


Abstract
Human mitochondrial DNA (h-mtDNA) codes for 13 subunits of the oxidative phosphorylation pathway, the essential route that produces ATP. H-mtDNA transcription and replication depends on the transcription factor TFAM, which also maintains and compacts this genome. It is well-established that TFAM activates the mtDNA promoters LSP and HSP1 at the mtDNA control region where DNA regulatory elements cluster. Previous studies identified still uncharacterized, additional binding sites at the control region downstream from and slightly similar to LSP, namely sequences X and Y (Site-X and Site-Y) (Fisher et al., Cell 50, pp 247–258, 1987). Here, we explore TFAM binding at these two sites and compare them to LSP by multiple experimental and in silico methods. Our results show that TFAM binding is strongly modulated by the sequence-dependent properties of Site-X, Site-Y and LSP. The high binding versatility of Site-Y or the considerable stiffness of Site-X tune TFAM interactions. In addition, we show that increase in TFAM/DNA complex concentration induces multimerization, which at a very high concentration triggers disruption of preformed complexes. Therefore, our results suggest that mtDNA sequences induce non-uniform TFAM binding and, consequently, direct an uneven distribution of TFAM aggregation sites during the essential process of mtDNA compaction.
INTRODUCTION
Human mitochondria contain hundreds of copies of a 16.5 kb circular genome (h-mtDNA) that encodes 13 out of the ∼80 subunits of the oxidative phosphorylation pathway (OXPHOS), the main source of ATP. Mutations, deletions, or misregulation of mtDNA impair the OXHPOS, which leads to considerable alterations of cell functions that are at the basis of rare diseases and syndromes (1), have been associated with aging (2) and can aggravate major diseases such as diabetes (2), cancer (2), obesity (3) and neurological disorders such as Alzheimer's and Parkinson's diseases (4). Therefore, mtDNA integrity must be well-preserved for correct cell function. Apart from OXPHOS proteins, tRNAs and rRNAs are encoded by both h-mtDNA strands, termed the heavy (HS) and light (LS) strands. Additionally, h-mtDNA contains a non-coding control region (CR) of 1.1 kb that harbors mtDNA control elements, including the heavy and light strand promoters (HSP and LSP, respectively) and the heavy strand origin of replication (OH), which is downstream from LSP (5,6). In addition, downstream from LSP there are three conserved sequence blocks named CSB- I (mtDNA coordinates nt 213–235), II (nt 299–315) and III (closest to LSP, nt 346–363) that are preserved in mammals (7) (Figure 1A). In addition to transcription, LSP together with CSB-II is further involved in mtDNA replication. Former studies showed that RNA fragments originated at LSP occasionally terminate downstream of CSB-II, creating a ∼120 nt RNA (R-loop) whose free 3′ ends prime the DNA polymerase so that RNA to DNA transitions are generated (6,8,9). This premature RNA termination depends on a poly-guanine sequence at CSB-II that forms a hybrid G-quadruplex DNA structure between the displaced HS and the transcribed RNA, which presumably destabilizes the RNA polymerase (mtRNAP) (10–13). Directly downstream of CSB-II is a six-adenine tract (A-tract, Figure 1A) whose mutations decrease the termination effect at CSB-II (11,13). This A-tract is at the centre of Site-X, a motif that overlaps 13 nucleotides from CSB-II and 15 nucleotides downstream (Figure 1A). Site-X together with Site-Y, which is right upstream of CSB-I and partially overlaps it (thus, both Site-X and Site-Y sequences lay between CSB-II and CSB-I Figure 1A), were both identified as sequences similar to the sites that are bound by the mitochondrial transcription factor A (TFAM or mtTFA) within promoters LSP and HSP1. From these latter, the protein recruits the transcription machinery (14), but both Site-X and Site-Y are not close to any transcription initiation site. TFAM is also involved in mtDNA compaction, and studies by in organello mtDNA methylation protection and hypersensitivity showed an organized phased binding of TFAM along the control region that was related to the compaction mechanism at this area (15). In particular, these studies specifically showed binding of TFAM to 28 bp precisely at Site-X (nt 276–303) and Site-Y (nt 233–260). In addition, recent ChIP-seq analysis in HeLa cells showed that the Site-Y sequence is a TFAM enriched site, whereas Site-X sequence is partially enriched (16). Interestingly, the slight similarity of Site-X and Site-Y to LSP (14,15) (Figure 1A and B) suggests a similar binding mechanism.
Figure 1.

Site-X and Site-Y sites and their complexes with TFAM. (A) The sequences of Site-Y, Site-X and LSP described by Fisher and collaborators (14) are indicated in turquoise. Conserved Sequence Block (CSB) I, II and III are framed and indicated. TFAM Hmg-box1 (HMG1) and HMG-box2 (HMG2) domains are represented on Site-Y (HMG1 in dark magenta, HMG2 in red), Site-X (HMG1 in blue, HMG2 in violet), and on LSP (HMG1 in orange, HMG2 in green). Note the inverted orientation of TFAM on Site-Y with respect to the other two sequences. Black arrowheads indicate the insertion sites. mtDNA sequence numbering is shown on the left, numbering of the DNA sequence, on the top. (B) Aligned sequences used for crystallization (corresponding to dsDNA) are shown from 5′ to 3′ (black arrow). mtDNA sequence numbering is indicated. Sites inserted by HMG-box1 and HMG-box2 are indicated by red and green arrowheads, respectively. In the right column, the orientation of the HMG boxes on the DNAs is represented. (C) TFAM/Site-X complex. Coloring as in panel (A); the domains Hmg-box1 (HMG1), HMG-box2 (HMG2), and linker are indicated, together with the N-and C-terminal ends. Leu58 and Leu182 side chains are depicted in green. The DNA sequence is indicated in one-letter code. (D) TFAM/Site-Y complex, representation as in (C). Note the DNA sequence assignment is tentative. (E) Superposition of crystal structures of TFAM/LSP (in gray), TFAM/Site-X (blue) and TFAM/Site-Y (red) by respective HMG-box1 domains. (F) Superimposition by HMG-box2.
The crystal structure of TFAM in complex with LSP shows that binding at this promoter occurs at a 22 bp sequence upstream of the transcription initiation site (14,17–19) (Figure 1A). The protein contains two HMG-box domains (HMG-box1 and HMG-box2, roughly 60 aa each) separated by a linker (30 aa) and followed by a flexible C-terminal tail. By means of a concerted mechanism between the HMG-boxes and linker (20), the HMG-boxes perform contacts with the phosphate backbone and base atoms and insert residues between base pairs (bp), strongly bending LSP in a U-turn (17,18), and bringing HMGbox2 close to the transcription initiation site to which it recruits mtRNAP and transcription factor TFB2 and form the initiation complex (19). In addition, HMG-box proteins recognize highly distorted DNA. TFAM, as such, shows in vitro high affinity for damaged or cisplatinated DNA (21), cruciforms (22), tRNAs and four-way junctions (23), or G-quadruplexes (24). The two TFAM activities, transcription initiation and mtDNA compaction, are balanced by mitochondrial TFAM levels, i.e. lower TFAM concentrations stimulate transcription, whereas high amounts lead to compaction (25–28).
TFAM binds to sequences that, albeit similar to LSP, are not involved in transcription activation but subjected to TFAM phased binding. This poses the question of whether the distortions induced by TFAM to LSP would also occur in Site-X and Site-Y during compaction at the mtDNA control region. To this end and gain insight into the DNA properties underlying dsDNA recognition by TFAM, we characterized Site-X and Site-Y free or in complex with the protein, by X-ray crystallography, molecular dynamics (MD) simulations, FRET and other biophysical methods. We compared these sites with binding to LSP, which is the best characterized binding site as shown by several X-ray structures, FRET, SAXS and mutational analyses (17,18,29). Our results show that sequence-dependent DNA conformation modulates TFAM binding, which does not always occur as predicted by sequence alignment. In addition, we show that a simple concentration effect of TFAM/DNA complexes induces their multimerization. Mutations at TFAM interfaces identified in the crystal structures disrupt this effect. These results suggest a mechanism of mtDNA compaction based on DNA-sequence dependent binding modes, and that multimerization of the complexes is mediated by protein-protein interactions involving distant surfaces.
MATERIALS AND METHODS
TFAM–DNA complex preparation
Mature full-length TFAM (residues 43–246; UniProtQ00059) in complex with double-stranded DNA (dsDNA) fragments was prepared as reported previously (18). Based on the alignment of the crystallized DNA sequence from the TFAM/LSP complex, oligonucleotides harboring 22 bp from both Site-X (5′-TAACAAAAAATTTCCACCAAAC-3′; 5′-TTTGGTGGAAATTTTTTGTTAG-3′, with overhanging ends) and SITE-Y (5′-TAACAATTGAATGTCTGCACAG-3′, and the complementary sequence that generated blunt ends) were purchased (Biomers) and annealed. TFAM:DNA complexes were assembled by mixing the protein and DNA in the gel filtration buffer (50 mM HEPES pH 7.5, 750 mM NaCl, 5 mM DTT) and the mixture was dialyzed stepwise at 4°C in buffer A (50 mM HEPES pH7.5, 500 mM NaCl, 5 mM DTT), buffer B (50 mM HEPES pH 7.5, 250 mM NaCl, 5 mM DTT) and buffer C (50 mM HEPES pH 7.5, 20 mM NaCl, 5 mM DTT). The resulting sample was concentrated and polydispersity was assessed to be low (<20%) by dynamic light scattering. The C246A mutant was generated by ‘round the horn PCR’ (30) with Herculase polymerase (Agilent). The HMGbox1 dimerization mutant gene (TFAM-Box1Mut) was a kind gift from David Chan's laboratory (31). The C-terminal tail deletion mutant gene (TFAM-CTΔ26) was purchased (Genscript) cloned in plasmid pET-28a(+). Proteins were expressed in pLysS Escherichia coli strain. Cultures were grown at 37°C to an OD of 0.75–0.8 and expression was induced with IPTG (1 mM) for 4 h. Lysis and purification procedures were the same as for WT TFAM (18), and included a proteolytic cleavage step with thrombin (1 U/100 μg protein) to get rid of the N-terminal his tag and a linker sequence. See Supplementary Figure S9 for quality of the samples.
TFAM/DNA complex crystallization and structure analysis
Sitting-drop vapor diffusion crystallization assays (using Cryschem plates from Hampton Research) yielded crystals of TFAM/Site-X complex by mixing 1 μl of complex solution (TFAM at 13 mg ml−1, and DNA at half molarity since previous titrations showed full shift of the DNA at this ratio (18), in 50 mM HEPES, 20 mM sodium chloride, 5 mM DTT, pH 7.5), and 1 μl of reservoir (18–24% PEG 3350, sodium potassium tartrate 0.1–0.25 M), at 20°C. TFAM/SITE-Y and TFAM/SITE-YBr crystals were obtained by mixing 1 μl of complex solution (TFAM at 12 mg ml−1 in 50 mM HEPES, 20 mM NaCl, 5 mM DTT, pH 7.5), and 1μl of reservoir (23–28% PEG 3350, 0.08–0.2 M ammonium acetate, and 0.1 M Bis–Tris pH 6.5 or HEPES pH 7.5), at 20°C. Crystals were cryoprotected by mixing the reservoir solution with 10–20% PEG400. Diffraction data were collected from single liquid-N2 flash-cryo-cooled crystals at 100 K (Oxford Cryosystems 700 series) at beamlines ID23-2 (native datasets) from the European Synchrotron Radiation Facility (ESRF) and at XALOC (bromine dataset, collected at λ = 0.919183 after an absorption near-edge scan XANES that confirmed the presence of Br) from the Spanish synchrotron ALBA. Crystals of TFAM/Site-X and TFAM/SITE-Y belonged to monoclinic C121 space group with four complexes in the asymmetric unit (a.u.), with respective Matthews’ coefficients of 3.31 and 2.59 Å3/Da. TFAM/SITE-YBr crystallized in the orthorhombic primitive P21212 space group and contained two complexes in the a.u. Diffraction data were integrated, scaled, merged, and reduced with XDS (32) and SCALA (33), the latter within the CCP4 suite (33). The phase problem of native data sets was solved by molecular replacement (MR) with Phaser (34) employing the TFAM/LSP structure (18) as a searching model. Crystallographic refinement with Phenix (35) and Buster (36) followed, which included TLS refinement, non-crystallographic symmetry, and Watson and Crick base pairing of the DNA. These were alternated with manual model building with COOT (37) until the models were completed. TFAM/Site-X includes residues Ser43 to Gln234 and TFAM/Site-Y Ser43 to Lys237 and respective DNA sequences. Structural superimpositions were performed with COOT by selecting specific regions (HMGbox1, box2, the linker or the DNA). The structures of TFAM in complex with Site-Y brominated at different thymine positions were solved by MR and refined as above. Anomalous difference maps were calculated (33) but no anomalous peak was detected even though XANES had confirmed the presence of Br in the crystal (see above).
Data access
Structures are deposited at the Protein Data bank with PDB codes 6HC3 (TFAM/Site-X complex) and 6HB4 (TFAM/Site-Y complex).
Computational analysis
Molecular dynamics (MD) simulations for the free sequences LSP, Site-X and Site-Y were performed starting from the B-DNA conformation (38) by using Amber14 package (39). All simulations were carried out using state-of-the-art simulation conditions (40,41), the newly developed parmbsc1 force field for DNA (42), an explicit TIP3P water model and sodium and chlorine ions to neutralize the systems and provide a realistic ionic environment (150 mM NaCl). All simulations were performed using a truncated octahedron periodic box, periodic boundary conditions and Particle-Mesh-Ewald method for long range electrostatics. In all three cases, we performed 500 ns long MD simulations. Collected trajectories were processed with the cpptraj (43) module of AMBERTOOLS 15 package (39). All the data can be found in the NAFlex server (44) that was also used for standard analysis.
The structural changes of DNA along MD simulations and the DNA structure in the crystals (TFAM/LSP, PDB code 3TQ6; TFAM/SITE-X and TFAM/SITE-Y; and TFAM/nsDNA, PDB code 4NNU, respectively) were studied using the DNA helical base-pair step parameters calculated by CURVES+ and CANAL programs (45). DNA base-pair step parameters consist of three translational (shift, slide and rise) and rotational (tilt, roll and twist) movements between each base-pair step. DNA deformability along these six directions can be described by pure stiffness constants (kshift, kslide, krise, ktilt, kroll and ktwist) that are extracted from the diagonal of the associated stiffness constant matrix (46). Total stiffness (Ktot) is obtained as a product of these six constants and provides a rough estimate of the flexibility of each base pair step.
The deformation energy was calculated using a mesoscopic energy model (47), which is based on a harmonic approximation to describe deformability along DNA helical parameters. The deformation energy is the energy required to reproduce the DNA conformation as found in the protein-bound DNA complex, starting from its free conformation. For each of the three cases, we took as reference structure the DNA conformation as found in the X-ray crystal complex, and the equilibrium geometry and stiffness force constants were extracted from a dataset built from long all-atoms MD simulations of short oligonucleotides in water using the parmbsc1 force field.
DNA intrinsic curvature calculation
The overall intrinsic curvature of the free DNAs and the bending of the DNA in the protein-bound structures were calculated using rotational base pairs parameters at each step by the equation:
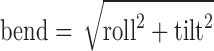 |
Isothermal titration calorimetry (ITC)
ITC experiments were performed at the Centres Científics i Tecnològics of the University of Barcelona using a VP-ITC microcalorimeter (MicroCal) with VPViewer2000 software for instrument control and data acquisition. Experiments were performed at 20°C and the heat of dilution, measured by the injection of titrant into the buffer solution, was subtracted for each titration to obtain the net reaction heat value. TFAM, Site Y, Site X and LSP (all DNAs from SIGMA, desalting purity) were dialyzed with a dialysis tubing pore of 3500 Da. To ensure identical buffer conditions, all dialyses were performed in the same vessel containing 150 mM NaCl, 50 mM HEPES pH 7.5, which was refreshed four times in 36 h at 4°C. In a typical experiment, TFAM (6–9 μM) was loaded in the sample cell at 20°C. Once the system reached equilibrium, 30 injections of 10 μl of a solution containing Site-Y or LSP were added to the cell solution, with intervals ranging from 180 to 300 s between injections to achieve complete equilibration and with an injection time of 20 s. Titration with Site-X involved 15 injections of 20 μl, 30 s/injection. The DNA concentrations were between 40 and 50 μM. Three independent measurements were performed for each TFAM/DNA complex and the best-fit values were averaged and reported. The complete binding isotherms were fit using a model consisting of one set of binding sites, by a nonlinear least squares algorithm implemented in the MicroCal Origin® (version 7.0) add-on ITC analysis software.
Electrophoretic mobility shift assays
Binding of TFAM to LSP, Site-X and SITE-Y was analyzed by non-denaturing PAGE, using 32P-labeled DNA or fluorescence. Affinity measurements of TFAM using 32PDNA were realized in triplicate with 1 nM DNA according to the protocol and calculation described in (24) in 10 μl reactions containing 25 mM Tris–HCl pH 7.0, 90 mM NaCl, 1 mM DTT, 3% glycerol and 0.01% Tween20 for 30 min at room temperature. TFAM serial protein dilutions were performed on ice in 5% glycerol, 700 mM NaCl, 20 mM Tris–HCl pH 7.0, 1 mM DTT and 0.01% Tween20. For the isotopic dilution of protein/DNA complexes (including WT, CT-Δ26 and Box1Mut TFAM proteins), 5′-fluorescein-labeled DNA was kept constant at 10 nM and complemented with non-labeled DNA to reach the indicated concentrations. Each protein was then added to the corresponding mixture and incubated for 45 min at room temperature before loading on the gel. The competition experiments used fluorescence detection of 5′-fluorescein-labeled DNAs. The protein and DNA were mixed in the gel filtration buffer and subsequently dialyzed in buffer A, buffer B (50 mM HEPES pH 7.5, 250 mM NaCl, 5 mM DTT) and a buffer D (50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM DTT). The dialyzed complexes were supplemented with a loading dye and the reactions mixtures were loaded onto 11% polyacrylamide native gel buffered with 0.5× TBE (0.5×). 8 × 10 cm gels were run on a Miniprotean II (Biorad) at 10 V/cm. The gels were next stained scanned for fluorescence with a Typhoon 8600 imager (GeHealthcare).
SEC-MALLS
Size exclusion chromatography in combination with multiangle laser light scattering (SEC-MALLS) was performed using a chromatographic system (1200 pump, Agilent Technologies) connected to a MALLS instrument (DAWN EOS, Wyatt Technology) and, in parallel, to a refracting index detector (Optilab rEX, Wyatt Technology). Protein, DNAs and protein/DNA complexes were analyzed by this technique. Protein concentration was previously estimated by the Bradford method. For TFAM/Site-Y complex, TFAM concentration was 351.5 μM (9 mg/ml); for TFAM/Site-X was 321.5 μM (8 mg/ml); and the two concentrations for TFAM/LSP were 218.7 μM (5.6 mg/ml) and 390.6 μM (10 mg/ml). In all cases, the protein:DNA ratio was 2:1. Samples with TFAM alone (97.6 μM, i.e. 2.5 mg/ml) and individual Site-Y, Site-X and LPS DNAs (200 μM each) were also analyzed. Samples were loaded onto a Superdex 75 10/300 (GE) size exclusion column equilibrated with 100 mM NaCl, 50 mM HEPES pH 7.5, 5 mM DTT, and the molecular weight (Mw) of the eluted species was estimated by employing ASTRA software (Wyatt Technology). ASTRA uses scattering data acquired across the entire sample peak and the 46 is determined with the Debye fitting method. A dn/dc value of 0.178 ml/g, 0.185 ml/g and 0.166 ml/g was used for the TFAM/DNA complexes (1:1), and protein and DNA alone, respectively. A calibration constant of 9.2 was obtained using ovalbumin. The experiments were performed at 0.5 and 0.3 ml/min fluxes, at RT.
Analytical ultracentrifugation analysis
TFAM/Site Y, Site X and LSP complexes were analyzed by sedimentation velocity (SV) using 4:1 and 2:1 protein:DNA concentration ratios. TFAM concentrations were 508 μM (13 mg/ml) and 429 μM (11 mg/ml) for the 2:1 and 4:1 ratios, respectively. TFAM alone was at 78 μM (2 mg/ml) whereas Site-Y, Site-X and LSP DNAs were at 200 μM. All samples were equilibrated in 100 mM NaCl, 50 mM HEPES pH 7.5, 2 mM TCEP. For an accurate analysis, the mixtures were incubated for long enough to reach equilibrium before starting the SV experiment. SV experiments were performed at 48 krpm and 20°C in a Beckman Optima XL-I analytical ultracentrifuge (Beckman–Coulter) equipped with absorbance and interference optics, using an An50Ti rotor. Absorbance scans (0.003 cm step size) were taken at 295 nm. Differential sedimentation coefficient distributions, c(s), were calculated by least squares boundary modeling of sedimentation velocity data using the program SEDFIT (48,49). The experimental values obtained from this analysis were corrected for solvent composition and temperature to obtain the corresponding standard S values (s20,w) using SEDNTERP software (50).
FRET measurements
Mutant C49A and HMGbox1-mut5 were subjected to buffer exchange (750 mM NaCl; 50 mM Tris pH 7.5; 2 mM TCEP) using a HiPrep 26/10 Desalting column (GE Healthcare) and labeled with Alexa 488 maleimide (A488, Molecular Probes) at a protein/dye ratio of ∼1:20 incubated o.n. at 4°C. The conjugate was separated from unreacted dye by HiPrep 26/10 Desalting column (GE Healthcare). Final concentration of total protein was determined by Bradford reaction (Bio-Rad), whereas labeled protein at A488 (ϵ0,495 = 73 000 M−1 cm−1) absorbance. Labeling efficiency (moles of dye/moles of protein) was over 95% in all cases.
FRET measurements were performed in a customized Photon Technology International (PTI) spectrofluorimeter at RT. A488-protein/DNA complexes were prepared using Site-X and Site-Y enlarged up to 28 bp (following the mtDNA sequence). Each DNA was labeled with Alexa 594 (A594) at one (5*) or at the other (3*) end annealed from single strand oligonucleotides purchased from IBA Life Sciences, namely Site-X5* (5′-A594-TATCATAACAAAAAATTTCCACCAAACA-3′ and complementary 5′-TGTTTGGTGGAAATTTTTTGTTATGATA-3′), Site-X3* (5′-TATCATAACAAAAAATTTCCACCAAACA-3′ and complementary 5′-A594-TGTTTGGTGGAAATTTTTTGTTATGATA-3′), Site-Y5* (5′-A594-TAGAGGCTGTGCAGACATTCAATTGTTA-3′ and complementary 5′-TAACAATTGAATGTCTGCACAGCCTCTA-3′) and Site-Y3* (5′-TAGAGGCTGTGCAGACATTCAATTGTTA-3′ and complementary 5′-A594-TAACAATTGAATGTCTGCACAGCCTCTA-3′). Initial absorbance measurements were done by preparing complexes at a protein/DNA ratio of 2:1 (see TFAM/DNA complex crystallization section) at respective concentrations 1.2 and 0.6 μM. This entailed mixing 20 μl of protein (12 μM in 750 mM NaCl; 50 mM Tris pH 7.5; 2 mM TCEP) with 20 μl of annealed oligos (6 μM in H2O). This was incubated for 2 h at RT with 20 μl of buffer R10X (100 mM Tris–HCl, 10 mM TCEP), 20 μl buffer D (50 mM Tris–HCl, 750 mM NaCl, 2 mM TCEP), 20 μl NP40 0.15%, 10 μl glycerol 10% and 90 μl H2O (total Vf = 200 μl). After absorbance measurements, each sample was diluted 6 times with the same buffer, and fluorescence data were collected from complexes with labeled (*) or non-labeled moieties, such as Protein*/DNA* (P*/D*), P*/D and P/D*.
Donor-derived spectra were collected by exciting P*/D* or P*/D at a wavelength of 495 nm and scans were recorded from 509 to 700 nm. In addition, P*/D* spectra excited at 590 were also scanned from 604 to 700 nm. Acceptor-derived spectra were collected by exciting P/D* at 590 nm and scans were recorded from 604 to 700 nm. The donor (or acceptor) peak values are characterized by the average from 511–525 (or 610–624) nm emission values. The energy transfer (ET) was calculated with the formula ET = [(S2 – S1·(D2/D1) – S4·(A3/A4))·b]/[S4 – A3/A4)]·a (51), see Supplementary Materials and Methods. Anisotropy of the donor was measured on the donor-only labeled complexes, while anisotropy of the acceptor was controlled on the double labeled complexes. For each complex, anisotropy values were below 0.2, so the κ2 = 2/3 value could be applied, allowing the calculation of distances from FRET efficiencies. To calculate the distances, the Förster R0 distance between A488 and A594 was set to 5.56 nm (52) and the equation E = 1/(1 + (r/R0)1/6) was used to calculate the distance r.
RESULTS
At the h-mtDNA control region, the sequences Site-X and Site-Y show a certain degree of conservation with LSP, and all three have been shown to be binding sites of TFAM (14,15). However, it is not known if there is a shared DNA recognition mechanism. To compare the DNA properties of these three sequences and the ability of TFAM to bind them, we followed the previous alignment (14) and restricted the length of the tested DNA molecules to the shortest DNA used for crystallisation, which is 22 bp for LSP (18,53) (Figure 1B). By doing this, we avoided any effect due to additional variability of sequences flanking the 22 bp binding site (54). Thus, we generated TFAM/Site-X and TFAM/Site-Y complexes and analysed them together with TFAM/LSP (18).
Structural flexibility of TFAM bound to Site-X and Site-Y
The crystal structures of TFAM/Site-X and TFAM/Site-Y show that respective asymmetric units (a.u.) contain four protein molecules (TFAM-A, -D, -G and -J) bound to respective DNA duplexes (strands BC, EF, HI and KL) (see Supplementary Table S1 for statistics; Figure 1C and D). In both cases, the electron density map is better defined in TFAM-A/BC. Thus, this complex will be taken as the reference, unless otherwise stated. TFAM contains two HMG boxes (HMG-box1 and HMG-box2) separated by a 30 aa linker. In general terms, HMG-box domains display an L-shape constituted of three α-helices, helices 1 and 2 form the short L-arm whereas helix 3, together with an elongated region from the N-terminus, shapes the long L-arm (e.g. see Figure 1C and D HMG-box2). The TFAM/DNA structures show that each HMG box contacts the DNA minor groove, separates the strands, and bends it by 90° toward the major groove (Figure 1C and D). Such a kink is facilitated by insertion of Leu58 from HMG-box1, or Leu182 from HMG-box2, within 10 bp separated DNA steps that consequently lose their stacking interactions (17,18). The two 90°-bent DNA regions are separated by one DNA turn, so that the DNA molecule adopts the overall shape of a U-turn (Figure 1C and D). The two HMG boxes are placed on different DNA sides and connected by the linker, which crosses the concavity of the U-turn and stabilises the folded DNA backbone (Figure 1C and D). After HMG-box2, a C-terminal tail loosely contacts the DNA major groove. The overall arrangement is similar to previous structures of TFAM bound to LSP sequences of 22 or 28 bp or longer (LSP22, PDB code 3TQ6; LSP28, 2TMM; longer DNA, 6ERP), to promoter HSP1 (6ERQ), or to a non-specific DNA molecule (nsDNA, 4NNU) (17–19,31). Please note that 6ERP and 6ERQ are at low resolution so they are not used here for detailed comparisons.
The overall superimposition of the four complexes in the a.u. (A/BC, D/EF, G/HI and J/KL) from crystal TFAM/Site-X, or TFAM/Site-Y, show structural rearrangements inside each crystal. Such divergences do not reside at the HMG-box domains, because the superimposition of the four HMG-box1 fragments from protein chains A, D, G, and J show negligible differences (maximum r.m.s.d. 0.87 Å, Supplementary Table S2). However, upon HMG-box1 superimposition, the relative orientation of HMG-box2 varies considerably from complex to complex, with concomitant deviations of DNA (Supplementary Table S2). The converse is also true: superimposition by HMG-box2 shows small intra-domain differences whereas the positions of HMG-box1 domains vary appreciably. The TFAM/LSP crystal shows the same variability between A and B molecules in the a.u. (Supplementary Table S2). Such divergences are also observed between crystals. The superimposition of molecule A HMG-box1 from Site-X, Site-Y, and LSP crystals shows reorientations between HMG domains (Figure 1E and F, Supplementary Table S2). To discern whether the different relative positions of HMG boxes is due to progressive distortion of the polypeptide or to a one-point hinge, superimpositions were performed locally. Unexpectedly, loops connecting the HMG-box domains with the linker show identical conformations at both main and side chains (Supplementary Figure S1), thus they do not function as hinges. Instead, the linker itself shows a slight progressive distortion that adds to a hinge midway between the linker helix, around Lys136-His137 (Supplementary Figure S2). At this point, the linker region that interacts with the DNA begins. This not only happens between these similar DNA sequences but also with nsDNA (31), which shows highly similar HMGbox1 (or HMGbox2) domains between molecules in the crystal or between crystals. However, their superimposition results in considerable HMGbox2 (or HMGbox1) reorientations concomitant with distortions at the linker (see Supplementary Table S2). Thus, structural U-turn variability is associated with the linker and depends on crystal packing but also, as we show below, depends on DNA properties.
Since Site-X and Site-Y sequences are similar to LSP (14,15) and the three are the same length, we expected that the insertions of amino acids would happen at equivalent DNA positions. In TFAM/LSP, Leu58 (from HMG-box1) inserts (↓) at T1A2A3↓C4A5, and Leu182 (HMG-box2) at C15↓A16 (31,53) (in bold, conserved bases between LSP, Site-X and Site-Y). However, whereas the insertions happened as predicted for Site-X, which presents a long A-tract, surprisingly for Site-Y the binding follows a completely different pattern.
TFAM bypasses a poly-adenine tract in Site-X
The TFAM/Site-X structure shows that HMG-box1 binds to the minor groove and inserts Leu58 (in helix 1) into the T1A2A3↓C4A5 region (base numbers from Chain C as in TFAM/Site-X structure; see Figure 1A and B for mtDNA coordinates). The insertion opens up step A3C4/G19T20, breaks the stacking interactions and induces a kink to this DNA region, which shows the highest roll angle (Figure 2). The following two neighbouring steps also show high roll angles, C4A5/T18G19 is partially inserted by Leu82 (in helix 2), and A5A6/T17T18 is slightly inserted by the loop between helices α1 and α2 at the tip of the short L arm. At the latter A5A6 pair the long A-tract A5A6A7A8A9A10T11T12T13 starts, which is located precisely between the two DNA kinks (Figure 1B and C). This sequence resumes two types of A-tracts, namely non-symmetric An (n ≥ 4, for the A5A6A7A8A9A10 part) and symmetric AnTn (n ≥ 2, A8A9A10T11T12T13). A-tracts have important, singular features: they are straight, relatively rigid and display a narrower minor groove (55). Comparison of minor groove width between U-turn structures shows that a minimum is always present at the B-DNA region between DNA kinks, yet in Site-X the narrowest part is longer, extends from A7 to T11, and reaches values <3.0 Å (minimum of 2.66 Å at step A8A9/T12T13; dark blue curve in Figure 3 and Supplementary Figure S3). The possibility that this reflects the properties of the free A-tract was corroborated by comparing the minor groove width of free Site-X, Site-Y and LSP DNA structures obtained by MD simulations (see below). Indeed, in both free and protein-bound conformations, Site-X shows the most extended narrow minor groove region (dark and light blue curves in Figure 3). Stiffness and narrowness of the minor groove in A-tracts precludes binding of HMG-box domains (56), but the TFAM/Site-X structure shows that TFAM overcomes the limitation by virtue of the long linker, which allows intertwining around the A-tract and binding of the HMG boxes at both flanking sequences.
Figure 2.

DNA parameters from complexes (from crystals) or naked (MD-derived) LSP (top), Site-X (middle) and Site-Y (bottom). Left column: the roll values (in degrees) for protein-bound (black) and naked DNA (gray) are shown. The high roll peaks in the crystal structures correspond to the insertion sites, indicated by the arrows (left arrow, Leu58 insertion site; right arrow Leu182 site). For Site-Y, the tentative orientation assigned in the crystal (in gray) and the previously predicted orientation (14) (in light blue) are shown. Right column, the stiffness (Ktot) of LSP, Site-X and Site-Y are shown along the sequences, which are aligned. The sites inserted by Leu58 and Leu182 in the X-ray structures are indicated by black vertical arrows. For Site-Y, the previously proposed insertion sites (14) are indicated by gray arrows.
Figure 3.
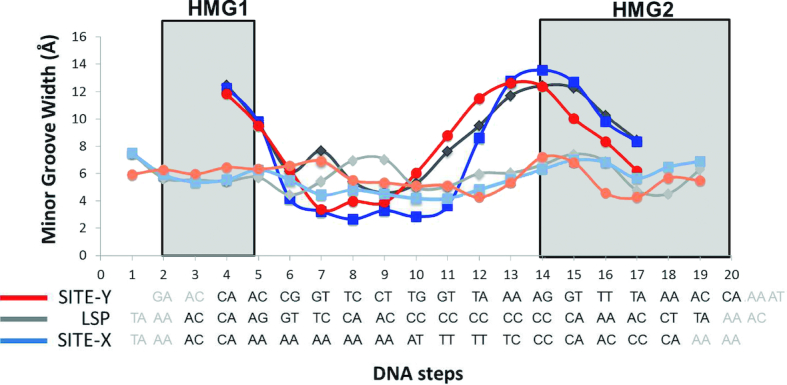
The minor groove width (Y axis) is shown for each DNA step (X axis, each DNA step is numbered). Below, the sequences are represented. Site-X is shown in light (naked) and dark (crystal) blue, Site-Y in orange and red, LSP in light and dark gray.
The linker contacts the minor groove between DNA kinks, at the side opposite the HMG-boxes. However, whereas the TFAM/LSP crystal structure shows that amino acids from the linker Lys139, Met143 and Lys147 penetrate the DNA minor groove, contact the phosphate backbone, and establish water bridges with base atoms (18), in TFAM/Site-X the extended narrowing of the minor groove prevents access of the aforementioned side chains and only Lys147 contacts the DNA phosphates (Supplementary Figure S5). Likewise, displacement of the linker is observed in the TFAM/nsDNA complex due to a narrower minor groove between the two inserted steps, which also show high roll. However, the narrowing is less extended compared to Site-X (Supplementary Figures S3–S5). Therefore, the linker is displaced outward from the minor groove and loses interactions with it, but it still stabilizes the U-turn independently of minor groove features.
Regarding HMG-box2, Leu182 (from helix 2) is inserted between DNA chainB C15↓A16, a dinucleotide that is also inserted in LSP. However, here Leu182 side chain has very weak electron density in all four complexes in the a.u. (A/BC to J/KL), which indicates structural variability. In addition, the loop between helix1 and helix2 (tip of the short L arm) in HMG-box2 shows weak density, and in chains D and G the loop between helices α2 and α3 (the L-elbow) displays a double conformation induced by crystallographic interactions. The DNA at this region (A16 to A20), which faces helices α2 and α3 and the L-elbow, also shows a second conformation that was only tentatively traced for DNA strand H (in complex G/HI). This second conformation of the DNA is also induced by a symmetrically related molecule. All these observations suggest high flexibility of HMG-box2 at Site-X.
The TFAM/Site-Y structure suggests an unpredicted protein orientation
In TFAM/Site-Y, the DNA sequence was initially assigned based on the alignment between Site-Y and LSP (14) and following the TFAM/LSP22 crystal structure (53). However, the traced bases did not fit well into the electron density map, which indicated an inaccurate sequence assignment. To find the actual DNA pattern, we substituted one by one thymines with brominated uracyl. This modified base generates a signal in the X-ray diffraction that unambiguously indicates the position of the uracyl and hence the DNA orientation. However, trials to substitute different thymine positions resulted in no crystals or did not yield any interpretable result, which suggested structural disorder. As an alternative, we refined the atom positions with all bases substituted by the smallest base, cytosine. This resulted in a difference mFo - DFc map that indicated deficient electron content at some positions, and thus suggested a purine for these sites instead of a pyrimidine. With this, we obtained a purine/pyrimidine pattern that was compared to that of the Site-Y sequence (Supplementary Figures S6 and S7), allowing a tentative DNA sequence assignment which, finally, rendered an acceptable model that was better refined. In this new orientation, Site-Y is inverted and shifted one bp compared to LSP (Figure 1A and B). However, the sequence assignment is not fully conclusive because the electron density at the DNA ends is not well-defined, which suggests that in the crystal some complexes may have continuous DNA at this position, i.e. the DNA is disordered in the crystal. Therefore, we indicate the tentative Site-Y sequence assignment in brackets. In this probable reverse orientation, Leu58 inserts between (T2↓G3, from the complementary chain) whereas Leu182 inserts at (C14↓A15), and not at A3C4 and C15T16, if following the initial alignment. Comparison of TFAM/SiteY and TFAM/LSP structures shows that the same amino acids from HMG1 and HMG2 are involved in DNA contacts. Loops between helices are tentatively traced in weak density, which indicates flexibility. Regarding the linker, it is not as close to the minor groove as in LSP but is displaced due to a repositioning of DNA strand C (Supplementary Figure S5), which narrows the minor groove and pushes the linker outward. However, neither the groove narrowing nor the displacement of the linker is as great as in Site-X. Consequently, from the triad Lys139, Met143 and Lys147, only the first penetrates the groove.
Free LSP, Site-X and Site-Y show specific structural features and flexibilities
The unexpected orientation of Site-Y in the crystal suggested that distinctive characteristics of the three sequences guided TFAM binding. This prompted us to characterize the physical properties of free LSP, Site-X, and Site-Y by MD and compare their states in the free and bound forms. The equilibrium conformation and the stiffness of each DNA sequence were calculated to analyze the sequence-dependent features of each binding site. Thus, we calculated the average of these base-pair parameters along the corresponding MD trajectories. We specifically focused our attention on the roll, because it is the feature that deviates most from an ideal B-DNA in the crystal structures, especially at the inserted sites, resulting in a bent DNA conformation. The results show that the naked sequences spontaneously deviate from ideal B-DNA conformation, thus they are intrinsically deformed (Figure 2). In addition, all three free sequences show that, at the region contacted by HMG-box2 in the crystal, the step inserted by Leu182 intrinsically has a high roll, thus it is already opened in the free DNA. Regarding Leu58, a high roll is only found at the insertion site in Site-Y. In the LSP-like orientation, this would correspond to a more closed step (light blue in the roll graph). Notably, in both LSP and Site-X, Leu58 insertion sites show low roll, and therefore the corresponding steps are closed.
The stiffness (Ktot) along the sequence of the three naked DNAs is shown in Figure 2. In LSP, flexible steps alternate with mildly stiff steps. Site-X is also rather flexible except at the A-tract, with highest stiffness at the ApT step. The crystal structures show that, in both LSP and Site-X, Leu58 inserts at the ApC/GpT step. In the free DNA, this step is slightly stiffer than the highly flexible CpA/TpG step inserted by Leu182 residue from either LSP or Site-X. Regarding Site-Y, the steps inserted by TFAM are both flexible. In addition, Site-Y presents two ApT rigid points, A6T7 and A11T12. HMG-box2 widens the minor groove of the former by introducing Arg157 deep into the groove, where its Nη1 and Nη2 (at the tip of the side chain) interact with O2 from T18 (complementary to A5). In LSP and Site-X, the arginine also interacts with base atoms deep into the minor groove. Therefore, this rigid spot in Site-Y does not prevent HMG-box2 from separating the strands and introducing the arginine as it does in Site-X and LSP. The second rigid region in Site-Y A11T12 is found at the B-DNA between DNA kinks.
In the comparison between the free DNAs sequence-dependent curvature and the protein-bound bending (Figure 2, Supplementary Figure S8) we found out that in all the three cases the central part of the DNA is straight and keeps almost the same conformation after protein binding. Between the base pairs 3–5 and 13–15, where the leucines insert, the DNA is heavily distorted in the bound configuration and roll contribution is the main factor in this bending. The protein-induced bending appears not to be the same at the two insertion points: at HMG-box2 the bending is localized at one base pair, precisely the most open and flexible one. At HMG-box1, the DNA bending is delocalized between adjacent base pairs, probably so as not to stress the DNA structure excessively. Interestingly, for all the sequences, there is a partial intrinsic curvature at the kink sites before protein binding, which probably facilitates the adaptation to the final bound conformation. This is particularly evident for Site-Y, where the main peaks of curvature correspond to the point where there is maximum bending in the protein complex (Supplementary Figure S8). We also observed that the protein has different effects on DNAs. At Site-X, the bending is almost equally imposed by the two HMG boxes, whereas at LSP and Site-Y, the HMG-box2 bends the DNA more than the HMG-box1. For a comparison, the TFAM/nsDNA structure show a very similar profile to the above (roll values are shown in Supplementary Figure S4). We only detected different behavior at the DNA region contacted by HMGbox1, in which nsDNA is distorted at the insertion site and neighboring step. The behavior of bending at the DNA region contacted by the HMGbox2 is highly similar in values and profile to TFAM/LSP and TFAM/Site-Y. These results suggest that, irrespective of the DNA sequence, TFAM imposes a U-turn and the local features of the U-turn arise from sequence-dependent properties, as described in our recent analysis of TFAM/LSP by FRET (20).
A final analysis consisted in the calculation of the deformation energy (ΔEdef) required to deform the DNA from the MD-derived equilibrium state conformation to the conformation in the corresponding crystal structures. To this end, we used the mesoscopic method previously developed and tested for the nucleosome core complex (57). Interestingly, LSP showed the highest energy cost (ΔEdef 8.4 kcal/mol·bp) to change its conformation when passing from its free form to the one in the crystal. Site-X requires less energy (6.3 kcal/ mol·bp) whereas, surprisingly, Site-Y requires the least (5.5 kcal/ mol·bp). In contrast, deformation of Site-Y into the conformation found in LSP (as initially traced in the electron density, see above) has the highest cost (8.7 kcal/ mol·bp).
Altogether, the above results indicate that free LSP, Site-Y and Site-X display divergent, sequence-dependent conformation and flexibility. Divergence includes the sites inserted by Leu58 and Leu182. In Site-Y, TFAM inserts both leucines into flexible and opened steps, while in both Site-X and LSP only Leu182 encounters such favorable conditions. In addition, Site-Y is the sequence that is easier to bend into the U-turn conformation. The question was then to experimentally assess this sequence-dependent variability, which was achieved in the following analyses.
TFAM shows differential binding dynamics to LSP, Site-X and Site-Y
A first experimental analysis to compare the TFAM binding to the different DNA sequences was by non-denaturing electrophoresis (electrophoretic mobility shift assay, EMSA). 32P-labeled DNA probes (1 nM) were titrated with increasing TFAM concentrations (Figure 4A). The intensity of the shifted bands was quantified and fitted to a modified Hill equation (see Materials and Methods). The apparent Kd for LSP was estimated as 9.2 nM (±0.98, n = 3) (Figure 4B), and thus was in agreement with the 1–10 nM range measured by other groups, including non-specific DNA (23,28,58,59). For Site-X, the apparent Kd was 13.6 nM (±2.89, n = 3) and for Site-Y it was 4.4 nM (±2.32, n = 3). The differences in Kd's are more evident if the values are expressed in logarithmic scale: 0.64 for Site-Y, 1.13 for Site-X and 0.97 for LSP. While the two latter are similar, the Site-Y Kd is lower by almost half a unit. At the tested concentration range, the binding of TFAM to any of the three ligands was detected as not cooperative (the Hill coefficient was close to 1). To further analyze the binding of TFAM to these sequences, competition assays were carried out by EMSA. TFAM bound to 5′ fluorescently labeled probes *Site-X or *Site-Y (0.8 μM) were put into competition with increasing amounts of unlabeled LSP (from 0.2 μM to 3.2 μM, Figure 5, upper panels). Strikingly, the competition revealed considerable differences between Site-Y compared to the other two sequences. The band of TFAM/*Site-X complex faded rapidly at low concentrations of LSP but not completely at the conditions tested. In contrast, the TFAM/*Site-Y complex was stable even at 4×-fold excess of competitor. Since LSP competed differently with Site-X and Site-Y, we analyzed the competition between each site for TFAM binding. The lower panel of Figure 5 shows that the non-labeled Site-Y strongly competed with *Site-X, whereas the non-labeled Site-X poorly displaced *Site-Y. Therefore, in these experiments TFAM shows a clear preference for Site-Y over both Site-X and LSP, which is explained by the higher affinity of Site-Y relative to Site-X and LSP. Nonetheless, the different properties of each sequence (see last and forthcoming sections) probably also influence the kinetics of complex formation, e.g. the time of residence of the protein on Site-Y, Site-X and LSP.
Figure 4.

Binding of TFAM to Site-Y, Site-X and LSP. In (A), gel-shift assays (EMSA) show TFAM-inducing equivalent DNA migration to all sequences. Lane ‘0’ contains free DNA (1 nM). In subsequent lanes from left to right, 32P-labeled DNA at 1 nM was titrated with TFAM at increasing amounts (as indicated above the corresponding lanes). The DNA shift corresponding to the complexes is indicated as ‘DNA+TFAM’. In (B), the measurements of TFAM binding to DNA are fitted to a modified Hill equation (see Materials and Methods). TFAM in complex with Site-X is shown with black circles; in complex with Site-Y with squares; and in complex with LSP with diamonds.
Figure 5.

Differential binding of TFAM to Site-X, Site-Y, and LSP. The upper left panel shows competition of TFAM-bound *Site-X (0.4 μM of DNA, 0.8 μM of TFAM) labeled with fluorescein) with increasing concentrations of unlabeled LSP (from 0.2 to 3.2μM), by EMSA. Right upper panel: similar competition of LSP, here against *Site-Y (also labeled). The lower panels show competition of TFAM-bound *Site-X (left) or *Site-Y (right) by increasing amounts (from 0.2 to 3.2 μM) of unlabeled Site-Y or Site-X, respectively. Lane 0 contains complexes with labeled DNA (0.4 μM *DNA + 0.8 μM TFAM). The input labeled *DNA control is shown at the far-left lane of the gels.
Thermodynamics points to complex formation variability depending on DNA sequence
Thermodynamic analysis of TFAM binding to LSP, Site-X and Site-Y was assessed by isothermal titration calorimetry (ITC) performed at the micromolar range (6–9 μM, see Materials and Methods). Protein titration with each DNA sequence (40–50 μM) showed that in all three cases, binding occurs with positive, unfavorable enthalpy (ΔH > 0) compensated by an important favorable entropy change (ΔS > 0) (Figure 6 and Table 1). Thus, a spontaneous entropically driven endothermic event occurs at the T at which we performed the experiments (20°C). This is consistent with previous analyses of TFAM binding to LSP, HSP1 and to non-specific DNA (59), which were also performed at 20°C, and it has been described for other HMG box proteins (60) and found for proteins that strongly distort the DNA (54). Interestingly, Site-Y showed a change in enthalpy 2-fold compared to Site-X and LSP, compensated by a higher entropy, pointing to an enthalpy-entropy compensatory effect (see Discussion). All experiments were perfectly fitted with a ‘one set of sites’ model, which indicates that the protein has a single type of DNA-binding site in all three sequences. A stoichiometry of 0.4 suggests the formation of a complex involving two TFAM molecules per DNA, as previously determined also at the micromolar range by others (59).
Figure 6.

Isothermal titration calorimetry of TFAM binding to LSP, Site-X and Site-Y. Top: representative isothermal titration calorimetry thermograms from the measurements done with LSP (left), Site-X (centre) and Site-Y (right). Below: fitting of the binding isotherms to a model with one binding site (red curve).
Table 1.
Values for the three TFAM/Site-X, TFAM/Site-Y and TFAM/LSP complexes derived from the ITC experiments are shown. The average from three measurements per sequence is shown
| K a (M-1) | N | ΔH (kcal.mol-1) | ΔS (kcal.mol-1.K-1) | Δ G (kcal.mol-1) | |
|---|---|---|---|---|---|
| LSP | 12 × 106 ± 1.2 × 106 | 0.40 ± 0.19 | 8369 ± 1829 | 61.0 ± 6.4 | −9505 ± 58 |
| Site-X | 23 × 106 ± 16.0 × 106 | 0.36 ± 0.05 | 7713 ± 857 | 59.3 ± 3.7 | −9662 ± 667 |
| Site-Y | 5.9 × 106 ± 4.4 × 106 | 0.37 ± 0.13 | 15460 ± 3376 | 83.0 ± 9.2 | −8869 ± 679 |
Note that the experiment was performed at concentrations at the micromolar range, above the dissociation constant (see main text).
Increasing concentration of TFAM/DNA complexes induces multimerization
The ITC-derived stoichiometry showed 2 proteins for 1 DNA, which contrasted with the single band shift of the aforementioned titrations in the nanomolar range and the crystal structures (in which one protein is bound to one DNA). In these EMSA, no second shift was apparent at increasing protein:DNA ratios, up to 160:1 (see Figure 5). In principle, a single EMSA shift is consistent with one single type of complex. Note that the ITC and EMSA experiments to measure the affinity were performed at different concentrations, which suggested that the multimerization of TFAM on the DNA detected by ITC could have been stimulated by a concentration effect, independent of the protein:DNA ratio. To test this possibility, we prepared TFAM/LSP complexes at increasing DNA and protein concentrations and keeping a constant ratio of TFAM:DNA 4:1, and analyzed the so-formed complexes by EMSA (Figure 7). At low concentration one single band appeared. Interestingly, when the concentration of the 4:1 components was increased, a second shift appeared at ≥0.8 μM (TFAM concentration). This indicated that higher-order species appeared depending on concentration. According to the crystal structures, the length of the DNA used (22 bp) is too short to allocate more than one protein. Therefore, the first shift should contain a TFAM molecule bound to the DNA, whereas the upper shift should contain a TFAM molecule that recognizes this initial complex. This second protein contact is stimulated by the concentration of the sample.
Figure 7.
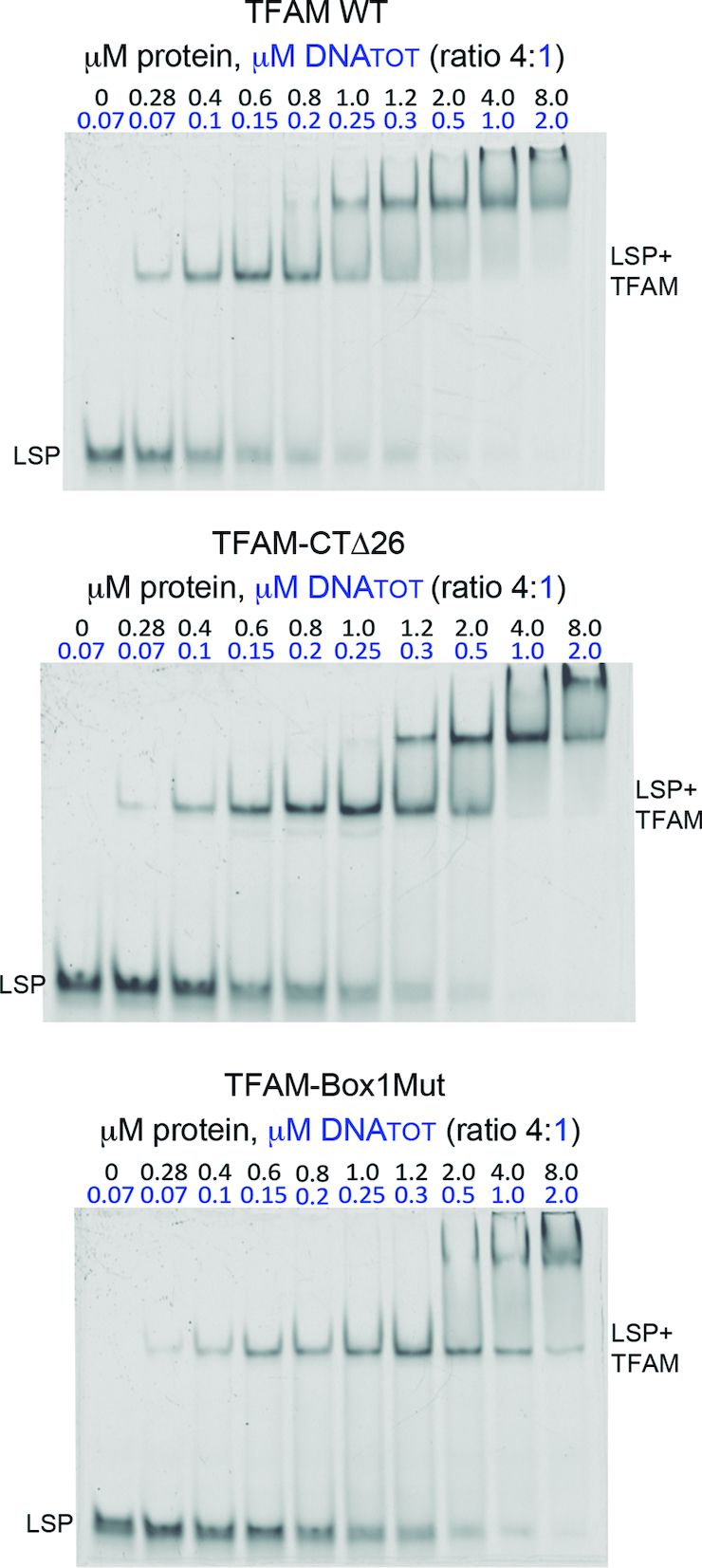
Multimerization of TFAM on the DNA depends on complex concentration. Non-denaturing polyacrylamide gels (10%) that contain, from left to right, increasing concentrations of a mixture of TFAM WT (top gel), TFAM-CTΔ26 (middle gel) and TFAM-Box1Mut (bottom gel) proteins mixed with LSP (22 bp used for crystallization) in a protein/DNA ratio of 4:1. DNATOT refers to total DNA, from which 10 nM was labeled. At low concentrations, the complex runs as a single band (LSP+TFAM) corresponding to a 1:1 protein:DNA ratio. At 0.8 μM of protein (0.2 μM DNA), an upper shift appears in TFAM-WT concomitant with progressive fading of both the first shift and the free DNA (LSP).
To investigate whether the second shift was due to dimerization, we performed the same EMSA using two previously reported dimerization mutants. In the crystals, one protein with one DNA (ratio 1:1) performs two types of contacts with two other complexes (Supplementary Figure S9A). Therefore, it shows two distant interfaces. One interface includes the loop contained within the last 26 residues (known as the C-terminal tail), which performs protein-protein contacts with the same loop from a second protein in the crystal (Supplementary Figure S9). These two proteins perform additional interactions through the N-terminal region of respective linkers. These two simultaneous contacts occur only in crystals of TFAM in complex with 22 bp DNA, whereas for longer DNAs, such as 28 bp, these contacts are not feasible (17). Contacts through the C-terminal tail are cancelled in the TFAM-Δ26 mutant (27,61), which lacks the last 26 aa (Supplementary Figure S9). A second mutant, TFAM-Box1Mut (31), harbors six mutations that cancel the large interaction seen in all crystal structures available, between two HMGbox1 domains (Supplementary Figure S9). Both mutants, TFAM-Δ26 and TFAM-Box1Mut, are stable in solution (31,61) (Supplementary Figure S9B to D). A qualitative analysis of our EMSAs shows that both mutants generate the first shift at the lowest 4:1 ratio concentration tested (0.28 μM TFAM:0.07 μM DNA, Figure 7). At increasing concentrations, TFAM Δ26 delays the second shift one point later in relation to WT (compare lanes Δ26 1.2 μM and WT 1.0μM). At the highest concentrations (TFAM 4 and 8 μM), the first shift disappears while the second band smears to higher bands or probable aggregates in both TFAM-Δ26 and WT. In contrast, TFAM-Box1Mut delays the second shift two points (2 μM), in a badly defined band that rapidly smears at higher concentrations, while the first shift does not disappear. The well-defined flat band of the second shift in the WT/LSP and TFAM-Δ26/LSP gels is consistent with non-aggregation of complexes. In addition, the gels suggest that whereas both mutants destabilise dimerization, TFAM-Box1Mut is more effective.
TFAM at high concentration multimerize on short DNAs
To further understand the protein:DNA stoichiometry of complexes formed at high concentration (mg/ml), we experimentally determined their Mw (Mwobs) by SEC-MALLS. Injection of 97.6 μM (2.5mg/ml) TFAM alone yielded two peaks corresponding to Mwobs of 57 ± 3 and 30 ± 3 kDa, which are consistent with a dimer and a monomer, respectively (Mw of TFAM construct, 25.6 kDa)(Supplementary Figure S10). This corroborated the previously reported monomer-dimer equilibrium for the protein alone (61). In complex with LSP, TFAM (390.6 μM, 10 mg/ml) yielded two separated species of 62±3 and 41±2 kDa, consistent with protein:DNA 2:1 (Mwcalc = 64.7 kDa) and 1:1 (Mwcalc = 39.1 kDa) stoichiometries, respectively (Figure 8). At lower TFAM/LSP concentration (218.7 μM; 5.6 mg/ml) a single peak of 41.5±2 kDa corresponding to the 1:1 ratio was observed (Figure 8). The existence of a concentration-dependent equilibrium of monomers and dimers of TFAM bound to short DNAs was thus confirmed. TFAM/Site-Y complex (351.5 μM TFAM concentration; 9 mg/ml) yielded two peaks of 62 ± 2 and 47 ± 1 kDa. Similar to LSP, the first peak corresponds to a 2:1 stoichiometry. However, the second peak could be a mixture of the 2:1 and 1:1 complexes, because of less efficient separation of these species. For Site-X at 321.5 μM (8 mg/ml) only the 47±3 kDa species appeared, which again suggests a mixture of 2.1 and 1:1 complexes.
Figure 8.

SEC-MALLS and AUC analyses of TFAM in complex with Site-X, Site-Y and LSP. Left graphs column: SEC-MALLS experiments are shown. The light scattering curve (LS) is shown in red, the differential refracting index curve (dRI-curve) in blue and the molecular weight peaks in green. The Y axis on the left reports the light scattering signal while the Y axis on the right shows the molecular weight. Central and right graphs columns correspond to analytical ultracentrifugation measurements (AUC) at Protein:DNA ratios of 2:1 and 4:1 respectively. In each graph, the continuous size distribution coefficient (S) is represented for the sedimentation coefficient (c(s)) values. First row corresponds to analyses of TFAM/Site-X complexes, second row to TFAM/Site-Y, and bottom row to TFAM/LSP. Peaks encircled as 1 and 2 in MALLS correspond to the same peaks in SV (AUC).
Since the MALLS experiments could not discern whether the 47 kDa species from Site-Y and Site-X could be a mixture of complex 1:1 and 2:1 or corresponded to a different species, we analyzed these complexes by sedimentation velocity (SV). This technique showed a higher resolution than SEC, with improved separation of species. Furthermore, this technique allowed the analysis of higher protein:DNA concentrations, as samples were not diluted during the experiment as in SEC. Site-X and Site-Y alone yielded main peaks in the range of 2.3–2.7 S, which were compatible with a dsDNA monomer (Mwcalc = 13.5 kDa) (Supplementary Figure S10). For LSP alone, an additional peak was observed that could correspond to a G-quadruplex (Supplementary Figure S10) (24)). TFAM alone showed a main peak at 1.8 ± 0.1 S (Supplementary Figure S10) corresponding to the monomer (Mwcalc = 25.6 kDa) and a small proportion of dimer (3.5 ± 0.1 S) and higher order oligomers (4.3 ± 0.1 S). Regarding the protein–DNA complexes, in all cases the different species could be separated and the stoichiometry elucidated by the combination of SV and SEC-MALLS techniques. In the analysis of 2:1 protein:DNA concentration ratio, peak at 3.3 ± 0.1 S is consistent with a 1:1 complex (Mwexp = 39.1 kDa,) and peak at 4.5 ± 0.2 S with a 2:1 complex (Mwexp = 64.8 kDa). Peak at 5.6 ± 0.1 S could correspond to a 4:1 complex (Mwexp of 116.1 kDa), (Figure 8) but this species was not detected by SEC-MALLS and could not be properly assigned. Strikingly, in the experiments at 4:1 protein:DNA concentration ratio, a peak compatible with free DNA appeared that was not present at the 2:1 ratio (or marginally present in TFAM/Site-Y 2:1 ratio). This suggests that the formation of higher order protein–DNA complexes is concomitant with some protein–DNA unbinding.
TFAM dimers bind Site-X and Site-Y in opposite orientations in solution
To confirm the orientation of the protein on Site-X and Site-Y, we performed bulk Förster resonance energy transfer (FRET) experiments in solution using four DNAs, namely Site-X with Alexa594 (A594) at one (Site-X5*) or the other end (Site-X3*), and Site-Y labeled similarly (Site-Y5* and Site-Y3*, respectively) (see Methods and Figure 9). As a donor, we used a C49A TFAM mutant (TFAM-C49A) that harbors only one cysteine at the end of the C-terminal tail, which was labeled with A488 (Supplementary Figure S11). Fluorescence measurements were performed at low concentration to avoid TFAM multimerization on the DNA. With this setup, we expected a higher FRET signal for both TFAM-C49A/Site-X3* and TFAM-C49A/Site-Y3* complexes than for the corresponding 5* counterparts. However, the FRET difference between labeled DNA ends was ambiguous for Site-X (Table 2 and Supplementary Figure S11), since Site-X5* FRET was unexpectedly slightly higher than for Site-X3* (0.85 ± 0.01 versus 0.79 ± 0.01). Instead, the Site-Y signal was as expected from our sequence assignment (stronger for Site-Y3*). Considering these results, we reasoned that the low differences in Site-X could reflect some sort of TFAM multimerization events. Therefore, we decided to perform FRET measurements with the dimer mutant that cancelled more efficiently the second shift in our EMSAs (see above): non-dimerizing TFAM-Box1Mut. This mutant was labeled at Cys49 (at HMGbox1), thus the dye (A488) was close to the N-terminus (Figure 9 and Supplementary Figure S11). We expected a higher FRET signal for Site-X5* and Site-Y5* than for the 3* DNAs. Indeed, for both 5* labels, the FRET was unambiguously higher than for 3*. The corresponding estimated distances (Table 2) were a bit higher than expected for Site-3*s, but still in a reasonable range according to the structure (3.1 nm). For Site-5*s, the estimated distances were in better agreement (5.1 nm). The higher values in solution are consistent with breathing of TFAM complexes (20). Therefore, our FRET experiments confirm the orientation of the protein on both Site-Y and Site-X.
Figure 9.

Confirmation of TFAM orientation on Site-X and Site-Y by FRET. (A) Design of the DNAs used for FRET assays. The 22 bp sequences from Site-X (blue sequence) and Site-Y (in red) are aligned based on the protein structure superposition. Note that the sequences follow opposite directions (compare with Figure 1B). Black squares Box 1 and 2 correspond to HMGbox1 and 2, respectively. The gray squares indicate the formerly predicted position of TFAM domains on Site-Y (note the inversion of the HMGboxes). Full-length TFAM is labeled with A488 at the C-terminal Cys246 (green C dot). The TFAM-Box1Mut non-dimerizing mutant is labeled at Cys49 (green N dot). Gray ‘N’ and ‘C’ dots correspond to the positions of labeled cysteines in case the orientation of TFAM on Site-Y followed the initial prediction (14). The red circle symbolizes the DNA 5′ end labeled with A594. Regarding the DNA sequences, the dots between bp indicate the insertion sites. In gray, the added bp of the mtDNA sequence to enlarge Site-X and Site-Y to 28 bp (upon superposition with the 28 bp TFAM/LSP structure (17)) is shown. Due to this enlargement, a new potential insertion site, TG-10bp-CA (in green), appeared in Site-Y, which was mutated to AG-10bp-CA. In addition, bases at the 5′ ends were mutated to Thy (underlined) to avoid stacking interactions with the dye. (B) Fluorescence emission spectra of single (TFAM-Box1Mut*/Site-X, dark blue curve) and double-labeled TFAM-Box1Mut*/Site-X5* (in green), and TFAM-Box1Mut*/Site-X3* (in sky blue) complexes excited at 495 nm (a.u., arbitrary units). (C) Same as in (B) but for complexes TFAM-Box1Mut*/Site-Y (dark red curve), TFAM-Box1Mut*/Site-Y5* (red curve) and TFAM-Box1Mut*/Site-Y3* (in violet).
Table 2.
FRET values for the C49A (label at the C-terminal tail, Cter) and Box1Mut (label at N-terminal HMGbox1 domain, Nt) mutants
| Acceptor position (*) on DNA sequences | TFAM-C49A Label Cter | Box1Mut Label Nter (distance) |
|---|---|---|
| Site-X5* | 0.85 ± 0.01 | 0.61 ± 0.01 (5.2 nm ± 0.4) |
| Site-X3* | 0.79 ± 0.01 | 0.38 ± 0.01 (6.1 nm ± 0.4) |
| Site-Y5* | 0.65 ± 0.01 | 0.74 ± 0.01 (4.7 nm –0.4, +0.5) |
| Site-Y3* | 0.84 ± 0.01 | 0.41 ± 0.01 (5.9 nm ± 0.4) |
In brackets, the calculated distances and errors (see Materials and Methods).
DISCUSSION
Decades ago, a sequence alignment of three TFAM binding sites at the mtDNA control region was reported that included LSP, Site-X, and Y (14). We determined the crystal structures of all the corresponding complexes (this study and (18)) and, based on the TFAM/LSP crystal structure, we assigned the DNA sequences in TFAM/Site-X and TFAM/Site-Y crystals. While this was valid for Site-X, unexpectedly Site-Y did not follow the predicted pattern. We attempted several Site-Y sequence assignments but none was fully conclusive. This suggests that, in addition to our best choice, other DNA orientations could be present in the crystal. However, our FRET studies clearly showed a preferred orientation of the protein on Site-Y that is consistent with that proposed for the structure. Our computational analysis indicated higher flexibility for Site-Y, a feature that explains the weak electron density map of this DNA.
Sequence-dependent DNA conformation and deformability modulates DNA binding and bending by TFAM. The analysis of the three naked DNA sequences shows a common pattern only at the site inserted by Leu182, a CpA/TpG base-pair step characterized by high flexibility and positive roll angle, thus it is a spontaneously flexible open step. This suggests that HMG-box2 introduces Leu182 into a step with an intrinsic conformation that appears more suitable for insertion, with a distortion that simply increases upon protein binding. In contrast, the HMG-box1 insertion site in both LSP and Site-X (but not in Site-Y) is a mildly flexible and closed ApC/GpT step. Interestingly, HMG-box1 has much higher affinity for and actually leads binding to DNA, whereas HMG-box2 barely contacts it (20,61). If the orientation of Site-Y was as predicted by Fisher in 1987 (14), two low roll angle steps (thus, closed) would be placed at the insertion sites. Instead, the Site-Y orientation positions two flexible steps at the right distance of 10 aa from each other, with a high roll angle (thus, open), which may explain the observed Site-Y orientation. These differences contribute to the lower energy required for DNA bending into a U-turn.
The particularities of each sequence were reflected experimentally during complex formation with TFAM. The ITC analysis showed spontaneous, endothermic and entropically driven complex formation at 20°C. During complex formation, the change in the Gibbs free energy is calculated from the enthalpy and entropy change, ΔG = ΔH – TΔS. A negative (exothermic) ΔH is interpreted as the result of formation of stronger intermolecular interactions in the complex, while positive ΔH may result from breakage of interactions with solvating water molecules, which are released to the bulk solvent. The entropy term (ΔS) usually decreases in complex formation because of the reduced mobility of interacting molecules. However, release of water molecules present in the sphere of solvation favorably increases the system entropy. In the case of high ΔH, the water release factor plays a major role in the enthalpy-entropy compensation that results in a favorable negative ΔG, but the interpretation of such an important phenomenon is difficult in every case (62). Entropy-driven binding reactions, in which unfavorable enthalpy is compensated by positive entropy change, have been described for protein/DNA complexes in which unfavorable enthalpy caused by destabilization of base stacking is compensated by favorable entropy caused by release of water molecules initially bound to the DNA (54,63). Endothermic and entropy-driven processes are typically found for proteins that bind to the minor groove and dramatically bend the DNA (63,64), which are the cases presented here. In addition, binding of the same protein to different DNA molecules may entail different enthalpy and entropy profiles with values that are mutually compensated, and result in similar Gibbs free energy (ΔG) change. This phenomenon is termed ‘isothermal enthalpy–entropy compensation’, as experiments with complexes different DNAs are carried out at a constant T (54). This was indeed the case for TFAM: binding to Site-Y resulted in a more positive enthalpy that was compensated by higher entropy than TFAM/LSP and TFAM/Site-X, while Gibbs free energy was similar for all sequences. For protein/DNA complexes, an increase in entropy difference has also been related to higher vibrational and conformational states that depend on the DNA sequence which, at the same time, lower the number of contacts and thus result in a more unfavorable enthalpy (54). The TFAM/Site-Y crystal shows a less ordered DNA molecule with associated higher overall B-factor, which is consistent with either a higher vibrational state or static disorder in the crystal, or both. Note that our experiments were performed at 20°. Interestingly, a recent publication showed that when the activity of the electron transport chain is fully functional, the T in the mitochondria is maintained near to 50°C (65). At this T, ΔH could reverse its sign, as shown for the HMG-box protein Sox-5 (66). Thermodynamic experiments at varying T showed that Sox-5 and other DNA-binding proteins (BamH1 endonuclease, GCN4 transcription activator or TATA box binding protein TBP) displayed a linear change in ΔS simultaneous to ΔH, which had a compensatory effect so that ΔG did not change substantially, i.e. ΔG is independent of both the T and the protein/DNA system (53,66). Recently, however, the compensatory effect has been questioned as an artefact derived from a high correlation between ΔH and ΔS, because it reflects the same phenomena. In any case, whether both a ΔH sign inversion and a compensatory effect occurs across T for TFAM/DNA complexes, in particular at T close to 50°C, is an aspect that needs to be further characterized. DNA competition assays showed that TFAM clearly prefers Site-Y over the other two sequences, which is consistent with previous results employing chromatin immunoprecipitation (16). These studies showed that Site-Y belongs to a sequence enriched with TFAM, whereas Site-X is only partially enriched (no data are available regarding LSP). The preference can be explained by the higher affinity at the nano-molar range. However, in almost all our analyses, Site-Y showed a different behavior compared to LSP and Site-X, and it is expected that the kinetics during complex formation also vary, e.g. contributing to a longer time of residence. This indicates that there is a non-systematic, uneven scenario at the mtDNA control region, in which the DNA sequence modulates TFAM binding, which predictably has implications for DNA regulation. Since most of the mtDNA molecule is covered by TFAM proteins, such variability in binding might extend to the rest of the genome.
At the control region, the three conserved sequences CSB-III, CSB-II and CSB-I are located downstream of LSP, CSB-III being the closest to LSP. Between CSB-II and CSB-I, Site-X overlaps with the former and Site-Y with the latter. On the other hand, according to our tentative DNA sequence assignment in TFAM/Site-Y, the TFAM HMG-boxes1 bound at Site-X and Site-Y are 20 bp apart and face each other. All available crystal structures show that, due to crystal packing, two HMG-box1 domains from different complexes contact each other precisely by this face, which is also shown to mediate mtDNA compaction (31). Such a contact between HMG1 domains could also happen if the DNA between Site-X and Site-Y melted upon protein binding, an event that was formerly considered to occur between complexes on the DNA (67). Overall mtDNA compaction is determined by TFAM concentration and achieved by cooperative interactions (25–27,31,68). Crystal structures of TFAM/DNA complexes present a protein:DNA ratio of 1:1. In addition, the crystal packing suggests that HMGbox1 and the C-terminal tail are distant regions that may form complementary protein interactions during mtDNA packaging (27,31). Our ITC, EMSA and MALLS experiments show ratios of 2 proteins bound to 1 very short DNA. It is likely that the second protein recognizes the complex since the DNA is too short to allocate two protein molecules. The crystal packing and our EMSAs with TFAM mutants suggest that the two distant regions, HMGbox1 surface and the C-terminal tail, may form complementary protein interactions during mtDNA packaging (27,31). In addition, the EMSA with increasing concentration of TFAM/DNA complexes show that, concomitant with the appearance of the second shift, the free DNA is reduced. This suggests that the second protein within the dimer can additionally bind DNA, and the complex dimer is, at highest concentrations, recruited to aggregates that consume all the DNA present in the solution. Of particular interest are the SV experiments, in which highly concentrated sample (hundreds of μM) is not diluted, but confined into a constant volume. These showed the existence of higher order complexes than the protein:DNA form 2:1, which is consistent with an aggregate of multiple proteins bound to the initial 1:1 or 2:1 complexes. In addition, SV shows that at high ratios and high concentration, free DNA is released. This implies that bigger aggregates have the potential to attract TFAM previously bound to DNA, disrupting protein/DNA complexes. Note that these experiments were performed with LSP. The affinity of TFAM for LSP, Site-X, Site-Y and nsDNA are at the nM range in vitro (59), and probably TFAM binding kinetics are modulated by the DNA sequence properties. In living cells, TFAM shows preferential binding to some sequences more than others (16). In the context of complete mtDNA compaction by TFAM, the DNA properties of potential binding sites are expected to modulate protein binding efficiency. An increase in protein ratios may promote strong nucleation at these particular sites, which would seed bigger aggregates and ultimately lead to nucleoid compaction.
DATA AVAILABILITY
Structures are deposited at the Protein Data bank with PDB codes 6HC3 (TFAM/Site-X complex) and 6HB4 (TFAM/Site-Y complex).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jose M. González de Cózar for his support in preparing TFAM. We thank the personnel at ESRF (Grenoble, France), ALBA (Cerdanyola del Vallès, Spain), SOLEIL (Gyves-sur-Yvette, France) synchrotrons and at the Automated Crystallography Platform (IBMB-CSIC) for their highly valuable support during X-ray data coollection.
Authors contributions: A.C., A.T.-S., E.R.-L. and A.R.-C. to cloning, protein production, and crystallization; S.L., A.C., Y.E., P.F.M. to electrophoretic analyses; R.P. and P.F.-M. to ITC; P.F.M., A.C. and M.S. to X-ray structure determination and refinement; F.B. and M.O. to molecular dynamics studies; A.C., A.T.-S., C.A. and G.R. to MALLS and ultracentrifugation. A.T.-S., G.I., M.P., K.T. and M.S. to FRET; M.S. designed and supervised the project. All authors participated in experimental designs, in manuscript writing and discussion, A.C., P.F., F.B. and M.S. to figure preparation. M.S., M.O., R.P., M.P. provided materials and infrastructure.
Notes
Present address: Anna Cuppari, ALBA Synchrotron Light Source, 08290 Cerdanyola del Vallès, Spain.
Present address: Pablo Fernández-Millán, Department of Biochemistry and Molecular Biology, Autonomous University of Barcelona (UAB), 08193 Cerdanyola del Vallès, Spain.
Present address: Sébastien Lyonnais, UMS 3725 CNRS – Université de Montpellier, Montpellier Cedex 05, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Science, Innovation and Universities, MINECO [BFU2012-33516, BFU2015-70645-R, RTI2018-101015-B-100 to M.S; BIO2015-64802-R, BFU2014-61670-EXP and BFU2014-52864-R to M.O., BIO2016-78006R to M.P.) Generalitat de Catalunya (2014-SGR-997 and 2017-SGR-1192 to M.S.; 2014-SGR-134 to M.O.), the Instituto de Salud Carlos III-Instituto Nacional de Bioinformática; the European Union (FP7-HEALTH-2010-261460, FP7-PEOPLE-2011-290246, FP7-HEALTH-2012-306029-2 to M.S.; H2020-EINFRA-2015-676556 to M.O.). The Biomolecular and Bioinformatics Resources Platform (ISCIII PT 13/000/0030 co-funded by the Fondo Europeo de Desarrollo Regional [FEDER]) [grants Elixir-Excelerate: 676559; BioExcel2:823830 and MuG: 676566] to M.O. P.F.-M. was awarded with FPI fellowship from MINECO, A.R.C. a JAE fellowship from Spanish Research Council (CSIC), an A. T-S. an FPU from Ministry of Education, Professional Formation (MEFP). A.T.-S. is doing his PhD thesis under the frame of the Doctorate Program Biochemistry, Molecular Biology and Biomedicine of the Autonomous University of Barcelona. The Structural Biology Unit at IBMB-CSIC is a “Maria de Maeztu” Unit of Excellence awarded by MINECO [MDM-2014-0435]. IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO. M.O. is an ICREA Academia researcher. Funding for open access charge: Spanish Ministry of Innovation, Science and Universities, formerly Spanish Ministry of Economy and Competitiveness [BFU2015-70645-R].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J.C., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C.. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007; 35:D823–D828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herst P.M., Rowe M.R., Carson G.M., Berridge M.V.. Functional mitochondria in health and disease. Front. Endocrinol. (Lausanne). 2017; 8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lopez-Lluch G. Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech. Ageing. Dev. 2017; 162:108–121. [DOI] [PubMed] [Google Scholar]
- 4. Burte F., Carelli V., Chinnery P.F., Yu-Wai-Man P.. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015; 11:11–24. [DOI] [PubMed] [Google Scholar]
- 5. Chang D.D., Clayton D.A.. Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell. 1984; 36:635–643. [DOI] [PubMed] [Google Scholar]
- 6. Kang D., Miyako K., Kai Y., Irie T., Takeshige K.. In vivo determination of replication origins of human mitochondrial DNA by ligation-mediated polymerase chain reaction. J. Biol. Chem. 1997; 272:15275–15279. [DOI] [PubMed] [Google Scholar]
- 7. Sbisa E., Tanzariello F., Reyes A., Pesole G., Saccone C.. Mammalian mitochondrial D-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene. 1997; 205:125–140. [DOI] [PubMed] [Google Scholar]
- 8. Chang D.D., Clayton D.A.. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. PNAS. 1985; 82:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pham X.H., Farge G., Shi Y., Gaspari M., Gustafsson C.M., Falkenberg M.. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J. Biol. Chem. 2006; 281:24647–24652. [DOI] [PubMed] [Google Scholar]
- 10. Wanrooij P.H., Uhler J.P., Shi Y., Westerlund F., Falkenberg M., Gustafsson C.M.. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012; 40:10334–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wanrooij P.H., Uhler J.P., Simonsson T., Falkenberg M., Gustafsson C.M.. G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. PNAS. 2010; 107:16072–16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hillen H.S., Parshin A.V., Agaronyan K., Morozov Y.I., Graber J.J., Chernev A., Schwinghammer K., Urlaub H., Anikin M., Cramer P. et al.. Mechanism of Transcription Anti-termination in Human Mitochondria. Cell. 2017; 171:1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan B.G., Wellesley F.C., Savery N.J., Szczelkun M.D.. Length heterogeneity at conserved sequence block 2 in human mitochondrial DNA acts as a rheostat for RNA polymerase POLRMT activity. Nucleic Acids Res. 2016; 44:7817–7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher R.P., Topper J.N., Clayton D.A.. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell. 1987; 50:247–258. [DOI] [PubMed] [Google Scholar]
- 15. Ghivizzani S.C., Madsen C.S., Nelen M.R., Ammini C.V., Hauswirth W.W.. In organello footprint analysis of human mitochondrial DNA: human mitochondrial transcription factor A interactions at the origin of replication. Mol. Cell Biol. 1994; 14:7717–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blumberg A., Danko C.G., Kundaje A., Mishmar D.. A common pattern of DNase I footprinting throughout the human mtDNA unveils clues for a chromatin-like organization. Genome Res. 2018; 28:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ngo H.B., Kaiser J.T., Chan D.C.. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat. Struc. Mol. Biol. 2011; 18:1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rubio-Cosials A., Sidow J.F., Jimenez-Menendez N., Fernandez-Millan P., Montoya J., Jacobs H.T., Coll M., Bernado P., Sola M.. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nat. Struc. Mol. Biol. 2011; 18:1281–1289. [DOI] [PubMed] [Google Scholar]
- 19. Hillen H.S., Morozov Y.I., Sarfallah A., Temiakov D., Cramer P.. Structural basis of mitochondrial transcription initiation. Cell. 2017; 171:1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubio-Cosials A., Battistini F., Gansen A., Cuppari A., Bernado P., Orozco M., Langowski J., Toth K., Sola M.. Protein flexibility and synergy of HMG domains underlie U-Turn bending of DNA by TFAM in solution. Biophys. J. 2018; 114:2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshida Y., Izumi H., Ise T., Uramoto H., Torigoe T., Ishiguchi H., Murakami T., Tanabe M., Nakayama Y., Itoh H. et al.. Human mitochondrial transcription factor A binds preferentially to oxidatively damaged DNA. Biochem. Biophys. Res. Commun. 2002; 295:945–951. [DOI] [PubMed] [Google Scholar]
- 22. Ohno T., Umeda S., Hamasaki N., Kang D.. Binding of human mitochondrial transcription factor A, an HMG box protein, to a four-way DNA junction. Biochem. Biophys. Res. Commun. 2000; 271:492–498. [DOI] [PubMed] [Google Scholar]
- 23. Brown T.A., Tkachuk A.N., Clayton D.A.. Mitochondrial Transcription Factor A (TFAM) Binds to RNA Containing 4-Way Junctions and Mitochondrial tRNA. PLoS One. 2015; 10:e0142436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lyonnais S., Tarres-Sole A., Rubio-Cosials A., Cuppari A., Brito R., Jaumot J., Gargallo R., Vilaseca M., Silva C., Granzhan A. et al.. The human mitochondrial transcription factor A is a versatile G-quadruplex binding protein. Sci. Rep. 2017; 7:43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Campbell C.T., Kolesar J.E., Kaufman B.A.. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta. 2012; 1819:921–929. [DOI] [PubMed] [Google Scholar]
- 26. Farge G., Mehmedovic M., Baclayon M., van den Wildenberg S.M., Roos W.H., Gustafsson C.M., Wuite G.J., Falkenberg M.. In vitro-reconstituted nucleoids can block mitochondrial DNA replication and transcription. Cell Rep. 2014; 8:66–74. [DOI] [PubMed] [Google Scholar]
- 27. Uchida A., Murugesapillai D., Kastner M., Wang Y., Lodeiro M.F., Prabhakar S., Oliver G.V., Arnold J.J., Maher L.J., Williams M.C. et al.. Unexpected sequences and structures of mtDNA required for efficient transcription from the first heavy-strand promoter. Elife. 2017; 6:e27283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramachandran A., Basu U., Sultana S., Nandakumar D., Patel S.S.. Human mitochondrial transcription factors TFAM and TFB2M work synergistically in promoter melting during transcription initiation. Nucleic Acids Res. 2017; 45:861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morozov Y.I., Temiakov D.. Human mitochondrial transcription initiation complexes have similar topology on the light and heavy strand promoters. J. Biol. Chem. 2016; 291:13432–13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doublie S. Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol. Biol. (Clifton, N.J.). 2007; 363:91–108. [DOI] [PubMed] [Google Scholar]
- 31. Ngo H.B., Lovely G.A., Phillips R., Chan D.C.. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014; 5:3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kabsch W. Xds. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al.. Overview of the CCP4 suite and current developments. Acta. Crystallogr. D, Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Afonine P.V., Grosse-Kunstleve R.W., Adams P.D.. The Phenix refinement framework. CCP4 Newsl. 2005; 42:http://www.phenix-online.org/papers/ccp4_july_2005_afonine.pdf. [Google Scholar]
- 36. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek P., Roversi P., Sharff A., Smart O., Vonrhein C. et al.. BUSTER Version 2.9. 2010; Cambridge: Global Phasing Ltd. [Google Scholar]
- 37. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D-Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 38. Arnott S., Hukins D.W.. Optimised parameters for A-DNA and B-DNA. Biochem. Biophys. Res. Commun. 1972; 47:1504–1509. [DOI] [PubMed] [Google Scholar]
- 39. Case D.A., Babin V., Berryman J.T., Betz R.M., Cai Q., Cerutti D.S., Cheatham T.E.I., Darden T.A., Duke R.E., Gohlke H.. AMBER. 2014; San Francisco: University of California. [Google Scholar]
- 40. Shields G.C., Laughton C.A., Orozco M.J.. Molecular dynamics simulations of the d(T·A·T) triple helix. J. Am. Chem. Soc. 1997; 119:7463–7469. [Google Scholar]
- 41. Perez A., Luque F.J., Orozco M.. Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc. 2007; 129:14739–14745. [DOI] [PubMed] [Google Scholar]
- 42. Ivani I., Dans P.D., Noy A., Perez A., Faustino I., Hospital A., Walther J., Andrio P., Goni R., Balaceanu A. et al.. Parmbsc1: a refined force field for DNA simulations. Nat. Methods. 2016; 13:55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Darden T., York D., Pedersen L.. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 44. Hospital A., Faustino I., Collepardo-Guevara R., Gonzalez C., Gelpi J.L., Orozco M.. NAFlex: a web server for the study of nucleic acid flexibility. Nucleic Acids Res. 2013; 41:W47–W55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K.. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009; 37:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Drsata T., Lankas F.. Multiscale modelling of DNA mechanics. J. Phys. Condens Matter. 2015; 27:323102. [DOI] [PubMed] [Google Scholar]
- 47. Portella G., Battistini F., Orozco M.. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput. Biol. 2013; 9:e1003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schuck P. Size-distribution analysis of proteins by analytical ultracentrifugation and lamm equation modeling. Biophys. J. 2000; 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schuck P., Perugini M.A., R. G.N., J. H.G., D. S.. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002; 82:1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laue T.M., Shah B.D., Ridgeway T.M., Pelletier S.L., Harding S.E., Rowe A.J., Horton J.C.. Interpretation of analytical sedimentation data for proteins. Analytical Ultracentrifugation in Biochemistry and Polymer Science. 1992; Cambridge: Royal Society of Chemistry; 90–125. [Google Scholar]
- 51. Toth K., Brun N., Langowski J.. Trajectory of nucleosomal linker DNA studied by fluorescence resonance energy transfer. Biochemistry. 2001; 40:6921–6928. [DOI] [PubMed] [Google Scholar]
- 52. Gansen A., Felekyan S., Kuhnemuth R., Lehmann K., Toth K., Seidel C.A.M., Langowski J.. High precision FRET studies reveal reversible transitions in nucleosomes between microseconds and minutes. Nat. Commun. 2018; 9:4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rubio-Cosials A., Sola M.. U-turn DNA bending by human mitochondrial transcription factor A. Curr. Opin. Struct. Biol. 2013; 23:116–124. [DOI] [PubMed] [Google Scholar]
- 54. Jen-Jacobson L., Engler L.E., Jacobson L.A.. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure. 2000; 8:1015–1023. [DOI] [PubMed] [Google Scholar]
- 55. Haran T.E., Mohanty U.. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009; 42:41–81. [DOI] [PubMed] [Google Scholar]
- 56. Chakraborty A., Lyonnais S., Battistini F., Hospital A., Medici G., Prohens R., Orozco M., Vilardell J., Sola M.. DNA structure directs positioning of the mitochondrial genome packaging protein Abf2p. Nucleic Acids Res. 2016; 45:951–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deniz O., Flores O., Battistini F., Perez A., Soler-Lopez M., Orozco M.. Physical properties of naked DNA influence nucleosome positioning and correlate with transcription start and termination sites in yeast. BMC Genomics. 2011; 12:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gangelhoff T.A., Mungalachetty P.S., Nix J.C., Churchill M.E.. Structural analysis and DNA binding of the HMG domains of the human mitochondrial transcription factor A. Nucleic Acids Res. 2009; 37:3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Malarkey C.S., Bestwick M., Kuhlwilm J.E., Shadel G.S., Churchill M.E.. Transcriptional activation by mitochondrial transcription factor A involves preferential distortion of promoter DNA. Nucleic Acids Res. 2012; 40:614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dragan A.I., Read C.M., Makeyeva E.N., Milgotina E.I., Churchill M.E., Crane-Robinson C., Privalov P.L.. DNA binding and bending by HMG boxes: energetic determinants of specificity. J. Mol. Biol. 2004; 343:371–393. [DOI] [PubMed] [Google Scholar]
- 61. Wong T.S., Rajagopalan S., Freund S.M., Rutherford T.J., Andreeva A., Townsley F.M., Petrovich M., Fersht A.R.. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009; 37:6765–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dragan A.I., Read C.M., Crane-Robinson C.. Enthalpy-entropy compensation: the role of solvation. Eur. Biophys J. 2017; 46:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Privalov P.L., Dragan A.I., Crane-Robinson C.. The cost of DNA bending. Trends Biochem. Sci. 2009; 34:464–470. [DOI] [PubMed] [Google Scholar]
- 64. Privalov P.L., Dragan A.I., Crane-Robinson C., Breslauer K.J., Remeta D.P., Minetti C.A.. What drives proteins into the major or minor grooves of DNA?. J. Mol. Biol. 2007; 365:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chrétien D., Bénit P., Ha H.H., Keipert S., El-Khoury R., Chang Y.T., Jastroch M., Jacobs H.T., Rustin P., Rak M.. Mitochondria are physiologically maintained at close to 50 °C. PLoS Biol. 2018; 16:e2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Privalov P.L., Jelesarov I., Read C.M., Dragan A.I., Crane-Robinson C.. The energetics of HMG box interactions with DNA: thermodynamics of the DNA binding of the HMG box from mouse sox-5. J. Mol. Biol. 1999; 294:997–1013. [DOI] [PubMed] [Google Scholar]
- 67. Farge G., Laurens N., Broekmans O.D., van den Wildenberg S.M., Dekker L.C., Gaspari M., Gustafsson C.M., Peterman E.J., Falkenberg M., Wuite G.J.. Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat. Commun. 2012; 3:1013. [DOI] [PubMed] [Google Scholar]
- 68. Kukat C., Davies K.M., Wurm C.A., Spahr H., Bonekamp N.A., Kuhl I., Joos F., Polosa P.L., Park C.B., Posse V. et al.. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. PNAS. 2015; 112:11288–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structures are deposited at the Protein Data bank with PDB codes 6HC3 (TFAM/Site-X complex) and 6HB4 (TFAM/Site-Y complex).