

Abstract
Down syndrome (DS) is a relatively common genetic condition caused by the triplication of human chromosome 21. No therapies currently exist for the rescue of neurocognitive impairment in DS. This review presents exciting findings showing that it is possible to restore brain development and cognitive performance in mouse models of DS with therapies that can also apply to humans. This knowledge provides a potential breakthrough for the prevention of intellectual disability in DS.
Keywords: Down syndrome, intellectual disability, brain abnormalities, mouse models, preventive therapies
Introduction
One of the most important consequences of trisomy 21 (Down syndrome, DS) is the delay in neurological development, which manifests progressively as microcephaly, hypotonia, and intellectual disability (Lydic and Steele, 1979; Schmidt-Sidor et al., 1990; Silva et al., 1996; Chapman and Hesketh, 2000; Rigoldi et al., 2011). Complex neurological function is the result of many molecular, cellular, and environmental events that must occur and be coordinated at precisely the right time. In mammals, many of these processes are either initiated or completed before birth. Because of this, the overall impact of the neurological deficits in DS may be lessened if the initial pathologic changes in the brain are prevented from occurring. Studies in human fetuses with DS demonstrate that the brain is significantly altered by the beginning of the second trimester. Therefore, the first window of opportunity for cognitive improvement occurs well before birth (Haydar et al., 1996, 2000; Guihard-Costa et al., 2006; Chakrabarti et al., 2007; Contestabile et al., 2007; Ishihara et al., 2010).
Most of the brain neurons are produced in the prenatal period, with the notable exception of those involved in the formation of the hippocampus, where neurogenesis continues postnatally and throughout life. Unlike neurogenesis, neuron maturation and establishment of brain wiring largely takes place in the perinatal period. Therefore, during this time, there is a unique opportunity to rescue a population of cells in the brains of fetuses with DS that would otherwise be permanently missing, thus allowing proper connectivity. In this review, we describe the changes known to occur in the brains of trisomic humans and mice and present several approaches currently under way to positively affect cognition by preventing prenatal brain alterations.
Cellular and molecular processes affecting development and function of the CNS in DS
The first major event in specifying brain size and complexity is development of the proper variety of the neural stem and progenitor cells that form the neurons and glia throughout the brain. In human forebrain development, this process is initiated at 6 weeks of gestation and lasts well into the second trimester (Bystron et al., 2008; Stiles, 2008). In several mouse models of DS during the equivalent developmental period, the radial glia stem cells of the cerebral cortex and hippocampus are found in reduced numbers and their cell division rate is slower than normal (Haydar et al., 2000; Chakrabarti et al., 2007; Contestabile et al., 2007; Ishihara et al., 2010). In addition, another type of neural precursor cell called the apical intermediate precursor cell is specifically underproduced in the prenatal neocortex (Tyler and Haydar, 2013). These two defects likely underlie the reductions in gray matter volume and the numbers of excitatory neurons in the maturing brain. These proliferation defects in mouse models have been recently supported by reports of similar reductions in stem cell proliferation in the hippocampi of human fetuses with DS (Guidi et al., 2008). However, although slower proliferation is found in many dividing cells in DS, increased cell production has also been found in specific brain regions. For example, in the Ts65Dn mouse model, whereas the number of excitatory neurons in the neocortex is reduced, the number of inhibitory neurons is increased compared with controls (Chakrabarti et al., 2010) and this alteration in the excitatory:inhibitory ratio has been attributed to triplication of the transcription factors OLIG1 and OLIG2. These supernumerary GABA-releasing inhibitory neurons are generated from neural precursors in the ganglionic eminences within the ventral telencephalon (Anderson et al., 2001).
After these changes in the fetal period, several postnatal abnormalities appear in multiple brain regions at both the systemic and cellular levels. One of the most robust changes is the slower growth in the cerebellum (Guihard-Costa et al., 2006), which begins to develop before birth in humans but after birth in mice. In both species, this delayed growth is due to slower proliferation of granule and Purkinje neuron precursors. This results in a simplified cerebellar morphology with reduced numbers of granule and Purkinje neurons (Baxter et al., 2000; Olson et al., 2004; Guidi et al., 2011; Starbuck et al., 2014). In addition, a paucity of excitatory synapses and an abundance of inhibitory synapses are evident in the mouse model forebrain, perhaps as a consequence of the alterations in prenatal neurogenesis of these two neuronal classes (Chakrabarti et al., 2007; Belichenko et al., 2009a; Perez-Cremades et al., 2010). In the neocortex, synapse-related structural changes, including alterations in dendritic spine morphology and density, have been found in humans (Marin-Padilla, 1976; Suetsugu and Mehraein, 1980; Takashima et al., 1981; Weitzdoerfer et al., 2001) and mouse models (Belichenko et al., 2004, 2009a; Villar et al., 2005; Haas et al., 2013) and potential molecular targets for these structural abnormalities have been suggested (Wang et al., 2012a). Reductions in white matter have also been described in the brains of children and adults with DS and may be due to alterations in numbers or in the function of oligodendroglia (White et al., 2003; Carducci et al., 2013; Powell et al., 2014). It is important to note that oligodendrocytes are generated in three waves, initially by prenatal neural precursor cells in the ventral telencephalon, followed postnatally by oligodendrocyte progenitors in the cortical parenchyma (Richardson et al., 2006).
Therefore, many cell classes in different parts of the brain are affected throughout the lifespan of people with DS, but changes first begin before birth during neural stem cell proliferation. These alterations in cell production lead to subsequent abnormalities in neuronal and glial cell allocation and to functional changes in the neural circuitry. Apart from the roles of DYRK1A (discussed below) and the OLIG genes on specific aspects of brain development, whether the prenatal and postnatal central nervous system defects are due to the overexpression of individual genes or to aneuploidy (i.e., the burden of segregating an additional chromosome during cell division) has not been conclusively determined for all of the abnormalities noted above. Nevertheless, it is clear that these prenatal changes may play a fundamental role in intellectual disability. By preventing them from occurring, we hypothesize that we will improve cognition and quality of life for people with DS.
Preventive therapies for cognitive disability in DS: the sooner the better
There is a consensus that the major causes underlying aberrant brain development and thus intellectual disability in DS are impaired ontogenetic neurogenesis, dendritic hypotrophy, spine density reduction, altered synaptic organization and function, and widespread alterations of various transmitter and receptor systems (for review, see Bartesaghi et al., 2011; Dierssen, 2012; Guedj and Bianchi, 2013; Gardiner, 2015). Although neonatal therapies may mainly shape the cerebellum and hippocampus, prenatal therapies may have a much larger impact on the trisomic brain. Below is a summary of the treatments to date that have been aimed at neonatal and prenatal intervention.
Neonatal treatments
In the 2-d-old Ts65Dn mouse model of DS, a single treatment with SAG, an activator of the mitogenic Sonic Hedgehog pathway, restored cerebellar granule cell production and improved learning and memory (Roper et al., 2006; Das et al., 2013). Based on evidence that the serotonergic system is altered in DS (Bar-Peled et al., 1991; Risser et al., 1997; Whitaker-Azmitia, 2001) and that serotonin is crucial for neurogenesis, a series of studies examined the effects of neonatal treatment with fluoxetine, a selective serotonin reuptake inhibitor (Wong et al., 1974), on hippocampal development. Previous studies showed that treatment with fluoxetine from postnatal day 3 (P3) to P15 resulted in long-term restoration of hippocampal neurogenesis, dendritic pathology, functional connectivity, and learning and memory in 45-d-old (Bianchi et al., 2010; Guidi et al., 2013; Stagni et al., 2013) and 90-d-old (Stagni et al., 2015) Ts65Dn mice, indicating that fluoxetine rescues many trisomy-linked developmental deficits. Fluoxetine, in addition to increasing serotonin availability, stimulates the production of the neurosteroid allopregnanolone (Pinna et al., 2009), a GABA-A receptor-positive allosteric modulator that has been shown to increase neurogenesis (Wang et al., 2010) and density of excitatory synapses (Shimizu et al., 2015). Fluoxetine binds to the σ-1 receptor that regulates Ca2+ signaling, ion channel activity, trophic factor signaling, cell survival, myelination, and synaptogenesis (Hayashi and Stahl, 2009). Fluoxetine also interacts with the mitochondrial voltage-dependent anion channel and protects against apoptotic cell death (Nahon et al., 2005). Therefore, these additional mechanisms may contribute to the positive effects of neonatal and embryonic (see below) treatment with fluoxetine on the trisomic brain.
Embryonic treatments
Administration of active fragments of neurotrophic factors during E8–E12 was found to prevent delay in the achievement of sensorimotor milestones in Ts65Dn pups (Toso et al., 2008) and to improve learning and memory in adults (Incerti et al., 2012). In a series of studies, choline (the acetylcholine precursor) was administered to Ts65Dn dams from conception until weaning. Choline supplementation was found to improve hippocampal neurogenesis and learning and memory in adult and aged trisomic offspring (Moon et al., 2010; Velazquez et al., 2013; Ash et al., 2014).
Oxidative stress appears to be involved in the pathogenesis of DS. Alpha-tocopherol, an antioxidant, when administered during gestation and postnatally (12 weeks), reduces lipid peroxidation and improves learning and memory in Ts65Dn mice (Shichiri et al., 2011). Particularly impressive results showing restoration of numerous DS brain phenotypes have been obtained with prenatal treatment with fluoxetine (Guidi et al., 2014). Pregnant Ts65Dn females were treated with fluoxetine from E10 to delivery. Although untreated Ts65Dn pups exhibited severe reduction in neurogenesis and hypocellularity throughout the forebrain, midbrain, and hindbrain, in embryonically treated Ts65Dn pups, neural precursor proliferation and cellularity were fully restored. The trisomic offspring of treated and untreated mothers were examined at postnatal day 45. Neurogenesis was still restored in the major postnatal brain neurogenic niches. In addition, total granule cell number and dendritic development of postnatally born granule neurons were normalized, with a full correction of the severe dendritic hypotrophy that characterizes the trisomic condition. The counterpart of this effect was restoration of presynaptic and postsynaptic terminals. Importantly, embryonically treated Ts65Dn mice at age 45 d exhibited restoration of cognitive performance, indicating that the positive impact of embryonic treatment on brain development was functionally effective in adulthood (Fig. 1).
Figure 1.
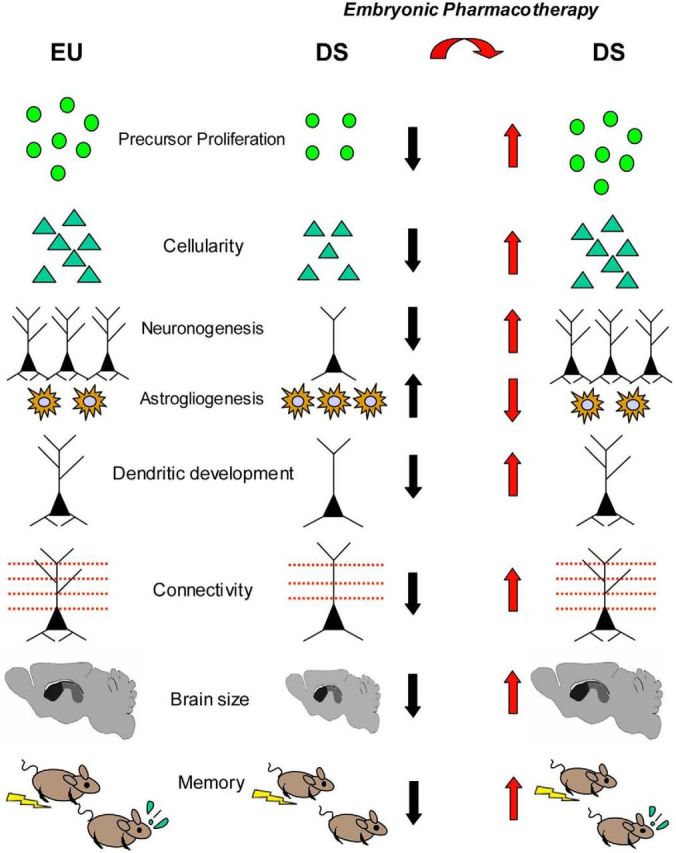
Summary of the effects of embryonic treatment with fluoxetine on brain development in Ts65Dn mice. Ts65Dn mice (DS) show impairment of proliferation, reduced cellularity, reduced generation of neurons, increased astrogliogenesis, dendritic hypotrophy, reduced connectivity, reduced brain size, and behavioral impairment. All of these defects are rescued by treatment with fluoxetine during the embryonic period.
Genomic approach to the identification of novel therapies for prenatal treatment of DS
The recent rapid rise in noninvasive prenatal testing for trisomy 21 (Bianchi, 2015), coupled with the known abnormalities in fetal brain development (Contestabile et al., 2007; Larsen et al., 2008; Guidi et al., 2011), creates a window of opportunity in humans for maternal treatment to improve fetal neurocognition as soon as DS has been diagnosed (Guedj and Bianchi, 2013; Guedj et al., 2013). Prenatal treatment of human fetuses with DS is associated with several unique challenges, including the fact that the (presumably healthy) mother will be treated simultaneously with the fetus. The first concern, therefore, is safety. Any proposed therapy cannot cause harm to the mother nor cause teratogenic effects to the growing and developing fetus. An additional challenge is achieving therapeutic drug levels across both the placental and blood–brain barriers.
With safety as the highest priority, investigators opted to identify novel therapies for DS using the Connectivity Map (CMap; Lamb, 2007). The goal of the CMap is to make “connections” among a disease, differentially regulated genes, and drugs. The CMap is a publicly available database of the gene expression patterns of a number of different cultured cell types before and after exposure to a large number (>1300) of US Food and Drug Administration (FDA)-approved drugs. The elegance of the CMap is that it uses expressed genes (or mRNA) as its common language. Therefore, any list of differentially regulated genes can be uploaded into the database to generate a list of drugs from which hypotheses regarding treatment can be tested.
With the exception of studies that have used cultured human fetal cells, few molecular research projects have analyzed biomaterial from living fetuses with DS. In 2009, investigators performed functional genomic analyses in fetuses with trisomy 21 versus gestational age- and sex-matched euploid controls (Slonim et al., 2009). Because cell culture can induce gene expression changes, they chose to use cell-free fetal mRNA obtained directly from uncultured amniotic fluid supernatant samples. Amniotic fluid is the only biofluid that can be safely analyzed in fetuses with a known karyotype. This mRNA is stable and derives from apoptotic cells. These investigators have shown previously that some of the transcripts in amniotic fluid map specifically to the fetal brain (Hui et al., 2012a, 2012b). In addition, fetuses with different chromosome abnormalities have completely different gene expression signatures that are consistent with the known pathophysiological abnormalities in these conditions (Zwemer and Bianchi, 2015).
Using Affymetrix gene expression microarrays, trisomy 21 and euploid mRNA samples were compared and 311 statistically significant differentially regulated genes were found. Only 5 of the genes (CLIC6, ITGB2, RUNX1, C21orf67, C21orf86) were physically located on human chromosome 21, suggesting that the majority of the phenotypic effects of DS were secondary due to genome-wide dysregulation (Slonim et al., 2009). A heat-map analysis showed distinct clustering by fetal genotype. In other words, there was clear evidence of a characteristic set of differentially expressed genes in all second-trimester human fetuses with trisomy 21. Further, an unbiased pathway analysis of the differentially regulated genes using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Dennis et al., 2003) demonstrated that the following functions were disrupted in fetuses with DS: oxidative stress, ion transport, G-protein signaling, immune and stress response, circulatory system functions, cell structure, sensory perception, and several developmental processes (Slonim et al., 2009). Similar pathway abnormalities have been found in E15.5 brains from a mouse model of DS, Ts1Cje (Guedj et al., 2015).
Applying a systems biology approach to fetuses with trisomy 21, investigators focused on oxidative stress as their first functional target for prenatal therapy. The CMap identified apigenin, a natural antioxidant and anti-inflammatory compound found in citrus fruit and green leafy vegetables, as a high-priority candidate molecule to reverse the gene expression pattern observed in second-trimester fetuses with DS. Preliminary data obtained by incubating cultured amniocytes from fetuses with trisomy 21 with different concentrations of apigenin have shown that concentrations of up to 2 μm are not toxic (i.e., they do not affect cell proliferation). This concentration statistically significantly reduced oxidative stress as measured by the single-cell gel electrophoresis assay (Guedj et al., 2013) In preliminary experiments, Ts1Cje dams were fed 200 mg/kg/d of apigenin with powdered laboratory chow from the time of conception throughout their pregnancies. Apigenin treatment normalized brain gene expression in some differentially regulated genes, shortened the time it took to achieve neonatal developmental milestones, and improved performance on the open field test. These preliminary data provide proof of principle that functional genomic analysis of the human fetal transcriptome can provide a rational basis for drug discovery in DS.
Targeting the excitation inhibition balance in DS
An imbalance of excitation and inhibition (E/I) is thought to underlie several neurological diseases, including autism (Rubenstein and Merzenich, 2003), Tourette syndrome (Singer and Minzer, 2003), and schizophrenia (Wassef et al., 2003). Cognitive deficits in DS have been proposed to result from an excess of inhibition. However, chromosome 21 genes responsible for such defects have not been clearly identified. Excessive inhibition in temporal lobe and hippocampal circuitry has also been observed in the Ts65Dn mouse (Kurt et al., 2000, 2004; Belichenko et al., 2007, 2009b; Perez-Cremades et al., 2010), which recapitulates the hallmarks of the DS phenotype, including serious cognitive impairment (Escorihuela et al., 1995; Reeves et al., 1995). Increased efficiency of GABA-A and GABA-B receptor-mediated neurotransmission has been reported for Ts65Dn mice (Kleschevnikov et al., 2012a, 2012b) and the GABA-B/GABA-A ratios evoked by stimulation within the stratum lacunosum moleculare of Ts65Dn hippocampus were found to be significantly altered (Best et al., 2012). This E/I imbalance may explain the alterations of LTP and LTD found in Ts5Dn mice (Siarey et al., 1997, 1999; Belichenko et al., 2009b). In view of the trisomy-dependent excessive inhibition, GABA receptor antagonists are considered a good therapeutic strategy for restoring memory in the Ts65Dn mouse. Growing evidence shows that GABA receptor antagonists restore LTP and memory in the Ts65Dn mouse (Kleschevnikov et al., 2004, 2012a; Fernández et al., 2007; Rueda et al., 2008; Colas et al., 2013; Martínez-Cué et al., 2013), suggesting their potential usefulness for cognitive improvement in DS.
In humans, altered copy number for segments of chromosome 21 that results in either deletion or duplication of DYRK1A can induce morphological defects and cognitive impairments (Delabar et al., 1993; Rahmani et al., 1998; Ronan et al., 2007; Oegema et al., 2010; van Bon et al., 2011). Phenotypic rescue experiments combining Ts65Dn mice, which have three copies of Dyrk1a, with mice monosomic for a chromosomal segment containing 33 genes including Dyrk1a (Ms1Rhr) or with mice heterozygous for invalidation of Dyrk1a produced progeny with a normal learning phenotype, indicating that duplication of this gene is necessary to produce a cognitive deficit (Morris water maze and contextual conditioning) (Belichenko et al., 2009a, 2009b; García-Cerro et al., 2014). Among the genes from this 33-gene region, Dyrk1a is an attractive candidate for inducing cognitive impairment phenotypes. It encodes a proline/arginine-directed serine/threonine kinase (Tejedor et al., 1995). Consistent with its etiological role in DS, DYRK1A targets proteins involved in neurodevelopment (Barallobre et al., 2014; Najas et al., 2015) and neuritogenesis (Murakami et al., 2009; Xie et al., 2012). Both in trisomic mice and in individuals with DS, brain levels of DYRK1A are increased ∼1.5-fold, indicating that this protein is overexpressed in a gene-dosage-dependent manner (Dowjat et al., 2007).
Molecular consequences of alterations in Dyrk1a dosage were assessed in mouse models with varying copy numbers of Dyrk1a: mBACtgDyrk1a, Ts65Dn and Dp(16)1Yey (with 3 gene copies) and Dyrk1a+/− (one functional copy). Increased expression of Dyrk1a in mBACtgDyrk1a-induced molecular alterations in synaptic plasticity pathways, particularly expression changes in GABAergic and DYRK1A glutaminergic-related proteins (Souchet et al., 2014). Similar alterations were observed in models with partial trisomy of MMU16, Ts65Dn and Dp(16)1Yey and were reversed in the Dyrk1a+/− model (Souchet et al., 2014). Dyrk1a overexpression produced an increased number and signal intensity of GAD67-positive neurons, indicating enhanced inhibition pathways in three different models: mBACtgDyrk1a, hYACtgDyrk1a, and Dp(16)1Yey. Functionally, Dyrk1a overexpression protected mice from PTZ-induced seizures related to GABAergic neuron plasticity (Souchet et al., 2014). Dyrk1a overexpression also affects pathways involved in synaptogenesis and synaptic plasticity and tips the E/I balance toward inhibition (Souchet et al., 2014).
Green tea contains a natural inhibitor of DYRK1A kinase activity: epigallocatechin gallate (EGCG; IC50 = 0.3 μm) (Bain et al., 2003). Control and transgenic mice overexpressing Dyrk1a were maintained on two different polyphenol-based diets from gestation to adulthood. The major features of the transgenic phenotype, including abnormal novel object recognition, were rescued in these mice (Guedj et al., 2009). Use of EGCG-containing extracts was also assessed at the adult stage. A 1-month treatment induced efficient rescue effects on the cognitive phenotypes of Ts65Dn, tgDyrk1a, and mBACtgDyrk1a mice (de la Torre et al., 2014). Investigators have discovered the molecular consequences of different long-term treatments (1 month) in adult mBACtgDyrk1a mice (Delabar et al., 2012). A major rescuing effect of a polyphenol extract (POL60; Sigma-Aldrich) was observed on GABAergic and glutamatergic pathways. A dose-effect experiment using a decaffeinated green tea extract (MGTE) similar to the extract used in clinical trials showed that the intermediate dose (60 mg/kg) acts both on components of GABAergic and glutamatergic pathways. The same dose was used to treat pregnant dams until weaning or adulthood (3 months) (Delabar et al., 2012). GAD67 protein dose and neuron density were rescued by the treatment. This rescue was maintained when the treatment was stopped after weaning. Controlling levels of active DYRK1A, possibly prenatally, is therefore a strong consideration for DS therapy (Fig. 2). It must be observed that EGCG, in addition to inhibit DYRK1A kinase, modulates numerous cellular pathways (Schroeter et al., 2007; Spencer, 2009; Kelsey et al., 2010; Wang et al., 2012b; Kim et al., 2014), suggesting that additional actions may take part in its positive effects on the brain.
Figure 2.

Action of EGCG on E/I balance in a T21 context.
Nonpharmacological approaches in combination with drug treatments
Trisomy of human chromosome 21 leads to intellectual disability by affecting CNS development and function, impairing cognition, and adaptive behavior (Dierssen, 2012). Insights into the neurobiological mechanisms of DS from mouse models and human studies have shown that alterations in neural plasticity mechanisms are related to cognitive impairment (Dierssen and Ramakers, 2006). This opens the possibility for the discovery of drugs for restoring cognitive function by pharmacologically targeting neural plasticity cascades that set the brain in a favorable state for cognitive function and could thus be disease-modifying treatments in individuals with DS (de la Torre and Dierssen, 2012). Overall scientific evidence supports a direct link between experience-dependent learning and changes in synaptic and neural plasticity. Cognitive training programs are an effective therapeutic interventional strategy in intellectual disability. Specifically, it was found that HSA21 candidate genes such as Dyrk1a are regulated in DS mouse models as a result of environmental enrichment (Pons-Espinal et al., 2013). Therefore, nonpharmacological therapeutic avenues can potentially play a key role as safe and effective coadjuvants for further enhancing the positive effects of experimental compounds. Two such avenues are cognitive training and noninvasive brain stimulation (NIBS), specifically transcranial direct current stimulation. These are emerging interventional techniques that have been shown to be safe and effective for ameliorating a wide range of cognitive and behavioral deficits in several pathological conditions [Parkinson's disease, Alzheimer's disease (AD)] and in children with mental disabilities (autism). Computerized cognitive training systems have been recognized as a powerful tool for cognitive enhancement (Green and Bavelier, 2008) in individuals with intellectual disability and neurodegenerative disorders (Anguera et al., 2013; Franceschini et al., 2013). Cognitive training can partially rescue atypical brain development and improve functioning by promoting structural reorganization in some brain regions. Effects have been demonstrated at different levels from gene regulation (Söderqvist et al., 2014), biochemical activity (McNab et al., 2009), and neuronal activity (Westerberg and Klingberg, 2007; Brehmer et al., 2011) to its effect on learning (Brehmer et al., 2011) and daily functioning (Klingberg et al., 2005). In children with DS, studies using Cogmed JM software (Pearson Education) suggest that computerized visuospatial memory training in a school setting is both feasible and effective (Bennett et al., 2013).
With the advent of novel technologies such as NIBS, new possibilities are also being proposed to treat intellectual disabilities. NIBS has been suggested to modulate cortical excitability (Wach et al., 2013) and reduce cortical inhibition (Hensch and Bilimoria, 2012), resetting the brain to a sensitive state. Because one of the main pathological features of DS is network overinhibition, NIBS may be a disease-modifying treatment to improve plasticity in DS. One important aspect to consider when optimizing outcomes using NIBS is that modulation of plasticity does not rely only on the modulation of excitability, but also on the state of the brain being stimulated. A key issue for an effective treatment will require an optimal orchestration of the internal processes of brain plasticity and therapeutic interventions.
Therapeutic approaches to delay the cognitive decline and degenerative processes in older mouse models of DS
A key phenotypic alteration in DS is the early appearance of AD-like pathology, including increased production of the β-amyloid peptide; presence of β-amyloid plaques, intracellular neurofibrillary tangles, and neuroinflammation; increases in oxidative stress; and neuron degeneration (Lott, 2012; Wilcock and Griffin, 2013). Ts65Dn mice, the most commonly used murine model of DS, also show some of these neuropathological hallmarks of AD such as increased levels of the APP protein, β-amyloid peptides, tau hyperphosphorylation, increased markers of oxidative stress, microglia activation, neuroinflammation, and cholinergic and noradrenergic neuron degeneration (Hunter et al., 2004b; Seo and Isacson, 2005; Shukkur et al., 2006; Lockrow et al., 2009, 2011; Netzer et al., 2010; Corrales et al., 2014). Recently, the administration of different compounds targeting some of these altered phenotypes has improved learning and reduced AD-related phenotypes in Ts65Dn mice. Among these drugs and hormones are DAPT (N-[N-(3,5-diflurophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester) (Netzer et al., 2010), minocycline (Hunter et al., 2004a), memantine (Costa et al., 2008; Rueda et al., 2010; Lockrow et al., 2011), vitamin E (Lockrow et al., 2009), and estrogens (Granholm et al., 2002, 2003).
Although the classic theory of AD proposes that the neuropathology and cognitive decline starts in this condition with the accumulation of amyloid plaques and neurofibrillary tangles, there is increasing evidence that other AD phenotypes such as oxidative stress and neuroinflammation might precede the other pathological hallmarks and lead to increases in β-amyloid load and tau phosphorylation (Varnum and Ikezu, 2012). Therefore, compounds targeting neuroinflammation and oxidative stress might be a promising strategy to delay the appearance of these alterations in the DS population.
Recent studies have demonstrated that chronic melatonin administration to adult Ts65Dn mice improves spatial learning; restores LTP, neurogenesis, and hippocampal cellularity; reduces oxidative stress; and protects against cholinergic neuron degeneration without affecting the levels of the APP protein or β-amyloid peptides, which implies that APP and β-amyloid load is not one of the mechanisms under the cognitive improvements induced by melatonin (Corrales et al., 2013, 2014). Investigators have made an exhaustive characterization of the different enzymes of the oxidative stress cascade. Their results suggest that melatonin produces its antioxidant effects by reducing lipid peroxidation, but does not have a significant effect on the levels of different antioxidant enzymes. They have also demonstrated that cellular senescence is enhanced in the subgranular zone of the dentate gyrus of Ts65Dn mice and this effect is completely rescued after melatonin administration. These results suggest that the positive effects of melatonin on the cognitive abilities of these mice might be due to their antioxidant effects and/or its ability to reduce cellular senescence.
However, other mechanisms such as the effects of melatonin and other drugs on neuroinflammation need to be explored. Neuroinflammation plays a key role in the development of AD neuropathology in both humans with DS and in Ts65Dn mice. Therefore, therapeutic approaches that reduce the activity of proinflammatory cytokines could also be a promising strategy to reduce AD-related phenotypes in DS. IL-17A is a proinflammatory cytokine that has a fundamental role mediating brain damage during neuroinflammatory processes because it acts as a modulatory factor in the induction of other cytokines (Zimmermann et al., 2013). Chronic administration of antibodies that block the IL-17 to animal models of brain damage reduce the infarcted area, where inflammatory activity is determinant, and improve the neurological status of the animals (Gelderblom et al., 2012). Investigators have evaluated the effect of chronic administration of an antibody against this cytokine in several altered phenotypes of aged Ts65Dn mice. Chronic administration of anti-IL17 to aged Ts65Dn mice enhanced spatial learning and memory, hippocampal proliferation, and mature neuronal density and reduced the levels of APP and of β-amyloid accumulation in these animals. These results provide further support for the theory that reducing neuroinflammation might delay or prevent the development of other AD neuropathological changes and the concomitant cognitive decline.
Conversely, several HSA21 genes are well known to be implicated in the AD-like pathology that appears in DS and in the Ts65Dn mouse. Among, them, the APP gene has been shown to have a preeminent role in cholinergic and noradrenergic degeneration, NGF retrograde transport, early endosomes, altered synaptic plasticity, and adult hippocampal neurogenesis (Cataldo et al., 2003; Salehi et al., 2006; Trazzi et al., 2013). Other genes that seem to play a role in the development of AD pathology in DS are SOD1 (Busciglio et al., 2002), RCAN1/DSCR1 (Ermak et al., 2001; Lott et al., 2006), ETS2 (Wolvetang et al., 2003; Helguera et al., 2005), and ITSN1 (Chang and Min, 2009). Morevover, the DYRK1A gene has been demonstrated to play a role in different AD phenotypes, including APP and tau phosphorylation and increases in β-amyloid accumulation (Ryoo et al., 2007, 2008; Wegiel et al., 2011). Recent studies provide evidence that reducing a copy of this gene in the Ts65Dn mouse leads to a reduction of APP and β-amyloid levels. Due to the well-known role of these pathological hallmarks of AD in cognitive deterioration, normalizing Dyrk1a gene dosage could improve or delay the cognitive deficits found in aged Ts65Dn mice. A recent report (García-Cerro et al., 2014) showed that normalizing Dyrk1a gene dosage in Ts65Dn mice partially rescues some phenotypes linked to cognition, such as learning, long-term potentiation, cell proliferation, and differentiation, while other phenotypes plausibly linked to cognition (density of mature hippocampal granule cells, the dentate gyrus volume and the subgranular zone area) were not modified by this genetic manipulation. Therefore, although the role of this gene on AD phenotypes needs to be further characterized, drugs such as EGCG that target DYRK1A could also be beneficial to delay or prevent the cognitive deterioration and the appearance of AD-related neurodegeneration in DS.
In summary, in view of the complexity and many facets of the AD-like phenotype, it has been possible to take advantage of different strategies to delay the cognitive decline and neurodegeneration in mouse models of DS, as demonstrated by the fact that many of the attempted therapies were effective. It can be speculated that a combination of therapies may be a strategic approach for a more effective improvement of AD pathology in DS individuals.
Conclusions
In this review, we show that there are multiple types of opportunities to rescue abnormalities in neurodevelopment and neurodgeneration in DS and equivalent mouse models. An important, but previously underappreciated, window of opportunity is during the prenatal period, when neuron maturation and brain wiring occurs. Here, we have highlighted three different approaches to rescue DS phenotypes using: (1) EGCG to target overexpression of Dyrk1a, among other effects; (2) selective serotonin-reuptake inhibitors such as fluoxetine; and (3) a systems biology approach to identifying key treatable pathways such as oxidative stress. Environmental enrichment and novel treatments such as NIBS have strong potential to amplify the pharmacologic approaches. Other therapies target prevention or delay of neurocognitive decline and AD pathology. These DS-associated brain alterations have long been considered to be irreversible. The demonstration that neurodevelopment can be improved in mouse models using multiple strategies provides proof of principle that intellectual disabilities in DS can be ameliorated.
References
- Anderson SA, Marín O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Anguera JA, Boccanfuso J, Rintoul JL, Al-Hashimi O, Faraji F, Janowich J, Kong E, Larraburo Y, Rolle C, Johnston E, Gazzaley A. Video game training enhances cognitive control in older adults. Nature. 2013;501:97–101. doi: 10.1038/nature12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash JA, Velazquez R, Kelley CM, Powers BE, Ginsberg SD, Mufson EJ, Strupp BJ. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol Dis. 2014;70:32–42. doi: 10.1016/j.nbd.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/bj20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barallobre MJ, Perier C, Bové J, Laguna A, Delabar JM, Vila M, Arbonés ML. DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson's disease. Cell Death Dis. 2014;5:e1289. doi: 10.1038/cddis.2014.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled O, Gross-Isseroff R, Ben-Hur H, Hoskins I, Groner Y, Biegon A. Fetal human brain exhibits a prenatal peak in the density of serotonin 5-HT1A receptors. Neurosci Lett. 1991;127:173–176. doi: 10.1016/0304-3940(91)90787-T. [DOI] [PubMed] [Google Scholar]
- Bartesaghi R, Guidi S, Ciani E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev Neurosci. 2011;22:419–455. doi: 10.1515/RNS.2011.037. [DOI] [PubMed] [Google Scholar]
- Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet. 2000;9:195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci. 2009a;29:5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J Comp Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504:329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Masliah E, Wu C, Takimoto-Kimura R, Salehi A, Mobley WC. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J Comp Neurol. 2009b;512:453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SJ, Holmes J, Buckley S. Computerized memory training leads to sustained improvement in visuospatial short-term memory skills in children with Down syndrome. Am J Intellect Dev Disabil. 2013;118:179–192. doi: 10.1352/1944-7558-118.3.179. [DOI] [PubMed] [Google Scholar]
- Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp Neurol. 2012;233:749–757. doi: 10.1016/j.expneurol.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi DW. Pregnancy: prepare for unexpected prenatal test results. Nature. 2015;522:29–30. doi: 10.1038/522029a. [DOI] [PubMed] [Google Scholar]
- Bianchi P, Ciani E, Guidi S, Trazzi S, Felice D, Grossi G, Fernández M, Giuliani A, Calzà L, Bartesaghi R. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. J Neurosci. 2010;30:8769–8779. doi: 10.1523/JNEUROSCI.0534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehmer Y, Rieckmann A, Bellander M, Westerberg H, Fischer H, Bäckman L. Neural correlates of training-related working-memory gains in old age. Neuroimage. 2011;58:1110–1120. doi: 10.1016/j.neuroimage.2011.06.079. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. 2002;33:677–688. doi: 10.1016/S0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- Bystron I, Blakemore C, Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci. 2008;9:110–122. doi: 10.1038/nrn2252. [DOI] [PubMed] [Google Scholar]
- Carducci F, Onorati P, Condoluci C, Di Gennaro G, Quarato PP, Pierallini A, Sarà M, Miano S, Cornia R, Albertini G. Whole-brain voxel-based morphometry study of children and adolescents with Down syndrome. Funct Neurol. 2013;28:19–28. [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci. 2003;23:6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Best TK, Cramer NP, Carney RS, Isaac JT, Galdzicki Z, Haydar TF. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat Neurosci. 2010;13:927–934. doi: 10.1038/nn.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Min KT. Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc Natl Acad Sci U S A. 2009;106:17117–17122. doi: 10.1073/pnas.0904397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev. 2000;6:84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Colas D, Chuluun B, Warrier D, Blank M, Wetmore DZ, Buckmaster P, Garner CC, Heller HC. Short-term treatment with the GABAA receptor antagonist pentylenetetrazole produces a sustained pro-cognitive benefit in a mouse model of Down's syndrome. Br J Pharmacol. 2013;169:963–973. doi: 10.1111/bph.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, Bartesaghi R, Ciani E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- Corrales A, Martínez P, García S, Vidal V, García E, Flórez J, Sanchez-Barceló EJ, Martínez-Cué C, Rueda N. Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J Pineal Res. 2013;54:346–358. doi: 10.1111/jpi.12037. [DOI] [PubMed] [Google Scholar]
- Corrales A, Vidal R, García S, Vidal V, Martínez P, García E, Flórez J, Sanchez-Barceló EJ, Martínez-Cué C, Rueda N. Chronic melatonin treatment rescues electrophysiological and neuromorphological deficits in a mouse model of Down syndrome. J Pineal Res. 2014;56:51–61. doi: 10.1111/jpi.12097. [DOI] [PubMed] [Google Scholar]
- Costa AC, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33:1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- Das I, Park JM, Shin JH, Jeon SK, Lorenzi H, Linden DJ, Worley PF, Reeves RH. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci Transl Med. 2013;5:201ra120. doi: 10.1126/scitranslmed.3005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, Dierssen M. Therapeutic approaches in the improvement of cognitive performance in Down syndrome: past, present, and future. Prog Brain Res. 2012;197:1–14. doi: 10.1016/B978-0-444-54299-1.00001-7. [DOI] [PubMed] [Google Scholar]
- de la Torre R, De Sola S, Pons M, Duchon A, de Lagran MM, Farré M, Fitó M, Benejam B, Langohr K, Rodriguez J, Pujadas M, Bizot JC, Cuénca A, Janel N, Catuara S, Covas MI, Blehaut H, Herault Y, Delabar JM, Dierssen M. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res. 2014;58:278–288. doi: 10.1002/mnfr.201300325. [DOI] [PubMed] [Google Scholar]
- Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, Noel B, Sinet PM. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet. 1993;1:114–124. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- Delabar J, Guedj F, Souchet B, Sahún I, Luilier S, Duchon A, Paly E, Janel N, Bizot J, Herault Y, Arbonés M, Dierssen M, Créau N. Excitation-inhibition balance and learning are modified by Dyrk1a gene dose and EGCG treatment. Soc Neurosci Abstr. 2012;38 348.14/M10. [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Dierssen M. Down syndrome: the brain in trisomic mode. Nat Rev Neurosci. 2012;13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5:48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- Dowjat WK, Adayev T, Kuchna I, Nowicki K, Palminiello S, Hwang YW, Wegiel J. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermak G, Morgan TE, Davies KJ. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem. 2001;276:38787–38794. doi: 10.1074/jbc.M102829200. [DOI] [PubMed] [Google Scholar]
- Escorihuela RM, Fernández-Teruel A, Vallina IF, Baamonde C, Lumbreras MA, Dierssen M, Tobeña A, Flórez J. A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neurosci Lett. 1995;199:143–146. doi: 10.1016/0304-3940(95)12052-6. [DOI] [PubMed] [Google Scholar]
- Fernández F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- Franceschini S, Gori S, Ruffino M, Viola S, Molteni M, Facoetti A. Action video games make dyslexic children read better. Curr Biol. 2013;23:462–466. doi: 10.1016/j.cub.2013.01.044. [DOI] [PubMed] [Google Scholar]
- García-Cerro S, Martínez P, Vidal V, Corrales A, Flórez J, Vidal R, Rueda N, Arbonés ML, Martínez-Cué C. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS One. 2014;9:e106572. doi: 10.1371/journal.pone.0106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner KJ. Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations. Drug Des Devel Ther. 2015;9:103–125. doi: 10.2147/DDDT.S51476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, Orthey E, Arumugam TV, Leypoldt F, Simova O, Thom V, Friese MA, Prinz I, Hölscher C, Glatzel M, Korn T, Gerloff C, Tolosa E, Magnus T. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793–3802. doi: 10.1182/blood-2012-02-412726. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Ford KA, Hyde LA, Bimonte HA, Hunter CL, Nelson M, Albeck D, Sanders LA, Mufson EJ, Crnic LS. Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiol Behav. 2002;77:371–385. doi: 10.1016/S0031-9384(02)00884-3. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O. Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus. 2003;13:905–914. doi: 10.1002/hipo.10130. [DOI] [PubMed] [Google Scholar]
- Green CS, Bavelier D. Exercising your brain: a review of human brain plasticity and training-induced learning. Psychol Aging. 2008;23:692–701. doi: 10.1037/a0014345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj F, Bianchi DW. Noninvasive prenatal testing creates an opportunity for antenatal treatment of Down syndrome. Prenat Diagn. 2013;33:614–618. doi: 10.1002/pd.4134. [DOI] [PubMed] [Google Scholar]
- Guedj F, Sébrié C, Rivals I, Ledru A, Paly E, Bizot JC, Smith D, Rubin E, Gillet B, Arbonés M, Delabar JM. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS One. 2009;4:e4606. doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj F, Hines D, Foley J, Haydon P, Bianchi D. Translating the transcriptome to develop antenatal treatments for fetuses with Down syndrome. Reproductive Sciences. 2013;20:64A. [Google Scholar]
- Guedj F, Pennings JL, Ferres MA, Graham LC, Wick HC, Miczek KA, Slonim DK, Bianchi DW. The fetal brain transcriptome and neonatal behavioral phenotype in the Ts1Cje mouse model of Down syndrome. Am J Med Genet A. 2015;167:1993–2008. doi: 10.1002/ajmg.a.37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Bonasoni P, Ceccarelli C, Santini D, Gualtieri F, Ciani E, Bartesaghi R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18:180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Ciani E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011;21:361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Stagni F, Bianchi P, Ciani E, Ragazzi E, Trazzi S, Grossi G, Mangano C, Calzà L, Bartesaghi R. Early pharmacotherapy with fluoxetine rescues dendritic pathology in the Ts65Dn mouse model of Down syndrome. Brain Pathol. 2013;23:129–143. doi: 10.1111/j.1750-3639.2012.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Stagni F, Bianchi P, Ciani E, Giacomini A, De Franceschi M, Moldrich R, Kurniawan N, Mardon K, Giuliani A, Calzà L, Bartesaghi R. Prenatal pharmacotherapy rescues brain development in a Down's syndrome mouse model. Brain. 2014;137:380–401. doi: 10.1093/brain/awt340. [DOI] [PubMed] [Google Scholar]
- Guihard-Costa AM, Khung S, Delbecque K, Ménez F, Delezoide AL. Biometry of face and brain in fetuses with trisomy 21. Pediatr Res. 2006;59:33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- Haas MA, Bell D, Slender A, Lana-Elola E, Watson-Scales S, Fisher EM, Tybulewicz VL, Guillemot F. Alterations to dendritic spine morphology, but not dendrite patterning, of cortical projection neurons in Tc1 and Ts1Rhr mouse models of Down syndrome. PLoS One. 2013;8:e78561. doi: 10.1371/journal.pone.0078561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Stahl S. The sigma-1 (delta-1) receptor and its role in the treament of mood disorders. Drugs of the Future. 2009;34:137. doi: 10.1358/dof.2009.034.02.1336115. [DOI] [Google Scholar]
- Haydar TF, Blue ME, Molliver ME, Krueger BK, Yarowsky PJ. Consequences of trisomy 16 for mouse brain development: corticogenesis in a model of Down syndrome. J Neurosci. 1996;16:6175–6182. doi: 10.1523/JNEUROSCI.16-19-06175.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Nowakowski RS, Yarowsky PJ, Krueger BK. Role of founder cell deficit and delayed neuronogenesis in microencephaly of the trisomy 16 mouse. J Neurosci. 2000;20:4156–4164. doi: 10.1523/JNEUROSCI.20-11-04156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, Busciglio J. ets-2 promotes the activation of a mitochondrial death pathway in Down's syndrome neurons. J Neurosci. 2005;25:2295–2303. doi: 10.1523/JNEUROSCI.5107-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK, Bilimoria PM. Re-opening windows: manipulating critical periods for brain development. Cerebrum. 2012;2012:11. [PMC free article] [PubMed] [Google Scholar]
- Hui L, Slonim DK, Wick HC, Johnson KL, Bianchi DW. The amniotic fluid transcriptome: a source of novel information about human fetal development. Obstet Gynecol. 2012a;119:111–118. doi: 10.1097/AOG.0b013e31823d4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Slonim DK, Wick HC, Johnson KL, Koide K, Bianchi DW. Novel neurodevelopmental information revealed in amniotic fluid supernatant transcripts from fetuses with trisomies 18 and 21. Hum Genet. 2012b;131:1751–1759. doi: 10.1007/s00439-012-1195-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann Neurol. 2004a;56:675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- Hunter CL, Bimonte-Nelson HA, Nelson M, Eckman CB, Granholm AC. Behavioral and neurobiological markers of Alzheimer's disease in Ts65Dn mice: effects of estrogen. Neurobiol Aging. 2004b;25:873–884. doi: 10.1016/j.neurobiolaging.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Incerti M, Horowitz K, Roberson R, Abebe D, Toso L, Caballero M, Spong CY. Prenatal treatment prevents learning deficit in Down syndrome model. PLoS One. 2012;7:e50724. doi: 10.1371/journal.pone.0050724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Amano K, Takaki E, Shimohata A, Sago H, Epstein CJ, Yamakawa K. Enlarged brain ventricles and impaired neurogenesis in the Ts1Cje and Ts2Cje mouse models of Down syndrome. Cereb Cortex. 2010;20:1131–1143. doi: 10.1093/cercor/bhp176. [DOI] [PubMed] [Google Scholar]
- Kelsey NA, Wilkins HM, Linseman DA. Nutraceutical antioxidants as novel neuroprotective agents. Molecules. 2010;15:7792–7814. doi: 10.3390/molecules15117792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Quon MJ, Kim JA. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014;2:187–195. doi: 10.1016/j.redox.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Faizi M, Jacobs LF, Htun K, Shamloo M, Mobley WC. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J Neurosci. 2012a;32:9217–9227. doi: 10.1523/JNEUROSCI.1673-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Gall J, George L, Nosheny R, Maloney MT, Salehi A, Mobley WC. Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2012b;45:683–691. doi: 10.1016/j.nbd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingberg T, Fernell E, Olesen PJ, Johnson M, Gustafsson P, Dahlström K, Gillberg CG, Forssberg H, Westerberg H. Computerized training of working memory in children with ADHD–a randomized, controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44:177–186. doi: 10.1097/00004583-200502000-00010. [DOI] [PubMed] [Google Scholar]
- Kurt MA, Davies DC, Kidd M, Dierssen M, Flórez J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 2000;858:191–197. doi: 10.1016/S0006-8993(00)01984-3. [DOI] [PubMed] [Google Scholar]
- Kurt MA, Kafa MI, Dierssen M, Davies DC. Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Res. 2004;1022:101–109. doi: 10.1016/j.brainres.2004.06.075. [DOI] [PubMed] [Google Scholar]
- Lamb J. The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer. 2007;7:54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- Larsen KB, Laursen H, Graem N, Samuelsen GB, Bogdanovic N, Pakkenberg B. Reduced cell number in the neocortical part of the human fetal brain in Down syndrome. Ann Anat. 2008;190:421–427. doi: 10.1016/j.aanat.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, Granholm AC. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp Neurol. 2009;216:278–289. doi: 10.1016/j.expneurol.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav Brain Res. 2011;221:610–622. doi: 10.1016/j.bbr.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–121. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Curr Alzheimer Res. 2006;3:521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- Lydic JS, Steele C. Assessment of the quality of sitting and gait patterns in children with Down's syndrome. Phys Ther. 1979;59:1489–1494. doi: 10.1093/ptj/59.12.1489. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome: a Golgi study. J Comp Neurol. 1976;167:63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- Martínez-Cué C, Martínez P, Rueda N, Vidal R, García S, Vidal V, Corrales A, Montero JA, Pazos Á, Flórez J, Gasser R, Thomas AW, Honer M, Knoflach F, Trejo JL, Wettstein JG, Hernández MC. Reducing GABAA alpha5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J Neurosci. 2013;33:3953–3966. doi: 10.1523/JNEUROSCI.1203-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, Varrone A, Farde L, Jucaite A, Bystritsky P, Forssberg H, Klingberg T. Changes in cortical dopamine D1 receptor binding associated with cognitive training. Science. 2009;323:800–802. doi: 10.1126/science.1166102. [DOI] [PubMed] [Google Scholar]
- Moon J, Chen M, Gandhy SU, Strawderman M, Levitsky DA, Maclean KN, Strupp BJ. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav Neurosci. 2010;124:346–361. doi: 10.1037/a0019590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami N, Bolton D, Hwang YW. Dyrk1A binds to multiple endocytic proteins required for formation of clathrin-coated vesicles. Biochemistry. 2009;48:9297–9305. doi: 10.1021/bi9010557. [DOI] [PubMed] [Google Scholar]
- Nahon E, Israelson A, Abu-Hamad S, Varda SB. Fluoxetine (Prozac) interaction with the mitochondrial voltage-dependent anion channel and protection against apoptotic cell death. FEBS Lett. 2005;579:5105–5110. doi: 10.1016/j.febslet.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Najas S, Arranz J, Lochhead PA, Ashford AL, Oxley D, Delabar JM, Cook SJ, Barallobre MJ, Arbonés ML. DYRK1A-mediated cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in down syndrome. EBioMedicine. 2015;2:120–134. doi: 10.1016/j.ebiom.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer WJ, Powell C, Nong Y, Blundell J, Wong L, Duff K, Flajolet M, Greengard P. Lowering beta-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS One. 2010;5:e10943. doi: 10.1371/journal.pone.0010943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema R, de Klein A, Verkerk AJ, Schot R, Dumee B, Douben H, Eussen B, Dubbel L, Poddighe PJ, van der Laar I, Dobyns WB, van der Spek PJ, Lequin MH, de Coo IF, de Wit MC, Wessels MW, Mancini GM. Distinctive phenotypic abnormalities associated with submicroscopic 21q22 deletion including DYRK1A. Mol Syndromol. 2010;1:113–120. doi: 10.1159/000320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, Reeves RH. Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Dev Dyn. 2004;230:581–589. doi: 10.1002/dvdy.20079. [DOI] [PubMed] [Google Scholar]
- Pérez-Cremades D, Hernández S, Blasco-Ibáñez JM, Crespo C, Nacher J, Varea E. Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down's syndrome. J Neural Transm. 2010;117:445–455. doi: 10.1007/s00702-010-0376-9. [DOI] [PubMed] [Google Scholar]
- Pinna G, Costa E, Guidotti A. SSRIs act as selective brain steroidogenic stimulants (SBSSs) at low doses that are inactive on 5-HT reuptake. Curr Opin Pharmacol. 2009;9:24–30. doi: 10.1016/j.coph.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons-Espinal M, Martínez de Lagran M, Dierssen M. Environmental enrichment rescues DYRK1A activity and hippocampal adult neurogenesis in TgDyrk1A. Neurobiol Dis. 2013;60:18–31. doi: 10.1016/j.nbd.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Powell D, Caban-Holt A, Jicha G, Robertson W, Davis R, Gold BT, Schmitt FA, Head E. Frontal white matter integrity in adults with Down syndrome with and without dementia. Neurobiol Aging. 2014;35:1562–1569. doi: 10.1016/j.neurobiolaging.2014.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani Z, Lopes C, Rachidi M, Delabar JM. Expression of the mnb (dyrk) protein in adult and embryonic mouse tissues. Biochem Biophys Res Commun. 1998;253:514–518. doi: 10.1006/bbrc.1998.9803. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–18. doi: 10.1038/nrn1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoldi C, Galli M, Mainardi L, Crivellini M, Albertini G. Postural control in children, teenagers and adults with Down syndrome. Res Dev Disabil. 2011;32:170–175. doi: 10.1016/j.ridd.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Risser D, Lubec G, Cairns N, Herrera-Marschitz M. Excitatory amino acids and monoamines in parahippocampal gyrus and frontal cortical pole of adults with Down syndrome. Life Sci. 1997;60:1231–1237. doi: 10.1016/S0024-3205(97)00067-2. [DOI] [PubMed] [Google Scholar]
- Ronan A, Fagan K, Christie L, Conroy J, Nowak NJ, Turner G. Familial 4.3 Mb duplication of 21q22 sheds new light on the Down syndrome critical region. J Med Genet. 2007;44:448–451. doi: 10.1136/jmg.2006.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, Baxter LL, Saràn NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci U S A. 2006;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda N, Flórez J, Martínez-Cué C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci Lett. 2008;433:22–27. doi: 10.1016/j.neulet.2007.12.039. [DOI] [PubMed] [Google Scholar]
- Rueda N, Llorens-Martín M, Flórez J, Valdizán E, Banerjee P, Trejo JL, Martínez-Cué C. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J Alzheimers Dis. 2010;21:277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, Kim IS, Cheon YH, Ahn YS, Chung SH, Song WJ. DYRK1A-mediated hyperphosphorylation of Tau: a functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282:34850–34857. doi: 10.1074/jbc.M707358200. [DOI] [PubMed] [Google Scholar]
- Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, Kim MJ, Son MY, Seo H, Chung SH, Song WJ. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. J Neurochem. 2008;104:1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Schmidt-Sidor B, Wisniewski KE, Shepard TH, Sersen EA. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin Neuropathol. 1990;9:181–190. [PubMed] [Google Scholar]
- Schroeter H, Bahia P, Spencer JP, Sheppard O, Rattray M, Cadenas E, Rice-Evans C, Williams RJ. (-)Epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J Neurochem. 2007;101:1596–1606. doi: 10.1111/j.1471-4159.2006.04434.x. [DOI] [PubMed] [Google Scholar]
- Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down's model mice. Exp Neurol. 2005;193:469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Shichiri M, Yoshida Y, Ishida N, Hagihara Y, Iwahashi H, Tamai H, Niki E. Alpha-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic Biol Med. 2011;50:1801–1811. doi: 10.1016/j.freeradbiomed.2011.03.023. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Ishizuka Y, Yamazaki H, Shirao T. Allopregnanolone increases mature excitatory synapses along dendrites via protein kinase A signaling. Neuroscience. 2015;305:139–145. doi: 10.1016/j.neuroscience.2015.07.079. [DOI] [PubMed] [Google Scholar]
- Shukkur EA, Shimohata A, Akagi T, Yu W, Yamaguchi M, Murayama M, Chui D, Takeuchi T, Amano K, Subramhanya KH, Hashikawa T, Sago H, Epstein CJ, Takashima A, Yamakawa K. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum Mol Genet. 2006;15:2752–2762. doi: 10.1093/hmg/ddl211. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z. Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology. 1997;36:1549–1554. doi: 10.1016/S0028-3908(97)00157-3. [DOI] [PubMed] [Google Scholar]
- Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38:1917–1920. doi: 10.1016/S0028-3908(99)00083-0. [DOI] [PubMed] [Google Scholar]
- Silva ML, Cieuta C, Guerrini R, Plouin P, Livet MO, Dulac O. Early clinical and EEG features of infantile spasms in Down syndrome. Epilepsia. 1996;37:977–982. doi: 10.1111/j.1528-1157.1996.tb00535.x. [DOI] [PubMed] [Google Scholar]
- Singer HS, Minzer K. Neurobiology of Tourette's syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev. 2003;25:S70–S84. doi: 10.1016/S0387-7604(03)90012-X. [DOI] [PubMed] [Google Scholar]
- Slonim DK, Koide K, Johnson KL, Tantravahi U, Cowan JM, Jarrah Z, Bianchi DW. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci U S A. 2009;106:9425–9429. doi: 10.1073/pnas.0903909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderqvist S, Matsson H, Peyrard-Janvid M, Kere J, Klingberg T. Polymorphisms in the dopamine receptor 2 gene region influence improvements during working memory training in children and adolescents. J Cogn Neurosci. 2014;26:54–62. doi: 10.1162/jocn_a_00478. [DOI] [PubMed] [Google Scholar]
- Souchet B, Guedj F, Sahún I, Duchon A, Daubigney F, Badel A, Yanagawa Y, Barallobre MJ, Dierssen M, Yu E, Herault Y, Arbonés M, Janel N, Créau N, Delabar JM. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol Dis. 2014;69:65–75. doi: 10.1016/j.nbd.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Spencer JP. The impact of flavonoids on memory: physiological and molecular considerations. Chem Soc Rev. 2009;38:1152–1161. doi: 10.1039/b800422f. [DOI] [PubMed] [Google Scholar]
- Stagni F, Magistretti J, Guidi S, Ciani E, Mangano C, Calzà L, Bartesaghi R. Pharmacotherapy with fluoxetine restores functional connectivity from the dentate gyrus to field CA3 in the Ts65Dn mouse model of Down syndrome. PLoS One. 2013;8:e61689. doi: 10.1371/journal.pone.0061689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagni F, Giacomini A, Guidi S, Ciani E, Ragazzi E, Filonzi M, De Iasio R, Rimondini R, Bartesaghi R. Long-term effects of neonatal treatment with fluoxetine on cognitive performance in Ts65Dn mice. Neurobiol Dis. 2015;74:204–218. doi: 10.1016/j.nbd.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Starbuck JM, Dutka T, Ratliff TS, Reeves RH, Richtsmeier JT. Overlapping trisomies for human chromosome 21 orthologs produce similar effects on skull and brain morphology of Dp(16)1Yey and Ts65Dn mice. Am J Med Genet A. 2014;164A:1981–1990. doi: 10.1002/ajmg.a.36594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J. The fundamentals of brain development: Integrating nature and nurture. Cambridge: MA, Harvard University; 2008. [Google Scholar]
- Suetsugu M, Mehraein P. Spine distribution along the apical dendrites of the pyramidal neurons in Down's syndrome: a quantitative Golgi study. Acta Neuropathol. 1980;50:207–210. doi: 10.1007/BF00688755. [DOI] [PubMed] [Google Scholar]
- Takashima S, Becker LE, Armstrong DL, Chan F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down's syndrome: a quantitative and qualitative Golgi study. Brain Res. 1981;225:1–21. doi: 10.1016/0006-8993(81)90314-0. [DOI] [PubMed] [Google Scholar]
- Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O. Minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- Toso L, Cameroni I, Roberson R, Abebe D, Bissell S, Spong CY. Prevention of developmental delays in a Down syndrome mouse model. Obstet Gynecol. 2008;112:1242–1251. doi: 10.1097/AOG.0b013e31818c91dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trazzi S, Fuchs C, Valli E, Perini G, Bartesaghi R, Ciani E. The amyloid precursor protein (APP) triplicated gene impairs neuronal precursor differentiation and neurite development through two different domains in the Ts65Dn mouse model for Down syndrome. J Biol Chem. 2013;288:20817–20829. doi: 10.1074/jbc.M113.451088. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tyler WA, Haydar TF. Multiplex genetic fate mapping reveals a novel route of neocortical neurogenesis, which is altered in the Ts65Dn mouse model of Down syndrome. J Neurosci. 2013;33:5106–5119. doi: 10.1523/JNEUROSCI.5380-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bon BW, Hoischen A, Hehir-Kwa J, de Brouwer AP, Ruivenkamp C, Gijsbers AC, Marcelis CL, de Leeuw N, Veltman JA, Brunner HG, de Vries BB. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin Genet. 2011;79:296–299. doi: 10.1111/j.1399-0004.2010.01544.x. [DOI] [PubMed] [Google Scholar]
- Varnum M, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez R, Ash JA, Powers BE, Kelley CM, Strawderman M, Luscher ZI, Ginsberg SD, Mufson EJ, Strupp BJ. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2013;58:92–101. doi: 10.1016/j.nbd.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ. Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12 Mamm Genome. 2005;16:79–90. doi: 10.1007/s00335-004-2428-7. [DOI] [PubMed] [Google Scholar]
- Wach C, Krause V, Moliadze V, Paulus W, Schnitzler A, Pollok B. Effects of 10 Hz and 20 Hz transcranial alternating current stimulation (tACS) on motor functions and motor cortical excitability. Behav Brain Res. 2013;241:1–6. doi: 10.1016/j.bbr.2012.11.038. [DOI] [PubMed] [Google Scholar]
- Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson R, Brinton RD. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:6498–6503. doi: 10.1073/pnas.1001422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhu JZ, Chang KT, Min KT. DSCR1 interacts with FMRP and is required for spine morphogenesis and local protein synthesis. EMBO J. 2012a;31:3655–3666. doi: 10.1038/emboj.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li M, Xu X, Song M, Tao H, Bai Y. Green tea epigallocatechin-3-gallate (EGCG) promotes neural progenitor cell proliferation and sonic hedgehog pathway activation during adult hippocampal neurogenesis. Mol Nutr Food Res. 2012b;56:1292–1303. doi: 10.1002/mnfr.201200035. [DOI] [PubMed] [Google Scholar]
- Wassef A, Baker J, Kochan LD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol. 2003;23:601–640. doi: 10.1097/01.jcp.0000095349.32154.a5. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Gong CX, Hwang YW. The role of DYRK1A in neurodegenerative diseases. Febs J. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzdoerfer R, Dierssen M, Fountoulakis M, Lubec G. Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J Neural Transm Suppl. 2001;61:59–70. doi: 10.1007/978-3-7091-6262-0_5. [DOI] [PubMed] [Google Scholar]
- Westerberg H, Klingberg T. Changes in cortical activity after training of working memory–a single-subject analysis. Physiol Behav. 2007;92:186–192. doi: 10.1016/j.physbeh.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res Bull. 2001;56:479–485. doi: 10.1016/S0361-9230(01)00615-3. [DOI] [PubMed] [Google Scholar]
- White NS, Alkire MT, Haier RJ. A voxel-based morphometric study of nondemented adults with Down Syndrome. Neuroimage. 2003;20:393–403. doi: 10.1016/S1053-8119(03)00273-8. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Griffin WS. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolvetang EW, Bradfield OM, Tymms M, Zavarsek S, Hatzistavrou T, Kola I, Hertzog PJ. The chromosome 21 transcription factor ETS2 transactivates the beta-APP promoter: implications for Down syndrome. Biochim Biophys Acta. 2003;1628:105–110. doi: 10.1016/S0167-4781(03)00121-0. [DOI] [PubMed] [Google Scholar]
- Wong DT, Horng JS, Bymaster FP, Hauser KL, Molloy BB. A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine. Life Sci. 1974;15:471–479. doi: 10.1016/0024-3205(74)90345-2. [DOI] [PubMed] [Google Scholar]
- Xie W, Adayev T, Zhu H, Wegiel J, Wieraszko A, Hwang YW. Activity-dependent phosphorylation of dynamin 1 at serine 857. Biochemistry. 2012;51:6786–6796. doi: 10.1021/bi2017798. [DOI] [PubMed] [Google Scholar]
- Zimmermann J, Krauthausen M, Hofer MJ, Heneka MT, Campbell IL, Müller M. CNS-targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PLoS One. 2013;8:e57307. doi: 10.1371/journal.pone.0057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwemer LM, Bianchi DW. The amniotic fluid transcriptome as a guide to understanding fetal disease. Cold Spring Harb Perspect Med. 2015;5 doi: 10.1101/cshperspect.a023101. pii:a023101. [DOI] [PMC free article] [PubMed] [Google Scholar]