

Abstract
While the structures of nearly every HIV-1 protein are known in atomic detail from X-ray crystallography and NMR spectroscopy, many questions remain about how the individual proteins are arranged in the mature infectious viral particle. Here, we report the three-dimensional structures of individual HIV-1 virus-like particles (VLPs) as obtained by electron cryotomography. These reconstructions revealed that while the structures and positions of the conical cores within each VLP were unique, they exhibited several surprisingly consistent features, including similarities in the size and shape of the wide end of the capsid (the “base”), uniform positioning of the base and other regions of the capsid 11 nm away from the envelope/MA layer, a cone angle that typically varied from 24° to 18° around the long axis of the cone, and an internal density (presumably part of the NC/RNA complex) cupped within the base. Multiple and nested capsids were observed. These results support the fullerene cone model for the viral capsid, indicate that viral maturation involves a free re-organization of the capsid shell rather than a continuous condensation, imply that capsid assembly is both concentration-driven and template-driven, suggest that specific interactions exist between the capsid and the adjacent envelope/MA and NC/RNA layers, and show that a particular capsid shape is favored strongly in-vivo.
Keywords: HIV, virus structure, tomography, electron cryomicroscopy, capsid
Introduction
While high-resolution structures of many viruses have now been determined by X-ray crystallography, such models will not be obtainable for the type 1 human immunodeficiency virus (HIV-1) because virions are individually unique. Instead, molecular models for HIV-1 must for now be obtained by combining high-resolution structural studies of isolated viral components together with lower-resolution reconstructions of intact virions generated by electron microscopy (EM). Thus, our understanding of HIV-1 structure has advanced stepwise with the development and application of ever more sophisticated EM methods. After the virus was first identified in the early 1980s, its gross structure and organization were quickly characterized using the traditional EM techniques of thin sectioning and immuno-staining or negative staining.1,2 Structural models of the mature virion have subsequently been refined using data from cryo-EM,3 STEM,4 and tomography.5,6
We now know that the mature HIV-1 virion has a roughly spherical outer lipid bilayer 120–200 nm in diameter,3 which is studded with trimeric clusters of the transmembrane Env protein.6 Inside the bilayer, there is a series of structural shells that organize the viral RNA genome and its associated enzymes for uncoating and replication in a new host cell. These different shells are assembled during the process of viral maturation, when the immature Gag polyprotein is processed proteolytically to produce the smaller MA, CA, and NC proteins (as well as several smaller peptides). The outermost shell of the mature virus (the matrix) is associated with the inner face of the bilayer and is composed of ~4000–5000 copies of the N-myristoylated MA protein. Inside the matrix is an unusual conical particle (the core), whose conical outer shell (the capsid) is composed of ~1000–1500 copies of the viral CA protein. Within the capsid is a ribonucleoprotein (RNP) particle composed of two copies of the positive-sense RNA genome, thousands of copies of the RNA-binding NC protein, and ~250 copies each of the reverse transcriptase (RT) and integrase (IN) enzymes.4
High-resolution structural models are now available for essentially all of the HIV-1 proteins or their constituent domains, including MA, CA, and NC.7 Moreover, molecular models for higher-order interactions within the viral matrix8–10 and capsid11 lattices have been proposed on the basis of structural analyses of recombinant MA and CA protein assemblies. In the case of the capsid, EM analyses of helical and two-dimensional crystals of HIV-1 CA have revealed that the protein assembles preferentially into p6-based lattices composed of CA hexamers.11,12 It has been proposed that the conical viral capsid assembles on a similar hexagonal CA net following the structural principles of a fullerene cone.13–16 In the fullerene cone model, the body of the conical capsid is composed of CA hexamers, and the two ends of the cone are allowed to close through the introduction of pentameric defects, with five pentons at the narrow end and seven pentons at the wide end. The fullerene cone model correctly predicts the observed quantization of the cone angles of synthetic capsid assemblies,15 and is supported strongly by a series of cryo-EM measurements made on authentic viral cores.3 However, all of these cone angle measurements were made with simple projection images, and this precluded a direct comparison of actual capsid cone angles to the quantized values predicted by the fullerene cone model, because the authentic capsids could adopt variable orientations with respect to the electron beam,3 and because the synthetic capsids were flattened and possibly distorted in other ways when dried onto carbon films.15
The recent development of electron cryotomography provides a new and more powerful way to investigate HIV-1 structure. This method offers the advantages inherent in cryo-EM, in which a sample in solution is spread into a thin film across an EM grid and plunged into liquid ethane.17 This procedure preserves the sample in a near-native, “frozen-hydrated” state, and allows the actual structure of the biological macromolecules to be imaged through their inherent contrast against an aqueous background. In tomography, a series of images is recorded of a specimen while it is tilted incrementally about some axis. Three-dimensional reconstructions of the specimen (tomograms) are then calculated by merging the images by back-projection in real space, or by Fourier interpolation in reciprocal space. While the potential advantages of electron cryotomography have been discussed for decades, only in the past few years has it become practically feasible and effective through the advent of high-quality CCD cameras, motorized and computer-controlled stages, programmable EMs, and development of automatic protocols to control specimen tracking, focusing, and exposures.18–20
Two tomographic analyses of HIV particles have been reported, yielding information on the structure of the virion,5 and on the shape and distribution of envelope glycoproteins on the virion surface.6 Both of these studies, however, utilized negative staining procedures that limited the accurate visualization of internal core structures. Here, we report the first application of electron cryotomography to determine the three-dimensional structure of several tens of individual, unfixed, unstained, whole HIV-1 virus-like particles (VLPs).
Results
Non-infectious HIV-1 VLPs were produced from a proviral expression construct with debilitating mutations in the RT and RNase H enzymes and a frameshift mutation that prevented Env protein expression. VLPs were produced in cultured human 293T cells, recovered from the supernatant, and purified by sucrose-density centrifugation. As expected, the HIV-1 CA, MA, and NC proteins were the major protein components in the sample as analyzed by SDS-PAGE (data not shown).
HIV-1 VLPs were plunge-frozen onto EM grids and imaged in a 300 kV FEG TEM. A variety of particles was seen, including many VLPs with mature cores, a few immature VLPs with spherical Gag shells, and some “empty” vesicles without any visible core-like internal density (Figure 1, top). Energy filtered, dual-axis tilt series were recorded of ten fields of VLPs at high defocus with the new “flip-flop” cryo-rotation stage, and three-dimensional reconstructions were calculated using the IMOD tomography package (Figure 1, middle).21 Inclusion of the second orthogonal tilt series improved the point-spread-function, as could be seen by the circular shape of the defocus fringe surrounding the gold fiducials in tomogram slices. A total of 76 mature VLPs were selected from the tomograms and denoised with non-linear anisotropic diffusion (examples in Figure 1, bottom).22 Despite dual-axis data collection, there is still a “missing pyramid” of data in reciprocal space, which causes a loss of resolution in the direction parallel with the electron beam. Thus, the shapes of the VLP capsids were best resolved when they were oriented with their long axes “vertically” in the thin “horizontal” layer of ice. These approximately one-third (26) clearest capsids and their corresponding envelopes were segmented in three dimensions using a semi-automatic procedure guided by manually identified landmarks. While the majority of the surface of each capsid was clearly recognizable, especially across the long, straight edges, there were also some gaps arising, presumably from noise, which in some unfavorable cases can actually be emphasized by the denoising algorithm. The structures of the capsid “tips” (narrow ends of the cones) were most difficult to detect, perhaps because their surfaces lay perpendicular to the electron beam (and are therefore smeared by the missing pyramid of data), but perhaps also because they are small and change abruptly. Following segmentation, the three principal axes of each segmented capsid were calculated so that the VLPs could be roughly aligned and compared.
Figure 1.
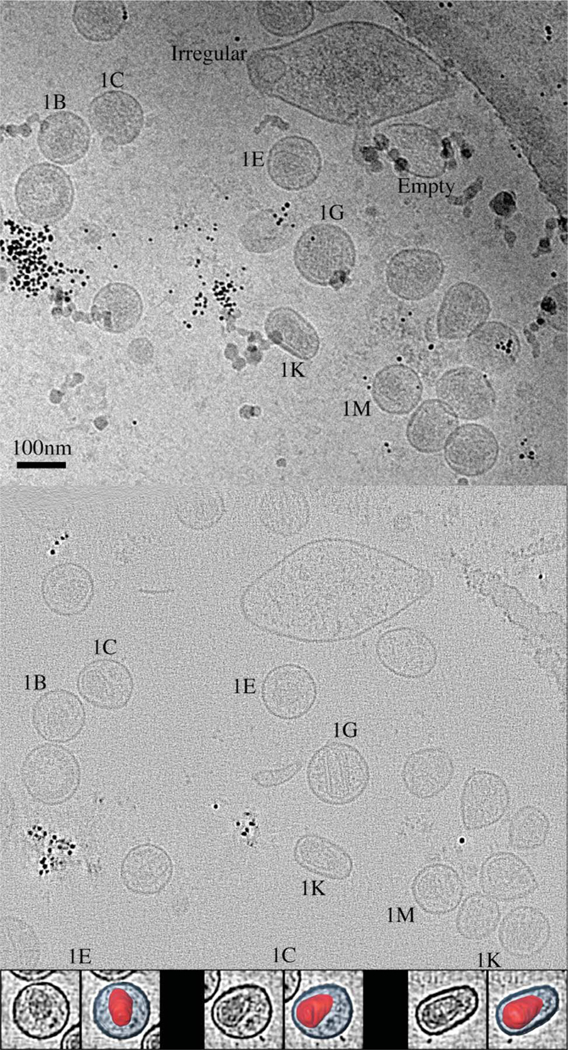
Primary data and processing steps. Top: purified HIV-1 virus-like particles were frozen in thin films across EM grids. Series of images were recorded of many such fields of frozen particles as the sample was tilted incrementally about one axis and then a second, orthogonal axis. One experimental image from one tilt series is shown. The roughly spherical objects are mostly HIV-1 VLPs (including those labeled 1B, 1C, 1E, 1G, 1K, and 1M), but some apparently empty liposomes are present in the sample (Empty) as well as other irregular forms (Irregular). The small black dots are 10 nm gold fiducials used to align the tilt series for reconstruction, and the large arc appearing across the upper right corner is the edge of a circular hole in a supporting carbon film. Middle: Three-dimensional reconstructions of the fields of VLPs were calculated from the various series of tilted images. A single section 1.34 nm thick through the middle of the reconstruction corresponding to the image above is shown. Because this is a single section, some objects that are seen in the projection image above (for instance many of the gold beads) do not appear. Bottom: Individual VLPs were selected from the reconstruction and denoised, and then the boundaries of the lipid bilayer and capsid were semi-automatically delineated (segmented). Three examples (1E, 1C, and 1K) are shown. In each pair, the left image shows a single section through the middle of an extracted, denoised VLP. The right image shows three-dimensional renditions of our interpretation of the capsids in red, shown in place within the denoised section, with the area enclosed by the lipid bilayer in blue.
Figure 2 shows a montage of all 26 segmented VLPs from the three orthogonal viewing directions. Of the 26, 16 exhibited the well-known conical capsid morphology. There were also several rodshaped capsids as well as another class of pleiomorphic shapes that, while non-uniform, nevertheless presented corners, edges, and angles similar to those seen in the conical capsids. In addition, two VLPs contained multiple capsids: 3g contained one conical and one rod-shaped capsid, and 1g actually had a total of five capsids. Surprisingly, two VLPs (10a and 1g) presented internal layers of density that appeared to be nested capsids, an organization that, to our knowledge, has not been reported previously (Figure 3). VLP envelopes were typically spheroidal with a mean largest diameter (the largest distance between any two points on the three-dimensional surface) of 143 nm (standard deviation 10.8 nm), but exhibited significant variability and in many cases (VLPs 1k; 6b, 1n, 2f, 3j, and 7g) followed the contours of the capsid a consistent distance of 11 nm away (peak-to-peak distance from middle of lipid envelope/MA layer to middle of CA layer) for a significant portion of their surfaces.
Figure 2.

Montage of all segmented HIV-1 VLPs. A total of 26 VLPs were segmented and aligned along their principal axes. Three views are shown for each VLP, presenting from left to right axial, sagittal, and coronal perspectives, respectively. Three-dimensional renditions of the capsid surfaces are shown in red for cones, orange for rods, and yellow for all others, placed within a denoised two-dimensional section through the middle of the VLP, with the area enclosed by the lipid bilayer in blue. Because each VLP was extracted as a cube from a larger three-dimensional reconstruction and then aligned to the others, the boundaries of the images appear as randomly oriented, clipped squares. Each capsid is identified by a number and a letter, where the number signifies which field of particles it came from. Capsids 3gR and 3gC are part of the same, double-capsid VLP. The edges of the boxes containing each VLP image here and in all other Figures represent 160 nm.
Figure 3.
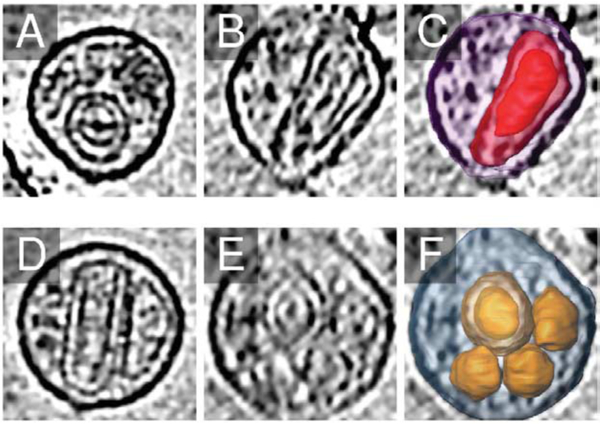
Nested capsids. Two VLPs, (a)–(c) 10a and (d)–(f) 1g, presented nested capsids. (a) and (d) show sagittal sections, (b) and (e) show coronal sections, and (c) and (f) show three-dimensional renditions as in Figure 2, except that the outer capsid surface has been made transparent to reveal the inner capsid, and in the case of VLP 1g ((f)), the surfaces of all five capsids present are rendered.
Knowledge of the three-dimensional shapes of the envelope/matrix and capsid layers allowed us to measure volumes and surface areas accurately. The mean external surface areas were 49,000 nm2 and 21,000 nm2 (standard deviations 5200 nm2 and 9000 nm2), respectively (Table 1). For this calculation, in those cases where VLPs presented multiple capsids (10a, 3g, and 1g), the surface areas of all the capsids were summed. The mean volumes of the VLPs and the conical cores were 980,000 nm3 and 190,000 nm3 (standard deviations 160,000 nm3 and 26,000 nm3), respectively. These small volumes imply that even single-copy proteins are present at appreciable concentrations (~9 μM) within viral cores, and that the concentrations of the viral RT and IN enzymes are quite high (~2 mM).
Table 1.
Surface area measurements
| VLP | Experimentally determined external surface areas (nm2) |
Calculated numbers of molecules |
||
|---|---|---|---|---|
| Envelope/matrix | Capsid | MA | CA | |
| 10a* | 53,000 | 27,000 | 4700 | 1600 |
| 10c | 45,000 | 18,000 | 3600 | 1100 |
| 1b | 46,000 | 17,000 | 3500 | 1000 |
| 1c | 48,000 | 17,000 | 4000 | 1000 |
| 1d | 47,000 | 16,000 | 3700 | 1000 |
| 1e | 52,000 | 19,000 | 4200 | 1200 |
| 1g* | 62,000 | 59,000 | 5100 | 3600 |
| 1k | 49,000 | 21,000 | 4300 | 1300 |
| 1m | 45,000 | 16,000 | 3400 | 900 |
| 2a | 48,000 | 18,000 | 3900 | 1100 |
| 3f | 52,000 | 17,000 | 4300 | 1000 |
| 3g* | 54,000 | 33,000 | 4200 | 2000 |
| 3i | 47,000 | 17,000 | 3900 | 1000 |
| 4a | 39,000 | 14,000 | 2800 | 900 |
| 4e | 46,000 | 16,000 | 3400 | 900 |
| 6c | 56,000 | 20,000 | 4500 | 1200 |
| 6e | 45,000 | 16,000 | 3400 | 1000 |
| 6b | 60,000 | 24,000 | 4900 | 1400 |
| 1n | 52,000 | 20,000 | 4400 | 1200 |
| 2b | 48,000 | 19,000 | 3700 | 1200 |
| 2f | 50,000 | 21,000 | 4000 | 1300 |
| 3j | 51,000 | 19,000 | 4000 | 1200 |
| 4k | 45,000 | 17,000 | 3600 | 1000 |
| 7g | 53,000 | 23,000 | 4200 | 1400 |
| 9e | 45,000 | 18,000 | 3300 | 1100 |
| Mean | 49,000 | 21,000 | 4000 | 1300 |
| Stddev | 5200 | 9000 | 530 | 540 |
The surface areas of each segmented envelope/matrix and capsid shell were measured and presented in the same order as they appear in Figure 2. Assuming each is a hexagonal net, and taking lattice spacings from the literature (see the text), the numbers of MA and CA monomers involved were estimated.
Asterisks mark VLPs with multiple capsids, where the entire capsid surface area was summed.
Previous studies have demonstrated that the virion diameter does not change upon maturation.3 Thus, to calculate the number of MA subunits in the matrix shell, the packing was assumed to be identical with a recent model for the packing of Gag in the immature virus: hexagonal, with six monomers per unit cell and center-to-center spacing of 8.0 nm, observed 12.5 nm from the outside edge of the lipid bilayer.4 We therefore contracted the boundaries of our segmented envelope/matrix layers by 12.5 nm normal to their surfaces at every point, and divided the contracted surface area by the presumed area per Gag monomer. For CA, we again assumed hexagonal packing, and used the measured center-to-center spacing of unit cells at the outside surface of 10.7 nm.11 The resulting averages of 4000 and 1300 monomers in the matrix and capsid shells agree well with previous estimates.4 This indicates that, in general, only about one-third of the available CA protein is involved in an organized capsid layer. In the extreme case of the five capsid VLP 1g, however, 71% of the total CA protein was incorporated into a capsid layer.
When aligned and compared in three dimensions, the majority class of capsids are more similar than suggested by the projection images found currently in the literature. Because the best current model for the organization of the capsid is a hexagonal net of CA protein arrayed following the principles of a fullerene cone,15 the cone angle, which in theory should adopt only certain quantized values (19.2°, 38.9°, 60°, etc.), is of particular interest. We measured the cone angle for all the conical capsids rigorously in three-dimensional space by considering the intersection of the capsid surface with planes containing its primary principal axis. In other words, we measured the cone angle in sections through the capsid ranging 180° from sagittal, past coronal, and back to sagittal. For each section, the tip and base of the capsid were ignored, and lines were fit to the long edges by minimizing squared errors. These two lines defined a cone angle, which was plotted as a function of rotation angle around the long axis of the cone (Figure 4). The plots show a pattern of cone angle progression from ~24° to ~18°, which agrees well with the predicted 19°, since in fact any real 5,7 fullerene cone with finite unit cells cannot be perfectly symmetric. Although the measured angles varied by a few degrees, depending on the precise boundaries used to exclude the tip and base of each capsid, the overall shape of each curve is insensitive to these parameters (data not shown). Most of the cones do tend to “flare” slightly (cone angle increases by a few degrees), however, towards their base. The observation that some capsids exhibit cone angles that appear more “flattened,” and reach as much as 30°, is robust to perturbations of the procedure. For comparison, the corresponding cone angles from three idealized fullerene cone models11,23 are plotted in Figure 4 (black lines). The mean angle across all rotations for the 16 actual capsids measured was 20.1°. The mean angle across all rotations for the idealized fullerene cone models was exactly the same: 20.1°.
Figure 4.
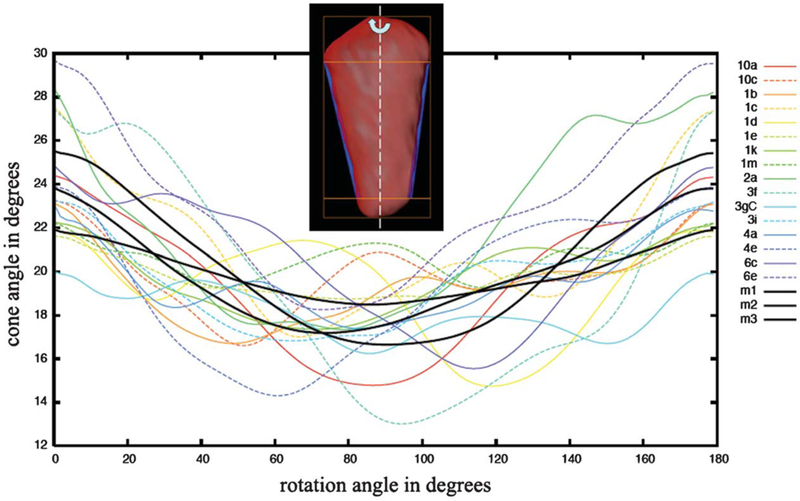
Cone angle variation in 3D. The cone angle (in degrees) for the 16 conical capsids is plotted as a function of rotation around the cone axis from 0–180°. The methodology is depicted in the inset. For each measurement, straight lines (blue in inset) were fit to the edges of the cone between the base and the tip. These lines defined the cone angle as the cone was rotated around its long axis. The resulting values were plotted by assigning the rotation position corresponding to the maximum cone angle as 0°. The unique VLP identifiers are shown in the legend. The three black curves are the corresponding cone angles for three different theoretical fullerene cones modeled to mimic HIV-1 capsids, as described.11,23
To compare the cores further and look for other conserved structural features, we averaged the VLP densities in three ways. First, guessing that important conserved structures within mature HIV-1 would be fixed with respect to the capsid, we aligned the VLPs by superposing their capsid principal axes as we had done before to measure cone angles, and calculated a three-dimensional average. Because the capsids vary in length, however, this produced confusing interferences. We next aligned the VLPs with respect to either the base or the tip of their capsids. Clear patterns in the resulting VLP averages then emerged (Figure 5). When bases were aligned, an inherent asymmetry became apparent, in that their most distal points appeared to the side of their cone axes (Figure 5(b)). The gross shape of the entire base was conserved, as was the 11 nm distance between the base of the capsid (green arrow in Figure 5(e)) and the envelope/matrix layer (magenta arrow in Figure 5(e)) surrounding it. When tips were aligned, the conserved cone angle and the same conserved distance of 11 nm between the tip and the envelope/matrix was again apparent (Figure 5(c) and (d)), but no clear density in the very top of the tip (hereafter referred to as the cap) was observed (yellow arrow in Figure 5(g) and (h)). This appears to be a hole and may very well be in many of the VLPs, although it might alternatively reflect variability in the shape of the cap or the smearing due to the missing pyramid of data. To clarify this ambiguity, we looked carefully at the non-denoised individual reconstructions in this region and observed that indeed there were many capsids without clear cap densities. In addition, the smearing effect of the missing pyramid would be expected to obscure the base and the tip regions equally, but the base regions and their averages uniformly showed strong density. Thus, we favor the interpretation that the cap region is variable, and in some cases is not even closed completely. This would not have been observed easily in projection images, because the lip of density surrounding the cap region would have appeared in that location. Finally, when the densities inside the capsids in the VLP averages are compared, we observe that more density appears within the base than within the tip. This density seems to be concentrated in a third concentric layer (yellow arrow in Figure 5(e)) cupped ~10 nm inside the capsid base.
Figure 5.
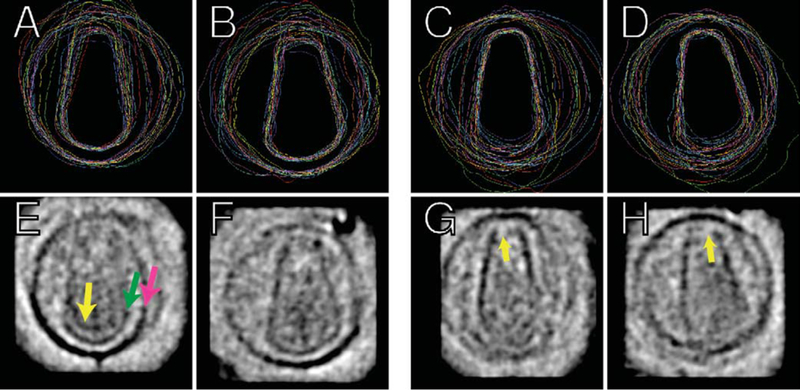
Conserved patterns of density in VLPs. The 16 VLPs with conical capsids were aligned using (a), (b), (e) and (f) the bases or (c), (d), (g) and (h) the tips. The top panels show (a) and (c) sagittal and (b) and (d) coronal sections through the capsids and lipid bilayer surfaces, with the same color code for each VLP as in Figure 4. The bottom panels show again (e) and (g) sagittal and (f) and (h) coronal sections through the corresponding three-dimensional average VLP. In (e), the magenta arrow points towards the edge of the average bilayer/matrix layer, the green arrow points towards the edge of the average capsid, and the yellow arrow points towards the presumed average internal RNP density. In (g) and (h), the yellow arrows point to the missing average cap density. Notable features include the surprisingly uniform, asymmetric capsid base ((b)); the uniform cone angles (particularly (b) and (d)); the uniform distance between the capsid base and the bilayer/matrix layer ((a) and (b)); the preponderance of internal density within the base as opposed to the tip of the average capsids ((e)–(h)); the conserved internal density lining the inside of the average capsid base ((e) in particular); and the lack of clear density in the cap of the average capsid tip ((g) and (h)). The very dark density in the upper right corner of (f) outside the VLP is an artefact due to gold fiducials in one of the reconstructions.
Discussion
The architecture of the layers of HIV-1 and their morphogenesis are intrinsically interesting from a structural point of view, and may lead to novel therapeutics that block assembly or maturation. Indeed, several small molecules have been reported to block structural transitions required for the assembly or maturation of several viruses, including HIV-1.24–26 Unfortunately, the architecture of HIV-1 has been difficult to study, because each virus is unique, and therefore traditional X-ray crystallographic and EM methods are not applicable. For unique specimens, the highest-resolution structural technique available today is electron tomography. In this study, we have applied state-of-the-art electron cryotomography to produce the first three-dimensional structures of whole, native HIV-1 VLPs.
Several lines of evidence indicate that the VLPs we imaged were indeed authentic structural homologs of wild-type HIV-1. Firstly, the point mutations introduced into the RT and RNase H proteins would not be expected to have any structural consequence at the low resolutions seen here. Similarly, the absence of Env protein is unlikely to impact the internal structure, since in a recent study Env trimers were not observed to be distributed uniformly or in any particular pattern.6 Moreover, our measurements of viral particle size and the general sizes and shapes of the viral core are in excellent agreement with recent cryo-EM measurements made on authentic, but chemically inactivated HIV-1 particles.3,4
The structures reported here support the fullerene cone model of the organization of the capsid. While the usual variety of capsid morphologies was seen ranging from cones to rods to other irregular shapes, all the capsids observed here, including those that were pleiomorphic, contain similarly shaped edges, vertices, and angles, giving the impression of objects built from the same underlying motifs. The majority were cones, and they could be aligned surprisingly well in three dimensions. The fullerene cone model predicts that for a cone with five pentons in the tip and seven in the base, the cone angle should be ~19° but, of course, this would be uniform around the long axis of the cone only if the distribution of pentons in the base and the tip were perfectly symmetric, which is physically impossible for a 5,7 fullerene cone. We observed a conserved progression of cone angles from ~24° to ~18°, consistent with the predicted 19° but indicating that the cones are flattened slightly and have roughly elliptical cross-sections. All of these measurements agreed very well with the corresponding cone angle parameters observed for idealized fullerene cones.
Our data provide strong support for the idea that HIV capsid assembly must involve a de novo assembly process rather than a concerted condensation of the immature Gag lattice. Specifically, we observed VLPs with multiple capsids, and even what we believe are nested capsids, both of which are incompatible with a concerted condensation mechanism. While it is possible that the most internal, concentric layers of density inside VLPs 10a and 1g are highly ordered RNP, we favor the interpretation that they are nested capsids, because they have the same width, shape, and density (contrast) as the other capsids. Furthermore, consistent with measurements made by others,4,27 we find that only about one-third of the available CA molecules assemble into capsids, which again precludes concerted condensation models (Table 1). Finally, we note that “simple condensation” would require the unreasonable proposition that pentagonal defects, which presumably are distributed throughout a spherical immature Gag lattice, could “migrate” through the hexagonal lattice to cause cone formation.
The mechanism of de novo capsid assembly is an important question, and two general models can be envisioned: (1) spontaneous, concentration-driven assembly pathways in which the shape of the viral capsid is ultimately determined by local inter-CA interactions; or (2) a template-driven mechanism in which other viral components nucleate assembly of the viral capsid. Our data indicate that both local inter-CA and templating interactions likely play important roles in HIV capsid assembly. Spontaneous assembly must be possible, because pure recombinant CA proteins can assemble into conical capsids in the absence of any other viral proteins,15 and because we observed the formation of nested capsids within VLPs (see Figure 3(a)–(c)). On the other hand, it is likely that the viral matrix and/or RNP layers play a role in helping to nucleate capsid assembly in vivo, because a simple concentration-driven model would not explain the observation that up to five total capsids can form in a single VLP (i.e. capsids can form even after the majority of free CA molecules have been incorporated into other capsids; see VLP 1g).4 Further support for a templated assembly mechanism is provided by our observations that there are defined spatial dispositions between the viral capsid and the adjacent matrix and RNP layers, and by the conserved structural features seen in different viral capsids. The latter two points are considered in greater detail below.
Evidence for specific interactions between the capsid and the adjacent envelope/matrix and RNP layers is provided by the fact that these different layers exhibit similar spacing in multiple VLP structures. The peak-to-peak distance between the matrix and the capsid was uniformly 11 nm along the base and the tip of the capsid (Figure 5), and this same spacing was sometimes observed along the side of the capsid (e.g. see Figure 2, VLP 1k, central image). This spacing is not an artefact of denoising, since the reconstructions used to calculate the average VLP (which document the consistency of the spacing) were not denoised, nor is it an artifact of defocus, because while defocus blurs edges, it does not change the separation between density peaks. At present, we cannot explain definitively why the matrix and capsid layers are spaced so consistently at either end of the capsid, though there must be, of course, some physical interaction that promotes this spacing. It is interesting to note that this distance matches very well the analogous spacing seen in immature virions (Wilk et al.,28 see Figure 2(b), measuring from the center of the lipid envelope/matrix layer to the middle of the presumed capsid peaks), and it may therefore be a remnant of that structure that is preserved during maturation. It is unlikely, however, that covalent MA–CA linkages are maintained in the mature virion, as Gag processing appears to proceed to completion and mature viral cores can be isolated away from the matrix/envelope layer.29–31 We did not observe any consistent “20 nm thin structural link,” previously termed the core-envelope linkage or CEL structure,5 between the cap of the viral core and the matrix layer. Similarly, we did not see regular “lateral bodies1” along the sides of the viral capsid, but instead typically saw scattered and only occasionally aggregated densities between the matrix and capsid layers. We suggest that these previous observations could have been artefacts of fixation and staining. Finally, we did not observe either an “irregular slit” or “cavities or holes...distributed primarily in the wide end” of the capsid structure, as reported.5
Others have observed a preponderance of density within the base (as opposed to the tip) of the core,3 and it is therefore likely that most of the RNP components reside in the wide end of the capsid. Consistent with these observations, we observed a third layer of density in the averaged VLP structures, which appeared just inside the capsid base (Figure 5(e), yellow arrow). We interpret this density as part of the RNP complex, and speculate that it might have served as a template to guide formation of the bases into their consistent shape and size. Interestingly, genetic studies also support the idea that the internal capsid surface may make functionally important interactions with the RNP complex and its associated enzymes. For example, subtle mutations in the internal MHR motif of Rous sarcoma virus (RSV) CA apparently do not compromise overall capsid assembly, but nevertheless prevent reverse transcription in the newly infected target cell.32 Similarly, point mutations that alter the stability of the HIV-1 capsid reduce the efficiency of DNA synthesis.33 Thus, it appears that the retroviral capsid performs an important functional role in helping to organize the viral RNP particle for successful uncoating and reverse transcription in a new host cell.
Several observations revealed that a special capsid shape is heavily favored in vivo. Firstly, while purified CA protein can form sheets, rods, and various cones in different ratios, depending on environmental conditions,15 much less variety was seen here in vivo, where the majority of capsids form cones of the 5,7 penton distribution type. If capsid assembly were not a directed process, then the hexagonal capsid net would self-assemble into lattices with pentameric disclinations at random positions. It has been shown, however, that the vast majority of such hypothetical self-assembling surfaces would not close, and it is clear that the vast majority of those that did would be roughly spherical.34 The number of cones that would emerge would be small, and those with a 5,7 penton distribution would be a special class among them. The frequency of finding a 5,7 cone with a particular size and shape in its base would be, we believe (though without specific calculation), essentially negligible. Nevertheless, the majority class of actual VLPs observed here does in fact share just such an improbable capsid structure, which argues strongly for the presence of local and/or templating interactions that promote this preferred capsid shape in vivo.
In this initial tomographic study we used imaging conditions chosen to highlight the gross structural features of HIV-1. Higher-resolution structures should be achievable in the near future through several improvements: the use of smaller defocus values, which enhance high-resolution features at the expense of low-resolution contrast; CTF-correction across the focal gradients present in the images;35 optimization of the process of merging dual-axis tilt series of frozen-hydrated samples;36,37 thinning of the ice and reduction of sucrose and other high-density, contrast-matching molecules in the buffer; and ultimately, detection of the underlying periodicity of the protein layers and subsequent “unit cell” averaging until atomic models can be reliably docked.
Materials and Methods
Preparation of VLP
HIV-1 VLPs were made non-infectious by inactivation of the essential Env, RT, and RNase H proteins. An HIV-1NL4–3 R9 proviral vector38 with a frameshift mutation in the env gene that blocked Env protein production was obtained as a gift from Dr Christopher Aiken (Vanderbilt University). Although Env deletion alone blocks viral infectivity, two additional mutations were added that independently block viral replication. First, the viral RT was inactivated with a D185A (RTD185A) mutation. D185 is an RT active-site residue that forms part of the catalytic triad. The mutation has been reported to block viral replication,39,40 and this was confirmed in our laboratory (data not shown). Second, the RNase H protein was inactivated with an H539A mutation (RNaseHH539A), which also blocks replication (our data, not shown).41 DNA sequencing confirmed the presence of all three inactivating mutations in the ΔEnv/RTD185A/RNaseHH539A R9 construct.
VLPs were produced by transfecting the ΔEnv/RTD185A/RNaseHH539A R9 proviral DNA construct into 293T cells. Lipofectomine (Invitrogen) transfections followed the manufacturer’s instructions (10cm×10cm plates; 10 μg of R9 DNA/plate), and VLP-containing supernatants (8 ml) were harvested 24 hours and 48 hours post-transfection. VLP particles were concentrated by ultracentrifugation (30,000 rpm for 1.5 hours in a Beckman Ti50.2 rotor, at 4 °C) through 20% (w/v) sucrose cushions (17.5 ml of culture samples overlaid on 8 ml cushions) in STE buffer (10 mM Tris (pH 7.4), 100 mM NaCl, 1 mM EDTA). Concentrated viral stocks were resuspended (250 μl in STE buffer), overlaid on 10 ml sucrose gradients (20%–60%, w/v), centrifuged (34,000 rpm for 16 hours in a Beckman SW-41 rotor), and harvested in 1 ml fractions. Viral fractions were assayed by SDS-PAGE, and the major bands made visible by staining with Coomassie brilliant blue were confirmed to be the viral MA, CA, and NC proteins by Western blotting. Four 1 ml VLP-containing fractions were combined, diluted with 2 ml of STE buffer, and VLPs were pelleted by ultracentrifugation (55,000 rpm for one hour in a tabletop ultracentrifuge). The concentrated pellets were then resuspended in ~100 μl of STE buffer for EM analyses.
Cryoelectron tomography
Purified HIV-1 VLPs were flash-frozen onto glow-discharged Quantifoil grids in liquid ethane using a Vitrobot. Gold fiducial markers (10 nm) were both dried onto the grids and mixed into the virus solutions before freezing. Frozen-hydrated grids were loaded into the new flip-flop tilt rotation holder, introduced into a 300 kV, FEG G2 Polara FEI TEM, and kept frozen by thermal contact with liquid nitrogen. Image tilt series were collected from approximately −60° to +60° with a 3° tilt step automatically using the predictive UCSF tomography package,42 and then the grid was rotated ~90° and a second tilt series was collected with the same tilt increment through approximately the same tilt range. All images were energy-filtered with an energy slit-width of 20 eV, recorded with a defocus of ~16 μm, with CCD pixels representing 0.67 nm on the specimen.
Image processing
Images were binned 2-fold, and then dual-axis tomographic reconstruction was performed using the IMOD package.21 Briefly, each single-axis tilt series was aligned independently and used to calculate a three-dimensional reconstruction in real space by standard methods. The second reconstruction was divided into cubes and aligned piecewise to the first by cross-correlation, and was then re-interpolated to compensate for potential distortions between the two tilt series. The first reconstruction and the adjusted second reconstruction were Fourier-transformed and merged in reciprocal space by simple averaging in the voxels for which structure factors were available from both tilt series. From the completed tomograms, individual VLPs were selected manually and denoised by 30 rounds of non-linear anisotropic diffusion.22 Capsids were selected and segmented semi-automatically along with their corresponding lipid envelopes using the Amira software package (Mercury Computer Systems, Inc.). Custom Amira modules were written to measure the capsid cone angle as a function of rotation about the cone axis and to align VLPs by either the tip or base of the capsid cone. The Figures were produced with Amira.
Acknowledgements
This work was supported by NIH grant PO1 GM66521 to W.I.S. and G.J.J., as well as gifts from the Ralph M. Parsons Foundation, the Agouron Institute, and the Gordon and Betty Moore Foundation to the California Institute of Technology. We are grateful to Uta von Schwedler and Kirsten Stray for assistance with VLP preparations, and we thank Simon Wain–Hobsen for suggesting that we estimate the concentrations of enzymes in the viral core.
Abbreviations used:
- HIV-1
type 1 human immunodeficiency virus
- EM
electron microscopy
- VLPs
virus-like particles
- RT
reverse transcriptase
- IN
integrase
References
- 1.Gelderblom HR, Hausmann EHS, Ozel M, Pauli G & Koch MA (1987). Fine-structure of human immunodeficiency virus (HIV) and immuno-localization of structural proteins. Micron Microsc. Acta, 18, 335–336. [DOI] [PubMed] [Google Scholar]
- 2.Goto T, Nakai M & Ikuta K (1998). The life-cycle of human immunodeficiency virus type 1. Micron, 29, 123–318. [DOI] [PubMed] [Google Scholar]
- 3.Briggs JAG, Wilk T, Welker R, Krausslich HG & Fuller SD (2003). Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briggs JAG, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM & Johnson MC (2004). The stoichiometry of Gag protein in HIV-1. Nature Struct. Mol. Biol. 11, 672–675. [DOI] [PubMed] [Google Scholar]
- 5.Hoglund S, Ofverstedt LG, Nilsson A, Lundquist P, Gelderblom H, Ozel M & Skoglund U (1992). Spatial visualization of the maturing HIV-1 core and its linkage to the envelope. AIDS Res. Hum. Retro-viruses, 8,1–7. [DOI] [PubMed] [Google Scholar]
- 6.Zhu P, Chertova E, Bess J, Lifson JD, Arthur LO, Liu J et al. (2003). Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl Acad. Sci. USA, 100, 15812–15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner BG & Summers MF (1999). Structural biology of HIV. J. Mol. Biol. 285, 1–32. [DOI] [PubMed] [Google Scholar]
- 8.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM & Stuart DI (1995). Crystal structure of SIV matrix antigen and implications for virus assembly. Nature, 378, 743–747. [DOI] [PubMed] [Google Scholar]
- 9.Hill CP, Worthylake D, Bancroft DP, Christensen AM & Sundquist WI (1996). Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl Acad. Sci. USA, 93, 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D & Summers MF (2004). Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl Acad. Sci. USA, 101, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S, Hill CP, Sundquist WI & Finch JT (2000). Image reconstructions of helical assemblies of the HIV-1CA protein. Nature, 407, 409–413. [DOI] [PubMed] [Google Scholar]
- 12.Mayo K, Huseby D, McDermott J, Arvidson B, Finlay L & Barklis E (2003). Retrovirus capsid protein assembly arrangements. J. Mol. Biol. 325, 225–237. [DOI] [PubMed] [Google Scholar]
- 13.Ge M & Sattler K (1994). Observation of fullerene cones. Chem. Phys. Letters, 220, 192–196. [Google Scholar]
- 14.Ebbesen T (1998). Cones and tubes: geometry in the chemistry of carbon. Accts Chem. Res. 31, 558–566. [Google Scholar]
- 15.Ganser BK, Li S, Klishko VY, Finch JT & Sundquist WI (1999). Assembly and analysis of conical models for the HIV-1 core. Science, 283, 80–83. [DOI] [PubMed] [Google Scholar]
- 16.Jin ZM, Jin L, Peterson DL & Lawson CL (1999). Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol. 286, 83–93. [DOI] [PubMed] [Google Scholar]
- 17.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW & Schultz P (1988). Cryo-electron microscopy of vitrified specimens. Quart. Rev. Biophys. 21, 129–228. [DOI] [PubMed] [Google Scholar]
- 18.Koster AJ, Grimm R, Typke D, Hegerl R, Stoschek A, Walz J et al. (1997). Perspectives of molecular and cellular electron tomography. J. Struct. Biol. 120, 276–308. [DOI] [PubMed] [Google Scholar]
- 19.Subramaniam S & Milne JLS (2004). Three-dimensional electron microscopy at molecular resolution. Annu. Rev. Biophys. Biomol. Struct. 33, 141–155. [DOI] [PubMed] [Google Scholar]
- 20.Sali A, Glaeser R, Earnest T & Baumeister W (2003). From words to literature in structural proteomics. Nature, 422, 216–225. [DOI] [PubMed] [Google Scholar]
- 21.Kremer JR, Mastronarde DN & McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76. [DOI] [PubMed] [Google Scholar]
- 22.Frangakis AS & Hegerl R (2001). Noise reduction in electron tomographic reconstructions using non-linear anisotropic diffusion. J. Struct. Biol. 135, 239–250. [DOI] [PubMed] [Google Scholar]
- 23.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C & Sundquist WI (2004). Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prevelige PE Jr, (1998). Inhibiting virus-capsid assembly by altering the polymerisation pathway. Trends Biotechnol. 16, 61–65. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C et al. (2003). PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl Acad. Sci. USA, 100, 13555–13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E et al. (2003). Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327, 1013–1020. [DOI] [PubMed] [Google Scholar]
- 27.Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M & Prevelige PE Jr, (2004). Key interactions in HIV-1 maturation identified by hydrogen–deuterium exchange. Nature Struct. Mol. Biol. 11, 676–677. [DOI] [PubMed] [Google Scholar]
- 28.Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R et al. (2001). Organization of immature human immunodeficiency virus type 1. J. Virol. 75, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welker R, Hohenberg H, Tessmer U, Huckhagel C & Krausslich HG (2000). Biochemical and structural analysis of isolated mature cores of human immuno-deficiency virus type 1. J. Virol. 74, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotov A, Zhou J, Flicker P & Aiken C (1999). Association of Nef with the human immuno-deficiency virus type 1 core. J. Virol. 73, 8824–88230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Accola MA, Ohagen A & Gottlinger HG (2000). Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag). J. Virol. 74, 6198–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cairns TM & Craven RC (2001). Viral DNA synthesis defects in assembly-competent Rous sarcoma virus CA mutants. J. Virol. 75, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forshey BM, von Schwedler U, Sundquist WI & Aiken C (2002). Formation of a human immuno-deficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76, 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manolopoulos DE, May JC & Down SE (1991). Theoretical studies of the fullerenes:C34 to C70. Chem. Phys. Letters, 181, 105–111. [Google Scholar]
- 35.Winkler H & Taylor KA (2003). Focus gradient correction applied to tilt series image data used in electron tomography. J. Struct. Biol. 143, 24–32. [DOI] [PubMed] [Google Scholar]
- 36.Mastronarde DN (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol. 120, 343–352. [DOI] [PubMed] [Google Scholar]
- 37.Penczek P, Marko M, Buttle K & Frank J (1995). Double-tilt electron tomography. Ultramicroscopy, 60, 393–410. [DOI] [PubMed] [Google Scholar]
- 38.Swingler S, Gallay P, Camaur D, Song J, Abo A & Trono D (1997). The Nef protein of human immuno-deficiency virus type 1 enhances serine phosphorylation of the viral matrix. J. Virol. 71, 4372–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyer PL, Ferris AL & Hughes SH (1992). Cassette mutagenesis of the reverse-transcriptase of human-immunodeficiency-virus type-1. J. Virol. 66, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaushik N, Rege N, Yadav PNS, Sarafianos SG, Modak MJ & Pandey VN (1996). Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry, 35, 11536–11546. [DOI] [PubMed] [Google Scholar]
- 41.Tisdale M, Schulze T, Larder BA & Moelling K (1991). Mutations within the RNase-H domain of human-immunodeficiency-virus type-1 reverse-transcriptase abolish virus infectivity. J. Gen. Virol. 72, 59–66. [DOI] [PubMed] [Google Scholar]
- 42.Zheng QS, Braunfeld MB, Sedat JW & Agard DA (2004). An improved strategy for automated electron microscopic tomography. J. Struct. Biol. 147, 91–101. [DOI] [PubMed] [Google Scholar]