

Abstract
Genome-wide association study identified prostate cancer risk variants explain only a relatively small fraction of its familial relative risk, and the genes responsible for many of these identified associations remain unknown. To discover novel prostate cancer genetic loci and possible causal genes at previously identified risk loci, we performed a transcriptome-wide association study in 79,194 cases and 61,112 controls of European ancestry. Using data from the Genotype-Tissue Expression Project, we established genetic models to predict gene expression across the transcriptome for both prostate models and cross-tissue models and evaluated model performance using two independent datasets. We identified significant associations for 137 genes at P < 2.61×10−6, a Bonferroni-corrected threshold, including nine genes that remained significant at P < 2.61×10−6 after adjusting for all known prostate cancer risk variants in nearby regions. Of the 128 remaining associated genes, 94 have not yet been reported as potential target genes at known loci. We silenced 14 genes and many showed a consistent effect on viability and colony-forming efficiency in three cell lines. Our study provides substantial new information to advance our understanding of prostate cancer genetics and biology.
Keywords: transcriptome-wide association study, genetic factors, prostate cancer, gene expression
Introduction
Prostate cancer is the most frequently diagnosed malignancy and the second leading cause of cancer mortality among males in the United States(1). Epidemiological studies provide strong evidence for a genetic predisposition to prostate cancer(2,3). Since 2006, genome-wide association studies (GWAS) have identified nearly 150 genetic loci harboring common, low-penetrance risk variants for prostate cancer(4-6). However, together these variants explain less than 30% of the familial relative risk of prostate cancer(4),6, leaving a substantial proportion of familial risk uncharacterized.
Many of the GWAS-identified disease risk variants are enriched in functional elements including promoters, enhancers, DNase I hypersensitive sites, and transcription factor binding sites, which may regulate the expression of genes causing diseases(7). It has been hypothesized that many of the genetic associations identified by GWAS may be mediated through the regulatory effects of risk variants on genes that are involved in the etiology of diseases(8-15). Specifically for prostate cancer, several recent studies using expression quantitative trait loci (eQTLs) analyses have shown that GWAS-identified risk variants may regulate the expression of certain genes that potentially play a role in prostate carcinogenesis(8,13,16). However, the causal genes for the large majority of the GWAS-identified prostate cancer risk loci remain unknown.
With a few exceptions, most common risk variants identified to date are only associated with diseases with modest effect sizes. It is possible that there are many risk variants in the genome that have not yet been identified. Because of their small effect size, these variants are difficult to identify in a typical GWAS, even with a very large sample size. Transcriptome-wide association studies (TWAS) can be used to systematically assess the association of genetically predicted gene expression levels with disease risk throughout the transcriptome, providing a powerful approach to identify novel disease risk genes and uncover possible causal genes at loci identified previously by GWAS(17-23). Instead of evaluating each specific genetic variant as conducted in GWAS, TWAS uses gene-based approaches that aggregate the effects of multiple SNPs into one testing unit and thus may increase power for identifying novel disease risk loci. Because it is expensive and often infeasible to profile the transcriptome of the target tissue in a large number of cases and controls, reference datasets containing both genotyping and gene expression data are used to establish genetic predictors for gene expression, which are then used to impute gene expression levels for subjects with genotype information available in a typical GWAS for association analyses of predicted gene expression with disease risk(18). By focusing on the genetically regulated component of gene expression, this approach can effectively overcome the potential influence of biases due to reverse causation and confounding effects on study results. Very recently, there has been a TWAS identifying new prostate cancer risk regions(24). This study, however, relies only on statistical inference and does not characterize potential function of the identified genes in prostate tumorigenesis using functional assays. Herein, we report results from another comprehensive TWAS of prostate cancer in which we used different strategies for modelling prostate gene expression and functionally characterized selected identified genes using in-vitro assays.
Methods
Building gene expression prediction models
We used transcriptome and high-density genotyping data from the Genotype-Tissue Expression (GTEx) study to establish gene expression prediction models using SNPs(25). In brief, genomic DNA samples obtained from study participants were genotyped using Illumina OMNI 2.5M or 5M SNP Array, and RNA samples from 51 tissue sites were sequenced to generate transcriptome profiling data. We used genotyping and prostate tissue transcriptome data from 73 European descendants to build prostate tissue gene expression prediction models. The genetic ancestry of GTEx subjects was determined based on the first two principal components, with reference to populations in the 1000 Genomes Project. Considering that the regulatory mechanisms for a large proportion of genes are similar across most human tissues(25-27), to increase the statistical power of building models that aim to capture genetic effects on gene expression of normal prostate tissue, we also generated cross-tissue models using gene expression data generated in all tissues from 369 GTEx participants of European descent(28). Genotyping data were processed according to the GTEx protocol (http://www.gtexportal.org/home/documentationPage). Briefly, SNPs having a call rate < 98%, with differential missingness between the 5M and 2.5M Array experiments, with Hardy-Weinberg equilibrium P-value < 10−6 (among subjects of European ancestry), or showing batch effects were excluded; also one participant diagnosed with Klinefelter disease, one participant with trisomy 17 mosaicism, and three related individuals were excluded. The genotype data were imputed in our study to the Haplotype Reference Consortium reference panel(29) using Minimac3 for imputation and SHAPEIT for prephasing(30,31). SNPs with high imputation quality (RSQR ≥ 0.8), minor allele frequency (MAF) ≥ 0.05, those that were included in HapMap Phase 2 for CEU population, and those on autosomal chromosomes were retained for the construction of gene expression prediction models. HapMap SNPs were used because it is expected that additional variants may increase noise without performance improvement, and such a strategy could generate stronger instruments because of fewer predicting SNPs being included in the models.
Detailed information of RNA-seq experiments and quality-control of the mRNA data performed as part of the GTEx project have been described in detail elsewhere(25,27). In brief, the same lab protocol was used to minimize batch effects on study results. Low quality samples and outlier samples were identified and removed. Gene-level read counts were produced using the following read-level filters: 1) reads were uniquely mapped; 2) reads were aligned in proper pairs; 3) the read alignment distance was ≤ 6; 4) reads were fully contained within exon boundaries. These data are available in dbGaP and were downloaded for model building in our study. For model building, the gene expression levels in reads per kilobase of transcript per million mapped reads (RPKM) units from RNA-SeQC was used(32). For prostate tissue models, genes with a median expression level of less than 0.1 RPKM across samples were removed. For the analysis of cross-tissue transcriptomic data, genes were retained when the mean expression levels were > 0.1 RPKM and expression levels were > 0 RPKM in at least 3 individuals. In both situations, for retained genes, the RPKM values were log2 transformed. Quantile normalization, to bring the expression profile of each sample to the same scale, and inverse quantile normalization, to map each set of expression values to a standard normal, were then performed. Further, adjustments were made for the top three principal components (PCs) derived from genotype data and the top 15 probabilistic estimation of expression residuals (PEER) factors(33) for prostate models, and the top three PCs, the top 35 PEER factors(33), and sex for cross-tissue models. The PEER analyses were used to further control for unmeasured determinant of gene expression variation, including batch effects(33).
In GTEx data, there are expression measurements in different tissues for each individual. A mixed effect model was used to decompose the expression level of a gene at a given tissue for individual i into a subject-specific cross-tissue component and a subject-by-tissue-specific component(28), as
Here YiCT represents the cross-tissue component, Z′i represents a vector of covariates (e.g., PEER factors, genetic ancestry, and sex) that have effects of β on the expression levels of the gene, and the subject-by-tissue-specific component was estimated as the difference between the expression levels and cross-tissue components (YiCT) given the lack of replicated measurement for a specific tissue/subject pair. The mixed effect model parameters were estimated using the lme4 package in R. Posterior models of the subject level random intercepts were used as estimates of the cross-tissue components. The whole tissue gene expression data of 6,124 GTEx tissue samples from 369 unique European ancestry individuals with genotyping data available were used.
Using both genotyping and gene expression data, an expression prediction model for each gene was built by applying the elastic net method as implemented in the glmnet R package, with α=0.5(18). The genetically regulated expression for each gene was estimated by including SNPs within the 2 MB flanking region of each gene, aligned with the biologic understanding that generally variants within this range may influence gene expression(34-36). For example, enhancers are known to increase gene transcription, and they can be located up to 1 Mbp away from the gene(34,35); it has also been found that megabase-sized local chromatin interaction domains are a common structure feature of the genome organization(36). Expression prediction models were built for protein coding genes, lncRNAs, microRNAs (miRNAs), processed transcripts, immunoglobulin genes, and T cell receptor genes, according to the Gencode V19 annotation file (http://www.gencodegenes.org/releases/19.html). Pseudogenes were not included due to concern for potentially inaccurate calling.(37) Ten-fold cross-validation was used to select the lambda parameter with which corresponding prediction models generated the smallest prediction error. The determined lambda was used in the whole dataset to generate the final models. The prediction R2 values (the square of the correlation between predicted and observed expression) were used to estimate the prediction performance of each of the finally established prediction models.
Evaluating performance of gene expression prediction models using Mayo Clinic and TCGA data
To further assess the external validity of the models we built using GTEx data, we performed external validation experiment using Mayo Clinic dataset comprising genetic data and gene expression data of fresh frozen normal prostate tissue obtained from patients with either radical prostatectomy or cystoprostatectomy (N=471)(8), and TCGA dataset comprising genetic data and gene expression data of tumour-adjacent normal prostate tissue from European-ancestry prostate cancer patients (N=45). Genotype data were imputed using the 1000 genomes phase 3 data as reference. Gene expression data were processed and normalized using a similar approach as described above. The predicted expression level for each gene was calculated using the models established using GTEx data and then compared with the observed level of that gene using the Spearman’s correlation.
Association analyses of predicted gene expression with prostate cancer risk
We used the following criteria to select prediction models with at least two predicting variants for the association analysis: 1) with a model prediction R2 of ≥ 0.01 in GTEx and a Spearman’s correlation coefficient of ≥ 0.1 between the predicted and measure gene expression in the external validation (Mayo Clinic or TCGA dataset), 2) with a prediction R2 of ≥ 0.04 in GTEx regardless of the performance in Mayo Clinic or TCGA dataset, 3) with a prediction R2 of ≥ 0.01 in GTEx but unable to be evaluated in Mayo Clinic or TCGA dataset. The second group of genes was selected because that the gene expression data of the Mayo Clinic dataset were derived from fresh frozen tissue obtained from patients with either radical prostatectomy or cystoprostatectomy, and it is expected that the expression patterns of some genes in these patients may be different from those in the healthy subjects included in GTEx; for TCGA, some gene expression levels might have changed in TCGA tumor-adjacent normal tissues, and thus it is anticipated that some genes may show low prediction performance in TCGA data due to the influence of tumor growth(38,39). Overall, 6,390 prostate tissue models and 12,779 cross-tissue models met the criteria and were used to evaluate for expression-trait associations.
To identify prostate cancer risk associated genes, the MetaXcan method (version 0.2.5), which has been described elsewhere, was used for the association analyses(17). Briefly, the formula:
was used to estimate the Z-score of the association between predicted gene expression and prostate cancer risk. Here wlg is the weight of SNP l for predicting the expression of gene g, and are the association regression coefficient and its standard error for SNP l in GWAS, and and are the estimated variances of SNP l and the predicted expression of gene g. The input variables for the MetaXcan analyses include the weights for gene expression predicting SNPs, GWAS summary statistics results, and correlations between predicting SNPs. For this study we estimated correlations between SNPs included in the prediction models using the phase 3, 1000 Genomes Project data focusing on European population.
We used the summary statistics data for the association of genetic variants with prostate cancer risk generated from 79,194 prostate cancer cases and 61,112 controls of European ancestry in the PRACTICAL consortium. Briefly, 46,939 prostate cancer cases and 27,910 controls were genotyped using OncoArray including 570,000 SNPs (http://epi.grants.cancer.gov/oncoarray/). Genotypes were phased and imputed to the cosmopolitan panel of the 1000 Genomes Project (1KGP; 2014 June release). Also included in the analysis were data from seven previous prostate cancer GWAS or high-density SNP panels of European ancestry imputed to 1KGP: UK stage 1 (1,854 cases/1,894 controls) and stage 2 (3,650 cases/3,940 controls); CaPS 1 (474 cases/482 controls) and CaPS 2 (1,458 cases/512 controls); BPC3 (2,068 cases/2,993 controls); NCI PEGASUS (4,600 cases/2,941 controls); and iCOGS (20,219 cases/20,440 controls). Logistic regression summary statistics were meta-analyzed using an inverse variance fixed effect approach using METAL. All participating studies were approved by their appropriate ethics review boards. The studies were conducted in accordance with Declaration of Helsinki. In each participating study, written informed consent was collected from the participants. This study was approved by the PRACTICAL/ELLIPSE Data Access Committee.
For our primary analyses, a Bonferroni corrected p threshold of 2.61 × 10−6 (0.05/19,169) was used to determine a statistically significant association. To determine whether the identified associations between genetically predicted gene expression and prostate cancer risk were influenced by association signals identified in GWAS, we conducted conditional analyses adjusting for all risk SNPs in the corresponding genomic region identified in GWAS or fine-mapping studies. Briefly, we performed GCTA-COJO analyses developed by Yang et al(40) (version 1.26.0) to calculate association betas and standard errors of SNPs with prostate cancer risk after adjusting for the index SNPs of interest. We then re-ran the MetaXcan analyses using the association statistics after conditioning on the index SNPs.
Prostate cancer cell lines
We performed cell viability and colony formation efficiency (CFE) assays to assess the functions of a selected set of candidate genes identified in our study. We used the human prostate cancer cell lines PC-3, DU-145, and LNCaP. These cell lines from American Type Culture Collection (ATCC, Manassas, VA) were cultured in RPMI 1640 medium (Gibco, cat#11875093) (DU145 and LNCaP cells) or Hams F-12K medium (Gibco, cat#21127022) (PC3 cells) supplemented with 2 mm l-glutamine (Gibco, cat# 25030081), 100 IU/ml penicillin-streptomycin (Gibco, cat#15140122), 1 mm sodium pyruvate (Sigma-Aldrich, cat#s8636), 10 mm Hepes (Gibco, cat#15630080), 1x nonessential amino acids (Gibco, cat# 11140076),, and 10% fetal bovine serum (Gibco, cat# 16000044) at 37°C in a humidified atmosphere with 5% CO2. All cell lines were authenticated by American Type Culture Collection (ATCC), and were checked for mycoplasma by MycoFluor™ Mycoplasma Detection Kit (Thermofisher).
Gene expression in prostate cancer cell lines
Total RNA was isolated from the three prostate cancer cell lines using the miRNeasy Mini Kit (Qiagen, cat# 217004). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific Inc, cat# 4368814). Real-time monitoring of PCR amplification of cDNA was performed using DNA primers and CFX384 Touch™ Real-Time PCR Detection System (Bio-Rad) with RT² SYBR Green qPCR Mastermix (Qiagen, cat# 330500). Target gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels in the respective samples as an internal standard, and the comparative cycle threshold (Ct) method was used to calculate relative amount of target mRNAs. The primer sequences are listed in Supplementary Table 1.
Short interfering RNA (siRNA) silencing
After performing transfection optimization, PC-3 and LNCaP cells were plated at 3,000 cells/well and DU145 cells at 4,000 cells/well in 96-well plates and reverse-transfected with siRNAs targeting genes of interest (GOI) purchased from Thermo Fisher Scientific and Integrated DNA Technologies, Inc. (IDT), a positive control siRNA (All Stars Hs Cell Death Control siRNA, Qiagen cat# 1027299) or a non-targeting (NT) control siRNA (All Stars Negative Control siRNA, Qiagen cat# 1027281) (Supplementary Tables 2 and 3) with RNAiMAX (Life Technologies, cat# 13778150) or lipofectamine2000 (Life Technologies, cat# 11668019) according to the manufacturer’s protocol. Verification of siRNA knockdown of gene expression of each GOI was done by qPCR 36 hours after transfection and compared to NT control. AllStars Negative Control siRNA has no homology to any known mammalian gene and has a minimal nonspecific effects, as validated using Affymetrix GeneChip arrays and a variety of cell-based assays (Qiagen).
Cell viability assays
Cell viability was determined using the Alamar blue (Thermo Fisher, cat# DAL1025) assay as previously performed for siRNA knockdowns(41). On day 5 following reverse-transfection of siRNAs Alamar blue was added to cell plates with fresh media (1:10 dilution), incubated for 2 hours, and fluorescence (ex570nm/em585nm) was measured using a plate reader (BioTek NEO) in the Vanderbilt High-Throughput Screening Facility. Percent relative viability was calculated as: (siGOI value / mean NT siRNA control value) × 100. For each cell line, each GOI siRNA experiment was conducted in quadruplicate each time and repeated for 3 times.
Colony formation assays
For colony formation assays, siRNA transfected cells (DU-145 and PC-3) were seeded in 6-well plates with a density of 1000 cells/well at 16 hours after transfection, and were cultured for two weeks. Colonies, as defined to consist of ≥50 cells, were fixed with methanol, stained with crystal violet (0.1% w/v) (Sigma-Aldrich, cat# C0775), scanned and counted using ImageJ as batch analysis by a self-defined plug-in Macro. Relative CFE % was calculated as: 100 +/− (relative CFE in indicated siRNA - CFE in NTC siRNA) / transfection efficiency (“+” if the GOI promotes colony formation (CF) and “-” if it inhibits CF). Two independent experiments were carried out for all siRNAs of each GOI siRNA in DU-145 and PC-3 cell lines. Due to a weak adherence ability of the LNCaP cells, we did not perform the colony formation experiments on the LNCaP cells.
Results
Gene expression prediction models
Of the prostate tissue models built for 11,172 genes, 7,893 demonstrated a prediction performance (R2) of at least 0.01 (≥ 10% correlation) (Supplementary Table 4). The cross-tissue models were built for 18,961 genes, of which 14,153 showed a prediction performance (R2) of at least 0.01 (Supplementary Table 4). We externally validated our models using Mayo Clinic and TCGA datasets. The correlations of two sets of R2s (external prediction performance and internal prediction performance) are shown in Supplementary Figures 1 and 2. Overall, models that predict gene expression well in GTEx data performed well in predicting gene expression in both Mayo Clinic and TCGA data sets, while models that predict gene expression poorly in GTEx showed lower external validity. The correlation coefficients between internal performance R2 of GTEx models and external performance R2 derived from the Mayo Clinic dataset were 0.60 for prostate tissue models (0.43 after removing outliers) and 0.68 for cross-tissue models (0.68 after removing outliers), which were higher than the corresponding correlation coefficients of 0.48 (0.28 after removing outliers) and 0.54 (0.43 after removing outliers) obtained using TCGA data for external validation. We prioritized 6,390 prostate-specific models and 12,779 cross-tissue models for association analyses based on their performance in GTEx, Mayo Clinic and TCGA datasets.
Association analyses of predicted gene expression with prostate cancer risk
Of the 19,169 models evaluated for the association analyses between predicted gene expression and prostate cancer risk, models for 137 genes showed a significant association at the Bonferroni-corrected threshold of p ≤ 2.61×10−6 (Tables 1-3, Supplementary Tables 5-6, Figure 1). Of them, 68 showed a positive association and 69 showed an inverse association. We conducted conditional analyses adjusting for all reported risk variants in the same genomic region identified in previous GWAS or fine-mapping studies to evaluate independency of the identified associations of the genes(40) (Tables 1-3; Supplementary Table 7). The associations for nine previously unreported genes in nine chromosome regions (six protein-coding genes and three long non-coding RNAs (lncRNAs)) remained statistically significant at p ≤ 2.61×10−6 even after conditioning on the known risk variants (Table 1), thus representing potential independent association signals. An association between higher predicted expression and increased prostate cancer risk was identified for KIAA0907 (1q22), HCG21 (6p21.33), RP11–103H7.5 (8q24.21), AGAP10 (10q11.22), and UQCC1 (20q11.22) (Table 1). Conversely, an association between lower predicted expression and increased prostate cancer risk was detected for LRRN2 (1q32.1), RP11-429J17.8 (8q24.3), USP28 (11q23.2) and EIF3K (19q13.2) (Table 1). Of the remaining 128 genes, 94 have not yet been previously implicated as genes responsible for association signals with prostate cancer risk through expression quantitative trait loci (eQTL) and/or functional studies, and they became insignificant at p ≤ 2.61×10−6 after conditioning on the known risk variants, indicating that these associations may be at least partially influenced by reported prostate cancer risk variants (Tables 2-3, Supplementary Table 5). Interestingly, 34 genes reported as potential causal genes at prostate cancer susceptibility loci identified through eQTL and/or functional studies were also found to be associated with prostate cancer risk in our agnostic search (Supplementary Table 6), substantially exceeding the number of genes (n = 1) expected by chance alone (p<0.0001).
Table 1.
Nine novel gene expression-trait associations independent of prostate cancer risk variants identified in GWAS or fine-mapping studies
| Region | Gene | Model | Typea | Z score |
P valueb | R2c | Index SNP(s)d | Distance to the index SNP (kb) |
P value after adjusting for index SNPse |
No. of SNPs in prediction models |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q22 | KIAA0907 | Prostate | Protein | 6.64 | 3.16 × 10−11 | 0.01 | rs1218582 | 1,049 | 2.41 × 10−6 | 4 |
| 1q32.1 | LRRN2 | Prostate | Protein | −5.08 | 3.86 × 10−7 | 0.06 | rs4245739 | 67 | 2.16 × 10−6 | 8 |
| 6p21.33 | HCG21 | Cross-Tissue | lncRNA | 6.61 | 3.76 × 10−11 | 0.21 | rs130067 | 196 | 9.55 × 10−10 | 31 |
| 8q24.3 | RP11-429J17.8 | Cross-Tissue | lncRNA | −5.27 | 1.37 × 10−7 | 0.06 | rs7837688 | 16,332 | 1.24 × 10−7 | 10 |
| 8q24.21 | RP11-103H7.5 | Prostate | lncRNA | 5.40 | 6.75 × 10−8 | 0.02 | rs12543663 | 355 | 4.89 × 10−15 | 9 |
| 10q11.22 | AGAP10 | Cross-Tissue | Protein | 4.79 | 1.66 × 10−6 | 0.01 | rs76934034 | 1,109 | 1.73 × 10−6 | 41 |
| 11q23.2 | USP28 | Cross-Tissue | Protein | −6.30 | 2.95 × 10−10 | 0.12 | rs11214775 | 61 | 1.04 × 10−6 | 87 |
| 19q13.2 | EIF3K | Cross-Tissue | Protein | −5.80 | 6.44 × 10−9 | 0.06 | rs12610267 | 365 | 1.95 × 10−6 | 39 |
| 20q11.22 | UQCC1 | Cross-Tissue | Protein | 5.02 | 5.28 × 10−7 | 0.28 | rs11480453 | 2,543 | 3.77 × 10−7 | 42 |
Type: lncRNA: long non-coding RNAs; Protein: protein coding genes
P value: derived from association analyses; associations with p≤2.61×10−6 considered statistically significant based on Bonferroni correction of 19,169 tests (0.05/19,169)
R2: prediction performance (R2) derived using GTEx data
Risk SNPs identified in previous GWAS or fine-mapping studies. The risk SNP closest to the gene is presented. A full list of all risk SNPs, and their distances to the genes are presented in the Supplementary Table 3
using COJO method(40)
Table 3.
Twenty-seven gene expression-trait associations with 2.61×10−6 < p < 0.05 after conditioning on reported prostate cancer risk variants for genes located at genomic loci within 500kb of previous GWAS-identified prostate cancer risk variants
| Region | Gene | Model | Typea | Z score | P valueb | R2c | Index SNP(s)d | Distance to the index SNP (kb) |
P value after adjusting for index SNPse |
No. of SNPs in prediction models |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q21.3 | CDC42SE1 | Prostate | Protein | −4.73 | 2.22 × 10−6 | 0.04 | rs17599629 | 365 | 4.75 × 10−4 | 74 |
| DCST2 | Cross-Tissue | Protein | −5.71 | 1.16 × 10−8 | 0.11 | rs4845695 | 79 | 0.03 | 9 | |
| RP11-307C12.11 | Cross-Tissue | lncRNA | −6.02 | 1.77 × 10−9 | 0.18 | rs4845695 | 106 | 0.003 | 40 | |
| 1q32.1 | PM20D1 | Cross-Tissue | Protein | 5.45 | 5.06 × 10−8 | 0.69 | rs1775148 | 39 | 0.005 | 73 |
| RP11-739N20.2 | Cross-Tissue | lncRNA | −5.26 | 1.47 × 10−7 | 0.06 | rs199774366 | 87 | 0.03 | 10 | |
| 6p21.32 | AGER | Cross-Tissue | Protein | −5.53 | 3.16 × 10−8 | 0.12 | rs3096702 | 40 | 0.03 | 32 |
| HLA-DPA1 | Cross-Tissue | Protein | 5.14 | 2.75 × 10−7 | 0.60 | rs9296068 | 44 | 0.007 | 129 | |
| 6p21.33 | PPP1R18 | Cross-Tissue | Protein | 5.78 | 7.35 × 10−9 | 0.03 | rs12665339 | 43 | 0.002 | 18 |
| HCP5 | Cross-Tissue | lncRNA | 5.28 | 1.27 × 10−7 | 0.02 | rs2596546 | 39 | 7.16 × 10−4 | 9 | |
| HCG22 | Cross-Tissue | lncRNA | 4.88 | 1.09 × 10−6 | 0.36 | rs130067 | 91 | 0.02 | 140 | |
| ATF6B | Cross-Tissue | Protein | 4.79 | 1.63 × 10−6 | 0.17 | rs3096702 | 96 | 0.002 | 34 | |
| APOM | Prostate | Protein | 5.49 | 3.93 × 10−8 | 0.02 | rs2596546 | 291 | 0.008 | 40 | |
| 6p22.1 | ZNRD1 | Cross-Tissue | Protein | 5.37 | 7.85 × 10−8 | 0.42 | rs7767188 | 41 | 2.06 × 10−5 | 215 |
| 9p22.2 | ADAMTSL1 | Prostate | Protein | −5.00 | 5.81 × 10−7 | 0.04 | rs1048169 | 145 | 5.28 × 10−6 | 74 |
| 10q24.32 | RP11-47A8.5 | Cross-Tissue | lncRNA | −5.49 | 4.01 × 10−8 | 0.05 | rs3850699 | 10 | 1.66 × 10−4 | 11 |
| 11q13.3 | CCND1 | Prostate | Protein | −9.02 | 1.94 × 10−19 | 0.12 | rs36225067 | 2 | 3.98 × 10−7 | 34 |
| Cross-Tissue | Protein | −6.06 | 1.37 × 10−9 | 0.04 | rs36225067 | 2 | 0.002 | 76 | ||
| RP11-554A11.9 | Prostate | lncRNA | 9.35 | 8.48 × 10−21 | 0.36 | rs11228565 | 51 | 0.001 | 47 | |
| Cross-Tissue | lncRNA | 7.98 | 1.51 × 10−15 | 0.65 | rs11228565 | 51 | 0.003 | 130 | ||
| RP11-554A11.5 | Prostate | lncRNA | 4.85 | 1.22 × 10−6 | 0.14 | rs11228565 | 206 | 0.04 | 38 | |
| MYEOV | Cross-Tissue | Protein | −12.55 | 4.09 × 10−36 | 0.04 | rs376592364 | 50 | 0.01 | 29 | |
| RP11-211G23.2 | Cross-Tissue | lncRNA | −7.20 | 6.19 × 10−13 | 0.02 | rs376592364 | 175 | 0.002 | 23 | |
| 12q13.11 | PFKM | Cross-Tissue | Protein | −5.63 | 1.77 × 10−8 | 0.05 | rs80130819 | 79 | 0.008 | 76 |
| 12q13.12 | RP11-386G11.10 | Cross-Tissue | lncRNA | −4.94 | 7.73 × 10−7 | 0.12 | rs56222401 | 131 | 0.03 | 47 |
| 18q21.2 | STARD6 | Cross-Tissue | Protein | 4.83 | 1.35 × 10−6 | 0.18 | rs8093601 | 78 | 0.007 | 71 |
| 18q21.33 | KDSR | Cross-Tissue | Protein | 4.86 | 1.16 × 10−6 | 0.16 | rs11381388 | 34 | 9.74 × 10−4 | 2 |
| 19p13.11 | MYO9B | Prostate | Protein | 5.51 | 3.50 × 10−8 | 0.07 | rs11666569 | inside the gene | 0.02 | 28 |
| 19q13.2 | AC006129.1 | Cross-Tissue | lncRNA | −8.39 | 4.74 × 10−17 | 0.03 | rs11672691 | 52 | 0.04 | 4 |
| 19q13.33 | SYT3 | Prostate | Protein | −6.02 | 1.77 × 10−9 | 0.08 | rs2659124 | 183 | 0.04 | 36 |
Protein: protein coding genes; lncRNA: long non-coding RNAs; transcript: processed_transcript
P value: nominal p value from association analysis; the threshold after Bonferroni correction of 19,169 tests (0.05/19,169 =2.61×10−6) was used
R2: prediction performance (R2) derived using GTEx data; NA: not available
Risk SNPs identified in previous GWAS or fine-mapping studies. The risk SNP closest to the gene is presented. A full list of all risk SNPs, and their distances to the genes are presented in the Supplementary Table 3
using COJO method(40); all index SNPs in the corresponding region were adjusted for
Figure 1. Manhattan plot of association results from the prostate cancer transcriptome-wide association study.
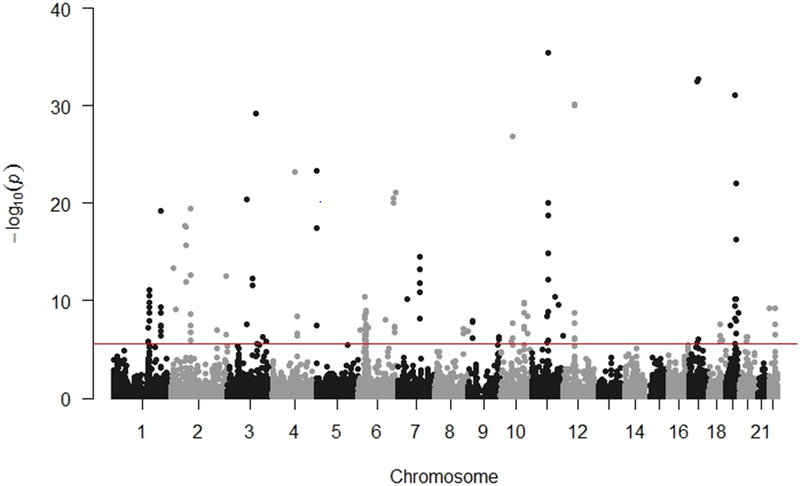
The red line represents P = 2.61 × 10−6 based on 19,169 tests. Each dot represents the genetically predicted expression of one specific gene by either prostate tissue or cross-tissue prediction models: the x axis represents the genomic position of the corresponding gene, and the y axis represents the negative logarithm of the association P-value. There are two associations with P < 1.00 × 10−40 not shown in this Figure.
Table 2.
Nineteen gene expression-trait associations that may be at least partially explained by prostate cancer risk variants identified in previous GWAS or fine-mapping studies for genes located at genomic loci at least 500kb away from any GWAS-identified prostate cancer risk variants
| Region | Gene | Model | Typea | Z score | P valueb | R2c | Index SNP(s)d | Distance to the index SNP (kb) |
P value after adjusting for index SNPse |
No. of SNPs in prediction models |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q21.2 | RP11-353N4.4 | Prostate | lncRNA | 4.74 | 2.19 × 10−6 | 0.03 | rs17599629 | 981 | 0.009 | 58 |
| 1q21.3 | RP11-98D18.3 | Cross-Tissue | lncRNA | −4.81 | 1.48 × 10−6 | 0.01 | rs17599629 | 1,078 | 2.87 × 10−6 | 12 |
| 2p11.2 | TMSB10 | Prostate | Protein | −4.88 | 1.08 × 10−6 | 0.04 | rs2028900 | 634 | 0.67 | 29 |
| 2p15 | MDH1 | Cross-Tissue | Protein | 7.11 | 1.19 × 10−12 | 0.14 | rs2430386 | 638 | 0.004 | 18 |
| 3q21.3 | EFCAB12 | Cross-Tissue | Protein | 4.73 | 2.28 × 10−6 | 0.09 | rs13062436 | 903 | 0.008 | 129 |
| 3q25.2 | DHX36 | Prostate | Protein | −5.05 | 4.42 × 10−7 | 0.03 | rs182314334 | 1,986 | 2.64 × 10−6 | 34 |
| 4q24 | RP11-710F7.2 | Prostate | lncRNA | 5.87 | 4.26 × 10−9 | 0.07 | rs7679673 | 787 | 0.45 | 39 |
| NPNT | Prostate | Protein | 5.08 | 3.75 × 10−7 | 0.06 | rs7679673 | 754 | 0.23 | 45 | |
| RP11-710F7.3 | Prostate | lncRNA | 5.19 | 2.07 × 10−7 | 0.03 | rs7679673 | 863 | 0.70 | 19 | |
| 5p15.33 | CTD-2589H19.6 | Prostate | lncRNA | −5.52 | 3.32 × 10−8 | 0.22 | rs2242652 | 603 | 2.37 × 10−4 | 68 |
| 6p24.2 | GCNT6 | Prostate | Protein | −5.32 | 1.06 × 10−7 | 0.03 | rs4713266 | 572 | 7.16 × 10−4 | 2 |
| 7p14.1 | MPLKIP | Prostate | Protein | −6.52 | 7.16 × 10−11 | 0.26 | rs17621345 | 701 | 0.18 | 49 |
| 10q11.22 | RP11-38L15.3 | Cross-Tissue | lncRNA | 4.74 | 2.10 × 10−6 | 0.01 | rs76934034 | 868 | 3.36 × 10−6 | 36 |
| 12q13.13 | RP1-288H2.2 | Prostate | transcript | 11.55 | 7.67 × 10−31 | 0.01 | rs902774 | 776 | NA | 4 |
| RP1-288H2.4 | Prostate | lncRNA | 11.53 | 8.97 × 10−31 | 0.04 | rs902774 | 788 | 0.34 | 6 | |
| 17q12 | PIP4K2B | Cross-Tissue | Protein | 4.90 | 9.78 × 10−7 | 0.02 | rs11263763 | 818 | 0.40 | 5 |
| CTC-268N12.2 | Cross-Tissue | lncRNA | −4.78 | 1.75 × 10−6 | 0.04 | rs8064454 | 692 | 0.12 | 28 | |
| 19q13.12 | CTD-3064H18.4 | Cross-Tissue | lncRNA | 4.72 | 2.34 × 10−6 | 0.17 | rs8102476 | 696 | 4.05 × 10−5 | 105 |
| 22q13.2 | RBX1 | Prostate | Protein | 5.12 | 3.08 × 10−7 | 0.03 | rs11704314 | 549 | 0.36 | 18 |
Protein: protein coding genes; lncRNA: long non-coding RNAs; transcript: processed_transcript
P value: nominal p value from association analysis; the threshold after Bonferroni correction of 19,169 tests (0.05/19,169 =2.61×10−6) was used
R2: prediction performance (R2) derived using GTEx data; NA: not available
Risk SNPs identified in previous GWAS or fine-mapping studies. The risk SNP closest to the gene is presented. A full list of all risk SNPs, and their distances to the genes are presented in the Supplementary Table 3
using COJO method(40); all index SNPs in the corresponding region were adjusted for
It is worth noting that, for some genes in Tables 2-3 and Supplementary Table 6, their associations were not too far from 2.61×10−6 after conditioning on reported prostate cancer risk variants. For these genes, it is possible that they may represent independent association signals, although the power of detecting them may be constrained by the available sample size in the current study.
For 56 of the 137 associated genes identified in this study, we were able to build both prostate tissue and cross-tissue prediction models that fulfill the inclusion criteria described in the method section. Thus, we could evaluate each of these genes for its predicted expression using both models with prostate cancer risk (Supplementary Table 8). Of these genes, 46 showed an association in the same direction using both models, including 14 with a p ≤ 2.61×10−6 in both models and an additional 21 with a p < 0.05 in both models (Supplementary Table 8). There were only two genes that showed a different direction of association at p < 0.05 (Supplementary Table 8).
In vitro functional assays using prostate cancer cells
We selected, for functional assays, 14 genes whose high predicted expression was associated with increased risk of prostate cancer using knockdown experiments in prostate cancer cells. These genes included 11 protein coding genes (KIAA0907, EFCAB12, UQCC1, DDX52, MYO9B, WDPCP, NPNT, VARS2, NUCKS1, HLA-DRB5, and TMEM180) and three lncRNAs (RP11-103H7.5, RP11-38L15.3, and AC092155.4). We searched The Human Protein Atlas website (http://www.proteinatlas.org) and noted that all 11 selected protein-coding genes were expressed in the prostate cancer cell line PC-3. We performed quantitative PCR (qPCR) on the three prostate cancer cell lines (LNCaP, PC-3 and DU-145) to analyze the expression levels of these genes (Supplementary Table 1). All 11 protein-coding genes and two lncRNAs (RP11-103H7.5 and RP11-38L15.3) were expressed in the three cell lines. The expression of AC092155.4 was undetectable in any of the three cell lines using the standard RT-PCR protocol. We used cell lines PC-3, DU-145, and LNCaP for the viability assay, and PC-3 and DU-145 for the colony formation assay. These genes were silenced using small short interfering RNA (siRNA) and the knockdown efficiency was calculated in each cell line for each siRNA. Through qPCR validation, robust knockdown of the gene of interests (GOI) was achieved with all the siRNAs for the 11 protein-coding genes and lncRNAs RP11-103H7.5 and RP11-38L15.3 (Supplementary Figure 3).
To assess the proliferation of cells following gene silencing, we quantified the relative viability of cells after knocking down genes of interest in comparison with that of cells treated with non-target control (NTC) siRNA (Figure 2). Except for MYO9B, VARS2, and NPNT, knocking down any of the other genes resulted in a significantly decreased cell viability in at least one of the three prostate cancer cell lines (LNCaP, PC-3 and DU-145) used in our experiments. These results were consistent with our hypothesis that silencing genes whose predicted high expression was associated with an increased prostate cancer risk should reduce cell viability. Interestingly, down-regulation of any of the three lncRNAs (RP11-103H7.5, RP11-38L15.3, and AC092155.4) resulted in significantly decreased cell viability in all three tested cell lines compared with control group. We further assessed the influence of silencing these genes on colony forming ability in PC-3 and DU-145 cells (Figure 3). With the exception of WDPCP, knockdown for any of the other 13 genes resulted in significant reduction in colony forming efficiency in DU-145 cells compared with the control. Experiments using PC-3 cells also showed, in general, reductions in colony forming efficiency, although the differences with controls were not statistically significant. These results were consistent with our a priori hypothesis as well.
Figure 2. Effects on cell viability in prostate cancer cells by gene silencing.
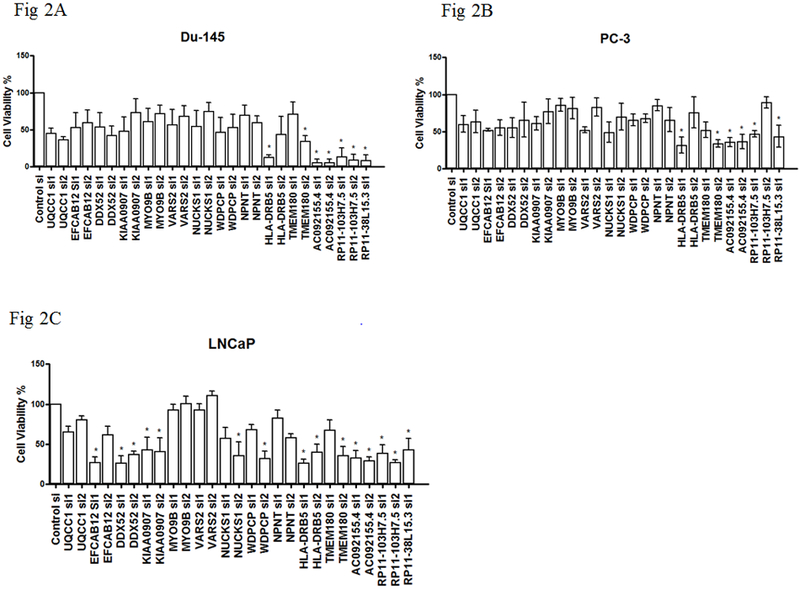
(A) DU-145, (B) PC-3 or (C) LNCaP cells were transfected with indicated siRNAs. On day 5, cell viability was determined using Alamar blue. Percent relative viability was calculated as: (siGOI value / mean NT siRNA control value) × 100. Error bars are from three independent experiments in quadruplicate, and represent standard deviation. P-values were determined by one-way ANOVA followed by Dunnett’s multiple comparisons test, which controlled for family-wise error-rate: *P-value < 0.05. NTC: non-target control.
Figure 3. Effects on colony formation efficiency (CFE) in prostate cancer cells by gene silencing.
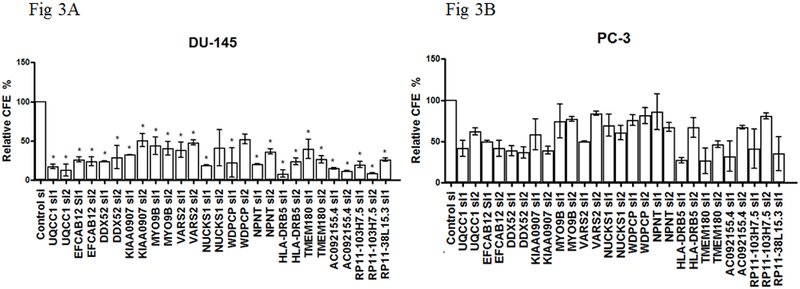
(A) DU-145 or (B) PC-3 cells were transfected with indicated siRNAs, then reseeded after 16 hours for colony formation (CF) assay. At day 14, colonies were fixed with methanol, stained with crystal violet, scanned and batch analyzed by ImageJ. Relative CFE % = 100 +/− (relative CFE in indicated siRNA - CFE in NTC siRNA) / transfection efficiency (“+” if the GOI promotes CF and “-” if it inhibits CF). Error bars are from two independent experiments in triplicate, and represent standard deviation. P-values were determined by Welch’s ANOVA followed by Dunnett’s multiple comparisons test, which controlled for family-wise error-rate: *P-value < 0.05. NTC: non-target control.
Discussion
This is the most comprehensive TWAS study to evaluate the associations of genetically predicted gene expression with prostate cancer risk throughout the human genome. We identified 137 genes demonstrating a statistically significant association after Bonferroni correction, including nine novel associations independent of any reported prostate cancer risk variants. Of the 128 remaining associated genes, 94 have not been reported previously as potential causal genes at GWAS-identified loci for prostate cancer risk. Based on The Human Protein Atlas, many of our identified genes show an enriched expression pattern in prostate or other cancers, and some even demonstrate potential prognostic significance in prostate or other cancers (Supplementary Table 9). For virtually all of the identified genes, at least one gene expression predicting SNPs showed a highly significant association with prostate cancer risk, and for many genes, multiple expression-predicting SNPs were associated with the risk of prostate cancer (Supplementary Tables 10 and 11). This study provides substantial novel information to improve the understanding of genetics and etiology for prostate cancer, the most common malignancy among men in most countries around the world.
Although TWAS-identified associations could be mediated by the expression level of the identified genes, it is also possible that such associations may be confounded via a linkage disequilibrium between expression predicting SNPs and a disease causal SNP acting through other mechanisms. To understand the functional importance of TWAS-identified associated genes, we silenced 14 genes whose predicted high levels of expression were associated with an increased prostate cancer risk in three prostate cancer cell lines, and assessed their influence on cell viability and colony forming efficiency. We observed that, interruption for many of these genes demonstrated an effect in the tested cell lines, especially on colony forming efficiency in DU-145 cells and on viability in LNCaP cells. Based on previous research, downregulation of one of the tested genes, KIAA0907, had no influence on cell proliferation or cell viability distribution in non-small cell lung cancer cells(42). This supports that KIAA0907 may not be an essential gene. Our observation that knocking down expression of KIAA0907 resulted in significantly decreased cell viability in LNCaP cells and significantly decreased colony forming efficiency in DU-145 cells thus support a potential role of KIAA0907 in prostate tumorigenesis. It is expected that some real biological effects may not be detected in all related cell lines, as each cell line has different characteristics and may not always accurately replicate the primary cells(43). We observed consistent and strong effects for the three lncRNAs evaluated in the experiments, RP11-103H7.5, RP11-38L15.3, and AC092155.4, although the expression and knockdown efficiency of AC092155.4 could not be detected in the three cell lines examined using the typical RT-PCR method. These results provide evidence for a potential causal role of these genes in the development of prostate cancer.
Some of the identified genes showing functional significance from our experiments have been previously reported to play important roles in the development of cancer. For example, MYO9B was found to be upregulated in prostate cancer cells with high metastatic potentials(44). Knockdown of MYO9B was found to increase stress fiber formation and directional persistence, and decrease 2D migration speed in prostate cancer cells(44). Another gene, NUCKS1, was identified as a putative oncogene and immunodiagnostic marker of hepatocellular carcinoma(45). Its overexpression was also identified as a prognostic marker for both colorectal cancer and cervical squamous cell carcinoma(46,47). Furthermore, NUCKS1 was found to be potentially involved in the etiology of lung cancer(48). Our study provided additional evidence that these two genes might play an oncogenic role in prostate cancer etiology.
In this large TWAS study we identified 103 associated genes which have not yet been implicated as potential causal genes at GWAS-identified loci for prostate cancer risk. Although we are not able to functionally characterize all of them in one single study, in vitro/in vivo studies or human studies have shown that some of these genes may play important roles in prostate tumorigenesis. For example, knockdown of CLIC1 exerts inhibitory effects on prostate cancer cell proliferation and migration(49). The USP39 gene has been suggested to play an oncogenic role in prostate tumorigenesis, and overexpression of this gene was associated with a poor prognosis for prostate cancer patients(50). Expressed only in normal prostate and prostate tumor tissues, ANO7 has been shown to play a role in promoting cell contact-dependent interactions of prostate cancer cells, and was a potential target for T cell-mediated immunotherapy of prostate cancer(51-53). PDLIM5 was identified to be overexpressed in prostate cancer cells compared with benign prostate tissue and noncancer prostate cells(54). These previous studies provide support of our findings regarding a potential role of these genes in prostate carcinogenesis.
Previous studies have shown that the gene expression prediction models are generally stable and can capture well the cis-regulatory effects of genetic variants on gene expression(18,19,55). Based on our external validation using both Mayo Clinic and TCGA data, the prostate tissue models and cross-tissue models built in this study demonstrated reasonable prediction performance, overall. The sample size for association analyses in this study was large, which provides high statistical power to detect a large number of prostate cancer susceptibility gene candidates. On the other hand, the sample size for building prostate tissue specific expression prediction models was relatively small (n=73), which may affect the precision of estimated model parameters. Given that the regulatory mechanisms for most genes are similar across most human tissues(25,26), we also built cross-tissue models using gene expression data generated in all tissues from 369 European descendants to increase the statistical power. The cross-tissue models are expected to have improved power for genes whose regulatory mechanisms are similar across most tissues. In comparison, prostate tissue models are likely to be more appropriate for genes whose regulatory mechanisms are specific to prostate tissue. With that being said, for genes that we could build both prostate tissue model and cross-tissue model, their associations with prostate cancer risk were, in general, consistent with each other (Supplementary Table 8). Not all genes could be evaluated in our study due to their various hereditary components in expression regulation. For example, previous studies suggested an important role of genes ASCL2(8), C10orf32(8,9), COL2A1(8), DBIL5P(8), EBF2(11), and GJB1(8) in the etiology of prostate cancer. However, expression of these genes cannot be predicted well using data currently available in the GTEx project which has precluded us from including them in the association analyses. With a large sample size and improved model building strategies, we expect that additional genes could be identified in relation to prostate cancer risk in future studies. As with most other in vitro experiments, we used cancer cell lines to evaluate the functional significance of associated genes identified in our study. Future studies could be conducted using normal prostate cell lines. In the current work we did not include negative controls in the in vitro experiments. However, it is difficult to identify negative control genes for which there is sufficient evidence supporting their irrelevance with prostate cancer. In addition, we did not build prediction models using data from other tissues, some of which could be relevant to prostate cancer etiology. Future studies using data from relevant tissues could be helpful in identifying additional candidate genes contributing to prostate cancer etiology.
In conclusion, in this large-scale TWAS study of prostate cancer, we identified a large number of novel genes in association with prostate cancer risk. The silencing experiments we performed suggest that many of the genes identified by TWAS are likely to mediate risk of prostate cancer by affecting viability or colony forming efficiency, two of the hallmarks of cancer. Further investigation of these genes will provide additional insight into the biology and genetic of prostate cancer.
Data availability
The GTEx data are publically available via dbGaP (www.ncbi.nlm.nih.gov/gap; dbGaP Study Accession: phs000424.v6.p1). The Mayo Clinic study data are available via dbGaP (Accession: phs000985.v1.p1). TCGA data are available via the National Cancer Institute’s Genomic Data Commons Data Portal (https://gdc.cancer.gov/). The OncoArray genotype data and relevant covariate information (i.e. ethnicity, country, principal components, etc.) for prostate cancer study are deposited into dbGAP (Accession #: phs001391.v1.p1). In total 47 of the 52 OncoArray studies, encompassing nearly 90% of the individual samples, are available. The previous meta-analysis summary results and genotype data currently are available in dbGaP (Accession #: phs001081.v1.p1).
Supplementary Material
Significance.
This study identifies novel prostate cancer genetic loci and possible causal genes, advancing our understanding of the molecular mechanisms that drive prostate cancer.
Acknowledgements
The authors thank Jing He, Wanqing Wen, Hui Cai and Bingshan Li of Vanderbilt University School of Medicine for their help with this study. The authors also would like to thank all the individuals for their participation in the parent studies and all the researchers, clinicians, technicians and administrative staff for their contribution to the studies. We are also grateful to Hae Kyung Im of University of Chicago for her help. The data analyses were conducted using the Advanced Computing Center for Research and Education (ACCRE) at Vanderbilt University. This project at Vanderbilt University Medical Center was supported in part by funds from Anne Potter Wilson endowment. Lang Wu was supported by NCI K99 CA218892 and the Vanderbilt Molecular and Genetic Epidemiology of Cancer (MAGEC) training program (U.S. NCI grant R25 CA160056). Joshua A. Bauer was supported by 1R50CA211206. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The GTEx data used for the analyses described in this manuscript were obtained from dbGaP accession number phs000424.v6.p1. A full description of funding and acknowledgments for PRACTICAL consortium, CRUK, BPC3, CAPS, PEGASUS are included in the Supplementary Note.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians 2019;69:7–34 [DOI] [PubMed] [Google Scholar]
- 2.Demichelis F, Stanford JL. Genetic predisposition to prostate cancer: Update and future perspectives. Urologic oncology 2015;33:75–84 [DOI] [PubMed] [Google Scholar]
- 3.Crawford ED. Epidemiology of prostate cancer. Urology 2003;62:3–12 [DOI] [PubMed] [Google Scholar]
- 4.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nature genetics 2014;46:1103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nature genetics 2013;45:385–91, 91e1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nature genetics 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thibodeau SN, French AJ, McDonnell SK, Cheville J, Middha S, Tillmans L, et al. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nature communications 2015;6:8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, Hazelett DJ, Wiklund F, Schumacher FR, Stram DO, Berndt SI, et al. Integration of multiethnic fine-mapping and genomic annotation to prioritize candidate functional SNPs at prostate cancer susceptibility regions. Human molecular genetics 2015;24:5603–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amin Al Olama A, Dadaev T, Hazelett DJ, Li Q, Leongamornlert D, Saunders EJ, et al. Multiple novel prostate cancer susceptibility signals identified by fine-mapping of known risk loci among Europeans. Human molecular genetics 2015;24:5589–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Stram A, Chen C, Kar S, Gayther S, Pharoah P, et al. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Human molecular genetics 2014;23:5294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H, Ahmed M, Zhang F, Yao CQ, Li S, Liang Y, et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nature genetics 2016;48:1142–50 [DOI] [PubMed] [Google Scholar]
- 13.Penney KL, Sinnott JA, Tyekucheva S, Gerke T, Shui IM, Kraft P, et al. Association of prostate cancer risk variants with gene expression in normal and tumor tissue. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2015;24:255–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du M, Tillmans L, Gao J, Gao P, Yuan T, Dittmar RL, et al. Chromatin interactions and candidate genes at ten prostate cancer risk loci. Scientific reports 2016;6:23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin HJ, Jung S, DebRoy AR, Davuluri RV. Identification and validation of regulatory SNPs that modulate transcription factor chromatin binding and gene expression in prostate cancer. Oncotarget 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gusev A, Shi H, Kichaev G, Pomerantz M, Li F, Long HW, et al. Atlas of prostate cancer heritability in European and African-American men pinpoints tissue-specific regulation. Nature communications 2016;7:10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature communications 2018;9:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nature genetics 2015;47:1091–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature genetics 2016;48:245–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature genetics 2016 [DOI] [PubMed] [Google Scholar]
- 21.Ferreira MA, Jansen R, Willemsen G, Penninx B, Bain LM, Vicente CT, et al. Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. The Journal of allergy and clinical immunology 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavlides JM, Zhu Z, Gratten J, McRae AF, Wray NR, Yang J. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome medicine 2016;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu L, Shi W, Long J, Guo X, Michailidou K, Beesley J, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nature genetics 2018;50:968–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nature communications 2018;9:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, Consortium GT, et al. Estimating the causal tissues for complex traits and diseases. Nat Genet 2017;49:1676–83 [DOI] [PubMed] [Google Scholar]
- 27.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, et al. Genetic effects on gene expression across human tissues. Nature 2017;550:204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheeler HE, Shah KP, Brenner J, Garcia T, Aquino-Michaels K, Consortium GT, et al. Survey of the Heritability and Sparse Architecture of Gene Expression Traits across Human Tissues. PLoS genetics 2016;12:e1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics 2016;48:1279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nature methods 2012;9:179–81 [DOI] [PubMed] [Google Scholar]
- 31.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 2012;28:1530–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nature protocols 2012;7:500–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annual review of genomics and human genetics 2006;7:29–59 [DOI] [PubMed] [Google Scholar]
- 35.Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nature reviews Genetics 2013;14:288–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo X, Lin M, Rockowitz S, Lachman HM, Zheng D. Characterization of human pseudogene-derived non-coding RNAs for functional potential. PloS one 2014;9:e93972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casbas-Hernandez P, Sun X, Roman-Perez E, D’Arcy M, Sandhu R, Hishida A, et al. Tumor intrinsic subtype is reflected in cancer-adjacent tissue. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2015;24:406–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang X, Stern DF, Zhao H. Transcriptional Profiles from Paired Normal Samples Offer Complementary Information on Cancer Patient Survival--Evidence from TCGA Pan-Cancer Data. Scientific reports 2016;6:20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nature genetics 2012;44:369–75, S1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauer JA, Ye F, Marshall CB, Lehmann BD, Pendleton CS, Shyr Y, et al. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast cancer research : BCR 2010;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mei YP, Liao JP, Shen J, Yu L, Liu BL, Liu L, et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene 2012;31:2794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaur G, Dufour JM. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012;2:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makowska KA, Hughes RE, White KJ, Wells CM, Peckham M. Specific Myosins Control Actin Organization, Cell Morphology, and Migration in Prostate Cancer Cells. Cell reports 2015;13:2118–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheong JY, Kim YB, Woo JH, Kim DK, Yeo M, Yang SJ, et al. Identification of NUCKS1 as a putative oncogene and immunodiagnostic marker of hepatocellular carcinoma. Gene 2016;584:47–53 [DOI] [PubMed] [Google Scholar]
- 46.Gu L, Xia B, Zhong L, Ma Y, Liu L, Yang L, et al. NUCKS1 overexpression is a novel biomarker for recurrence-free survival in cervical squamous cell carcinoma. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 2014;35:7831–6 [DOI] [PubMed] [Google Scholar]
- 47.Kikuchi A, Ishikawa T, Mogushi K, Ishiguro M, Iida S, Mizushima H, et al. Identification of NUCKS1 as a colorectal cancer prognostic marker through integrated expression and copy number analysis. International journal of cancer 2013;132:2295–302 [DOI] [PubMed] [Google Scholar]
- 48.Shen H, Wang L, Ge X, Jiang CF, Shi ZM, Li DM, et al. MicroRNA-137 inhibits tumor growth and sensitizes chemosensitivity to paclitaxel and cisplatin in lung cancer. Oncotarget 2016;7:20728–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian Y, Guan Y, Jia Y, Meng Q, Yang J. Chloride intracellular channel 1 regulates prostate cancer cell proliferation and migration through the MAPK/ERK pathway. Cancer biotherapy & radiopharmaceuticals 2014;29:339–44 [DOI] [PubMed] [Google Scholar]
- 50.Huang Y, Pan XW, Li L, Chen L, Liu X, Lu JL, et al. Overexpression of USP39 predicts poor prognosis and promotes tumorigenesis of prostate cancer via promoting EGFR mRNA maturation and transcription elongation. Oncotarget 2016;7:22016–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cereda V, Poole DJ, Palena C, Das S, Bera TK, Remondo C, et al. New gene expressed in prostate: a potential target for T cell-mediated prostate cancer immunotherapy. Cancer immunology, immunotherapy : CII 2010;59:63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das S, Hahn Y, Nagata S, Willingham MC, Bera TK, Lee B, et al. NGEP, a prostate-specific plasma membrane protein that promotes the association of LNCaP cells. Cancer research 2007;67:1594–601 [DOI] [PubMed] [Google Scholar]
- 53.Bera TK, Das S, Maeda H, Beers R, Wolfgang CD, Kumar V, et al. NGEP, a gene encoding a membrane protein detected only in prostate cancer and normal prostate. Proceedings of the National Academy of Sciences of the United States of America 2004;101:3059–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guyon I, Fritsche H, Choppa P, Yang LY, Barnhill S A four-gene expression signature for prostate cancer cells consisting of UAP1, PDLIM5, IMPDH2, and HSPD1. UroToday Int J 2009;2:3834–44 [Google Scholar]
- 55.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature genetics 2016;48:481–7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GTEx data are publically available via dbGaP (www.ncbi.nlm.nih.gov/gap; dbGaP Study Accession: phs000424.v6.p1). The Mayo Clinic study data are available via dbGaP (Accession: phs000985.v1.p1). TCGA data are available via the National Cancer Institute’s Genomic Data Commons Data Portal (https://gdc.cancer.gov/). The OncoArray genotype data and relevant covariate information (i.e. ethnicity, country, principal components, etc.) for prostate cancer study are deposited into dbGAP (Accession #: phs001391.v1.p1). In total 47 of the 52 OncoArray studies, encompassing nearly 90% of the individual samples, are available. The previous meta-analysis summary results and genotype data currently are available in dbGaP (Accession #: phs001081.v1.p1).