

Abstract
Over the last decade, deep learning (DL) methods have been extremely successful and widely used to develop artificial intelligence (AI) in almost every domain, especially after it achieved its proud record on computational Go. Compared to traditional machine learning (ML) algorithms, DL methods still have a long way to go to achieve recognition in small molecular drug discovery and development. And there is still lots of work to do for the popularization and application of DL for research purpose, e.g., for small molecule drug research and development. In this review, we mainly discussed several most powerful and mainstream architectures, including the convolutional neural network (CNN), recurrent neural network (RNN), and deep auto-encoder networks (DAENs), for supervised learning and nonsupervised learning; summarized most of the representative applications in small molecule drug design; and briefly introduced how DL methods were used in those applications. The discussion for the pros and cons of DL methods as well as the main challenges we need to tackle were also emphasized.
Keywords: artificial intelligence, artificial neural networks, big data, deep learning, drug discovery
INTRODUCTION
In March 2016, AlphaGo knocked out Lee Sedol, one of the best Go players in the world, bringing artificial intelligence (AI) back into public attention overnight, spurring extensive interest (1). Compared to the Deep Blue, the chess-playing computer developed by IBM that beat the world champion for the first time back to the 1990s, AlphaGo integrated an advanced and innovative architecture called the convolutional neural network (CNN), which is one of the most successful implementations of the deep learning (DL) algorithms in neural networks (NNs) (2). Benefiting from the rise of big data analysis and the development of large-scale computing capabilities, especially the developing of graphics processing unit (GPU) computing (3), using deep learning architectures has emerged as the first attempted technologies to address AI challenges (4).
Deep learning is the rebranding of a traditional machine learning (ML) algorithm called artificial neural network (ANN), which is a network system consisting of connected artificial neurons in order to mimic the human central neural system (CNS) (5). In early times, ANNs were not ‘deep’ but ‘shallow’; these ANNs were composed of one input layer, one output layer, and one hidden layer in between (Fig. 1). The input layer received input data directly by putting a feature into each node. Then, each node in the hidden layer received a weighted linear combination as input from all the units in the input layer and then used an activation function to perform a nonlinear transformation. The output layer did similar work to the hidden layer. It received signals from the hidden layer and then used an activation function to produce an outcome. With a data stream following this process, those ANNs could be considered as feedforward neural networks (FNNs) (5). The optimizations of these “shallow” NNs systems were achieved through a process which first calculated the error between the output result and the actual value using the back propagation (BP) algorithm (6), and then modified the internal adjustable parameters (weights) to minimize the errors through gradient descent (7). The universal approximation theorem states that shallow NNs, with only one hidden layer containing a finite number of nodes, could approximate any continuous function (8). Models with such architectures may be susceptible to overfitting when the number of adjustable parameters, such as number of nodes with adjustable weight connections in the hidden layer increases. By careful training of shallow networks, especially when regularization is applied, overfitting can be minimized (9). Nevertheless, more hidden layers could be designed to recognize more abstract patterns from input data, with lower layers learning basic patterns and upper layers learning higher-level patterns. However, adding more hidden layers and nodes could greatly increase the computation task. And those multilayer NNs with many hidden layers may suffer from gradient vanishing problem (10), resulting in the difficulty of changing weights to optimize the model training. To overcome these situations, in the development of DL models, GPU acceleration is commonly applied (3) to tremendously improve the computing power. Meanwhile, the network architectures were modified to optimize the initialization and the updating of weights, and different transfer functions and regularization techniques were adopted to minimize overfitting (11). Examples of those architectures included deep belief network (DBN) (12), CNN, and recurrent neural network (RNN) (13). Moreover, in the era of big data, DL has a major advantage compared to other traditional shallow ML algorithms, such as linear regression, logistic regression (14), support vector machine (SVM) (15), naive Bayesian methods (16), and decision tree or random forest algorithm (17). Those algorithms are also considered to be shallow in their capability of learning compared to DL algorithms (18). Those traditional algorithms have difficulty in processing naturalistic data of raw forms, and therefore, hand-engineered features must be extracted to represent the input data, which is crucial but often intractable, and requires expertise in the specific area of input data. Deep learning algorithms, on the other hand, belong to the representation learning class, which has the capability of handling raw data and automatically extracting useful features as the representations needed for further detection or classification (7).
Fig. 1.
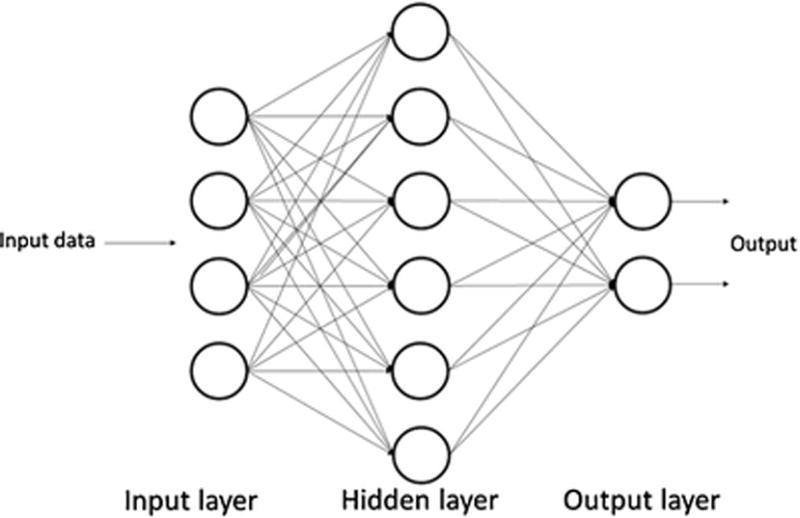
Architecture of artificial neural networks
In modern computer-aided small molecular drug discovery and development, ML methods, especially traditional learning methods, were widely used for building predictive models such as quantitative structure-activity relationship (QSAR) models, quantitative structure-property relationship (QSPR) models, and so on (17,19–22). In recent years, the new DL techniques have been adopted in drug discovery and development, opening a new door to computational decision making in pharmaceutical science (Fig. 2). The success of DL techniques benefits from the rapid development of the DL algorithms, the progress in high-performance computing technique, as well as the explosion of chemical information in chemical databases (Fig. 3) (23).
Fig. 2.
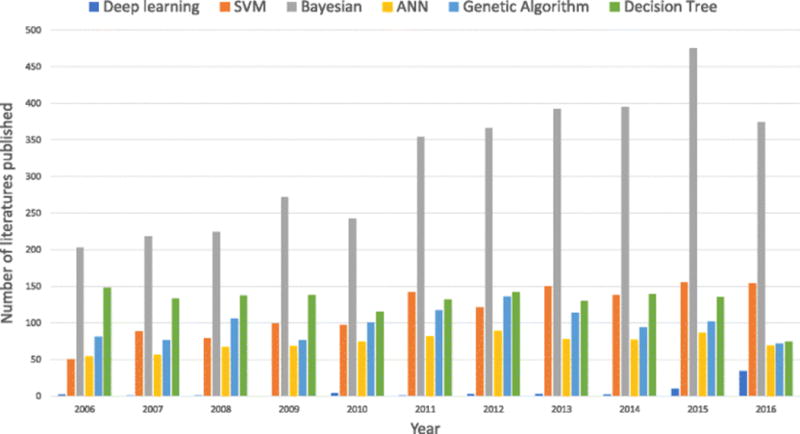
Comparison of number of publications using different machine learning methods in small molecule drug discovery in recent one decade (SVM: support vector machine, ANN: artificial neural network, Bayesian: Bayesian methods including naive Bayes classifier and Bayesian network)
Fig. 3.
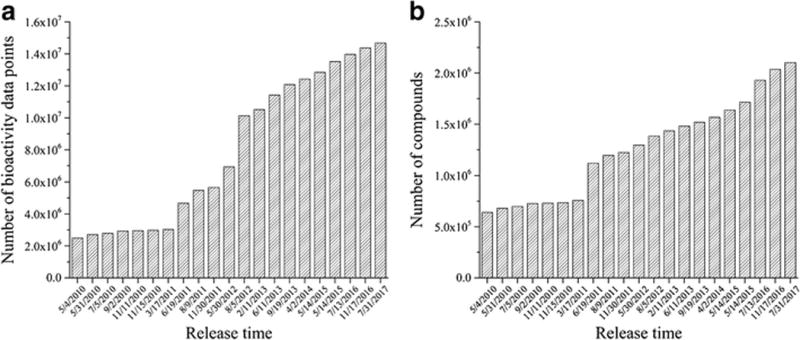
The explosive growth of published bioactivity data (a) and chemicals (b) for small molecule drug discovery in ChEMBL database (based on ChEMBL database releases from 2010 to 2017)
The purpose of our review is to help readers to gain an insight into DL applications in the field of computational chemistry and chemoinformatics, so that they can use DL in their research. As such, the article mainly summarized how those DL applications were built, what the architectures of the deep neural network (DNN) models were, and what input features they adopted. We also compared DL with traditional ML algorithms and discussed the future perspective of DL. Despite the importance of predictive performance of mathematical models, the validation and parallel comparisons for those DL applications were not emphasized in this review, for the following reasons. One is that accuracy and precision vary with datasets, model architectures, hyperparameter configurations, and evaluation methods. For example, many researches used coefficient of determination (R2) or (RMSE) to assess the performances of their models, while other researchers believed that the standard error of prediction (SEP) for a test set might be more reasonable (24). Secondly, there have been several review articles discussing the performance of DL models, as well as comparing them to models generated using traditional ML algorithms (4,18,25–31).
PRINCIPLE OF DEEP LEARNING
The Development of Deep Learning
The origin of DL can be traced back to the neural network (NN) model proposed by Warren McCulloch and Walter Pitts in the 1940s, and the invention of perceptron by Frank Rosenblatt (32), both of which were designed to mimic the excitation of neurons in the human brain by analogizing the activation of a binary logic gate in the NN. The main idea of the early ANN was to define an algorithm to learn the weight vector w, which was used as the coefficient of an eigenvalue. Then, an activation function inside the neuron, such as Heaviside Step Function or Sigmoid Function, was used to determine whether the neuron was activated or not (5,32). Later on, the development of the BP algorithm (33) for ANN modeling brought the boom of those statistics-based ML methods for supervised learning. The practical framework of DL was proposed by Geoffrey Hinton, Yann LeCun, and other scientists in 2006, opening the revolutionary waves of DL and new AI, not only in academia but also in industry (7). They developed a novel architecture for multilayer NNs to introduce feature learning into DL for abstracting the essentials of the data. Through feature learning, DL methods could automatically extract features from input data with raw format, then transform and distribute them into more abstract levels (7). Meanwhile, the rapid development in parallel computing techniques and computing hardware, especially the emerging application-specific integrated circuit designed for DL study, such as the tensor processing unit (TPU) technique (34), ensured that the tremendous computing workload of DNNs may no longer be an inaccessible domain (35).
Common Deep Learning Architectures Used in Small Molecular Drug Discovery
There are different types of DL architectures, each of which can recognize patterns and extract high-level features in distinct ways based on the structure of the training data. In this review, we mainly discussed the mainstream architectures, including the CNN, RNN, and the generative networks (7). We briefly introduced how they were used in DL applications for small molecule drug design and development.
Convolutional Neural Network
CNN is one of the most representative architectures in DL and is widely adopted in many fields such as image and voice recognition, as well as natural language processing (NLP). The modern CNN came from the development of the neocognitron by Fukushima in the 1980s, which was inspired by the research of receptive field in a cat’s visual cortex by Hubel and Wiesel (36,37). When processing visual signals, local neuron patterns take responsibility for perceiving particular regions in the sensory space (38) and CNN mimics its traits by developing two main characters in the convolutional layers: sparse connectivity and shared weights. In the convolutional layer k (Fig. 4a), there are two feature maps (A and B), either of which shares the same weight (wa or wb). Every pixel in each feature map of the hidden layer k comes from the convolution of weight matrix and the local pixel cluster of the layer k-1 (30).
Fig. 4.
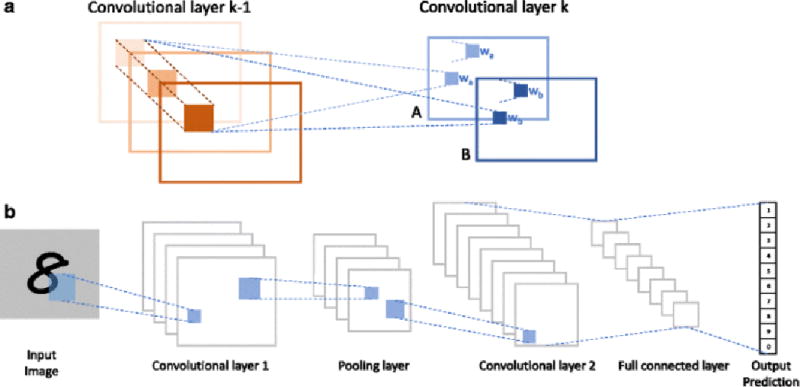
a Structure of convolutional layer. b Architecture of convolutional neural network (LeNet-5)
Furthermore, the increase of robustness achieved by pooling layers and the integration of dropout technique for regularization make the CNN even more sophisticated (7). For those complicated signaling processes, in which the input data have a gigantic number of input features and extremely abstract connections, the adoption of CNN could circumvent the headache of feature selection by directly importing the input data into the model. There are three types of layers commonly used in CNN: the convolutional layer, the pooling layer, and the full connection layer (Fig. 4b). Those layers were carefully selected and arranged to form the multilayer network (39,40). Depending on the input data modality, different forms of layers can be considered. For example, for sequence signals such as language, layers can be formed with 1D arrays; for images or audios, layers can be formed with 2D arrays; and for videos, layers formed with 3D arrays can be applied (7).
Recurrent Neural Network
RNN is another representative type of architecture in DL. Specially aiming for handling sequence data, RNN has been widely used and achieved great success in NLP. RNN is different from regular FNNs which follow the feedforward architecture. In regular FNNs, there is no connection between hidden nodes in the same layer, but only between nodes in adjacent layers (Fig. 1). One of the major shortages of FNNs is that they cannot handle sequence problems, because the output is related to not only the current input information, but also prior information, for example machine translation. However, RNN can process sequential information by (1) introducing directed cycles into its network; (2) affiliating the adjacent hidden nodes with each other; (3) capturing the calculated information from preceding time slices; and (4) storing it for the subsequent procedure (7,13) (Fig. 5). The one-way data flow stream from input units to output units, going through each sequential hidden unit. St represents the transition states of the step t, which stands for the memorial units in the network containing all the extracted information from the prior data in the sequence. The output from the output units in that step (t) is only correlated with the transition state at that moment (St). In the RNN, each hidden layer with directed cycles could be unfolded and processed as a traditional NN sharing the same weight matrices U, V, W in every same layer.
Fig. 5.
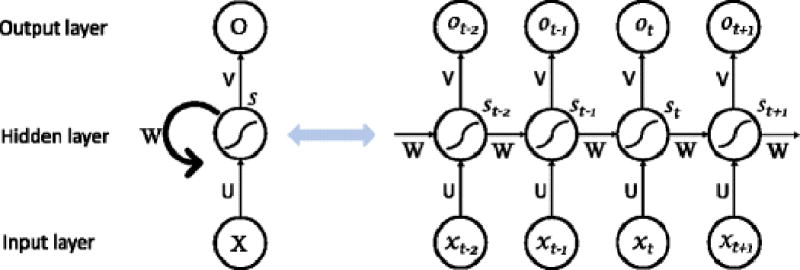
Framework of basic recurrent neural network. Recurrent neural network consists of input units (x, the vector representing the matrix of input data) and hidden units (s, the vector representing the matrix in the hidden layer), and output units (o, the vector representing the matrix of output data). U, V, and W are the weight matrixes for the transition from x to s, s to s, and s to o, respectively
There are plenty of variations of RNNs. The most common ones are gated recurrent unit recurrent neural network (GRURNN) (41), long short-term memory (LSTM) network (42), and clockwork RNN (CW-RNN) (43). Among those RNN architectures, LSTM is currently the most popular and widely used one in NLP. In NLP, LSTM is often combined with distributed representation of word embedding, which is achieved by checking the statements and part-of-speech tagging (7,35). Using a specialized function to compute the transition state in the hidden layer, the LSTM network is powerful when capturing long-term dependencies compared to regular RNNs. In addition, LSTM is also as popular and successful as CNN in the image retrieval domain and is usually combined with CNNs for the automatic generation of image description in AI (7).
Generative Deep Neural Network
DNNs are not only for processing labeled data in supervised learning, but also for analyzing nonlabeled data in unsupervised learning. Deep auto-encoder network (DEAN) is one of the most common generative network architectures for unsupervised learning (25,30). DEAN consists of an encoder and a decoder, which are two symmetric DBNs, a DNN proposed by Hilton et al. in 2006 (12). Those two DBNs are usually composed of several restricted Boltzmann machines (RBMs) (44), a bipartite network that contains one visible layer and one invisible layer. In RBM, there are symmetric connections between every two nodes from different layers, and no connection between nodes from the same layer. The function of a simple auto-encoder can be regarded as the compression of data which can then be decompressed and recovered based on a BP algorithm with a minimal loss of information (33). Thus, DAEN is also considered as the method for dimensionality reduction because of its capacity of reducing the redundancy. In this case, DAEN can be used specifically for feature extraction, in order that the reduced features can be used to train a classification model using supervised learning algorithms (45). This paradigm may be valuable in the future development of DL applications.
More recently, generative adversarial networks (GANs), another type of DL algorithms for unsupervised learning, have been developed and widely used in the image synthesis, image-to-image translation, and super-resolution (46). It was motivated by the underlying probability density or probability mass function of observation data. Generator (G) is responsible for making nonrealistic images from random vectors to confuse the other network which known as discriminator (D). When D receives both forgeries and real (authentic) images, it will tell them apart. In that module, G and D compete with each other and are trained simultaneously until both of them find the optimal parameters. Under those parameters, the G maximizes its classification accuracy and D maximizes its discrimination accuracy. The networks can be implemented by multilayer networks consisting of fully connected GANs, convolutional GANs, conditional GANs, GANs with inference models, and adversarial auto-encoder (AAE).
Regularization and Dropout
Since over-fitting is a serious problem in multilayer DNNs, a broad range of techniques for regularizing have been developed to minimize the over-fitting problem. Dropout is one of the common ways to regularize NNs by dropping out units (hidden and visible) in NNs (47). The key idea of dropout is to add noise to its hidden units randomly; therefore, preventing over-fitting and improving test performance. Those DNNs which adopt dropout techniques can be trained through stochastic gradient descent (SGD) apparently like regular DNNs. Similarly, each hidden unit in an NN adopted dropout must learn to work with a randomly chosen sample of other units, which makes them more robust rather than relying on other hidden units to correct its mistakes.
Bayesian regularized artificial neural network (BRANN) is another development that introduced regularization into NN architecture. By using ridge regression in the mathematical process of model training, nonlinear regression can be converted into a “well-posed” statistical problem in the BRANN (48). By using BRANN, the cross-validation step for assessing the model, which is usually tedious and time-consuming in DL modeling, may also be omitted. Automatic relevance determination (ARD) of the input features can be applied in BRANN to help calculate several effective network parameters or weights, which will cause the removal of parameters with smaller weights. In such way, those indices which are irrelevant or highly correlated are neglected, and variables which are the most important for modeling are highlighted. Those two characteristics are very beneficial for chemoinformatics and QSAR/QSPR researches, because there are usually too many features to describe one molecule.
RESOURCES USED FOR DEVELOPING DEEP LEARNING APPLICATION
With the rapid development of the DL technique, many open source packages and libraries for developing DL framework are available for individual developers and small groups to explore the DL—they may not need to develop their own DL platform. Most of these packages have well-established built-in codes for GPU computing with detailed tutorials and annotations. We have listed and briefly summarized the representative packages with their link on Table I.
Table I.
Summary of Current Deep Learning Packages
| Package Name | Platform/API | Resources |
|---|---|---|
| TensorFlow | Python | https://www.tensorflow.org/ |
|
|
||
| Torch | Lua | http://torch.ch/ |
|
|
||
| Theano | Python | http://deeplearning.net/software/theano/ |
|
|
||
| Caffe | C++/Python | http://caffe.berkeleyvision.org/ |
|
|
||
| DL4J | Java | https://github.com/deeplearning4j/deeplearning4j |
|
|
||
| Paddle | Python | http://paddlepaddle.org/ |
|
|
||
| Keras | Python | https://keras.io/ |
|
|
||
| CNTK | C++/Python | https://www.microsoft.com/en-us/cognitive-toolkit/ |
|
|
||
| MxNet | R/Python/Julia | http://mxnet.io/ |
|
|
||
| AlexNet | MATLAB | https://www.mathworks.com/products/matlab.html |
|
|
||
| PyTorch | Python | http://pytorch.org/ |
|
|
||
| DeepChem | Python | https://deepchem.io/ |
In addition to the packages and tools, the dataset, especially the benchmark dataset, is another essential part of constructing a model. The development of DL benefited from the breakthrough of CNN in computer vision, which was mainly facilitated by the benchmark dataset ImageNet and the annual competition ImageNet Large Scale Visual Recognition Challenge (ILSVRC) (49). In the drug discovery field, the Merck Kaggle challenge using a Merck-activity dataset, as well as the Tox21 challenge using its benchmark datasets greatly speeded up the application of ML methods in the QSAR/QSPR studies (50,51). Compared to traditional ML methods, DL methods have the capacity of processing “big data.” Therefore, the need for large standardized datasets for DL modeling is dire. Recently, Wu et al. introduced their large-scale benchmark package, MoleculeNet, for molecular ML study (52). The MoleculeNet dataset integrated multiple public molecular datasets, covering quantum mechanics data, physical chemistry data, biophysics data, and physiology data. In addition, all the datasets, established metrics for model evaluation, and implementations for calculated molecular features were packaged together with the DL modeling toolkits in their python library called DeepChem. Besides, Lenselink et al. published their benchmark bioactivity dataset generated from ChEMBL database (53), which could be another choice of a standardized dataset for developing DL models (54).
APPLICATIONS USING DEEP LEARNING IN SMALL MOLECULE DRUG DESIGN
DL models have been reported in three major areas in computational chemistry—predicting the drug-target interactions (DTIs), generating novel molecules, and predicting absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties for translational researches (55). Like other ML algorithms, DL undergoes more and more successful applications in building QSAR/QSPR models (Table II). As early as 2012, Hilton’s group won the Merck Kaggle challenge (https://www.kaggle.com/c/MerckActivity) using their DL models, opening a new chapter of applications using DL methods on predicting chemical compound activity and property. Similarly, Wang and Zeng published their DTI-discriminative model using RBM, the commonly recognized first generation of DNNs (56). In the following year, Dahl et al. from Hilton’s group and Google Inc. published several papers on DL-based QSAR modeling. They tried multiple tasks and different features using DNNs with various hyper-parameters and started to use GPUs for a benchmark test (57–59). In 2014, Wang et al. reported their DIT-predictive model using pairwise-input NNs, offering a new reasonable idea of adding target information into the model (60). To mimic the interactions between compounds and proteins, separated groups of weights were assigned to the compound features and protein features, and then fed into the first hidden layer, respectively. In 2015, Wallach et al. introduced their DL models, AtomNet, to predict binding affinity for selecting active compounds for drug discovery (61). AtomNet was claimed to be the first DL model adopting CNN for small molecular binding affinity prediction. In AtomNet, a novel approach to combine both ligand and target structure information was used. However, AtomNet required the 3D structures for both ligand and target protein containing the location of each atom involved in the interaction at the binding site of the target. Recently, Wan and Zeng published their new model for compound-protein interaction prediction using DL methods, in which they adopted a widely used technique in NLP studies called feature embedding (62). In their model, both the ligand information (molecular fingerprints) (63) and protein sequence were embedded into multidimensional vectors. Following the embedding process, a sequence of fully connected layers which consisted of rectified linear units (ReLUs) was constructed (64).
Table II.
Summary of recent representative applications of DL in small molecular drug discovery
| Method | Prediction | Dataset | Features | Reference |
|---|---|---|---|---|
| RNN | Compound aqueous solubility | Multiple dataset with size of 1144, 1026, 74, 125 | Molecular graph | Lusci et al. (65) |
| DNN | Drug target interactions | 2710 ligands & 836 targets | Molecular descriptors and protein features | Wang et al. (60) |
| DNN | Drug target interactions | MATADOR dataset and STITCH dataset | Molecular descriptors | Wang and Zeng (56) |
| DNN | permeability | 663 + 209 chemical compounds | Molecular descriptors | Shin et al. (66) |
| DNN | Toxicity | Tox21 dataset | Molecular fingerprints and descriptors | Unterthiner et al. (67) |
| RNN | Compound protein interaction | ChEMBL database; BindingDB | Molecular fingerprints and protein sequence | Wang et al. (62) |
| CNN | Biological Activity | ChEMBL database, DUDE dataset | Molecular Graph (AtomNet) | Wallach et al. (61) |
| CNN | Virtual Screening | DUDE dataset | Molecular Graph and docking result | Pereira et al. (68) |
| Auto-Encoder | Virtual Screening | 6252 | Fingerprints | Kadurin et al. (75) |
| CNN | Biological Activity/Toxicity | 41193/8014 | 2D chemical structure image | Goh et al. (73, 74) |
| RNN | Biological Activity | 756 | SMILES | Bjerrum (71) |
| DNN | Biological Activity | ChEMBL database | Molecular descriptors and fingerprints | Lenselink et al. (54) |
| RNN | Generating focused molecular libraries | ChEMBL database | SMILES | Segler et al. (77) |
| RNN | Generating novel molecules | ChEMBL database | SMILES | Olivecrona et al. (78) |
| Gan | Generating novel molecules | Multiple datasets | SMILES | Guimaraes et al. (79) |
| CNN | the Kohn-Sham kinetic energy | 3D electron density | Fingerprints | Yao and Parkhill (70) |
Besides predicting target selectivity and DTIs, DL methods have been adopted to predict ADMET properties. In 2013, Lusci et al. reported their model for predicting aqueous solubility using DL architecture (65). They segmented small molecules into atoms and bonds to build a digraph by sequencing those atoms and linking them using their corresponding bonds, and then put the contracted graph into an RNN model. In 2015, Shin et al. published their model developed using DL method to predict the absorption potential of small molecules (66). In vitro permeability data of 663 small molecules from the human colorectal carcinoma cell line (Caco-2) were used as training data and 209 molecular descriptors were calculated using CDK toolkits based on their 2D structures (http://www.rguha.net/code/java/cdkdesc.html). Without using any specialized architecture, four layers of fully connected neural networks were generated to extract and transform the input information and finally classify the absorption potential of the input compound. DL methods were also effective in predicting the toxicity of small molecules in the Tox21 Data Challenge launched by the NIH, EPA, and FDA. Mayr and colleagues reported their DL-based models for toxicity prediction in 2015 (67). Multiple types of molecular features, such as different fingerprints and chemical properties, were tested and compared in their study. Forty thousand input features and a huge number of hidden layers were adopted in their models. The average performance of their DL-based models was good in multitask testing, showing that overall the DL algorithm was quite robust regarding training data, parameters, and tasks. Recently, Pereira et al. proposed their DL-based protocol for docking-based virtual screening (68). In their model, they used both ligand information and the interactive amino acids from docking to optimize the docking results. The input data were the distributed representation (69) of the compound-protein complexes generated using embedding technique, followed by a three-layer convolutional neural network.
A lot of the earlier DL attempts in the drug discovery field had been using human-engineered features like molecular descriptors and fingerprints. In such cases, the characteristic of DL as representation learning, which allows DL to automatically engineer molecular features directly from data, is largely missing. Yet, that is possibly the most important aspect that distinguishes DNNs from traditional ML algorithms. It is nice to see that more recent publications have demonstrated that learning directly on “unprocessed” chemical data may also be a viable strategy. A work using “unprocessed” chemical data on convolutional neural networks was published by Yao and Parkhill (70). Notably, they used the electron density from the 3D small molecules, rather than 2D molecular fingerprints or physical chemical properties, as the input data and developed a 3D convolutional neural network model to predict the Kohn-Sham kinetic energy of hydrocarbons. Bjerrum reported his study on generating a DL model using LSTM-cell-based NN (71). The innovative part of his research was that he used SMILES (72) enumeration, a single-line text uniquely representing one molecule, as the raw input data in the model. Another research from Goh et al. tried to use 2D molecule drawing images of molecules as the input data of a CNN model to predict chemical properties (73,74). They also compared their method to a CNN model using conventional molecular features as the input features, giving the result that the model constructed using their image-based input features slightly outperformed conventional molecular features.
More recently, with the development of unsupervised learning and generative NNs, the application of those generative models using DL algorithms has seen progress. Kadurin et al. developed a seven-layer generative AAE model for screening compounds (75). Different from a regular screening method using the QSAR model, their model extracted features from the input molecular fingerprints of 6252 training molecules and generated new fingerprint vectors for potential selective compounds using a nonsupervised generative model. Then, they screened those selected output vectors against a large library of 72 million compounds from PubChem (76) and predicted 320 compounds as potential compounds, in which 69 were identified as true hits experimentally. Besides selecting novel compounds using auto-encoders, there were several attempts generating novel compounds using other deep generative networks. Segler et al. introduced their generative models for designing novel focused library using RNNs, achieving a satisfied performance to complete the de novo drug design cycle (77). Similar methods were developed for the de novo library design by Olivecrona et al., with the novelty of adding reinforcement learning (RL) (35) into the method (78). Guimaraes et al. adopted GANs, as well as RL to construct a generative model for generating different types of molecules using their SMILES data, giving a novel idea of design novel compounds using state-of-the-art unsupervised DL methods (79).
DISCUSSION AND FUTURE PROSPECTIVE
Deep Learning Versus Traditional Machine Learning
As the state-of-the-art ML algorithms, DL algorithms have been challenged by comparing to other shallow ML algorithms (18). Winkler et al. recently reported their comparison between their Bayesian regularized neural network (BNN) models and the DL models generated by Ma et al. using the same KAGGLE dataset from Merck (57). They showed that shallow NNs with one single hidden layer could perform as well as DNNs with more hidden layers, given sufficient training data in QSAR or QSPR modelling (11). A similar conclusion was generated from Capuzzi et al. from the comparison using Tox21 data (80). It appears that those results were consistent with the universal approximation theorem (8), inferring that DL algorithms may not have superiority over regular shallow NNs. Those results may overturn our preconception that novel DL should be better than traditional shallow ML methods. In fact, for supervised learning with the final purpose of classification or regression, both DL and shallow learning have their own places (11,22).
Schmidhuber et al. suggested that the primary deficiency of most traditional ML methods is that they have a limited ability to simulate a complicated approximation function and generalize to an unseen instance (35). NNs have advances in QSAR/QSPR modeling (4), and the universal approximation theorem proves its advanced capacity on approximation. Shallow NNs can generalize to new data very well in most cases, given sufficient diverse data. Given the same descriptors and training data, both types of NN generate similar quality models. However, deep NNs can generate complex abstractions of the descriptors. As mentioned, the essential features of DL methods that distinguish them from shallow NNs are not only the emphasis on the depth of the network, but also the emphasis on feature learning. Compared to the shallow NNs that need to “manually” select the features, DL methods can learn features from data by constructing nonlinear network models to extract latent information of the big data. In the early QSAR/QSPR studies, descriptors were designed manually, which did not capture all the features impacting the QSAR/QSPR response surface (11). As a result, a tiny change in the values of those descriptors could lead to a significant change in the activity. Such phenomena are called activity cliff (81), which is a very common concern in QSAR modeling. The presence of activity cliffs is also highly correlated with the distribution of the activity responding surface used for training the model, which is referring to not only small molecular feature learning, but also protein target feature extraction. Researches have been done to show that the addition of protein features makes the DL model perform better (31,54). From the aspect of DL modeling, both the choice of different DL architectures and the configuration of hyper-parameters are very important for achieving good performance.
Besides, other differences between DL methods and traditional shallow ML methods were explored by other researchers. Lenselink et al. found that DL methods and traditional shallow ML methods performed similarly on randomly split data; however, they had significant differences when the data were split by congeneric chemical series (such as by the nature of publishing) (54). They thought that compounds published together were usually very similar in chemical structure and splitting in such a way could make the validation more in line with the experiments performed.
The Limitation of Deep Learning and Future Perspective
Because of the advance of feature learning, DL can reach a high accuracy of identification under the premise that the training set should contain a tremendous amount of data. With very limited data, the DL techniques cannot achieve an unbiased estimate of the generalization so that they may not be as practical as some traditional shallow ML methods (11,35). Also, with the rapid increase of time complexity because of the complication of the network architecture, stronger hardware facilities and advanced programming skills are required to grant the feasibility and effectiveness of DL methods. In addition, although DL methods usually have outstanding performance in practice, the tuning of the hyper-parameters in DL modeling is often tricky. Also, it is hard to know how many hidden layers and nodes could be enough to establish the best simulation without redundancy for a specific DL modeling. Finally, the strategy for unsupervised learning in DL is inspiring but still falling far behind (35). In the real-world application, especially in drug discovery, most of the data are nonlabeled data, with plenty of information contained. Exploring and developing novel unsupervised learning methods using DL methods, as well as mining useful information from those data are still difficult.
Although DL methods have been successfully applied in many areas, the adaptation of the algorithms is still a problem for the chemistry-centric modeling in small molecule drug discovery, especially for RNNs and CNNs, which are powerful but have higher restrictions on the format of input data. On the other hand, DL systems are considered as a “black box” systems; thus, they are hard for interpretation and have limited power to engage in logical reasoning. Those factors limit the application and approbation of DL in many domains such as clinical data analysis. In such cases, the interpretation of a structure-activity relationship (SAR) study is more practical from the descriptor perspective. However, regular features commonly used by the traditional ML models in current chemoinformatics studies to describe the small molecules, such as molecular fingerprints (21,63,82,83), physicochemical properties, topological properties, and thermodynamics properties (70), are not fully appropriate to be used in DL architecture (84). Thus, the development of more interpretable descriptors is dire. Specifically, since DL methods belong to representation learning thT can automatically abstract features from raw data, there are two very important problems to DL modeling: (1) how to optimize DL architectures to abstract useful features and (2) how to interpret those features. As discussed above, several recent studies started to use chemical data in a raw format to construct their DL models, indicating that conventional feature engineering may no longer be necessary for chemistry.
Beyond that, compared to the amount of big data for training the DL models such as the AlphaGo, the size of chemoinformatics databases for DL modeling is far behind. In spite of the size of the major database, like ChEMBL, which has reached the magnitude of a million, the actual available data for building a specific model is still limited (53). An increasing number of researchers are changing their strategies from chemistry-centric modeling to combined methods, which not only consider the chemical features of the small molecules, but also include target protein information, as well as other types of data, such as the DTI network (59,85,86).
Overall, small molecule drug discovery will become more and more complex. Designed for intricate simulation, DL should have the capability to handle that complexity. Also, with DL methods, we should not restrict ourselves in the traditional predictions on biological activities, ADMET properties, or pharmacokinetic simulations, but it may also be possible to integrate all the data and information systematically and achieve a new level of AI in drug discovery.
Acknowledgments
The authors thank Dr. Yuanqiang Wang, Nan Wu, and Yubin Ge in the CCGS Center at the University of Pittsburgh (Pitt) for carefully reviewing the manuscripts and providing helpful comments for revision. Thanks to all the students and faculty in the CDAR Center, School of Pharmacy at Pitt for their help and supports. The authors also acknowledge the funding support to our laboratory from NIH NIDA (P30DA035778) and DOD (W81XWH-16-1-0490).
References
- 1.Artificial intelligence: Google’s AlphaGo beats Go master Lee Se-dol. Technology BBC NEWS. 2016 Mar 12; http://www.bbc.com/news/technology-35785875#. Accessed 15 Dec 2017.
- 2.Silver D, Huang A, Maddison CJ, Guez A, Sifre L, van den Driessche G, et al. Mastering the game of Go with deep neural networks and tree search. Nature. 2016;529(7587):484–9. doi: 10.1038/nature16961. [DOI] [PubMed] [Google Scholar]
- 3.Ma C, Wang L, Xie X-Q. GPU accelerated chemical similarity calculation for compound library comparison. J Chem Inf Model. 2011;51(7):1521–7. doi: 10.1021/ci1004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskin II, Winkler D, Tetko IV. A renaissance of neural networks in drug discovery. Expert Opin Drug Discov. 2016;11(8):785–95. doi: 10.1080/17460441.2016.1201262. [DOI] [PubMed] [Google Scholar]
- 5.McCulloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys. 1943;5(4):115–33. [PubMed] [Google Scholar]
- 6.Rumelhart DE, Hinton GE, Williams RJ. Learning representations by back-propagating errors. Nature. 1986;323:533–6. [Google Scholar]
- 7.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–44. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 8.Gao B, Xu Y. Univariant approximation by superpositions of a sigmoidal function. J Math Anal Appl. 1993;178(1):221–6. [Google Scholar]
- 9.Lawrence S, Giles CL. Overfitting and neural networks: conjugate gradient and backpropagation. Neural Networks, 2000. IJCNN 2000, Proceedings of the IEEE-INNS-ENNS International Joint Conference; Como, Italy. 2000. pp. 114–19. [Google Scholar]
- 10.Hochreiter S. The vanishing gradient problem during learning recurrent neural nets and problem solutions. Int J Uncertainty Fuzziness Knowledge Based Syst. 1998;6(2):107–16. [Google Scholar]
- 11.Winkler DA, Le TC. Performance of deep and shallow neural networks, the universal approximation theorem, activity cliffs, and QSAR. Mol Inf. 2017;36(1–2):1600118. doi: 10.1002/minf.201600118. [DOI] [PubMed] [Google Scholar]
- 12.Hinton GE, Osindero S, Teh YW. A fast learning algorithm for deep belief nets. Neural Comput. 2006;18:1527–54. doi: 10.1162/neco.2006.18.7.1527. [DOI] [PubMed] [Google Scholar]
- 13.Olurotimi O. Recurrent neural network training with feedforward complexity. IEEE Trans Neural Netw. 1994;5(2):185–97. doi: 10.1109/72.279184. [DOI] [PubMed] [Google Scholar]
- 14.Cox DR. The regression-analysis of binary sequences. J R Stat Soc Ser B Stat Methodol. 1958;20(2):215–42. [Google Scholar]
- 15.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20(3):273–97. [Google Scholar]
- 16.Domingos P, Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Mach Learn. 1997;29(2–3):103–30. [Google Scholar]
- 17.Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP. Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Comput Sci. 2003;43:1947–58. doi: 10.1021/ci034160g. [DOI] [PubMed] [Google Scholar]
- 18.Goh GB, Hodas NO, Vishnu A. Deep learning for computational chemistry. J Comput Chem. 2017;38(16):1291–307. doi: 10.1002/jcc.24764. [DOI] [PubMed] [Google Scholar]
- 19.Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inf. 2010;29(6–7):476–88. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 20.Chen B, Sheridan RP, Hornak V, Voigt JH. Comparison of random forest and Pipeline Pilot naïve Bayes in prospective QSAR predictions. J Chem Inf Model. 2012;52:792–803. doi: 10.1021/ci200615h. [DOI] [PubMed] [Google Scholar]
- 21.Myint KZ, Xie X-Q. Ligand biological activity predictions using fingerprint-based artificial neural networks (FANN-QSAR) Methods Mol Biol (Clifton, NJ) 2015;1260:149–64. doi: 10.1007/978-1-4939-2239-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma C, Wang L, Yang P, Myint KZ, Xie XQ. LiCABEDS II. Modeling of ligand selectivity for G-protein coupled cannabinoid receptors. J Chem Inf Model. 2013;53(1):11–26. doi: 10.1021/ci3003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015;43(D1):D1079. doi: 10.1093/nar/gku1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander DL, Tropsha A, Winkler DA. Beware of R(2): simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J Chem Inf Model. 2015;55(7):1316–22. doi: 10.1021/acs.jcim.5b00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bengio Y. Learning deep architectures for AI. Found Trends® Mach Learn. 2009;2(1):1–127. [Google Scholar]
- 26.Ekins S. The next era: deep learning in pharmaceutical research. Pharm Res. 2016;33(11):2594–603. doi: 10.1007/s11095-016-2029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gawehn E, Hiss JA, Schneider G. Deep learning in drug discovery. Mol Inf. 2016;35(1):3–14. doi: 10.1002/minf.201501008. [DOI] [PubMed] [Google Scholar]
- 28.Hamet P, Tremblay J. Artificial intelligence in medicine. Metabolism. 2017;69(Supplement):S36–40. doi: 10.1016/j.metabol.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 29.Mamoshina P, Vieira A, Putin E, Zhavoronkov A. Applications of deep learning in biomedicine. Mol Pharm. 2016;13(5):1445–54. doi: 10.1021/acs.molpharmaceut.5b00982. [DOI] [PubMed] [Google Scholar]
- 30.Pastur-Romay AL, et al. Deep artificial neural networks and neuromorphic chips for big data analysis: pharmaceutical and bioinformatics applications. Int J Mol Sci. 2016;17(8):E1313. doi: 10.3390/ijms17081313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Westen GJP, Wegner JK, IJzerman AP, van Vlijmen HWT, Bender A. Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. Med Chem Comm. 2011;2(1):16–30. [Google Scholar]
- 32.Rosenblatt F. The perceptron, a perceiving and recognizing automaton project para. Buffalo: Cornell Aeronautical Laboratory; 1957;85:460–61. [Google Scholar]
- 33.Kelley HJ. Gradient theory of optimal flight paths. Ars J. 1960;30(10):947–54. [Google Scholar]
- 34.Google supercharges machine learning tasks with TPU custom chip. 2016 [cited 2017 May 20th]. [Google Scholar]
- 35.Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw. 2015;61:85–117. doi: 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Hubel DH, Wiesel TN. Receptive fields, binocular interaction and functional architecture in the cat’s visual cortex. J Physiol. 1962;160(1):106–54. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubel DH, Wiesel TN. Receptive fields of single neurones in the cat’s striate cortex. J Physiol. 1959;148(3):574–91. doi: 10.1113/jphysiol.1959.sp006308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeiler MD, Fergus R. European conference on computer vision 2014 Sep 6. Cham: Springer; 2014. Visualizing and understanding convolutional networks; pp. 818–33. [Google Scholar]
- 39.Lecun Y, Jackel LD, Bottou L, Brunot A, Cortes C, Denker JS, et al. Comparison of learning algorithms for handwritten digit recognition. In: Fogelman F, Gallinari P, editors. International conference on artificial neural networks. Paris: EC2 & Cie; 1995. pp. 53–60. [Google Scholar]
- 40.LeCun Y, et al. Gradient-based learning applied to document recognition. Proc IEEE. 1998;86(11):2278–324. [Google Scholar]
- 41.Cho K, Van Merriënboer B, Gulcehre C, Bahdanau D, Bougares F, Schwenk H, Bengio Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. Conference on empirical methods in natural language processing, Doha, Qatar. 2014;1:1724–34. [Google Scholar]
- 42.Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput. 1997;9(8):1735–80. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 43.Si S, Hsieh C, Dhillon I. Proceedings of the 31st international conference on machine learning. 2014 [Google Scholar]
- 44.Hinton GE, Salakhutdinov RR. Reducing the dimensionality of data with neural networks. Science. 2006;313(5786):504–7. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Lin Z, Zhao X, Wang G, Gu Y. Deep learning-based classification of hyperspectral data. IEEE J Sel Topics Appl Earth Observ Remote Sens. 2014;7(6):2094–107. [Google Scholar]
- 46.Goodfellow I, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, Bengio Y. Generative adversarial nets. Advances in neural information processing systems. 2014:2672–80. [Google Scholar]
- 47.Srivastava N, et al. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15:1929. [Google Scholar]
- 48.Burden F, Winkler D. Bayesian regularization of neural networks. Methods Mol Biol. 2008;458:25–44. doi: 10.1007/978-1-60327-101-1_3. [DOI] [PubMed] [Google Scholar]
- 49.Russakovsky O, Deng J, Su H, Krause J, Satheesh S, Ma S, et al. ImageNet large scale visual recognition challenge. Int J Comput Vis. 2015;115(3):211–52. [Google Scholar]
- 50.Unterthiner T, Mayr A, Klambauer G, Steijaert M, Wegner JK, Ceulemans H, Hochreiter S. Deep learning as an opportunity in virtual screening. Proceedings of the deep learning workshop at NIPS. 2014 Dec 8;27:1–9. [Google Scholar]
- 51.Casey W. Tox21 overview and update. In Vitro Cell Dev Biol Anim. 2013;49:S7–8. [Google Scholar]
- 52.Wu Z, Ramsundar B, Feinberg EN, Gomes J, Geniesse C, Pappu AS, Leswing K, Pande V. MoleculeNet: a benchmark for molecular machine learning. Chem Sci. 2018;9(2):513–30. doi: 10.1039/c7sc02664a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(D1):D1100–7. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lenselink EB, ten Dijke N, Bongers B, Papadatos G, van Vlijmen HWT, Kowalczyk W, et al. Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J Cheminform. 2017;9(1):45. doi: 10.1186/s13321-017-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rubio DM, Schoenbaum EE, Lee LS, Schteingart DE, Marantz PR, Anderson KE, et al. Defining translational research: implications for training. Acad Med: J Assoc Am Med Coll. 2010;85(3):470–5. doi: 10.1097/ACM.0b013e3181ccd618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Zeng J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics. 2013;29(13):i126–34. doi: 10.1093/bioinformatics/btt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure–activity relationships. J Chem Inf Model. 2015;55(2):263–74. doi: 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- 58.Dahl GE, Jaitly N, Salakhutdinov R. Multi-task neural networks for QSAR predictions. arXiv preprint in Machine Learning (stat ML) arXiv:1406.1231. 2014 Jun 4; [Google Scholar]
- 59.Ramsundar B, Kearnes S, Riley P, Webster D, Konerding D, Pande V. Massively multitask networks for drug discovery. arXiv preprint in Machine Learning (stat.ML). arXiv. 2015 Feb 6;1502:02072. [Google Scholar]
- 60.Wang C, Liu J, Luo F, Tan Y, Deng Z, Hu QN. 2014 IE International Conference on Bioinformatics and Biomedicine (BIBM) IEEE; 2014. Nov 2, Pairwise input neural network for target-ligand interaction prediction; pp. 67–70. [Google Scholar]
- 61.Wallach I, Dzamba M, Heifets A. Atomnet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv preprint in Learning (cs.LG). arXiv. 2015 Oct 10;1510:02855. [Google Scholar]
- 62.Wan F, Zeng J. Deep learning with feature embedding for compound-protein interaction prediction. bioRxiv. 2016 doi: 10.1101/086033. [DOI]
- 63.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50(5):742–54. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 64.Nair V, Hinton GE. Rectified linear units improve restricted boltzmann machines. Proceedings of the 27th international conference on machine learning (ICML-10) 2010:807–14. [Google Scholar]
- 65.Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. 2013;53(7):1563–75. doi: 10.1021/ci400187y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shin M, Jang D, Nam H, Lee KH, Lee D. Predicting the absorption potential of chemical compounds through a deep learning approach. IEEE/ACM Trans Comput Biol Bioinform. 2016;PP(99):1–1. doi: 10.1109/TCBB.2016.2535233. [DOI] [PubMed] [Google Scholar]
- 67.Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: Toxicity Prediction using Deep Learning. Front Environ Sci. 2016;3(80) doi: 10.3389/fenvs.2015.00080. . [DOI] [Google Scholar]
- 68.Pereira JC, Caffarena ER, Dos Santos CN. Boosting docking-based virtual screening with deep learning. J Chem Inf Model. 2016;56(12):2495–506. doi: 10.1021/acs.jcim.6b00355. [DOI] [PubMed] [Google Scholar]
- 69.Hinton GE, McClelland JL, Rumelhart DE. Parallel distributed processing: explorations in the microstructure of cognition. 3. Vol. 1. Cambridge: MIT Press; 1986. Distributed representations; pp. 77–109. [Google Scholar]
- 70.Yao K, Parkhill J. Kinetic energy of hydrocarbons as a function of electron density and convolutional neural networks. J Chem Theory Comput. 2016;12(3):1139–47. doi: 10.1021/acs.jctc.5b01011. [DOI] [PubMed] [Google Scholar]
- 71.Bjerrum EJ. Smiles enumeration as data augmentation for neural network modeling of molecules. arXiv preprint in Learning (cs LG) arXiv:1703.07076. 2017 Mar;:21. [Google Scholar]
- 72.Weininger D. Smiles, a chemical language and information-system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28(1):31–6. [Google Scholar]
- 73.Goh GB, Siegel C, Vishnu A, Hodas NO, Baker N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed qsar/qspr models. arXiv preprint in Machine Learning (stat ML) arXiv:1706.06689. 2017 Jun 20; [Google Scholar]
- 74.Goh GB, Siegel C, Vishnu A, Hodas NO, Baker N. How much chemistry does a deep neural network need to know to make accurate predictions? arXiv preprint in Machine Learning (stat.ML) arXiv:1710.02238. 2017 Oct 5; [Google Scholar]
- 75.Kadurin A, Aliper A, Kazennov A, Mamoshina P, Vanhaelen Q, Khrabrov K, et al. The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget. 2017;8(7):10883–90. doi: 10.18632/oncotarget.14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202–13. doi: 10.1093/nar/gkv951. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Central Science. 2018;4(1):120–31. doi: 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. Journal of Cheminformatics. 2017;9(1):48. doi: 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lima Guimaraes G, Sanchez-Lengeling B, Cunha Farias PL, Aspuru-Guzik A. Objective-reinforced generative adversarial networks (ORGAN) for sequence generation models. arXiv preprint in Machine Learning (stat ML) arXiv:1705.10843. 2017 May [Google Scholar]
- 80.Capuzzi SJ, et al. QSAR modeling of Tox21 challenge stress response and nuclear receptor signaling toxicity assays. Front Environ Sci. 2016;4(3):45. [Google Scholar]
- 81.Maggiora GM. On outliers and activity cliffs—why QSAR often disappoints. J Chem Inf Model. 2006;46(4):1535. doi: 10.1021/ci060117s. [DOI] [PubMed] [Google Scholar]
- 82.Myint K-Z, Wang L, Tong Q, Xie XQ. Molecular fingerprint-based artificial neural networks QSAR for ligand biological activity predictions. Mol Pharm. 2012;9(10):2912–23. doi: 10.1021/mp300237z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L, Ma C, Wipf P, Liu H, Su W, Xie XQ. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013;15(2):395–406. doi: 10.1208/s12248-012-9449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kearnes S, McCloskey K, Berndl M, Pande V, Riley P. Molecular graph convolutions: moving beyond fingerprints. J Comput Aided Mol Des. 2016;30(8):595–608. doi: 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hughes TB, Dang NL, Miller GP, Swamidass SJ. Modeling reactivity to biological macromolecules with a deep multitask network. ACS Cent Sci. 2016;2(8):529–37. doi: 10.1021/acscentsci.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hughes TB, Miller GP, Swamidass SJ. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent Sci. 2015;1(4):168–80. doi: 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]