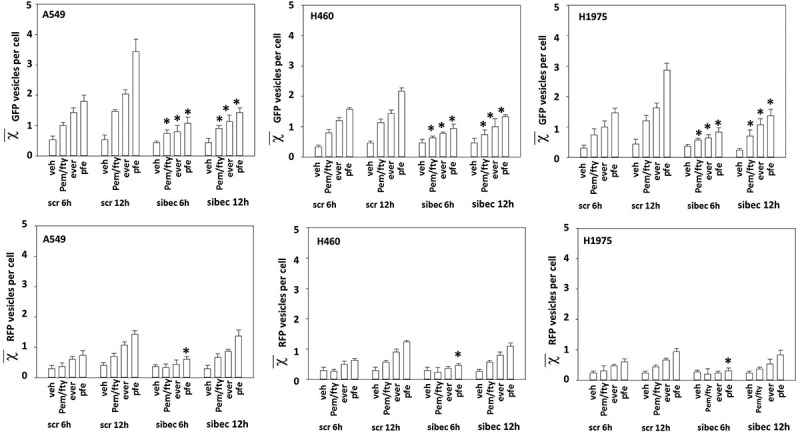
Down-regulation of Beclin1 delays but does not prevent autolysosome formation. NSCLC cells were transfected with a scrambled siRNA or an siRNA to knock down the expression of Beclin1. In parallel, the cells were transfected with a plasmid to express LC3-GFP-RFP. Twenty-four h later, cells were treated with vehicle control, [pemetrexed (500 nM) + fingolimod (200 nM)], everolimus (50 nM) or the three drugs in combination for 6h or 12h. At each time point the mean number of intense staining GFP+ and RFP+ vesicles in the cells was determined (at least 40 cells per condition were counted) (n = 120 cells ± SEM). * p < 0.05 less than corresponding vehicle control value.
Importantly, everolimus markedly enhanced the lethality of [pemetrexed + fingolimod] in NSCLC cells (Figure 5A). In afatinib-resistant H1975 cells we discovered that the three-drug combination exhibited a greater degree of cell killing when compared to the lethality of the combination in the wild type control clones (Figure 5B). Knock down of ATG5, Beclin1 or ULK1 suppressed these drug combination-induced cell deaths, with ATG5 knock down again being significantly more capable of reducing cell killing when compared to Beclin1 or ULK1 knock down (Figure 6A). In contrast to the mechanisms of killing caused by [pemetrexed + fingolimod], killing by [pemetrexed + fingolimod + everolimus] was suppressed by over-expression of the caspase 8/10 inhibitor c-FLIP-s, suggesting that everolimus was facilitating death receptor signaling or auto-activation of these caspase enzymes (Figure 6B). As was observed in Figure 3, expression of activated mTOR, activated AKT or activated STAT3 significantly reduced cell killing by the three-drug combination, whereas expression of activated MEK1 was less protective (Figure 6B).
Figure 5.
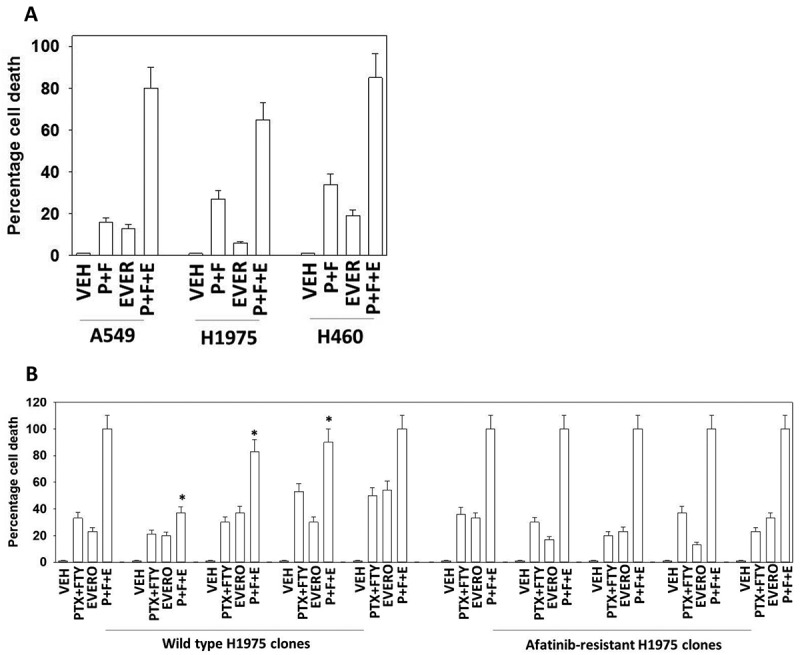
Everolimus enhances [pemetrexed + fingolimod] lethality. A. NSCLC cells were treated with vehicle control, [pemetrexed (500 nM) + fingolimod (200 nM)], everolimus (50 nM) or the three drugs in combination for 12h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). # p < 0.05 greater value than [pemetrexed + fingolimod] treatment alone. B. Wild type parental H1975 clones and afatinib-resistant H1975 clones were treated with vehicle control, [pemetrexed (500 nM) + fingolimod (200 nM)], everolimus (50 nM) or the three drugs in combination for 12h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). * p < 0.05 less than 100%.
Figure 6.
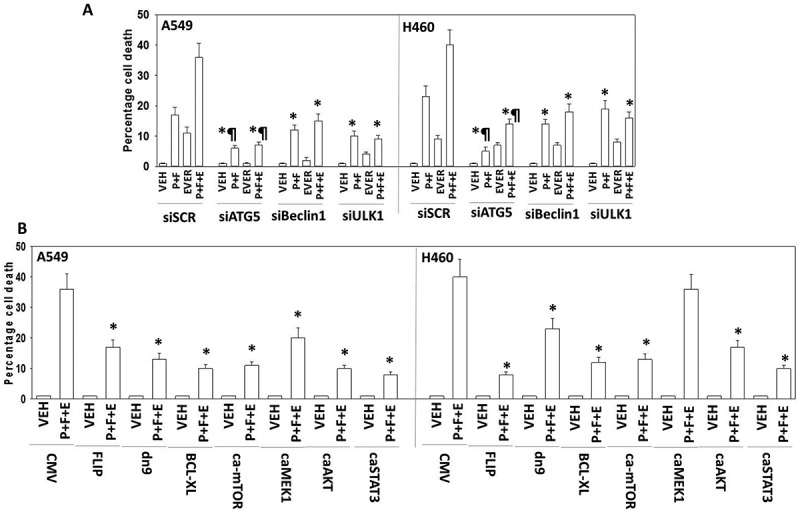
[Pemetrexed + fingolimod + everolimus] is ATG5-dependent and blocked by inhibition of caspases 8/10. A. NSCLC cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of ATG5, Beclin1 or ULK1. Twenty-four h after transfection cells were treated with vehicle control, [pemetrexed (500 nM) + fingolimod (200 nM)], everolimus (50 nM) or the three drugs in combination for 24h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). * p < 0.05 less than corresponding value in siSCR transfected cells; ¶ p < 0.05 less than corresponding value in siBeclin1 cells. B. NSCLC cells were transfected with the following plasmids to express: CMV/empty vector; c-FLIP-s; BCL-XL; dominant negative caspase 9; activated AKT; activated MEK1; activated mTOR; activated STAT3. Twenty-four h after transfection cells were treated with vehicle control, [pemetrexed (500 nM) + fingolimod (200 nM)], everolimus (50 nM) or the three drugs in combination for 24h. Cells were then isolated and viability determined via a live/dead assay (n = 3 ± SEM). * p < 0.05 less than corresponding value in control CMV transfected cells.
Discussion
The present studies were focused on new drug combinations for treatment of NSCLC, and in particular, afatinib-resistant NSCLC, where pemetrexed is an approved drug. After their tumors have become resistant to RTK inhibitors, this NSCLC patient population has few therapeutic options, e.g. these tumors are generally ‘cold’ to checkpoint immunotherapy approaches. The present studies were performed to define whether pemetrexed and the S1P signaling down-regulator fingolimod interact to kill NSCLC cells, and then determined the mechanisms of killing.
We discovered that combination of Pemetrexed with the S1PR1 modulator fingolimod interacted in a greater than additive manner to kill NSCLC cells. This effect was evident in numerous NSCLC cell lines with different oncogenic mutations as well as afatinib-resistance. Although both early nucleation and mid-stage elongation autophagosome formation played a role in killing by this drug combination, as judged by knock down of Beclin1 and ATG5, respectively, the induction of mid-stage autophagosome formation, dependent on ATG5, and enhanced autolysosome levels, played a more significant role in the killing process than early-stage autophagosomes. The relevance of autolysosome formation to the cell killing process was also affirmed by our data demonstrating that knock down of cathepsin B or BID, but not over-expression of c-FLIP-s, prevented tumor cell death.
We also found that pemetrexed combined with fingolimod can stimulate autophagy, in part, through activation of an ATM-AMPK pathway that leads to mTOR inactivation and ULK1 activation. First, knock down of either ATM or AMPKα significantly reduced drug combination lethality. Second, this drug combination enhanced AMPKα T172 phosphorylation and this was associated with increased activating ULK1 S317 phosphorylation.21 Moreover, in parallel, expression and phosphorylation of mTOR declined and was associated with reduced inhibitory ULK1 S757 phosphorylation and increased ATG13 S318 phosphorylation. These effects would then a priori be predicted to play a key driving role in the formation of toxic autophagosomes/autolysosomes (Figure 7).
Figure 7.
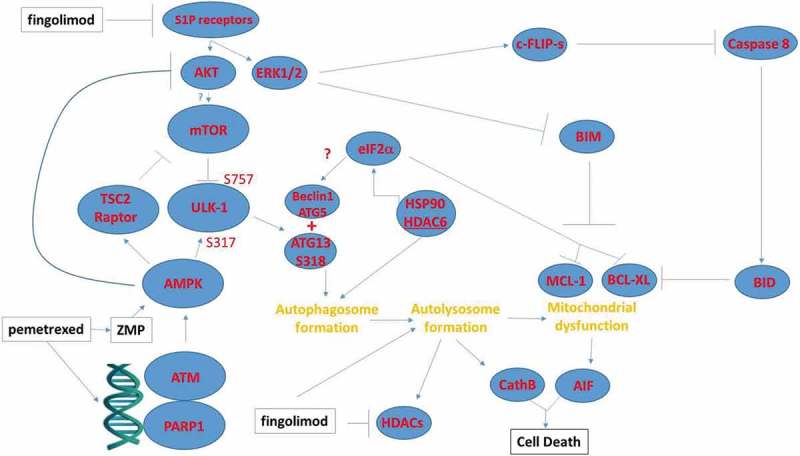
Possible mechanisms by which pemetrexed and fingolimod/FTY720 interact to kill NSCLC cells. A schematic representing the signaling pathways and biology being examined in this manuscript. This includes inhibition of upstream signaling at the level of receptors; the activation of a DNA damage and ATM-AMPK response; and the regulatory pathways influencing autophagosome formation, autolysosome formation and the induction of tumor cell death.
However, in H460 and A549 cells the protective effect of ULK1 knock down was modest and similar to the protection observed when Beclin1 was knocked down. In other words, despite the pro-autophagy changes in AMPK, mTOR and ULK1 activity, these changes only play a partial role in facilitating autophagosome formation. Thus, although the drug-induced production of toxic autophagosomes in part requires ULK1 and Beclin1, the majority of the toxic signals from the drug combination act at the elongation stage of autophagosome formation.
Downstream of autophagic and lysosomal events, our data demonstrated that the promotion of mitochondrial dysfunction played a key role in the execution of the tumor cells by [pemetrexed + fingolimod]. Over-expression of BCL-XL almost abolished drug combination lethality. Downstream of the mitochondrion inhibition of caspase 9 signaling reduced cell killing by 30–50% whereas knock down of apoptosis inducing factor (AIF) reduced killing by 60–75%. These findings argue that reduced ATP levels in cells after drug exposure, that are required for caspase 9 activation, may only play a modest inhibitory role in the execution of tumor cells. In A549 cells and to a lesser extent in H460 cells, knock down of rest in peace 1 (RIP-1) also prevented cell killing, which argues for necroptotic processes being engaged.23 Collectively these findings suggest that this drug combination kills through both autophagic and necroptotic processes.
As mTOR/ULK1 regulation does not appear to be the primary regulator of the toxic autophagy process, we reasoned that additional suppression of mTOR activity could result in a further enhancement in the lethality of [pemetrexed + fingolimod]. Inhibitors of mTOR function approved for clinical use include temsirolimus (IV administration) and everolimus (PO administration).24,25 Temsirolimus is approved for the treatment of renal cell carcinoma and everolimus approved for the treatment of breast, RCC and astrocytomas. Everolimus tends to have less negative sequelae than temsirolimus in patients. Indeed, we found that everolimus enhanced [pemetrexed + fingolimod] lethality in vitro and in vivo. Unexpectedly, the ability of everolimus to enhance [pemetrexed + fingolimod] killing required activation of caspases 8/10 as judged by the ability of c-FLIP-s over-expression to prevent death. The ability of everolimus to enhance killing was blocked by knock down of Beclin1 and of ULK1, arguing that everolimus was acting in an on-target fashion, inhibiting mTORC1. However, as noted in our assays measuring autophagosome and autolysosome levels, whereas knock down of Beclin1 prevented everolimus from enhancing autophagosome levels, it only delayed and did not alter the increase in autolysosome levels. These data argue that mTOR signaling is acting downstream of ULK1/ATG13/Beclin1 to facilitate autophagosome and autolysosome formation. These data suggest that in addition to autophagic cell death and necroptosis, this triple drug combination also effectively enhances killing by apoptosis.
In conclusion, the lethality of the approved NSCLC therapeutic pemetrexed against NSCLC cells and ovarian cancer cells is significantly enhanced by the multiple sclerosis drug fingolimod.26 Cell killing is stimulated through promoting the elongation process in autophagosome formation. Further stimulation of autophagosome formation using the mTOR inhibitor everolimus enhanced [pemetrexed + fingolimod] lethality. Our present findings strongly argue for the performance of clinical trials in ovarian cancer and in NSCLC patients who have failed anti-ERBB1 maintenance therapy with the addition of fingolimod to the established NSCLC and ovarian cancer therapeutic pemetrexed.
Materials and methods
Materials
Pemetrexed and everolimus were purchased from Selleckchem (Houston, TX). Fingolimod (FTY720) was purchased from Sigma-Aldrich (St. Louis MO). Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). Other reagents and performance of experimental procedures were as described.20,21,27–31 Antibodies used: AIF (5318), BAX (5023), BAK (12105), BAD (9239), BIM (2933), BAK1 (12105), Beclin1 (3495), cathepsin B (31718), CD95 (8023), FADD (2782), eIF2α (5324), P-eIF2α S51 (3398), ULK-1 (8054), P-ULK-1 S757 (14202), P-AMPK S51 (2535), AMPKα (2532), P-ATM S1981 (13050), ATM (2873), and ATG5 (12994). TSC2 (4308), P-TSC2 T1462 (3617), Raptor (2280), P-Raptor S792 (2083), mTOR (2983), P-mTOR S2448 (5536), P-mTOR S2481 (2974), ATG13 (13468), MCL-1 (94296), BCL-XL (2764), P-AKT T308 (13038), P-ERK1/2 (5726), P-STAT3 Y705 (9145), P-p65 S536 (3033), p62 (23214), LAMP2 (49067) all from Cell Signaling Technology; P-ULK-1 S317 (3803a) from Abgent; P-ATG13 S318 (19127) from Novus Biologicals (Figure 8).
Figure 8.

Control siRNA knock down images for the proteins whose expression was manipulated in this manuscript.
Methods
Cell culture and exposure to drugs
All “H” series NSCLC lines and established ovarian cancer cell lines were purchased from the ATCC. Cells were re-purchased every ~ 6 months. ADOR cells were a gift to the Dent lab from a female NSCLC patient. Spiky ovarian cancer cells were kindly provided by Dr. Karen Paz (Champions Oncology, NJ). All cell lines were cultured at 37 °C (5% (v/v CO2) in RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short term cell killing assays and immune-staining studies, cells were plated at a density of 3 × 103 per cm2 and 24h after plating treated with various drugs, as indicated. In vitro drug treatments were generally from a 100 mM stock solution of each drug and the maximal concentration of Vehicle carrier (VEH; DMSO) in media was 0.02% (v/v).
Transfection of cells with sirna or with plasmids
Validated short hairpin RNA molecules used to knock down specific target proteins were purchased from Qiagen (Valencia, CA): Figure 11: siSCR (SI03650318), ATM (SI00604737), cathepsin B (1027416), BAX (GS581), BAK (GS578); AMPKα (GS5562), BIM (GS10018), BAD (GS572), Beclin1 (GS8678), ATG5 (GS9474), CD95 (GS355), AIF (GS9131), eIF2α (GS83939), FADD (GS8772), ULK-1 (GS8408), ATG13 (GS9776). Cells in serum-free media were transfected with specific siRNAs or scrambled control siRNA (siControl) using Hiperfect (Qiagen) according to the supplier’s guidelines as we previously described in [26–28]; 24 h later, cells were cultured in DMEM containing 10% serum. For protein overexpression, cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen) following the manufacturer’s guidelines. After 4 h, serum-free transfection media was replaced with fresh media containing 10% serum as previously described.20,21,27–31
Determination of cell viability, protein expression and protein phosphorylation by immuno-fluorescence using a hermes wiscan machine
http://www.idea-bio.com/, Cells, (4,000) are plated into the wells of a 96 well plate, and are allowed to grow for an additional ~ 18h [26–28]. Cells are then genetically manipulated and 24 h afterwards are exposed to drugs. For immunofluorescence studies cells are fixed in place and staining performed using primary antibodies and red/green fluorescent secondary antibodies. For live/dead assays, each plate is cyto-spun to associate dead cells (for live-dead assays) with the base of each well. Cells are incubated with live-dead reagent (Thermo Fisher Scientific, Waltham MA) with imaging of the green/red/yellow cells the Hermes instrument at 10X magnification.20,21,27–31
Assessment of autophagy
Cells were transfected with a GFP-LC3-RFP plasmid to express LC3 fused to green and red fluorescent proteins for 24 h. Cells were then treated with drugs and GFP and RFP‐positive vesicles in cells were enumerated with a Zeiss Axiovert fluorescent microscope (× 40 objective). At least 40 cells per condition were counted in triplicate for each condition.
Data analysis
Comparison of the effects of various treatments (performed in triplicate three times) was using one-way analysis of variance and a two tailed Student’s t-test. Statistical examination of in vivo animal survival data utilized both a two tailed Student’s t-test and log rank statistical analyses between the different treatment groups. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Abbreviations
- ERK
extracellular regulated kinase
- PI3K
phosphatidyl inositol 3 kinase
- ca
constitutively active
- dn
dominant negative
- ER
endoplasmic reticulum
- AIF
apoptosis inducing factor
- AMPK
AMP-dependent protein kinase
- mTOR
mammalian target of rapamycin
- JAK
Janus Kinase
- STAT
Signal Transducers and Activators of Transcription
- MAPK
mitogen activated protein kinase
- PTEN
phosphatase and tensin homologue on chromosome ten
- ROS
reactive oxygen species
- CMV
empty vector plasmid or virus
- si
small interfering
- SCR
scrambled
- IP
immunoprecipitation
- VEH
vehicle
- PTX
pemetrexed
- FTY
FTY720, also known as fingolimod and Gilenya
- EVERO
everolimus
- HDAC
histone deacetylase
- NSCLC
non-small cell lung cancer.
Acknowledgments
Support for the present study was funded from philanthropic funding from Massey Cancer Center, the Universal Inc. Chair in Signal Transduction Research and PHS R01-CA192613 (PD) and R01GM043880 (SS). Thanks to Dr. H.F. Young and the Betts family fund for support in the purchase of the Hermes Wiscan instrument.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Arteaga CL. EGF receptor mutations in lung cancer: from humans to mice and maybe back to humans. Cancer Cell. 2006;9:421–423. doi: 10.1016/j.ccr.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Jr RR. ErbB/HER protein-tyrosine kinases: structures and small molecule inhibitors. Pharmacol Res. 2014;87:42–59. doi: 10.1016/j.phrs.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Jr RR. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Dhomen NS, Mariadason J, Tebbutt N, Scott AM. Therapeutic targeting of the epidermal growth factor receptor in human cancer. Crit Rev Oncog. 2012;17:31–50. [DOI] [PubMed] [Google Scholar]
- 5.Reinmuth N, Brandt B, Kunze WP, Junker K, Thomas M, Achatzy R, Scheld HH, Semik M. Ploidy, expression of erbB1, erbB2, P53 and amplification of erbB1, erbB2 and erbB3 in non-small cell lung cancer. Eur Respir J. 2000;16:991–996. [DOI] [PubMed] [Google Scholar]
- 6.Gao JW, Zhan P, Qiu XY, Jin JJ, Lv TF, Song Y. Erlotinib-based doublet targeted therapy versus erlotinib alone in previously treated advanced non-small-cell lung cancer: a meta-analysis from 24 randomized controlled trials. Oncotarget. 2017;8:73258–73270. doi: 10.18632/oncotarget.18319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kazandjian D, Keegan P, Suzman DL, Pazdur R, Blumenthal GM. Characterization of outcomes in patients with metastatic non-small cell lung cancer treated with programmed cell death protein 1 inhibitors past RECIST version 1.1-defined disease progression in clinical trials. Semin Oncol. 2017;44:3–7. doi: 10.1053/j.seminoncol.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Mengoli MC, Barbieri F, Bertolini F, Tiseo M, Rossi G. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: report of two cases and review of the literature. Lung Cancer. 2016;93:55–58. doi: 10.1016/j.lungcan.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Piva S, Ganzinelli M, Garassino MC, Caiola E, Farina G, Broggini M, Marabese M. Across the universe of K-RAS mutations in non-small-cell-lung cancer. Curr Pharm Des. 2014;20:3933–3943. [DOI] [PubMed] [Google Scholar]
- 10.Ihle NT, Jr LR, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–150. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carón RW, Yacoub A, Zhu X, Mitchell C, Han SI, Sasazuki T, Shirasawa S, Hagan MP, Grant S, Dent P. H-RAS V12-induced radioresistance in HCT116 colon carcinoma cells is heregulin dependent. Mol Cancer Ther. 2005;4:243–255. [PubMed] [Google Scholar]
- 12.Carón RW, Yacoub A, Li M, Zhu X, Mitchell C, Hong Y, Hawkins W, Sasazuki T, Shirasawa S, Kozikowski AP, et al. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Ther. 2005;4:257–270. [PubMed] [Google Scholar]
- 13.Booth L, Roberts JL, Tavallai M, Webb T, Leon D, Chen J, McGuire WP, Poklepovic A, Dent P. The afatinib resistance of in vivo generated H1975 lung cancer cell clones is mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling. Oncotarget. 2016;7:19620–19630. doi: 10.18632/oncotarget.7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Booth L, Roberts JL, Poklepovic A, Dent P. NEDD4 over-expression regulates the afatinib resistant phenotype of NSCLC cells. Oncology Signaling. doi: 10.1016/j.onsig.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruickshanks N, Roberts JL, Bareford MD, Tavallai M, Poklepovic A, Booth L, Spiegel S, Dent P. Differential regulation of autophagy and cell viability by ceramide species. Cancer Biol Ther. 2015;16:733–742. doi: 10.1080/15384047.2015.1026509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azuma H, Takahara S, Ichimaru N, Wang JD, Itoh Y, Otsuki Y, Morimoto J, Fukui R, Hoshiga M, Ishihara T, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410–1419. [PubMed] [Google Scholar]
- 17.Nagahashi M, Hait NC, Maceyka M, Avni D, Takabe K, Milstien S, Spiegel S. Sphingosine-1-phosphate in chronic intestinal inflammation and cancer. Adv Biol Regul. 2014;54:112–120. doi: 10.1016/j.jbior.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagahashi M, Yamada A, Katsuta E, Aoyagi T, Huang WC, Terracina KP, Hait NC, Allegood JC, Tsuchida J, Yuza K, et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation and breast cancer metastasis. Cancer Res. 2018. January 19. doi: 10.1158/0008-5472.CAN-17-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Booth L, Roberts JL, Tavallai M, Chuckalovcak J, Stringer DK, Koromilas AE, Boone DL, McGuire WP, Poklepovic A, Dent P. [Pemetrexed + Sorafenib] lethality is increased by inhibition of ERBB1/2/3-PI3K-NFκB compensatory survival signaling. Oncotarget. 2016;7:23608–23632. doi: 10.18632/oncotarget.8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8:1449–1468. doi: 10.18632/oncotarget.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima S, Takabe K, Newton J, Saurabh K, Young MM, Leopoldino AM, Hait NC, Roberts JL, Wang HG, Dent P, et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy. 2018;25:1–50. doi: 10.1080/15548627.2018.1429875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–349. doi: 10.1016/j.devcel.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia CA, Wu S. Attributable risk of infection to mTOR inhibitors everolimus and temsirolimus in the treatment of cancer. Cancer Invest. 2016;34:521–530. doi: 10.1080/07357907.2016.1242009. [DOI] [PubMed] [Google Scholar]
- 25.Abdel-Rahman O, Fouad M. Risk of oral and gastrointestinal mucosal injury in patients with solid tumors treated with everolimus, temsirolimus or ridaforolimus: a comparative systematic review and meta-analysis. Expert Rev Anticancer Ther. 2015;15:847–858. doi: 10.1586/14737140.2015.1047350. [DOI] [PubMed] [Google Scholar]
- 26.Ziemssen T, Medin J, Couto CA, Mitchell CR. Multiple sclerosis in the real world: A systematic review of fingolimod as a case study. Autoimmun Rev. 2017;16:355–376. doi: 10.1016/j.autrev.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Newton J, Hait NC, Maceyka M, Colaco A, Maczis M, Wassif CA, Cougnoux A, Porter FD, Milstien S, Platt N, et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017;31:1719–1730. doi: 10.1096/fj.201601041R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hait NC, Wise LE, Allegood JC, O’Brien M, Avni D, Reeves TM, Knapp PE, Lu J, Luo C, Miles MF, et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat Neurosci. 2014;17:971–980. doi: 10.1038/nn.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hait NC, Avni D, Yamada A, Nagahashi M, Aoyagi T, Aoki H, Dumur CI, Zelenko Z, Gallagher EJ, Leroith D, et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERα expression and enhances hormonal therapy for breast cancer. Oncogenesis. 2015;4:e156. doi: 10.1038/oncsis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Booth L, Roberts JL, Poklepovic A, Dent P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol Ther. 2017;18:705–714. doi: 10.1080/15384047.2017.1362511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. 2017;8:83155–83170. doi: 10.18632/oncotarget.17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.