
Figure 1.
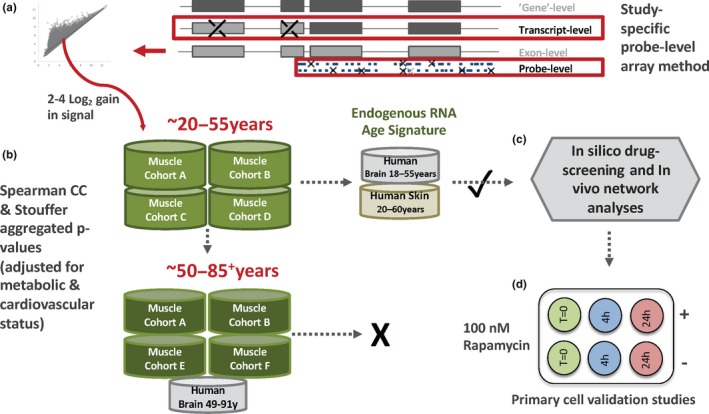
A schematic representation of the study analysis strategy. (a) For the HTA 2.0 or exon arrays, the 25‐mer array probes were realigned to the current genome; “single match” probes were GC content‐adjusted and study‐specific expression confirmed (low signal/variance filtering) before creating the template for combining probes into a transcript signal (selected from ensembl, ENST, Figure S1). (b) Linear modeling for “age” versus RNA was conducted using independent cohorts of human muscle profiles from physiologically characterized “healthy” drug‐free humans (n = 330 biopsies for decades third to sixth, n = 247 for decades sixth to ninth). The clinical data originate from our studies: Cohort A (Timmons et al., 2018), Cohort B (Phillips et al., 2013), Cohort C (AbouAssi et al., 2015), Cohort D (Phillips et al., 2017), Cohort E (Slentz et al., 2016), and Cohort F (Hangelbroek et al., 2016). The pattern of muscle age‐related transcript expression was confirmed in human brain (n = 299) and skin (n = 59), relying on published exon array data and our optimized transcript detection protocol. (c) An age‐related protein‐coding transcriptome was identified, adjusting for metabolic and aerobic capacity, and this provided a robust framework for characterization of the biology of age‐regulated lncRNAs, which are largely of unknown function, using network analysis and an age signature for in silico cMAP database drug screening. (d) The results of in silico drug screening were validated primary muscle cell studies