

Abstract
Background
Viruses have evolved to create a cellular environment permissive for viral replication in susceptible hosts. Possibly both enabling and resulting from these virus-triggered changes, infected hosts undergo a dramatic transcriptional reprogramming, the analysis of which can shed light on the molecular processes underlying the outcome of virus-host interactions. The study of the transcriptional changes triggered by the plant DNA viruses geminiviruses is potentially hampered by the low representation of infected cells in the total population, a situation that becomes extreme in those cases, like that of Tomato yellow leaf curl virus (TYLCV), in which the virus is restricted to phloem companion cells.
Results
In order to gain insight into how different the transcriptional landscapes of TYLCV-infected cells or whole tissues of TYLCV-infected plants might be, here we compare the transcriptional changes in leaf patches infected with TYLCV by agroinfiltration or in systemic leaves of TYLCV-infected plants in Nicotiana benthamiana. Our results show that, in agreement with previous works, infection by TYLCV induces a dramatic transcriptional reprogramming; the detected changes, however, are not equivalent in local and systemic infections, with a much larger number of genes differentially expressed locally, and some genes responding in an opposite manner. Interestingly, a transcriptional repression of the auxin signalling pathway and a transcriptional activation of the ethylene signalling pathway were detected in both local and systemically infected samples. A transcriptional activation of defence was also detectable in both cases. Comparison with the transcriptional changes induced by systemic infection by the geminivirus Tobacco curly shoot virus (TbSV) shows common subsets of up- and down-regulated genes similarly affected by both viral species, unveiling a common transcriptional repression of terpenoid biosynthesis, a process also suppressed by the geminivirus Tomato yellow leaf curl China virus.
Conclusions
Taken together, the results presented here not only offer insight into the transcriptional changes derived from the infection by TYLCV in N. benthamiana, but also demonstrate that the resolution provided by local and systemic infection approaches largely differs, highlighting the urge to come up with a better system to gain an accurate view of the molecular and physiological changes caused by the viral invasion.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5842-7) contains supplementary material, which is available to authorized users.
Keywords: Geminivirus, Transcriptome, Nicotiana benthamiana, TYLCV, Agroinoculation
Background
As intracellular parasites, viruses have evolved to create a cellular environment permissive for viral replication in susceptible hosts; for this purpose, viruses induce a re-wiring of the host’s physiology and development concomitant to the establishment of a successful infection. In plants, these virus-induced changes can be easily visualized and quantified, and infection by different viruses frequently produces some of a common array of symptoms, including stunting, chlorosis, and leaf curling. Possibly both enabling and resulting from these virus-triggered changes, infected hosts undergo a dramatic transcriptional reprogramming; the analysis of the modifications in the transcriptional landscape of the host upon the viral infection can shed light on the molecular processes underlying the outcome of virus-host interactions. Such transcriptional studies have proliferated in the past decade, possibly owing to technical advances allowing for in-depth sequencing, availability of genomic information, and the increased affordability of these approaches.
Geminiviruses are insect-transmitted DNA viruses causing severe diseases in crops worldwide, and currently pose a serious threat to food security; however, our understanding of the molecular basis of the infection is still partial, which limits the development of effective anti-geminiviral strategies for crop protection. The transcriptional changes triggered by the infection by geminiviruses have been studied in a number of plant-virus interactions [1–8]. When comparing the results obtained in these studies, few commonalities arise: plant hormone signaling pathways, especially those for jasmonates (JA) and brassinosteroids (BR), frequently appear as altered, although the direction of the change is not consistent [4, 5, 8, 9]; and cell cycle-related genes, which need to be reactivated in the virus-infected terminally differentiated cells to allow for viral DNA replication, are detected as differentially expressed in a couple of cases only [2, 9]. In an attempt to unveil the molecular basis of tolerance or recovery, Chen et al. (2013) and Gongora-Castillo et al. (2012) [3, 4] compared the transcriptome of susceptible or tolerant tomato cultivars infected with Tomato yellow leaf curl virus (TYLCV) and that of recovered and symptomatic leaves of pepper infected with Pepper golden mosaic virus (PepGMV), respectively, by RNA-seq; however, and somewhat surprisingly, only limited differences were detected in both cases.
One factor frequently neglected in these transcriptional studies of geminivirus-infected plants is the low representation of infected cells in the total population: in an average infection, only some cells will be supporting viral replication at a given time. This situation becomes extreme in those cases, like that of TYLCV, in which the virus is restricted to phloem companion cells. The global transcriptome of infected plants will be the average of that in all cells, infected and non-infected, and all tissues: this not only creates a serious dilution issue, but also averages the potential transcriptional responses in infected and systemic, uninfected cells, which could be opposite, hence generating potentially misleading results difficult to interpret. However, and since to date no transcriptional profile of isolated infected cells is available, the extent to which this could be different to that obtained from complete aerial organs of an infected plant is unclear.
In order to gain insight into how different the transcriptional landscapes of TYLCV-infected cells or whole tissues of TYLCV-infected plants might be, here we compare the transcriptional changes in leaf patches infected with TYLCV by agroinfiltration or in systemic leaves of TYLCV-infected plants in Nicotiana benthamiana by RNA-seq. Strikingly, our results show that, as expected and in agreement with previous works, infection by TYLCV induces a dramatic transcriptional reprogramming; the detected changes, however, are not equivalent in local and systemic infections, with a much larger number of genes differentially expressed locally, and some genes responding in an opposite manner in local and systemic samples. Interestingly, a transcriptional repression of the auxin signaling pathway and a transcriptional activation of ethylene signaling and defence responses were detected both in local and systemic infections. Despite more limited changes detected in the systemically infected samples, comparison with the transcriptional changes induced by systemic infection by the geminivirus Tobacco curly shoot virus (TbSV) [5] unveiled common subsets of up- and down-regulated genes similarly affected by both viral species. Among the common biological processes potentially affected by the transcriptional changes we find terpenoid biosynthesis as transcriptionally repressed; notably, another geminivirus species, Tomato yellow leaf curl China virus (TYLCCN), has been shown to suppress terpenoid biosynthesis [10], making it tempting to speculate that depletion of terpenoids might be a requirement for geminiviruses to establish a successful infection in nature. Taken together, the results presented here not only shed light on the transcriptional changes derived from the infection by TYLCV in N. benthamiana, but also demonstrate that the resolution provided by local and systemic infection approaches largely differs, highlighting the urge to come up with a better system to gain an accurate view of the molecular and physiological changes caused by the viral invasion.
Results
Transcriptional changes upon local infection by TYLCV in N. benthamiana
From the observation that only a fraction of cells support active replication by a geminivirus at a given time [11, 12] logically follows the idea that an accurate study of the cellular changes triggered by the viral invasion will require the isolation of the infected cells specifically, and their comparison with similar cells from an uninfected sample. Analysis of whole organs of systemically infected plants, which is the common practice due to the lack of a more precise approach, would presumably result in potential dilution and masking issues. In order to test this idea, we decided to compare the transcriptional changes detectable by RNA sequencing (RNA-seq) upon local or systemic infection in the model plant N. benthamiana by the geminivirus TYLCV.
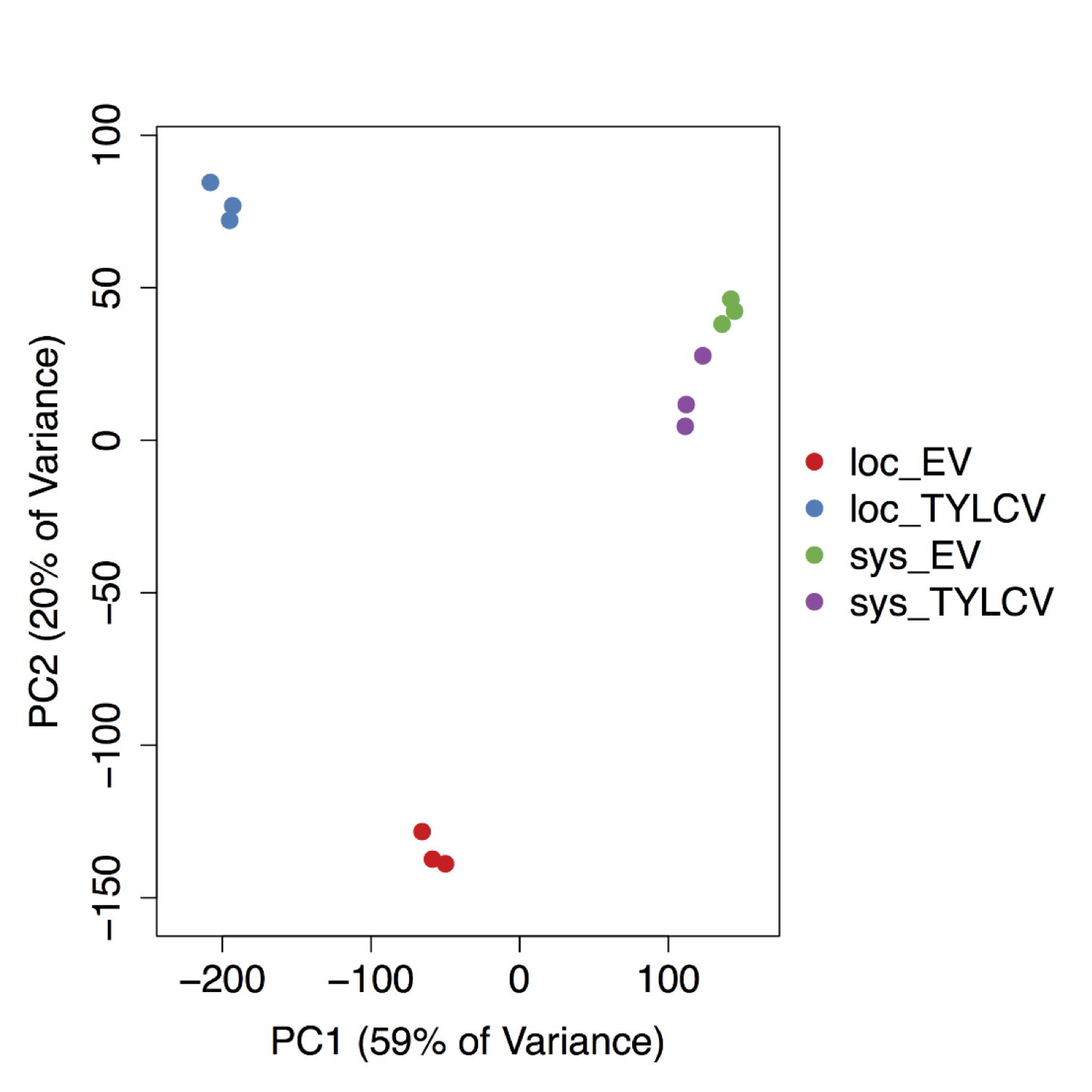
The leaf patch infection results in the viral replication in most cells [11], which the virus must effectively manipulate to generate a permissive environment, hence serving as a good surrogate system to study the viral infection. For the local infection, we performed agroinfection in leaf patches of four-week-old N. benthamiana leaves and took samples at 6 days post-infiltration (dpi) (Fig. 1a), when the virus is still actively replicating. In order to exclude the potential effect of the bacteria on plant transcription, N. benthamiana leaves inoculated with an Agrobacterium clone containing the empty vector were used as control; three independent biological replicates were used in each case. RNA-seq was performed by Illumina sequencing as indicated in the methods section. The raw HiSeq reads were filtered and trimmed, and between 34 and > 51 million clean pair-end reads were obtained per sample; these clean reads were mapped to the N. benthamiana draft genome (v1.0.1) from the Sol Genomics Network (ftp://ftp.solgenomics.net/genomes/Nicotiana_benthamiana/assemblies/)) with a mapping rate between 89 and > 98% (Additional file 6 and Table 1). The PCA analysis of the three biological replicates for TYLCV and control samples is shown in Additional file 1: Figure S1.
Fig. 1.
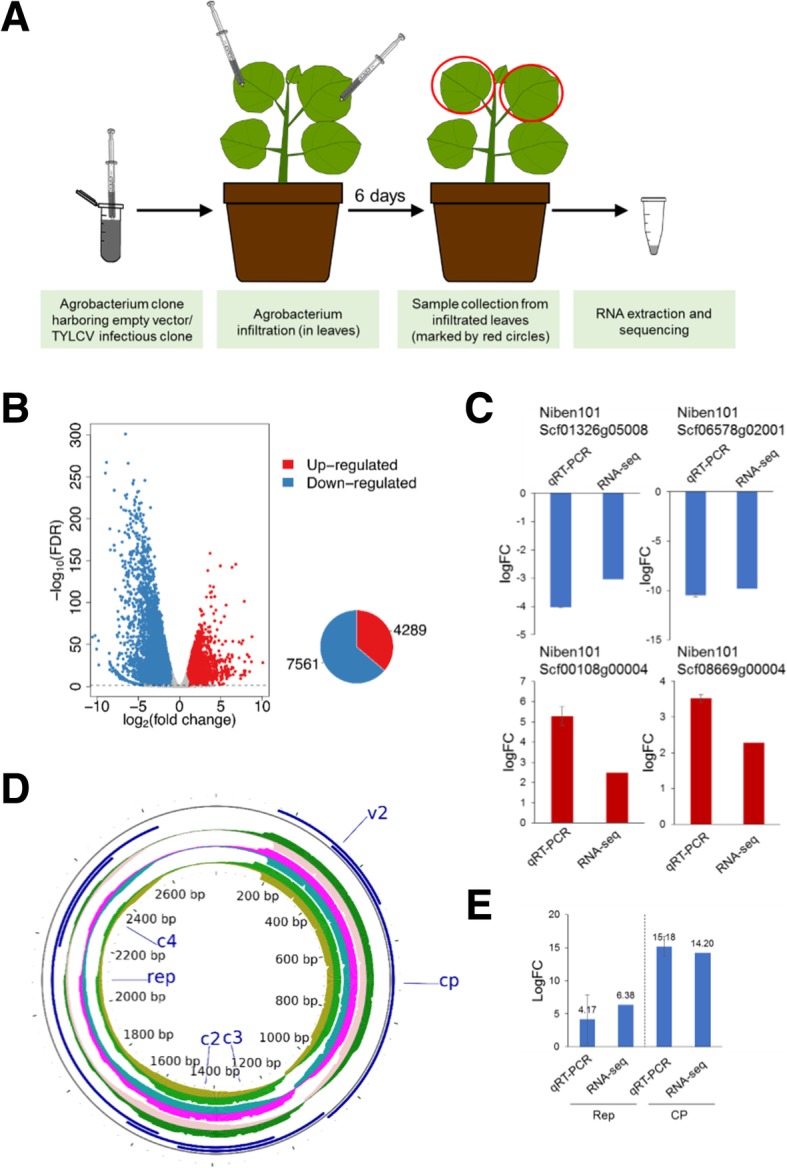
Transcriptional changes upon local infection by TYLCV in N. benthamiana. a Schematic representation of the experimental design. b Differentially expressed genes in the local infection are shown by volcano plot. The x-axis shows the log2 transformed gene expression fold change between infected and control samples. The y-axis indicates the negative log10 transformed adjusted p-values (FDR) of the differential expression test calculated by R package edgeR. The up-regulated and down-regulated genes are represented by red and blue dots, respectively. Pie chart shows the number of up−/down-regulated genes. c Validation of selected DEGs by qPCR. Values are the average of three biological replicates, relative to mock. NbACT was used as the normalizer. d Mapping of viral reads to the TYLCV genome. Three biological replicates (inner concentric circles) are represented; the upper side of each circle represents the virion (+) strand; the lower side of each circle represents the complementary (−) strand. ORFs are depicted in blue. Please note that the accumulated reads in the area containing the CP and V2 ORFs have been trimmed; the numbers of total reads are shown in Additional file 7: Table S2. e Validation of the expression of Rep and CP by qPCR. Expression values are relative to NbACT
Table 1.
Summary of the RNA-seq results
| Infection | Sample | Replicate | Raw reads | Clean reads | Mapped reads | Mapping rate |
|---|---|---|---|---|---|---|
| local | EV | 1 | 56,095,920 | 49,607,102 | 48,784,974 | 98.34% |
| local | EV | 2 | 40,783,494 | 35,461,778 | 34,878,028 | 98.35% |
| local | EV | 3 | 59,215,650 | 50,797,194 | 49,957,654 | 98.35% |
| local | TLCV | 1 | 46,414,226 | 42,273,896 | 38,070,811 | 90.06% |
| local | TYLCV | 2 | 40,583,678 | 34,799,420 | 31,263,478 | 89.84% |
| local | TYLCV | 3 | 42,441,872 | 36,642,412 | 32,897,690 | 89.78% |
| systemic | EV | 1 | 45,831,882 | 42,069,696 | 41,242,974 | 98.03% |
| systemic | EV | 2 | 40,432,870 | 36,748,216 | 35,955,596 | 97.84% |
| systemic | EV | 3 | 48,885,308 | 45,262,860 | 44,405,978 | 98.11% |
| systemic | TYLCV | 1 | 47,349,296 | 43,508,220 | 42,484,166 | 97.65% |
| systemic | TYLCV | 2 | 55,797,434 | 51,761,828 | 50,618,532 | 97.79% |
| systemic | TYLCV | 3 | 49,035,824 | 45,797,334 | 44,821,767 | 97.87% |
A total of 7561 and 4289 down- and up-regulated genes, respectively, were identified in these locally infected samples (Fig. 1b; Additional file 6: Table S1). The RNA-seq results were validated by qPCR analysis of selected genes (Fig. 1c).
The clean reads were also mapped to the TYLCV genome, with an average of 88,550 reads per million (RPM) (Fig. 1d; Additional file 7: Table S2). Unexpectedly, reads for both strands of the virus were detected throughout the viral genome with uneven and non-perfectly symmetrical distribution, and not restricted to the described open reading frames (ORFs); the number of reads was much higher in the region of the genome containing the V2 and CP (late) genes (Additional file 7: Table S2). The accumulation of viral reads was confirmed by qPCR to detect expression of the viral genes encoding the Rep (Replication-associated protein) and the CP (capsid protein), contained in the complementary and the virion strand of the viral genome, respectively (Fig. 1e).
Transcriptional changes upon systemic infection by TYLCV in N. benthamiana
In order to analyze the transcriptional changes detectable during the systemic infection by TYLCV, we performed agroinfection of two-week-old N. benthamiana plants as described in [12], and took samples at 14 days post-infection (dpi) (Fig. 2a), when the virus is actively replicating in the apical leaves and the first symptoms have already appeared (Additional file 2: Figure S2). Apical leaves of N. benthamiana plants inoculated with an Agrobacterium clone containing the empty vector were used as control; three independent biological replicates were used in each case. RNA-seq was performed by Illumina sequencing as indicated in the methods section. The raw HiSeq reads were filtered and trimmed, and between 36 and > 52 million clean pair-end reads were obtained per sample; these clean reads were mapped to the N. benthamiana draft genome (v1.0.1) from the Sol Genomics Network (ftp://ftp.solgenomics.net/genomes/Nicotiana_benthamiana/assemblies/) with a mapping rate between 97 and > 98% (Additional file 6: Table S1). The PCA analysis of the three biological replicates for TYLCV and control samples is shown in Additional file 1: Figure S1.
Fig. 2.
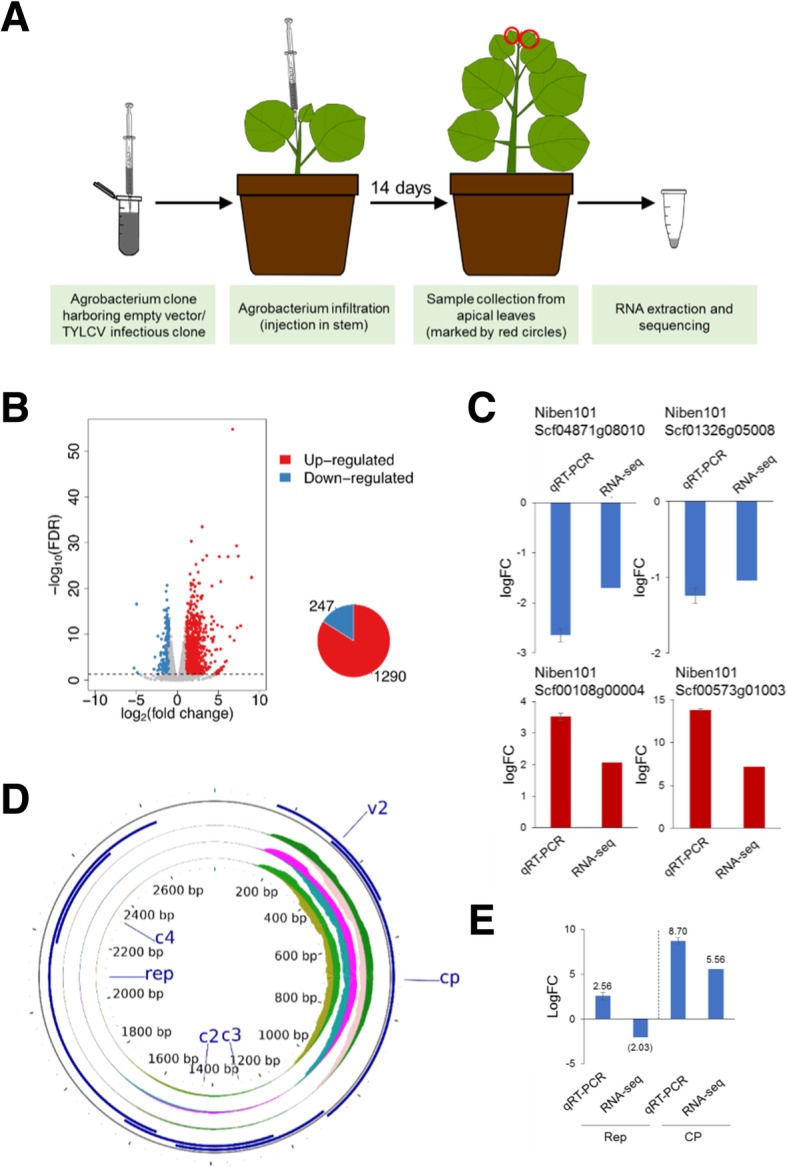
Transcriptional changes upon local infection by TYLCV in N. benthamiana. a Schematic representation of the experimental design. b Differentially expressed genes in the systemic infection are shown by volcano plot. The x-axis shows the log2 transformed gene expression fold change between infected and control samples. The y-axis indicates the negative log10 transformed adjusted p-values (FDR) of the differential expression test calculated by R package edgeR. The up-regulated and down-regulated genes are represented by red and blue dots, respectively. Pie chart shows the number of up−/down-regulated genes. c Validation of selected DEGs by qPCR. Values are the average of three biological replicates, relative to mock. NbACT was used as the normalizer. d Mapping of viral reads to the TYLCV genome. Three biological replicates (inner concentric circles) are represented; the upper side of each circle represents the virion (+) strand; the lower side of each circle represents the complementary (−) strand. ORFs are depicted in blue. The number of total reads are shown in Additional file 7: Table S2. e Validation of the expression of Rep and CP by qPCR. Expression values are relative to NbACT
A total of 247 and 1290 down- and up-regulated genes, respectively, were identified in these systemically infected samples (Fig. 2b; Additional file 8: Table S3). The RNA-seq results were validated by qPCR analysis of selected genes (Fig. 2c).
The clean reads were also mapped to the TYLCV genome, with an average of 3344 reads per million (RPM) (Fig. 2d; Additional file 7: Table S2). Also in this case, reads for both strands of the virus were detected throughout the viral genome with uneven and non-perfectly symmetrical distribution, and not restricted to the described ORFs; as observed in the locally infected samples, the number of reads was notably higher in the region of the genome containing the V2 and CP (late) genes (Fig. 2d; Additional file 7: Table S2). The accumulation of viral reads was confirmed by qPCR to detect expression of the viral genes encoding the Rep and the CP (Fig. 2e).
Distinct landscapes of transcriptional changes detectable in local and systemic infections by TYLCV in N. benthamiana
In both the locally and the systemically infected samples used, the virus is actively replicating, and therefore those putative transcriptional changes underlying successful viral multiplication must have been established. The vast difference in the number of differentially expressed genes (DEGs) between these infected samples raises the possibility that the larger proportion of infected cells in the agroinfected leaf patches might provide an increase in resolution, which may result from a lower dilution of infection-induced transcriptional changes due to a higher infected-to-uninfected cell ratio and/or negligible masking from potential non-cell-autonomous plant responses to the viral infection.
In order to gain further insight into the differences between locally and systemically infected samples, we set out to compare the subsets of induced or repressed genes in each case. Strikingly, as shown in Fig. 3a, both the up- and the down-regulated genes in systemic infections show only a partial overlap with those in local infections: only 56.7% of the repressed genes are also repressed in local infections, while, surprisingly, 6% are induced; among the induced genes in systemic infections, only 24% are also up-regulated in local infections, with a 23.8% down-regulated in these samples. Hierarchical clustering (Fig. 3b) shows that the systemically infected samples cluster closer to their control than to the locally infected samples. Taken together, these results clearly indicate that the differences detected between datasets go beyond a higher sensitivity in locally infected samples, which would explain the quantitative differences in DEGs, but not the apparent opposite behavior of some of them.
Fig. 3.
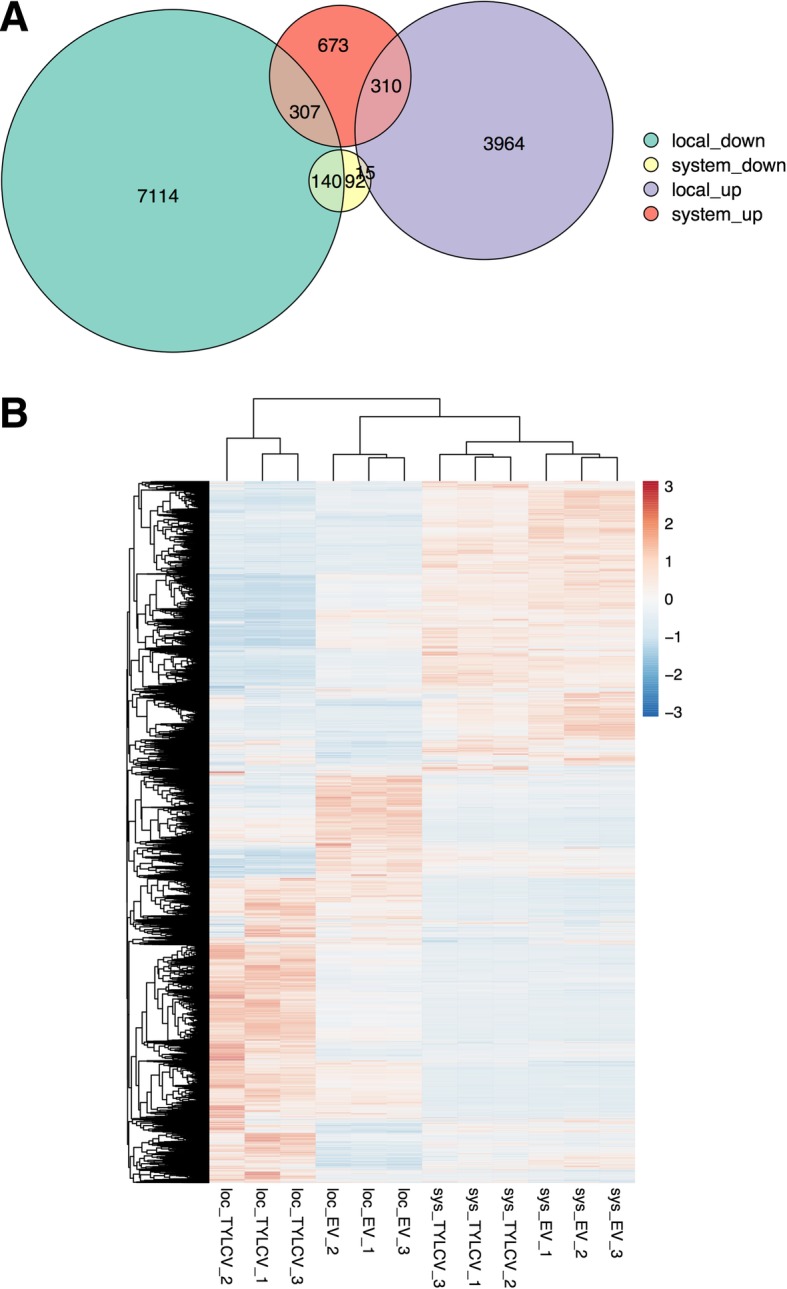
Different transcriptional changes are detected upon local and systemic infections by TYLCV in N. benthamiana. a Venn diagram of DEGs in local and systemic infections. b The differential gene expressions of local and systemic infection samples are shown in heatmap with hierarchical clustering, which is drawn by R package pheatmap. The values in the gene expression matrix are centered and scaled in the row direction. The color bar indicates the Z-score. Loc: local infections; sys: systemic infections. EV: empty vector
With the aim of determining whether the distinct transcriptional landscapes of local and systemic infections may nevertheless result in similar functional outputs, we performed functional enrichment analysis using Gene Ontology and KEGG pathways annotations. As shown in Figs. 4 and 5 and Table 2, the overlap in over-represented GO categories (Biological Process Ontology) or KEGG pathways for systemic and local infections is only marginal. Common over-represented functional categories in both subsets of DEGs include trehalose biosynthetic process and defence response among the induced genes, and mitotic nuclear division and cellulose biosynthetic process among the repressed genes (Table 2).
Fig. 4.
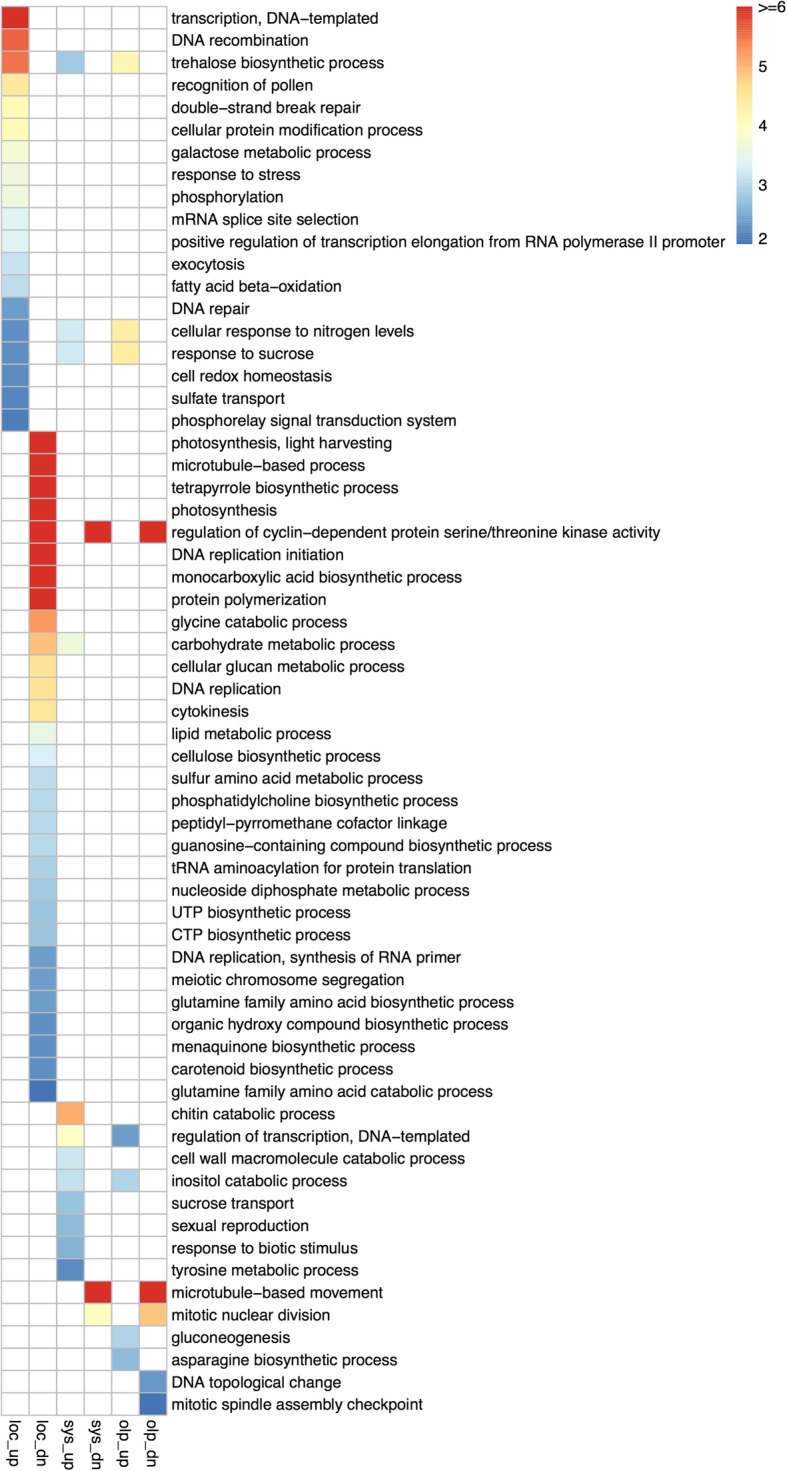
Functional enrichment analysis of differentially expressed genes in local and systemic TYLCV infections. Heatmap shows the Gene Ontology terms of biological processes that are significantly enriched (p < 0.01) in the up−/down-regulated genes in the local infection, the systemic infection, and their overlaps. Each square is colored according to the value of –log10(p), where p is the p-value for the significance of GO term enrichment. The color bar indicates the –log10(p). loc: local infection; sys: systemic infection; olp: overlapping between local and systemic infections. up: up-regulated; dn: down-regulated
Fig. 5.
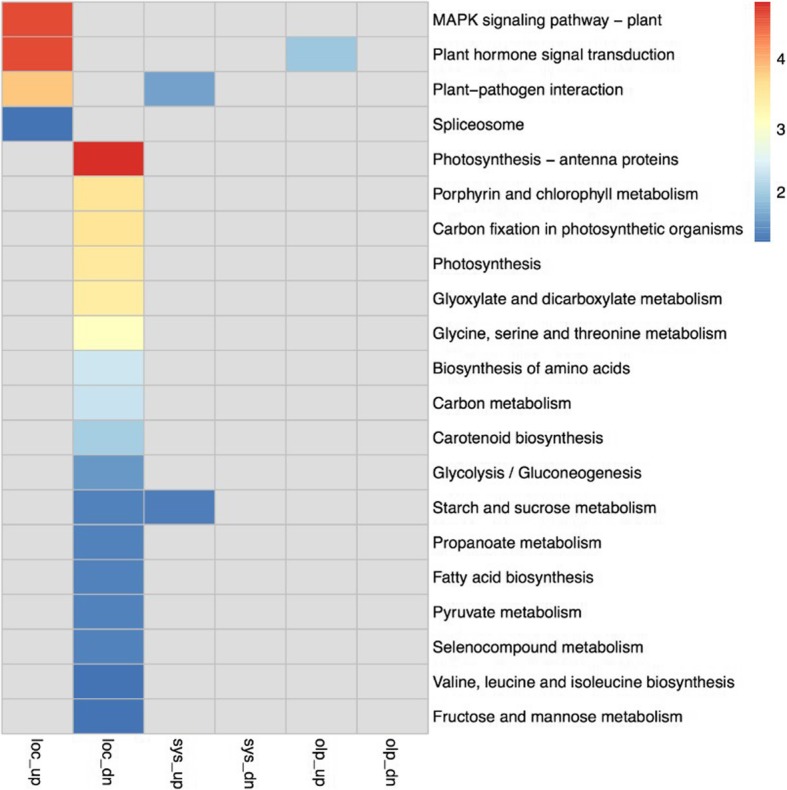
KEGG pathways of differentially expressed genes in local and systemic TYLCV infections. Heatmap shows the KEGG pathways that are significantly enriched (p < 0.05) in the up−/down-regulated genes in the local infection, the systemic infection, and their overlaps. Each square is colored according to the value of –log10(p), where p is the p-value for the significance of KEGG pathway enrichment. The color bar indicates the –log10(p). up: up-regulated; dn: down-regulated
Table 2.
Significantly enriched GO terms (Biological Process Ontology) in the subsets of differentially expressed genes (p-value < 0.05)
| GO.ID | Term | Annotated | Significant | Expected | p-value |
|---|---|---|---|---|---|
| Overrepresented in the subset of up-regulated in local infection | |||||
| GO:0006351 | transcription, DNA-templated | 2046 | 271 | 151.48 | 1.8E-22 |
| GO:0006310 | DNA recombination | 47 | 15 | 3.48 | 2.5E-06 |
| GO:0005992 | trehalose biosynthetic process | 34 | 12 | 2.52 | 3.1E-06 |
| GO:0048544 | recognition of pollen | 69 | 16 | 5.11 | 0.000034 |
| GO:0006302 | double-strand break repair | 16 | 7 | 1.18 | 0.000076 |
| GO:0006464 | cellular protein modification process | 2490 | 237 | 184.35 | 0.00008 |
| GO:0006012 | galactose metabolic process | 23 | 8 | 1.7 | 0.00016 |
| GO:0006950 | response to stress | 873 | 108 | 64.63 | 0.00022 |
| GO:0016310 | phosphorylation | 2148 | 196 | 159.03 | 0.00023 |
| GO:0006376 | mRNA splice site selection | 6 | 4 | 0.44 | 0.0004 |
| GO:0032968 | positive regulation of transcription elongation from RNA polymerase II promoter | 3 | 3 | 0.22 | 0.00041 |
| GO:0006887 | exocytosis | 72 | 14 | 5.33 | 0.00071 |
| GO:0006635 | fatty acid beta-oxidation | 7 | 4 | 0.52 | 0.00087 |
| GO:0006281 | DNA repair | 271 | 38 | 20.06 | 0.00415 |
| GO:0043562 | cellular response to nitrogen levels | 2 | 2 | 0.15 | 0.00548 |
| GO:0009744 | response to sucrose | 2 | 2 | 0.15 | 0.00548 |
| GO:0045454 | cell redox homeostasis | 229 | 28 | 16.95 | 0.00608 |
| GO:0008272 | sulfate transport | 31 | 7 | 2.3 | 0.00653 |
| GO:0000160 | phosphorelay signal transduction system | 102 | 15 | 7.55 | 0.00799 |
| GO:0006334 | nucleosome assembly | 59 | 10 | 4.37 | 0.01074 |
| GO:0009298 | GDP-mannose biosynthetic process | 7 | 3 | 0.52 | 0.0113 |
| GO:0070966 | nuclear-transcribed mRNA catabolic process, no-go decay | 3 | 2 | 0.22 | 0.01562 |
| GO:0070481 | nuclear-transcribed mRNA catabolic process, non-stop decay | 3 | 2 | 0.22 | 0.01562 |
| GO:0071025 | RNA surveillance | 3 | 2 | 0.22 | 0.01562 |
| GO:0015706 | nitrate transport | 3 | 2 | 0.22 | 0.01562 |
| GO:0010167 | response to nitrate | 3 | 2 | 0.22 | 0.01562 |
| GO:0007064 | mitotic sister chromatid cohesion | 8 | 3 | 0.59 | 0.01709 |
| GO:0006032 | chitin catabolic process | 31 | 6 | 2.3 | 0.02419 |
| GO:0035434 | copper ion transmembrane transport | 9 | 3 | 0.67 | 0.02425 |
| GO:0050832 | defense response to fungus | 4 | 2 | 0.3 | 0.02972 |
| GO:0042742 | defense response to bacterium | 4 | 2 | 0.3 | 0.02972 |
| GO:0007050 | cell cycle arrest | 10 | 3 | 0.74 | 0.03277 |
| GO:0006529 | asparagine biosynthetic process | 11 | 3 | 0.81 | 0.04263 |
| GO:0006020 | inositol metabolic process | 11 | 3 | 0.81 | 0.04263 |
| Overrepresented in the subset of down-regulated in local infection | |||||
| GO:0009765 | photosynthesis, light harvesting | 74 | 61 | 12.92 | 1E-30 |
| GO:0007017 | microtubule-based process | 254 | 105 | 44.35 | 4.6E-19 |
| GO:0033014 | tetrapyrrole biosynthetic process | 53 | 32 | 9.25 | 3.3E-12 |
| GO:0015979 | photosynthesis | 387 | 169 | 67.57 | 4.9E-12 |
| GO:0000079 | regulation of cyclin-dependent protein serine/threonine kinase activity | 48 | 29 | 8.38 | 3.4E-11 |
| GO:0006270 | DNA replication initiation | 18 | 16 | 3.14 | 7.7E-11 |
| GO:0072330 | monocarboxylic acid biosynthetic process | 171 | 61 | 29.85 | 8.1E-09 |
| GO:0051258 | protein polymerization | 86 | 34 | 15.01 | 8.8E-09 |
| GO:0006546 | glycine catabolic process | 11 | 9 | 1.92 | 5.8E-06 |
| GO:0005975 | carbohydrate metabolic process | 1345 | 320 | 234.82 | 0.000013 |
| GO:0006073 | cellular glucan metabolic process | 142 | 56 | 24.79 | 0.000026 |
| GO:0006260 | DNA replication | 119 | 55 | 20.78 | 0.000027 |
| GO:0000910 | cytokinesis | 24 | 12 | 4.19 | 0.000033 |
| GO:0006629 | lipid metabolic process | 928 | 234 | 162.02 | 0.00028 |
| GO:0030244 | cellulose biosynthetic process | 62 | 22 | 10.82 | 0.00051 |
| GO:0000096 | sulfur amino acid metabolic process | 50 | 18 | 8.73 | 0.00084 |
| GO:0006656 | phosphatidylcholine biosynthetic process | 4 | 4 | 0.7 | 0.00093 |
| GO:0018160 | peptidyl-pyrromethane cofactor linkage | 4 | 4 | 0.7 | 0.00093 |
| GO:1901070 | guanosine-containing compound biosynthetic process | 17 | 9 | 2.97 | 0.00094 |
| GO:0006418 | tRNA aminoacylation for protein translation | 95 | 29 | 16.59 | 0.00125 |
| GO:0009132 | nucleoside diphosphate metabolic process | 135 | 40 | 23.57 | 0.00155 |
| GO:0006228 | UTP biosynthetic process | 15 | 8 | 2.62 | 0.00171 |
| GO:0006241 | CTP biosynthetic process | 15 | 8 | 2.62 | 0.00171 |
| GO:0006269 | DNA replication, synthesis of RNA primer | 5 | 4 | 0.87 | 0.00399 |
| GO:0045132 | meiotic chromosome segregation | 5 | 4 | 0.87 | 0.00399 |
| GO:0009084 | glutamine family amino acid biosynthetic process | 31 | 12 | 5.41 | 0.00416 |
| GO:1901617 | organic hydroxy compound biosynthetic process | 23 | 14 | 4.02 | 0.00531 |
| GO:0009234 | menaquinone biosynthetic process | 3 | 3 | 0.52 | 0.00532 |
| GO:0016117 | carotenoid biosynthetic process | 14 | 7 | 2.44 | 0.00536 |
| GO:0009065 | glutamine family amino acid catabolic process | 12 | 6 | 2.1 | 0.00997 |
| GO:0046168 | glycerol-3-phosphate catabolic process | 6 | 4 | 1.05 | 0.01031 |
| GO:0015671 | oxygen transport | 6 | 4 | 1.05 | 0.01031 |
| GO:0006801 | superoxide metabolic process | 23 | 9 | 4.02 | 0.01161 |
| GO:0007076 | mitotic chromosome condensation | 13 | 6 | 2.27 | 0.01582 |
| GO:0006166 | purine ribonucleoside salvage | 4 | 3 | 0.7 | 0.01849 |
| GO:0006096 | glycolytic process | 114 | 29 | 19.9 | 0.02014 |
| GO:0046274 | lignin catabolic process | 33 | 11 | 5.76 | 0.02045 |
| GO:0042549 | photosystem II stabilization | 7 | 4 | 1.22 | 0.02075 |
| GO:0042026 | protein refolding | 14 | 6 | 2.44 | 0.02366 |
| GO:0019307 | mannose biosynthetic process | 2 | 2 | 0.35 | 0.03047 |
| GO:0007155 | cell adhesion | 2 | 2 | 0.35 | 0.03047 |
| GO:0045038 | protein import into chloroplast thylakoid membrane | 2 | 2 | 0.35 | 0.03047 |
| GO:0048478 | replication fork protection | 2 | 2 | 0.35 | 0.03047 |
| GO:0007623 | circadian rhythm | 2 | 2 | 0.35 | 0.03047 |
| GO:0010027 | thylakoid membrane organization | 2 | 2 | 0.35 | 0.03047 |
| GO:0006450 | regulation of translational fidelity | 2 | 2 | 0.35 | 0.03047 |
| GO:0030259 | lipid glycosylation | 15 | 6 | 2.62 | 0.03373 |
| GO:0009082 | branched-chain amino acid biosynthetic process | 23 | 8 | 4.02 | 0.03537 |
| GO:0006168 | adenine salvage | 5 | 3 | 0.87 | 0.04023 |
| GO:0010207 | photosystem II assembly | 5 | 3 | 0.87 | 0.04023 |
| GO:0006353 | DNA-templated transcription, termination | 5 | 3 | 0.87 | 0.04023 |
| GO:0006662 | glycerol ether metabolic process | 68 | 18 | 11.87 | 0.04094 |
| Overrepresented in the subset of up-regulated in systemic infection | |||||
| GO:0006032 | chitin catabolic process | 31 | 7 | 0.78 | 9.1E-06 |
| GO:0006355 | regulation of transcription, DNA-templated | 1764 | 69 | 44.11 | 0.00012 |
| GO:0005975 | carbohydrate metabolic process | 1345 | 63 | 33.64 | 0.00022 |
| GO:0043562 | cellular response to nitrogen levels | 2 | 2 | 0.05 | 0.00062 |
| GO:0009744 | response to sucrose | 2 | 2 | 0.05 | 0.00062 |
| GO:0016998 | cell wall macromolecule catabolic process | 29 | 5 | 0.73 | 0.00069 |
| GO:0019310 | inositol catabolic process | 8 | 3 | 0.2 | 0.00079 |
| GO:0005992 | trehalose biosynthetic process | 34 | 5 | 0.85 | 0.00146 |
| GO:0015770 | sucrose transport | 3 | 2 | 0.08 | 0.00184 |
| GO:0019953 | sexual reproduction | 11 | 3 | 0.28 | 0.00221 |
| GO:0009607 | response to biotic stimulus | 43 | 6 | 1.08 | 0.00271 |
| GO:0006570 | tyrosine metabolic process | 5 | 2 | 0.13 | 0.00594 |
| GO:0006094 | gluconeogenesis | 8 | 2 | 0.2 | 0.01581 |
| GO:0006952 | defense response | 112 | 8 | 2.8 | 0.01862 |
| GO:0006541 | glutamine metabolic process | 23 | 3 | 0.58 | 0.01898 |
| GO:0000272 | polysaccharide catabolic process | 25 | 3 | 0.63 | 0.02376 |
| GO:0006662 | glycerol ether metabolic process | 68 | 5 | 1.7 | 0.02756 |
| GO:0003333 | amino acid transmembrane transport | 46 | 4 | 1.15 | 0.02758 |
| GO:0006529 | asparagine biosynthetic process | 11 | 2 | 0.28 | 0.02956 |
| GO:0008283 | cell proliferation | 11 | 2 | 0.28 | 0.02956 |
| GO:0044264 | cellular polysaccharide metabolic process | 148 | 7 | 3.7 | 0.03961 |
| GO:0008272 | sulfate transport | 31 | 3 | 0.78 | 0.04163 |
| GO:0006857 | oligopeptide transport | 33 | 3 | 0.83 | 0.04874 |
| Overrepresented in the subset of down-regulated in systemic infection | |||||
| GO:0007018 | microtubule-based movement | 129 | 11 | 0.7 | 1.2E-10 |
| GO:0000079 | regulation of cyclin-dependent protein serine/threonine kinase activity | 48 | 7 | 0.26 | 7.2E-09 |
| GO:0007067 | mitotic nuclear division | 51 | 5 | 0.28 | 0.0001 |
| GO:0016114 | terpenoid biosynthetic process | 30 | 2 | 0.16 | 0.0116 |
| GO:0006265 | DNA topological change | 30 | 2 | 0.16 | 0.0116 |
| GO:0007094 | mitotic spindle assembly checkpoint | 3 | 1 | 0.02 | 0.0163 |
| GO:0006952 | defense response | 112 | 3 | 0.61 | 0.0235 |
| GO:0009813 | flavonoid biosynthetic process | 6 | 1 | 0.03 | 0.0323 |
| GO:0030244 | cellulose biosynthetic process | 62 | 2 | 0.34 | 0.0452 |
| Overrepresented in the subset of up-regulated genes common to local and systemic infections | |||||
| GO:0043562 | cellular response to nitrogen levels | 2 | 2 | 0.01 | 0.000041 |
| GO:0009744 | response to sucrose | 2 | 2 | 0.01 | 0.000041 |
| GO:0005992 | trehalose biosynthetic process | 34 | 4 | 0.22 | 0.000066 |
| GO:0006094 | gluconeogenesis | 8 | 2 | 0.05 | 0.0011 |
| GO:0019310 | inositol catabolic process | 8 | 2 | 0.05 | 0.0011 |
| GO:0006529 | asparagine biosynthetic process | 11 | 2 | 0.07 | 0.0022 |
| GO:0006355 | regulation of transcription, DNA-templated | 1764 | 21 | 11.37 | 0.0043 |
| GO:0007205 | protein kinase C-activating G-protein coupled receptor signaling pathway | 24 | 2 | 0.15 | 0.0104 |
| GO:0006659 | phosphatidylserine biosynthetic process | 3 | 1 | 0.02 | 0.0192 |
| GO:0042742 | defense response to bacterium | 4 | 1 | 0.03 | 0.0255 |
| GO:0006562 | proline catabolic process | 4 | 1 | 0.03 | 0.0255 |
| GO:0050832 | defense response to fungus | 4 | 1 | 0.03 | 0.0255 |
| GO:0006570 | tyrosine metabolic process | 5 | 1 | 0.03 | 0.0318 |
| GO:0003333 | amino acid transmembrane transport | 46 | 2 | 0.3 | 0.0355 |
| GO:0009064 | glutamine family amino acid metabolic process | 57 | 3 | 0.37 | 0.0453 |
| Overrepresented in the subset of down-regulated genes common to local and systemic infections | |||||
| GO:0007018 | microtubule-based movement | 129 | 10 | 0.43 | 1.4E-11 |
| GO:0000079 | regulation of cyclin-dependent protein serine/threonine kinase activity | 48 | 7 | 0.16 | 2.1E-10 |
| GO:0007067 | mitotic nuclear division | 51 | 5 | 0.17 | 0.000014 |
| GO:0006265 | DNA topological change | 30 | 2 | 0.1 | 0.0045 |
| GO:0007094 | mitotic spindle assembly checkpoint | 3 | 1 | 0.01 | 0.0099 |
| GO:0030244 | cellulose biosynthetic process | 62 | 2 | 0.21 | 0.0181 |
| GO:0006606 | protein import into nucleus | 12 | 1 | 0.04 | 0.0392 |
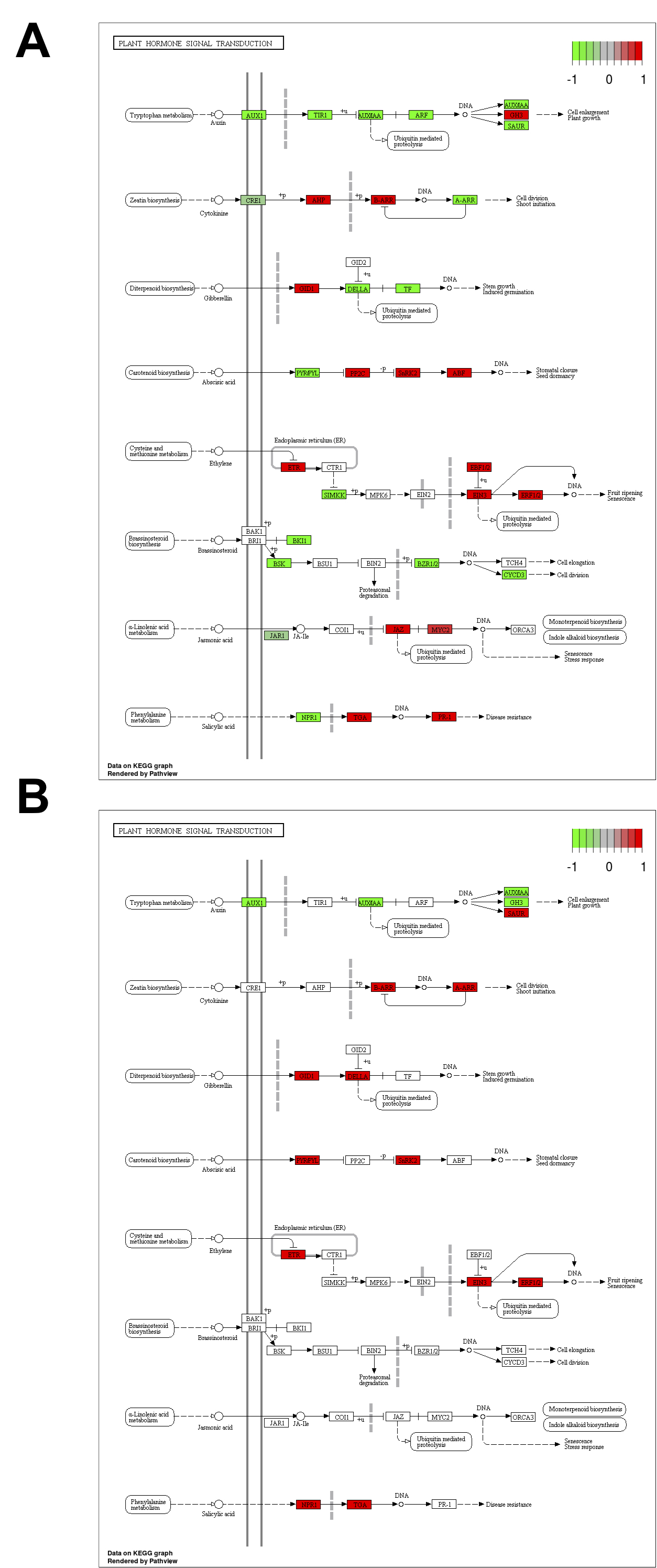
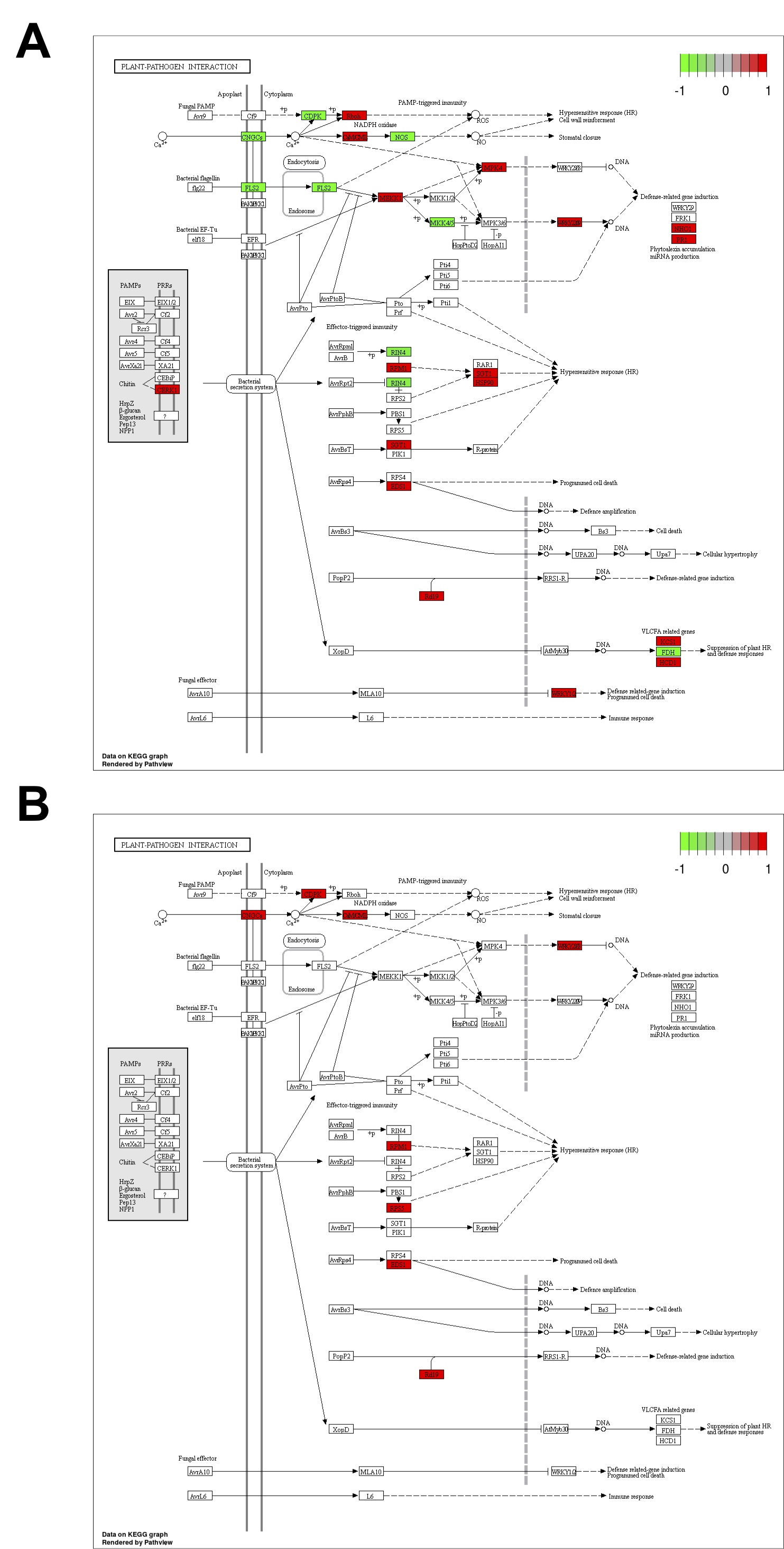
Three KEGG pathways from Fig. 5 were selected to be graphically displayed, allowing for visualization and easy comparison of the transcriptional regulation of their components: MAPK signaling pathway (Additional file 3: Figure S3), Plant hormone signal transduction (Additional file 4: Figure S4), and Plant-pathogen interaction (Additional file 5: Figure S5). As observed in Additional file 3: Fig. S3 and Additional file 4: Figure S4, although not statistically significant in all cases, both local and systemic infections have an impact on these pathways, with the local infections having the strongest effect. As previously mentioned, the presence of the virus seems to activate plant defence responses (Additional file 3: Figures S3; Additional file 5: Figure S5). Notably, both types of infection trigger a detectable transcriptional repression of auxin signaling and a transcriptional activation of ethylene signaling, while only local infections resulted in a repression of the brassinosteroid signaling pathway (Additional file 4: Figure S4).
TYLCV and TbSCV modify the expression of a set of common genes upon systemic infection in N. benthamiana
In an attempt to identify potential central targets of the transcriptional geminiviral manipulation and/or effectors of the plant anti-geminiviral response, we decided to compare the transcriptional changes triggered by systemic infections by TYLCV and the geminivirus TbSCV, in combination with or without its associated satellite, in N. benthamiana [5]. Remarkably, a proportion of DEG were commonly affected by both TYLCV and TbSCV (12.6% of up-regulated genes by TYLCV infection, and 9.7% of down-regulated genes by TYLCV infection) (Fig. 6a); the proportion of induced, but not repressed, genes largely increased (to 28.7% of up-regulated genes by TYLCV infection) when TbSCV was inoculated in combination with its satellite (Fig. 6b), suggesting that some of the virulence functions provided by this ancillary molecule result in the activation of host genes and are already encoded in the TYLCV genome. Functional enrichment analysis of the genes commonly activated or repressed by TYLCV and TbSCV revealed the existence of a number of GO categories over-represented in these subsets (Table 3), including trehalose biosynthetic process and defence responses (in the common up-regulated gene set), and terpenoid biosynthetic process (in the common down-regulated gene set). Interestingly, a third geminiviral species, TYLCCN, has been proven to suppress terpenoid biosynthesis and release, and this effect in turn improves performance of its insect vector, the whitefly Bemisia tabaci [10]. Considering this negative regulation by three different geminivirus species, it is tempting to speculate that depletion of terpenoids is a requirement for geminiviruses to establish a successful infection in nature, perhaps at least partly through an indirect effect on favouring the insect vector-virus mutualism.
Fig. 6.
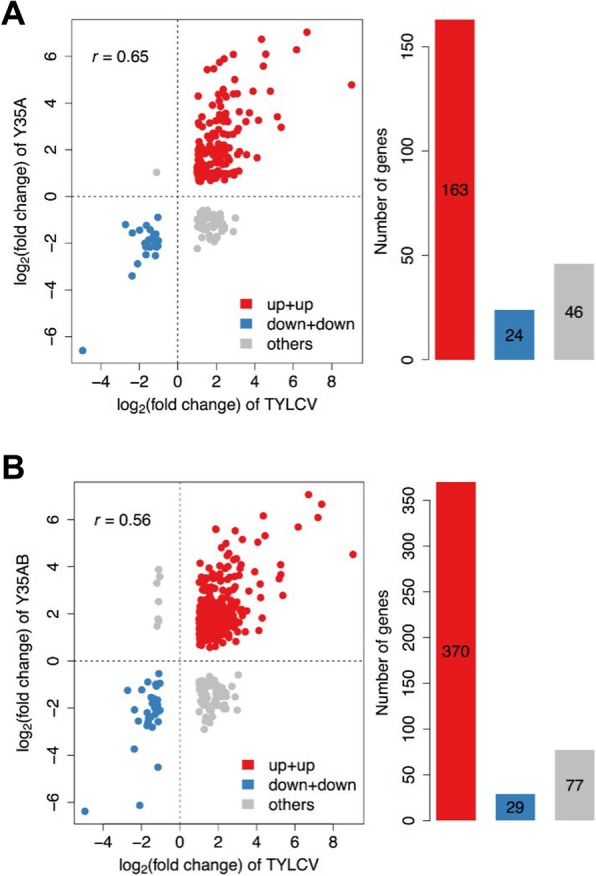
Systemic infection by TYLCV and TbSCV in N. benthamiana trigger partially overlapping transcriptional changes. The transcriptional changes triggered by the systemic infection by TYLCV were compared to those triggered by the geminivirus TbSCV, without (a) or with (b) its associated satellite, in N. benthamiana; the TbSCV data are from Li et al., 2018
Table 3.
GO enrichment of DEGs common to systemic infections by TYLCV and TbSCV in N. benthamiana. Over-represented GO categories (Biological Process Ontology) in the different subsets of DEGs represented in Fig. 6
| GO.ID | Term | Annotated | Significant | Expected | p value |
|---|---|---|---|---|---|
| Y35A_up+up | |||||
| GO:0006108 | malate metabolic process | 36 | 3 | 0.13 | 0.00027 |
| GO:0046470 | phosphatidylcholine metabolic process | 19 | 2 | 0.07 | 0.00197 |
| GO:0000272 | polysaccharide catabolic process | 25 | 2 | 0.09 | 0.00341 |
| GO:0016998 | cell wall macromolecule catabolic process | 29 | 2 | 0.1 | 0.00457 |
| GO:0006032 | chitin catabolic process | 31 | 2 | 0.11 | 0.00521 |
| GO:0045454 | cell redox homeostasis | 229 | 4 | 0.8 | 0.00835 |
| GO:0050832 | defense response to fungus | 4 | 1 | 0.01 | 0.01386 |
| GO:0042742 | defense response to bacterium | 4 | 1 | 0.01 | 0.01386 |
| GO:0046373 | L-arabinose metabolic process | 5 | 1 | 0.02 | 0.0173 |
| GO:1902358 | sulfate transmembrane transport | 7 | 1 | 0.02 | 0.02413 |
| GO:0019953 | sexual reproduction | 11 | 1 | 0.04 | 0.03767 |
| Y35A_down+down | |||||
| GO:0016114 | terpenoid biosynthetic process | 30 | 2 | 0.02 | 0.00028 |
| GO:0009813 | flavonoid biosynthetic process | 6 | 1 | 0 | 0.00498 |
| GO:0006012 | galactose metabolic process | 23 | 1 | 0.02 | 0.01897 |
| Y35AB_up+up | |||||
| GO:0006355 | regulation of transcription, DNA-templated | 1764 | 27 | 11.74 | 0.000032 |
| GO:0006529 | asparagine biosynthetic process | 11 | 2 | 0.07 | 0.0023 |
| GO:0006952 | defense response | 112 | 4 | 0.75 | 0.0067 |
| GO:0046470 | phosphatidylcholine metabolic process | 19 | 2 | 0.13 | 0.007 |
| GO:0006571 | tyrosine biosynthetic process | 3 | 1 | 0.02 | 0.0198 |
| GO:0005992 | trehalose biosynthetic process | 34 | 2 | 0.23 | 0.0215 |
| GO:0005975 | carbohydrate metabolic process | 1345 | 19 | 8.95 | 0.0215 |
| GO:0006979 | response to oxidative stress | 257 | 5 | 1.71 | 0.0288 |
| GO:0006421 | asparaginyl-tRNA aminoacylation | 5 | 1 | 0.03 | 0.0328 |
| GO:0046373 | L-arabinose metabolic process | 5 | 1 | 0.03 | 0.0328 |
| GO:0009607 | response to biotic stimulus | 43 | 2 | 0.29 | 0.0332 |
| Y35AB_down+down | |||||
| GO:0016114 | terpenoid biosynthetic process | 30 | 2 | 0.03 | 0.00032 |
| GO:0009813 | flavonoid biosynthetic process | 6 | 1 | 0.01 | 0.00529 |
Discussion
In this work, we describe and compare the genome-wide transcriptional changes detectable by RNA-seq occurring in N. benthamiana upon local or systemic infection by the geminivirus TYLCV. Our results show that, as expected and in agreement with previous works, infection by TYLCV causes a strong transcriptional reprogramming in the host; however, the detectable changes are more dramatic in our local infection system. It is possible that the local infection offers higher resolution owing to lower dilution of the infected cells; however, the opposite behavior of some DEGs in local and systemic samples suggests that the absence of transcriptional changes coming from non-infected cells and masking local changes also plays a role in the differences detected. Additionally, we cannot rule out that some differences between the two infected samples could be due to differences in the stage of the infection: although in both cases the virus is actively replicating in infected cells, in the leaf patch assay the infection is supposed to be more or less synchronized, while in the systemic infection a mixture of cells at different phases of the infection is most likely present in the samples. Another potentially relevant difference between local and systemic samples is the presence of Agrobacterium in the former; nevertheless, Agrobacterium is also present in the respective control. It is relevant to note that, if local and systemic samples show different results due to dilution and masking issues, these problems would potentially not only affect the study of transcriptional changes, but also the analysis of other molecular or physiological alterations resulting from the viral infection.
Functional enrichment analyses can help us gain insight into the biological processes underlying infection by TYLCV (Tables 2 and 3). In locally infected samples, over-represented GO categories included DNA recombination among the up-regulated genes, and cytokinesis among the down-regulated genes. These processes are expected to be connected to the viral manipulation of DNA replication and cell cycle, and might not be detectable in the systemically infected samples as a result of dilution. Photosynthesis also appears as transcriptionally down-regulated; photosynthetic shut-down seems to be a common outcome in viral infections ([8, 13–16], among others). A transcriptional negative regulation of BR signaling can also be detected in locally infected samples; these results are in agreement with a recent work by Seo et al. (2018), in which suppression of BR signaling was shown to underpin symptom development triggered by TYLCV in tomato.
Among those categories over-represented in both local and systemic infections, we can find cellulose biosynthetic process as transcriptionally repressed, suggesting that the viral infection might be affecting cell wall composition; changes in cell wall dynamics have been recently shown as triggered by Potato virus Y [17] and Rice tungro spherical virus [18], and in the latter case they have been proposed to correlate with virus-induced stunting. Intriguingly, both viruses negatively impact the cellulose biosynthetic machinery; whether impaired cellulose biosynthesis is a general plant response to the viral invasion is an idea that will require further investigation. Auxin signaling is also transcriptionally repressed upon the viral infection; these changes may also mediate or modulate the impact of the viral infection on plant development.
The identification of common subsets of up- and down-regulated genes in the systemic infections by TYLCV and TbSV [5] indicates that geminiviral manipulation of the cell and/or plant defence responses to geminiviral infection follow common transcriptional routes in different geminivirus/N. benthamiana interactions. Perhaps of particular interest is the finding that defence responses are activated in response to the viral infection; this observation reveals that these geminiviruses are being efficiently perceived as non-self by the plant, which in turn triggers a defence response. Although the activated plant defence reponses are not sufficient to fend off the virus, since the infection is established successfully, the fact that the plant is capable of detecting these pathogens in the first place paves the way for future engineering of the host, potentially boosting defences downstream of perception of the virus and hence tilting the balance in favour of the plant. Another pathway that emerges as a potential valuable target for engineering anti-geminiviral resistance is terpenoid biosynthesis, which is transcriptionally repressed by TYLCV and TbSV and has been proven to be suppressed by TYLCCN [10], raising the idea that its down-regulation might underpin a successful geminiviral infection.
Our results support the idea that the leaf patch assay entails great potential for the study of geminiviruses. Not only does this system result in a relatively synchronic infection and provides high resolution to detect virus-induced changes, but it also allows for the study of mutant viruses. All TYLCV null mutants for single genes, with the exception of those mutated in Rep, are capable of replicating their genome, but unable to infect the plant systemically; the use of local infections makes it possible to analyze the differences between the cellular changes triggered by wild-type and mutant viruses, therefore providing insight into the function of the viral genes in the context of the infection. An inherent limitation of this surrogate system, however, is the inevitable loss of cell type specificity: while TYLCV naturally infects phloem companion cells exclusively, in a leaf patch assay the virus is forced to replicate in mesophyll cells. Another obvious shortcoming is the impossibility of studying those mechanisms involved in cell-to-cell or long-distance transport, or the interactions between virus and vector.
All things considered, and while the analysis of systemic and local infections has provided and will continue to provide useful insight into the molecular events underlying the infection by geminiviruses, both approaches are imperfect for a number of reasons, as mentioned above. In order to considerably deepen our view, offering a substantial leap in our understanding of the molecular and physiological changes occurring during the plant-geminivirus interaction, isolating those infected cells, ideally based on the stage of the infection, will be crucial. Several approaches would enable the isolation of geminivirus infected cells: the use of transgenic plants harbouring a replicon-based system to label those cells sustaining active viral replication (like those described in [11]) could be combined with Fluorescence Activated Cell Sorting (FACS) or Laser Capture Microdissection (LCM), leading to the separation of infected from non-infected cells; high-throughput single-cell sequencing would also allow the unbiased identification and analysis of those cells containing the virus in the context of the infected plant. The increase in precision and resolution provided by the isolation of infected cells and their comparison to uninfected cells in the same plant will foreseeably result in an unprecedented view of the molecular landscaping triggered by the viral invasion.
Conclusions
Our results show that TYLCV induces a dramatic transcriptional reprogramming in N. benthamiana, the detection of which largely differs in local and systemic infections. Nevertheless, some responses, including a transcriptional repression of the auxin signaling pathway and a transcriptional activation of defence, can be commonly detected.
Comparison with the transcriptional changes induced by systemic infection by the geminivirus TbSV shows common subsets of up- and down-regulated genes similarly affected by both viral species, among which the suppression of terpenoid biosynthesis might be a general change triggered by geminiviruses. Taken together, our results not only provide insight into the transcriptional changes resulting from the infection by TYLCV in N. benthamiana, but also highlight the need to come up with an optimized system to gain a precise overview of the molecular and physiological changes caused in the host by the viral invasion.
Methods
Plant material and growth conditions
Wild-type N. benthamiana plants were grown in a controlled growth chamber in long day conditions (16 h light/8 h dark) at 25 °C.
Viral infections
The TYLCV infectious clone is described in [19, 20]; it contains a partial dimer of the TYLCV genome (AJ489258; [21]) in the pGWB501 vector [22]. Agrobacterium tumefaciens GV3101 strain was used for the delivery of TYLCV infectious clone and empty pGWB501 vector. Agrobacterium cells carrying these constructs were liquid-cultured in LB with appropriate antibiotics at 28 °C overnight. Bacterial cultures were centrifuged at 4000 g for 10 min and resuspended in the infiltration buffer (10 mM MgCl2, 10 mM MES pH 5.6, 150 μM acetosyringone) to an OD600 = 0.5. Bacterial suspensions were incubated in the buffer at room temperature and in the dark for 4 h before using them to infiltrate 4-week-old N. benthamiana for leave patch assays (local infections) and three-week-old N. benthamiana for systemic infection as described in [12].
RNA extraction
Total RNA was extracted from 8 mm leaf discs using the RNeasy plant mini kit (Qiagen) following the manufacturer’s instructions.
RNA sequencing
Transcriptome analyses were performed at the Genomic Core Facility, Shanghai Center for Plant Stress Biology, CAS. Three biological replicates were used. Total RNA (1 μg) from each sample was used for library preparation with NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England BioLabs, E7420L) following the manufacturer’s instructions. Prepared libraries were assessed for quality using NGS High-Sensitivity kit on a Fragment Analyzer (AATI) and for quantity using Qubit 2.0 fluorometer (Thermo Fisher Scientific). All libraries were sequenced in paired-end 125 bases protocol (PE125) on an Illumina HiSeq sequencer.
Quantitative RT-PCR
First-strand cDNA synthesis was performed with the iScriptTM cDNA Synthesis Kit (Bio-Rad #1708890) according to the manufacturer’s instructions. For qPCR reactions, the reaction mixture consisted of cDNA first-strand template, primers (500 nM each) and iTaqTM Universal SYBRR Green Supermix (Bio-Rad, #1725120). qPCR was performed in a BioRad CFX96 real-time system. Expression result was determined using the comparative Ct method (2-ΔΔCt). Primers used are described in Additional file 9: Table S4. NbACT was used as the reference gene, using primers described in [23].
Preprocessing of RNA-Seq data
We cleaned the paired-end reads by Trimimomatic [24] (version 0.36). After trimming the adapter sequence, removing low quality bases and filtering short reads, clear read pairs were retained for further analysis.
Mapping and quantification of TYLCV reads
Cleaned reads were mapped to TYLCV DNA (GenBank: AJ489258.1) and its six ORFs by HISAT [25] (version 2.1.0) with default parameters. The RPM (Reads per Million) was used to quantify the expression level of each ORF and the whole viral genome. The read coverage of each base on the reference DNA was calculated by samtools [26] (version 1.5) with maximum coverage depth 8000 (−d 8000) and normalized to RPM. The expression level and read coverage were calculated for forward and reverse strand, respectively. The circular viral genome and read coverage of RNA-Seq data were visualized by CGView [27].
Reads mapping and quantification of N. benthamiana genes
The N. benthamiana draft genome sequence [28] (v1.0.1) was downloaded from the Sol Genomics Network (ftp://ftp.solgenomics.net/genomes/Nicotiana_benthamiana/assemblies/). Cleaned reads were mapped to the genome sequence by HISAT with default parameters. Number of reads that were mapped to each N. benthamiana gene was calculated with the htseq-count script in HTSeq [28].
Differential gene expression analysis
EdgeR [29] was used to identify genes that were differentially expressed between control and experiment samples. Genes with at least two-fold change in expression and had a FDR < 0.05 were considered differentially expressed genes (DEGs).
GO enrichment analysis
The Gene Ontology (GO) terms assigned to N. benthamiana genes were extracted from annotation file downloaded from the SGN (ftp://ftp.solgenomics.net/genomes/Nicotiana_benthamiana/annotation/Niben101/). GO enrichment analysis of DEGs was implemented by topGO [30] with custom gene-to-GOs mapping annotations.
KEGG pathway enrichment analysis
Since N. benthamiana is not supported by KEGG, we searched its ortholog genes in Arabidopsis by BLAST+ (version 2.5.0, evalue = 1e-5). The ortholog genes of the DEGs were used to perform KEGG enrichment analysis by clusterProfiler [31]. The enriched KEGG pathways were visualized with DEGs by Pathview [32].
Additional files
{kind=link}
Figure S1. PCA analysis of the RNA-seq datasets (local and systemic TYLCV infections) (JPG 529 kb)
{kind=link}
Figure S2. Pictures of plants systemically infected by TYLCV at the time of sample collection (JPG 299 kb)
{kind=link}
Figure S3. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the MAPK signaling pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 919 kb)
{kind=link}
Figure S4. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the plant hormone signal transduction pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 759 kb)
{kind=link}
Figure S5. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the plant-pathogen interaction pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 640 kb)
Table S1. DEGs in local TYLCV infections (XLSX 1067 kb)
Table S2. Reads mapping to the viral genome in local and systemic infections (XLSX 10 kb)
Table S3. DEGs in systemic TYLCV infections (XLSX 146 kb)
Table S4. Primers used in this work (DOCX 14 kb)
Acknowledgements
The authors would like to thank the PSC Genomics Core Facility; Eduardo R Bejarano and Araceli G Castillo for useful discussions; Laura Medina-Puche for valuable advice on figure design; and Xinyu Jian, Aurora Luque, and Yujing (Ada) Liu for technical assistance.
Authors’ contributions
R-LD and XF conceived the project; MW, XD, and XF performed and analysed data; R-LD wrote the manuscript, with input from all authors. All authors read and approved the manuscript.
Funding
This work is funded by the Shanghai Center for Plant Stress Biology, Chinese Academy of Sciences (CAS); the 100 Talent program from CAS (to RL-D); and the Chinese Academy of Sciences Strategic Pilot Science and Technology Special (b) Funding XDB27040206 (to RL-D). The funders had no role in study design, data collection and analysis, decision to publish, or preparations of the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mengshi Wu and Xue Ding contributed equally to this work.
Contributor Information
Mengshi Wu, Email: mswu@psc.ac.cn.
Xue Ding, Email: dingxue@sibs.ac.cn.
Xing Fu, Email: xfu@sibcb.ac.cn.
Rosa Lozano-Duran, Email: lozano-duran@sibs.ac.cn.
References
- 1.Allie F, Pierce EJ, Okoniewski MJ, Rey C. Transcriptional analysis of south African cassava mosaic virus-infected susceptible and tolerant landraces of cassava highlights differences in resistance, basal defense and cell wall associated genes during infection. BMC Genomics. 2014;15:1006. doi: 10.1186/1471-2164-15-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascencio-Ibanez JT, Sozzani R, Lee TJ, Chu TM, Wolfinger RD, Cella R, Hanley-Bowdoin L. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 2008;148(1):436–454. doi: 10.1104/pp.108.121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen T, Yuanda LV, Zhao T, Nan L, Yang Y, Yu W, He X, Liu T, Zhang B. Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus. PLoS One. 2013;8(11):e80816. doi: 10.1371/journal.pone.0080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Góngora-Castillo E, Ibarra-Laclette E, Trejo-Saavedra DL, Rivera-Bustamante RF. Transcriptome analysis of symptomatic and recovered leaves of geminivirus-infected pepper (Capsicum annuum) Virol J. 2012;9:295. doi: 10.1186/1743-422X-9-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li K, Wu G, Li M, Ma M, Du J, Sun M, Sun X, Qing L. Transcriptome analysis of nicotiana benthamiana infected by tobacco curly shoot virus. Virol J. 2018;15(1):138. doi: 10.1186/s12985-018-1044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis B, Rey C. Resistance gene analogs involved in tolerant cassava–geminivirus interaction that shows a recovery phenotype. Virus Genes. 2015;51(3):393–407. doi: 10.1007/s11262-015-1246-1. [DOI] [PubMed] [Google Scholar]
- 7.Naqvi AR, Sarwat M, Pradhan B, Choudhury NR, Haq QM, Mukherjee SK. Differential expression analyses of host genes involved in systemic infection of tomato leaf curl New Delhi virus (ToLCNDV) Virus Res. 2011;160(1–2):395–399. doi: 10.1016/j.virusres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Seo JK, Kim MK, Kwak HR, Choi HS, Nam M, Choe J, Choi B, Han SJ, Kang JH, Jung C. Molecular dissection of distinct symptoms induced by tomato chlorosis virus and tomato yellow leaf curl virus based on comparative transcriptome analysis. Virology. 2018;516:1–20. doi: 10.1016/j.virol.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Pierce EJ, Rey ME. Assessing global transcriptome changes in response to south African cassava mosaic virus [ZA-99] infection in susceptible Arabidopsis thaliana. PLoS One. 2013;8(6):e67534. doi: 10.1371/journal.pone.0067534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luan JB, Yao DM, Zhang T, Walling LL, Yang M, Wang YJ, Liu SS. Suppression of terpenoid synthesis in plants by a virus promotes its mutualism with vectors. Ecol Lett. 2013;16(3):390–398. doi: 10.1111/ele.12055. [DOI] [PubMed] [Google Scholar]
- 11.Morilla G, Castillo AG, Preiss W, Jeske H, Bejarano ER. A versatile transreplication-based system to identify cellular proteins involved in geminivirus replication. J Virol. 2006;80(7):3624–3633. doi: 10.1128/JVI.80.7.3624-3633.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lozano-Duran R, Rosas-Diaz T, Luna AP, Bejarano ER. Identification of host genes involved in geminivirus infection using a reverse genetics approach. PLoS One. 2011;6(7):e22383. doi: 10.1371/journal.pone.0022383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bengyella L, Waikhom SD, Allie F, Rey C. Virus tolerance and recovery from viral induced-symptoms in plants are associated with transcriptome reprograming. Plant Mol Biol. 2015;89(3):243–252. doi: 10.1007/s11103-015-0362-6. [DOI] [PubMed] [Google Scholar]
- 14.Kyselakova H, Prokopova J, Naus J, Novak O, Navratil M, Safarova D, Spundova M, Ilik P. Photosynthetic alterations of pea leaves infected systemically by pea enation mosaic virus: a coordinated decrease in efficiencies of CO (2) assimilation and photosystem II photochemistry. Plant Physiol Biochem. 2011;49(11):1279–1289. doi: 10.1016/j.plaphy.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Lei R, Jiang H, Hu F, Yan J, Zhu S. Chlorophyll fluorescence lifetime imaging provides new insight into the chlorosis induced by plant virus infection. Plant Cell Rep. 2017;36(2):327–341. doi: 10.1007/s00299-016-2083-y. [DOI] [PubMed] [Google Scholar]
- 16.Perez-Bueno ML, Rahoutei J, Sajnani C, Garcia-Luque I, Baron M. Proteomic analysis of the oxygen-evolving complex of photosystem II under biotec stress: studies on nicotiana benthamiana infected with tobamoviruses. Proteomics. 2004;4(2):418–425. doi: 10.1002/pmic.200300655. [DOI] [PubMed] [Google Scholar]
- 17.Otulak-Kozieł Katarzyna, Kozieł Edmund, Lockhart Benham. Plant Cell Wall Dynamics in Compatible and Incompatible Potato Response to Infection Caused by Potato Virus Y (PVYNTN) International Journal of Molecular Sciences. 2018;19(3):862. doi: 10.3390/ijms19030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budot BO, Encabo JR, Ambita ID, Atienza-Grande GA, Satoh K, Kondoh H, Ulat VJ, Mauleon R, Kikuchi S, Choi IR. Suppression of cell wall-related genes associated with stunting of oryza glaberrima infected with rice tungro spherical virus. Front Microbiol. 2014;5:26. doi: 10.3389/fmicb.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Tan H, Wu M, Jimenez-Gongora T, Tan L, Lozano-Duran R. Dynamic virus-dependent subnuclear localization of the capsid protein from a geminivirus. Front Plant Sci. 2017;8:2165. doi: 10.3389/fpls.2017.02165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosas-Diaz T, Zhang D, Fan P, Wang L, Ding X, Jiang Y, Jimenez-Gongora T, Medina-Puche L, Zhao X, Feng Z, et al. A virus-targeted plant receptor-like kinase promotes cell-to-cell spread of RNAi. Proc Natl Acad Sci U S A. 2018;115(6):1388–1393. doi: 10.1073/pnas.1715556115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morilla G, Janssen D, Garcia-Andres S, Moriones E, Cuadrado IM, Bejarano ER. Pepper (Capsicum annuum) is a dead-end host for tomato yellow leaf curl virus. Phytopathology. 2005;95(9):1089–1097. doi: 10.1094/PHYTO-95-1089. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Suzuki T, Murata S, Nakamura S, Hino T, Maeo K, Tabata R, Kawai T, Tanaka K, Niwa Y, et al. Improved gateway binary vectors: high-performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci Biotechnol Biochem. 2007;71(8):2095–2100. doi: 10.1271/bbb.70216. [DOI] [PubMed] [Google Scholar]
- 23.Viczián O, Künstler A, Hafez YM, Király L. Catalases may play different roles in influencing resistance to virus-induced hypersensitive necrosis. Acta Phytopathologica et Entomologica Hungarica. 2014;49(2):189–200. doi: 10.1556/APhyt.49.2014.2.5. [DOI] [Google Scholar]
- 24.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21(4):537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- 28.Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB. A draft genome sequence of nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant-Microbe Interact. 2012;25(12):1523–1530. doi: 10.1094/MPMI-06-12-0148-TA. [DOI] [PubMed] [Google Scholar]
- 29.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R package version. 2009;1162.
- 31.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo W, Brouwer C. Pathview: an R/bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PCA analysis of the RNA-seq datasets (local and systemic TYLCV infections) (JPG 529 kb)
Figure S2. Pictures of plants systemically infected by TYLCV at the time of sample collection (JPG 299 kb)
Figure S3. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the MAPK signaling pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 919 kb)
Figure S4. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the plant hormone signal transduction pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 759 kb)
Figure S5. Differentially expressed genes of local (A) and systemic (B) TYLCV infections in the plant-pathogen interaction pathway. The log2 transformed expression fold changes of DEGs are converted to pseudo colors using default limit (e.g. from − 1 to 1), and mapped to KEGG pathway by R package pathview. The up- and down- regulated genes are labeled in red and green, respectively. (JPG 640 kb)
Table S1. DEGs in local TYLCV infections (XLSX 1067 kb)
Table S2. Reads mapping to the viral genome in local and systemic infections (XLSX 10 kb)
Table S3. DEGs in systemic TYLCV infections (XLSX 146 kb)
Table S4. Primers used in this work (DOCX 14 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files).