

Abstract
Intracerebral injection of amyloidogenic α-synuclein (αS) has been shown to induce αS pathology in the CNS of nontransgenic mice and αS transgenic mice, albeit with varying efficiencies. In this study, using wild-type human αS transgenic mice (line M20), we demonstrate that intracerebral injection of recombinant amyloidogenic or soluble αS induces extensive αS intracellular inclusion pathology that is associated with robust gliosis. Near the injection site, a significant portion of αS inclusions are detected in neurons but also in astrocytes and microglia. Aberrant induction of expression of the intermediate filament protein peripherin, which is associated with CNS neuronal injury, was also observed predominantly near the site of injection. In addition, many pSer129 αS-induced inclusions colocalize with the low-molecular-mass neurofilament subunit (NFL) or peripherin staining. αS inclusion pathology was also induced in brain regions distal from the injection site, predominantly in neurons. Unexpectedly, we also find prominent p62-immunoreactive, αS-, NFL-, and peripherin-negative inclusions. These findings provide evidence that exogenous αS challenge induces αS pathology but also results in the following: (1) a broader disruption of proteostasis; (2) glial activation; and (3) a marker of a neuronal injury response. Such data suggest that induction of αS pathology after exogenous seeding may involve multiple interdependent mechanisms.
Keywords: alpha-synuclein, amyloid, neurofilament, Parkinson's disease, pathology, peripherin
Introduction
α-Synucleinopathies, such as Parkinson's disease (PD), are a spectrum of neurodegenerative disorders characterized by intracellular amyloidogenic α-synuclein (αS) inclusions (Goedert, 1997; Cookson, 2005; Waxman and Giasson, 2009). αS is normally a soluble presynaptic protein that aggregates in disease states (Goedert, 1997; Cookson, 2005; Waxman and Giasson, 2009). The causal role of αS with neurodegenerative disease is directly supported by the identification of PD-linked genetic alterations in the αS gene (SNCA); these alterations can promote protein aggregation by altering the primary sequence of αS or increasing levels of its expression through duplication or triplication of the SNCA locus (Polymeropoulos et al., 1997; Farrer et al., 1999; Krüger et al., 2000; Singleton et al., 2003; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Kiely et al., 2013; Lesage et al., 2013; Proukakis et al., 2013).
Recent studies have been interpreted as supportive of a “prion-like” spread mechanism in which exogenous amyloidogenic αS serves as a conformational template to drive the conversion of soluble αS to a conformationally altered, aggregated form (Kordower et al., 2008; Li et al., 2008, 2010; Volpicelli-Daley et al., 2011; Luk et al., 2012a,b; Polymenidou and Cleveland, 2012; Guo et al., 2013; Jucker and Walker, 2013; Sacino et al., 2013a,b, 2014). In several studies examining the effects of brain injections of amyloidogenic αS into A53T αS transgenic (Tg) mice (line M83; Luk et al., 2012b) or nontransgenic (nTg) mice (Luk et al., 2012a; Masuda-Suzukake et al., 2013), the spread of pathology was primarily tracked with an antibody against the αS pSer129 phospho-epitope. After the intrahippocampal injection of amyloidogenic αS into nTg mice or E46K human αS Tg mice (line M47), we observed limited αS pathology mostly confined to the site of injection (Sacino et al., 2014). In comparison, we observed both local and distal induction of αS pathology in A53T human αS Tg mice (line M83), but a caveat of these mice is that they are intrinsically prone to form αS inclusion pathology throughout the neuroaxis (Giasson et al., 2002; Sacino et al., 2014).
To further study the mechanism of inclusion pathology induced by intracerebral injection of αS, we used human αS Tg mice (line M20) that never intrinsically develop αS pathology (Giasson et al., 2002; Emmer et al., 2011). We find that intrahippocampal injection of aggregated αS can induce the formation of αS inclusion pathology at the site of injection. It also induces the following: (1) significant gliosis; (2) inclusions comprising the low-molecular-mass neurofilament subunit (NFL) and p62; and (3) expression of the neuronal injury response marker peripherin, which also colocalizes with inclusions. We also observed inclusion pathology distal to the injection site, but this pathology included both αS aggregates and p62-positive/αS-negative inclusions. Thus, in addition to conformational templating, neuroimmune activation, injury response, and altered proteostatic mechanisms may contribute to induction and spread of the αS proteinopathy.
Materials and Methods
Expression and purification of recombinant αS protein.
The pRK172 cDNA construct expressing N-terminal truncated wild-type human αS (with a Met codon added before amino acid 21) or with a deletion of amino acid residues 71–82 in the middle hydrophobic region (Δ71–82) were described previously (Giasson et al., 1999; Waxman and Giasson, 2010, 2011b). αS proteins were expressed in Escherichia coli BL21 (DE3) and purified to homogeneity by size exclusion (Superdex 200 gel filtration) and ion exchanged (Mono Q) chromatographies as described previously (Giasson et al., 2001; Greenbaum et al., 2005).
Fibril preparation of recombinant αS for mouse brain injection.
Human 21–140 αS fibril (hfib) was assembled into filaments by incubation at 37°C at 5 mg/ml in sterile PBS (Invitrogen) with continuous shaking at 1050 rpm (Thermomixer R; Eppendorf). αS amyloid fibril assembly was monitored as described previously with K114 fluorometry (Crystal et al., 2003; Waxman et al., 2009). αS fibrils were diluted to a concentration of 2 mg/ml in sterile PBS and treated at room temperature by mild water bath sonication for 2 h, which results in shorter fibrils (Luk et al., 2009; Waxman and Giasson, 2010; Sacino et al., 2014). These fibrils were tested for induction of intracellular amyloid inclusion formation as described previously (Waxman and Giasson, 2010, 2011a; Sacino et al., 2013b).
Mice husbandry and stereotactic injections.
All procedures were performed according to the National Institutes of Health Guide for the Care and Use of Experimental Animals and were approved by the University of Florida Institutional Animal Care. M20 Tg mice expressing human wild-type αS that do not develop any intrinsic phenotype or αS pathology and M83 Tg mice expressing human A53T αS were described previously (Giasson et al., 2002; Emmer et al., 2011). For stereotactic intracerebral injections of exogenous αS, mice of either sex at 2 months of age were bilaterally stereotaxically injected with 2 μl of 2 mg/ml hfib 21–140 αS or Δ71–82 αS, or 2 μl PBS in the hippocampus (coordinates from bregma: anteroposterior, −1.7 mm; lateral, ±1.6 mm; dorsoventral, −2.0 mm). The inoculum was injected at a rate of 0.2 μl/min (total volume of 2 μl/hemisphere) with the needle in place for 15 min at each site. For all cohorts, n = 4 mice (Table 1).
Table 1.
Summary of intracerebral injections of mice
| Strain | Inoculum | Time after injection (months) | Number of mice | Pathological findings | Induced pSer129/81A staining | Detection by multiple markersa |
|---|---|---|---|---|---|---|
| M20 (WT αS) | 2 μl of PBS | 1 | 4 | 1 of 4 mice shows rare puncta at the injection site | Yes | No |
| M20 (WT αS) | 2 μl of PBS | 2 | 2 | No inclusions | No | No |
| M20 (WT αS) | 2 μl of PBS | 4 | 5 | No inclusions | No | No |
| M20 (WT αS) | Δ71–82 αS (2 μl of 2 mg/ml) | 1 | 4 | 3 of 4 mice show rare puncta in the cortex and at the injection site | Yes | No |
| M20 (WT αS) | Δ71–82 αS (2 μl of 2 mg/ml) | 2 | 4 | 4 of 4 mice show sparse puncta in the cortex and at the injection site | Yes | No |
| M20 (WT αS) | Δ71–82 αS (2 μl of 2 mg/ml) | 4 | 6 | 5 of 6 mice show moderate pathology spreading beyond the injection siteb | Yes | Yes |
| M20 (WT αS) | hfib αS (2 μl of 2 mg/ml) | 1 | 4 | 4 of 4 mice show moderate pathology at the injection sitec | Yes | Yes |
| M20 (WT αS) | hfib αS (2 μl of 2 mg/ml) | 2 | 4 | 4 of 4 mice show moderate pathology in the cortex and the injection sitec | Yes | Yes |
| M20 (WT αS) | hfib αS (2 μl of 2 mg/ml) | 4 | 8 | 8 of 8 mice show abundant, widespread pathologyc | Yes | Yes |
| M83 (A53T αS) | hfib αS (2 μl of 2 mg/ml) | 2 | 4 | 4 of 4 mice show abundant, widespread pathology | Yes | Yes |
Antibodies.
pSer129, also known as clone 81A, is a mouse monoclonal antibody that reacts with αS phosphorylated at Ser129 (Waxman et al., 2008). N52 is a phosphorylation-independent anti-heavy-molecular-mass neurofilament subunit (NFH) antibody (Sigma-Aldrich), and NN18 is a phosphorylation-independent anti-middle-molecular-mass neurofilament subunit (NFM) antibody (Sigma-Aldrich). SMI31 and SMI34 (EMD Millipore) are phosphorylation-dependent anti-NFH antibodies. Rabbit polyclonal antibodies to peripherin and α-internexin and chicken and rabbit polyclonal antibodies for NFL were from EnCor Biotechnology. NR4 is a mouse monoclonal antibody specific for NFL (Sigma-Aldrich). Rabbit anti-NFL monoclonal antibody C28E10 was purchased from Cell Signaling Technology. Paired helical filament-1 (generously provided by Dr. Peter Davies, Albert Einstein University, New York, NY) is specific toward phosphorylation sites Ser396 and Ser404 in tau (Otvos et al., 1994). AT100 (Thermo Fisher Scientific) is specific toward phosphorylation sites Ser212 and Thr214 in tau (Zheng-Fischhöfer et al., 1998). SNL-4 and SNL-1 are rabbit polyclonal antibodies raised against synthetic peptides corresponding to amino acids 2–12 and 104–119 of αS, respectively (Giasson et al., 2000). SNL-1 reacts equally with murine and human αS (Giasson et al., 2000). Syn506 and Syn505 are conformational anti-αS mouse monoclonal antibodies that preferentially detect αS in pathological inclusions (Duda et al., 2002; Waxman et al., 2008). HuA is a rabbit polyclonal antibody raised against full-length αS (Giasson et al., 2000). Syn211 is a mouse monoclonal antibody specific for human αS (Giasson et al., 2000). Syn214 is a mouse monoclonal antibody that specifically reacts with the C terminus of αS (Giasson et al., 2000). Anti-p62 (SQSTM1; Proteintech), anti-glial fibrillary acidic protein (GFAP; Promega), anti-ionized calcium-binding adaptor molecule 1 (IBA-1; Dako), and anti-TAR DNA-binding protein 43 (TDP-43; Proteintech) are rabbit polyclonal antibodies. Anti-TDP-43 rabbit polyclonal antibody was purchased from Proteintech. Rabbit anti-phospho-Ser409/410 TDP-43 antibody is from Cosmo Bio USA. Anti-actin (clone C4) is an affinity-purified monoclonal antibody that reacts with all vertebrate isoforms of actin (Millipore).
Immunoblotting analysis.
The tissue from different brain regions of nTg or M20 Tg mice were lysed in 2% SDS and 50 mm Tris, pH 6.8, with sonication and heated to 100°C for 10 min. Protein concentrations were quantified using the bicinchoninic acid assay and bovine serum albumin as a standard (Pierce Biotechnology). Protein was resolved by SDS-PAGE on 13% polyacrylamide gels, followed by electrophoretic transfer onto nitrocellulose membranes. Membranes were blocked in Tris-buffered saline (TBS) with 5% dry milk and incubated with primary antibodies in TBS/5% dry milk overnight. After washes, membranes were incubated with a goat anti-mouse antibody conjugated to horseradish peroxidase (HRP) or a goat anti-rabbit antibody conjugated to HRP. Protein bands were detected using chemiluminescent reagent (PerkinElmer Life and Analytical Sciences) and a FluorChem E and M Imager (Proteinsimple).
Immunohistochemical analysis.
Mice were killed by CO2 overdose and perfused with PBS/heparin. The brain and spinal cord were then removed and fixed with 70% ethanol/150 mm NaCl for at least 48 h. As described previously, tissues were dehydrated at room temperature through a series of ethanol solutions, followed by xylene, and then were infiltrated with paraffin at 60°C (Duda et al., 2000). The tissues were then embedded into paraffin blocks, which were cut into 7 μm sections. Immunostaining of the sections was performed using previously described methods (Duda et al., 2000) using the avidin–biotin complex (ABC) system (Vectastain ABC Elite kit; Vector Laboratories), and immunocomplexes were visualized with the chromogen 3,3′-diaminobenzidine. Sections were counterstained with hematoxylin. All slides were scanned using an Aperio ScanScope CS (40× magnification; Aperio Technologies), and images of representative areas of αS pathology were taken using the ImageScopeTM software (40× magnification; Aperio Technologies).
Double-labeling immunofluorescence analysis of mouse brain tissue.
Paraffin-embedded tissue sections were deparaffinized and hydrated through a series of graded ethanol solutions, followed by 0.1 m Tris, pH 7.6. The sections were blocked with 5% dry milk/0.1 m Tris, pH 7.6, and were incubated simultaneously with combinations of primary antibodies diluted in 5% dry milk/0.1 m Tris, pH 7.6. After extensive washing, sections were incubated with secondary antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 488 (Invitrogen). Sections were postfixed with formalin, incubated with Sudan Black, and stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). The sections were coverslipped with Fluoromount-G (Southern Biotechnology Associates) and visualized using an Olympus BX51 microscope mounted with a DP71 Olympus digital camera to capture images.
Results
To further investigate the induction of αS inclusion pathology using exogenous αS, we conducted intracerebral injection of soluble (Δ71–82) αS or fibrillar αS in M20 Tg mice that do not intrinsically develop αS pathology (Giasson et al., 2002; Emmer et al., 2011). In these mice, expression of wild-type human αS is driven by the mouse prion protein promoter, and, as expected from this promoter (Borchelt et al., 1996), we observed relative equivalent expression throughout different regions of the brain, including the hippocampus (Fig. 1). For these studies, we performed bilateral injections with 21–140 hfib αS in the hippocampi. We used amino-truncated 21–140 αS, because fibrils comprising this protein can seed αS similarly to the full-length protein in cultured cells (Luk et al., 2009; Waxman and Giasson, 2010, 2011a; Sacino et al., 2013b), and it provides the ability to definitively assess aggregation of the endogenous αS by detection with N-terminal-specific αS antibodies. Using the pSer129/81A antibody, at 1 month after injection of hfib αS, we observed αS inclusion pathology predominantly at the site of injection in the hippocampus; however, at 2 and 4 months after injection, pSer129/81A-positive inclusion pathology increased in the hippocampus and had progressed in many brain areas. These inclusions were especially prominent in the entorhinal cortex area and sparsely distributed in other cortical areas, the striatal region, the midbrain region, and the brainstem (Fig. 2). In comparison, at 1 and 2 months after injection of soluble Δ71–82 αS protein, which has a deletion in the middle of the hydrophobic region of αS that lacks the ability to form or seed αS amyloid in vitro and in cultured cells (Giasson et al., 2001; Luk et al., 2009; Waxman et al., 2009; Sacino et al., 2013b), we observed punctated staining at the injection site only with the pSer129/81A antibody. However, at 4 months after injection of soluble Δ71–82 αS, mature αS inclusion pathology was induced and spread to the cortex, amygdala, thalamus, and hypothalamus (Fig. 3).
Figure 1.
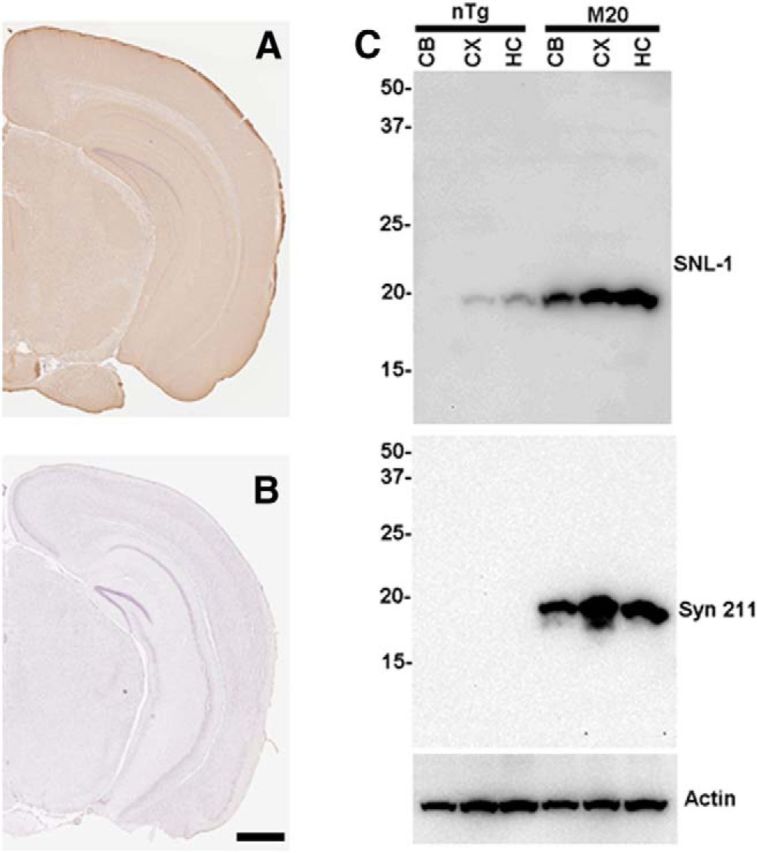
Human αS expression in the hippocampus of M20 Tg mice. Immunocytochemical analysis showing the widespread expression of human αS throughout the hippocampus of M20 Tg mice using anti-human αS antibody Syn 211 (A) and the lack of staining in brain sections from an nTg mouse (B). C, Equal amount of total proteins from the cerebellum (CB), cortex (CX), and hippocampus (HC) of nTg and M20 Tg mice were loaded onto SDS polyacrylamide. Immunoblotting with antibody SNL-1, which reacts equally with mouse and human αS, demonstrating the overexpression of αS in the brain of M20 Tg mice relative to nTg mice. Immunoblotting with human αS antibody Syn211 showing the expression of human αS in all three of these brain regions in M20 Tg mice. Immunoblotting for actin is shown as a loading control. The relative mobility of molecular mass markers in kilodaltons are shown to the left of the blots. Scale bar, 150 μm.
Figure 2.
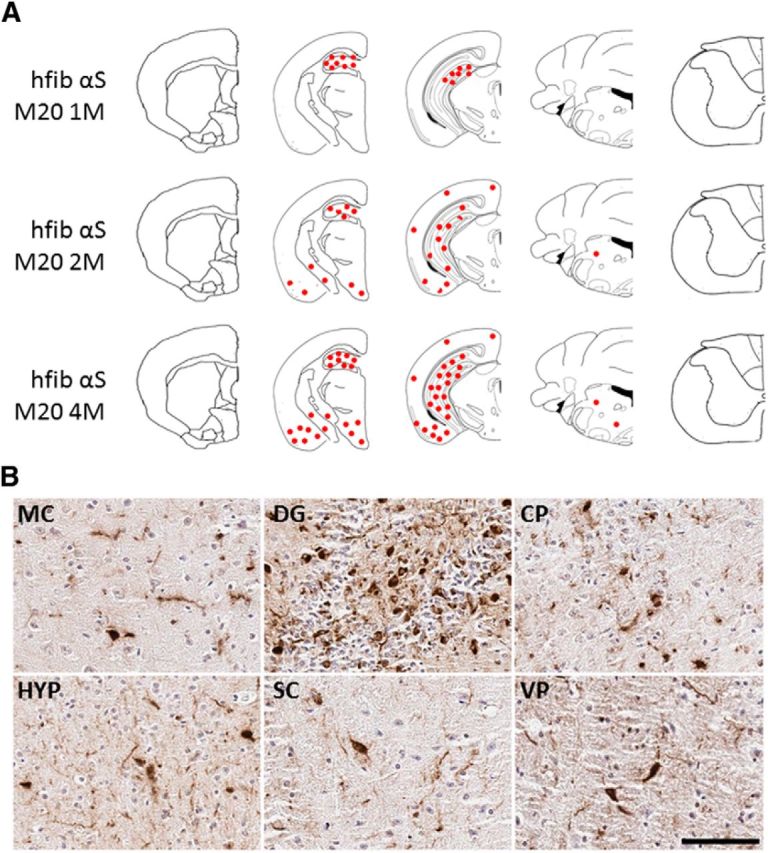
Induction of pSer129/81A-stained inclusions in M20 Tg mice after intrahippocampal injection of 21–140 hfib αS. A, Schematic map showing rostrocaudal distribution of inclusions detected with pSer129/81A antibody in M20 Tg mice at 1, 2, and 4 months after injection. Similar density and distribution of inclusion pathology was seen bilaterally. Inclusions were mainly localized around the site of injection (hippocampus) at 1 month after injection. At 2 and 4 months after injection, more abundant inclusion formation was observed in the hippocampus, and it had progressed to the cortex, striatum, midbrain, and brainstem. B, Representative images of immunohistochemistry showing regions with pSer129/81A-immunoreactive inclusions at 4 months after injection. Perinuclear, intracellular aggregates and neuritic pathology extending into processes are shown in the motor cortex (MC), hippocampus (dentate gyrus; DG), caudate–putamen (CP), hypothalamus (HYP), superior colliculus (SC), and ventral pons (VP). Tissue sections were counterstained with hematoxylin. Scale bar: MC, 100 μm; DG, CP, HYP, SC, and VP, 50 μm.
Figure 3.

Induction of αS inclusions in M20 Tg mice after intrahippocampal injection of soluble Δ71–82 αS. A, Intrahippocampal injection of PBS did not result in the formation of αS inclusions at 4 months after injection. B, C, Intrahippocampal injection of Δ71–82 αS resulted in punctated staining at the site of injection, only detectable by pSer129/81A at 1 and 2 months after injection. D, Intrahippocampal injection of Δ71–82 αS resulted in the formation of αS inclusion pathology throughout the hippocampus, cortex, amygdala, thalamus, and hypothalamus at 4 months after injection. E, αS inclusion pathology was detected by pSer129/81A, Syn506, and p62. Scale bar (A–C, E), 50 μm.
Because we have shown that pSer129/81A immunoreactivity can be misleading in tracking αS pathology because it cross-reacts with NFL phosphorylated at Ser473 (Sacino et al., 2014), we assessed the immunoreactivity of the induced aggregates with other αS antibodies. In the hippocampus, the vast majority of pSer129/81A-positive inclusions could be readily detected with multiple phosphorylation-independent αS antibodies, although some inclusions could also be detected with NFL antibodies (Figs. 4, 5). Quantitative analysis of pSer129/81A-positive inclusions at the injection site of M20 Tg mice revealed that 97 ± 4 and 92 ± 5% of these inclusions were also labeled with anti-αS antibodies SNL-4 or HuA, respectively. Near the site of injection, 49 ± 8% of the pSer129/81A-positive inclusions were labeled with NFL, whereas 23 ± 5% colocalized with peripherin staining. However, in many cortical regions away from the injection site, a significant proportion of pSer129/81A-stained inclusions were not detected with many other αS pathology antibodies, including Syn505, Syn506, SNL-1, SNL-4, HuA, Syn211, and Syn214 (Figs. 5). Staining for NFL showed that only a small subset of these αS-negative pSer129 inclusions contain NFL (Fig. 5); therefore, they are likely comprised of another phospho-protein(s) that can cross-react with the pSer129/81A antibody. These inclusions were not detected with antibodies to tau, TDP-43, NFM, or NFH.
Figure 4.

Immunohistochemical characterization of spatially divergent pSer129/81A inclusion pathology induced by intrahippocampal injection of 21–140 hfib αS after 4 months in M20 Tg mice. Schematic map showing the neuroanatomical distribution of pSer129/81A-stained inclusions mainly around the site of injection in the hippocampus and also in the cortex and midbrain region. A, In the hippocampus at the site of injection, αS inclusions are readily detected by Syn506 in addition to pSer129/81A antibody staining. B, However, in the entorhinal cortex, away from the site of injection, hfib αS injection-induced pSer129/81A inclusion pathology shows very restricted detection with other αS antibodies, such as Syn506. Tissue sections were counterstained with hematoxylin. Scale bar, 100 μm.
Figure 5.
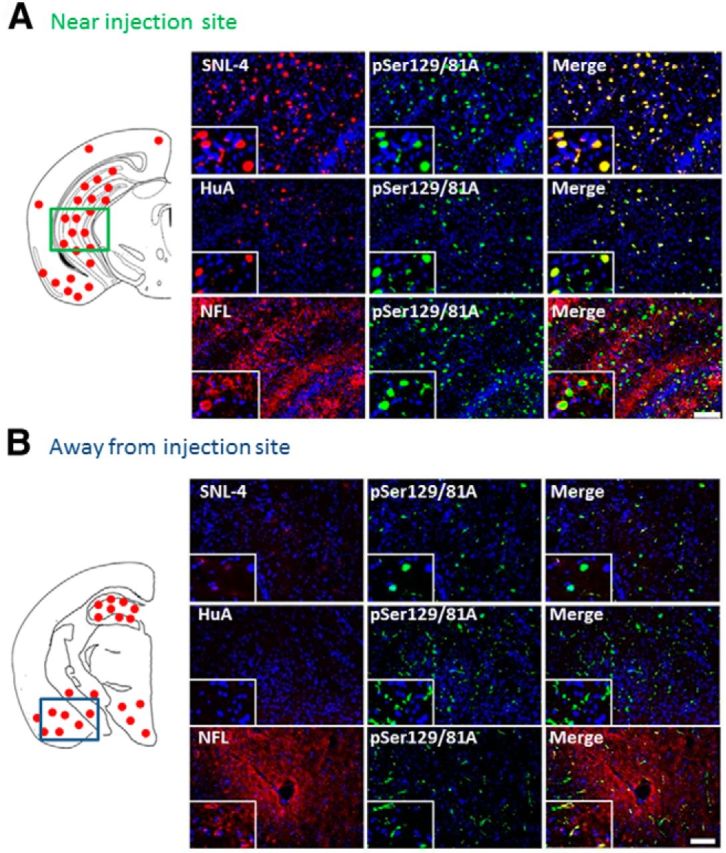
Immunofluorescence analysis of spatially divergent pSer129/81A-stained inclusion pathology induced by intrahippocampal injection of 21–140 hfib αS after 4 months in M20 Tg mice. Schematic map showing the neuroanatomical distribution of pSer129/81A-stained inclusions mainly at the site of injection in the hippocampus and also in the cortex and midbrain region. Double-immunofluorescence analysis at 4 months after injection at sites near (A) and away from (B) the injection of αS fibrils. A, In the hippocampus at the site of injection, pSer129/81A colocalized with both anti-αS antibodies (SNL-4 and HuA) and occasionally with antibodies to NFL. B, In the entorhinal cortex, away from the injection site, pSer129/81A staining (green) rarely colocalized with anti-N-terminal αS antibody SNL-4 (red; top row) or the polyclonal anti-αS antibody HuA (red; middle row); however, there are some pSer129/81A inclusions that contains NFL. Cell nuclei were stained with DAPI. Scale bars: 100 μm; insets, 25 μm.
To further investigate general induction of protein inclusion formation, we stained sections with antibodies to p62/sequestosome, which is a robust general marker of protein aggregation (Kuusisto et al., 2003). Near the site of injection, the vast majority of p62-stained inclusions were also reactive with both the pSer129/81A and Syn506 (anti-αS antibody) antibodies, and many also stained positive for NFL (Fig. 6); however, in areas distal from the injection site, the composition of inclusion pathology was more complex. Many pSer129/81A- or Syn506-stained inclusions were colabeled with p62. However, a significant proportion of p62-labeled inclusions were not pSer129/81A reactive (Fig. 7A), nor were they labeled with the anti-αS antibody Syn506 (Fig. 7B), indicating that some of these inclusions did not comprise αS. The majority of the distal p62-reactive inclusions also were not stained for NFL (Fig. 7C), indicating that they comprised an additional unknown protein or less likely a modified form of αS no longer immunoreactive with multiple antibodies.
Figure 6.

Most anti-p62-reactive inclusions at the site of injection are composed of αS and/or NFL. Immunofluorescence analysis of the hippocampal region at 4 months after injection of 21–140 hfib αS in M20 Tg mice with anti-p62 (red; A–C) and pSer129/81A, Syn506, and anti-NFL (green; A–C, respectively). Most anti-p62-stained inclusions colocalized with pSer129/81A, Syn506, and NFL staining, indicating at the injection site that the majority of the induced inclusions were composed of αS and/or NFL. Cell nuclei were stained with DAPI. Scale bar: 100 μm; insets, 25 μm.
Figure 7.

Many anti-p62-reactive inclusions distal to the site of injection do not contain αS or NFL. Immunofluorescence analysis of cortical gray matter region away from the site of injection with anti-p62 (red; A–C) and pSer129/81A, Syn506, and anti-NFL antibodies (green; A–C, respectively) in an M20 Tg mouse 4 months after hippocampal injection of 21–140 hfib αS injection. Although some inclusions detected with anti-p62 antibody colocalized with either pSer129/81A or Syn506 staining, many of these inclusions do not colocalize with these αS markers, especially Syn506. There also was no colocalization with anti-NFL (white arrow indicates white matter tract). Cell nuclei were stained with DAPI. Scale bar: 100 μm; insets, 25 μm.
Because a significant proportion of protein inclusions near the site of injection were labeled with NFL antibodies, we investigated the presence of other neuronal intermediate filament proteins. We did not observe the accumulation of α-internexin, a protein that can form prominent inclusions in the frontotemporal lobar degenerative disease called neuronal intermediate filament inclusion disease (Cairns et al., 2004; Armstrong et al., 2011). However, in M20 Tg mice injected with hfib αS, robust induction of the neuronal injury response intermediate filament protein peripherin was observed proximal to the injection site; this peripherin staining sometimes colocalized with pSer129/81A and p62 staining (Fig. 8A,E). The aberrant expression of peripherin was not observed in M20 Tg mice at 4 months after injection of Δ71–82 αS (Fig. 8C) or away from the site of injection (in the entorhinal cortex) at 4 months after injection of 21–140 hfib αS (Fig. 8D). Peripherin expression was not unique to the M20 Tg line because we also observed a similar pattern of expression in the M83 Tg mouse line expressing the human PD-linked A53T mutant (Giasson et al., 2002) at 2 months after intrahippocampal injection of 21–140 hfib αS in the presence of robust αS inclusion pathology (Fig. 8B,F; Sacino et al., 2014). The induction of peripherin staining is intriguing because this neuronal intermediate filament protein is typically predominantly expressed in the peripheral nervous system, but its expression in the adult CNS can be induced by injury (Beaulieu et al., 2002).
Figure 8.
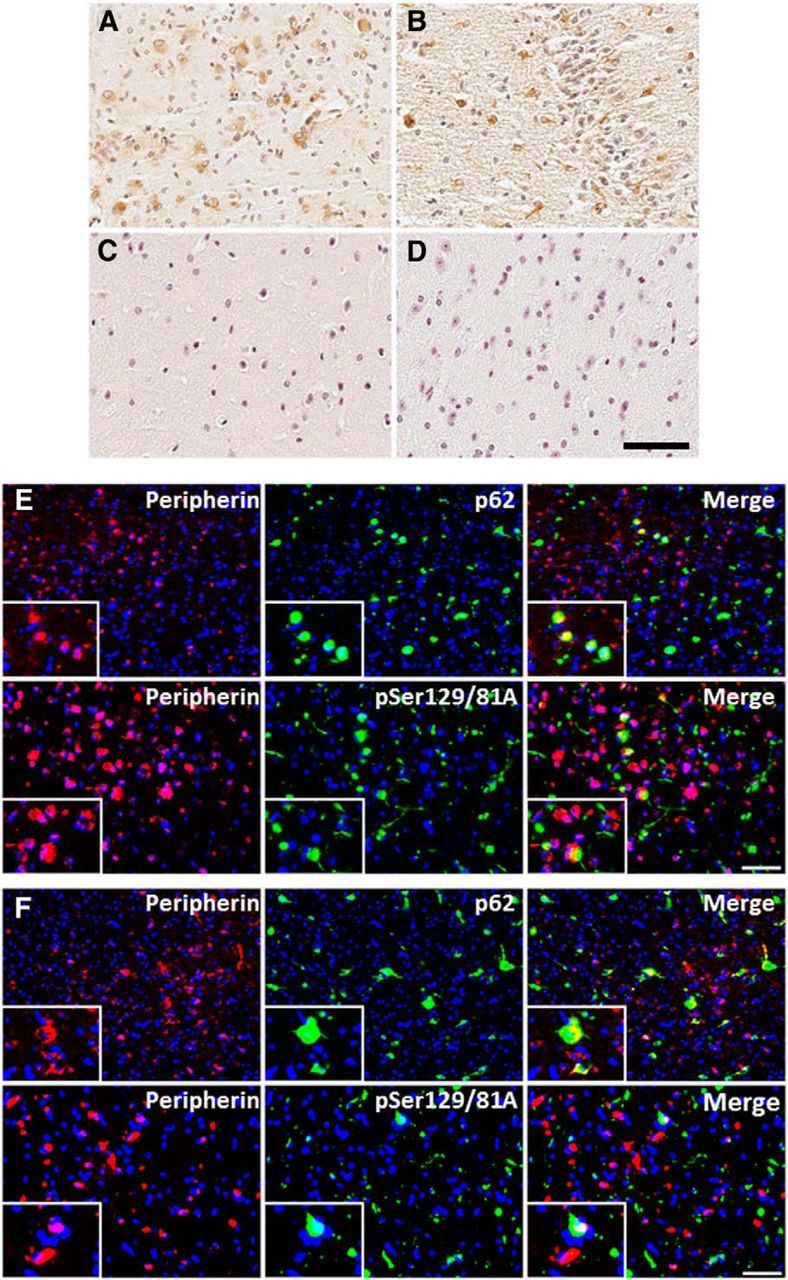
Peripherin immunoreactivity induced by 21–140 hfib αS 4 months after injection in M20 Tg mice and 2 months after injection in M83 Tg mice. Immunohistochemistry with anti-peripherin shows the expression of cytoplasmic peripherin in both M20 Tg mice (A) and M83 Tg mice (B). Aberrant expression of peripherin was not observed in M20 Tg mice at 4 months after injection of Δ71–82 αS (C) or away from the site of injection (in the entorhinal cortex) at 4 months after injection of 21–140 hfib αS (D). Immunofluorescence analysis of the hippocampus with anti-peripherin and anti-p62 or anti-pSer129/81A in M20 Tg mice 4 months after 21–140 hfib αS injection (E) or in M83 Tg mice 2 months after 21–140 hfib αS injection (F) shows that some cells containing p62 inclusions or pSer129 inclusion pathology also express peripherin; however, some neurons demonstrating peripherin immunoreactivity did not have these inclusions. Cell nuclei were stained with DAPI. Scale bars: 250 μm; insets, 62.5 μm.
It has been shown previously by our group that M20 Tg mice injected neonatally with αS fibrils developed generalized massive astrogliosis and moderate microgliosis (Sacino et al., 2013a); therefore, we assessed for neuroinflammation in adult injected M20 Tg mice. Tissue sections from all M20 Tg mice injected intrahippocampally with 21–140 hfib αS were stained with GFAP (astrocyte marker) and IBA-1 (microglia marker). All M20 Tg mice analyzed at 4 months after injection developed both massive astrogliosis and microgliosis in the hippocampus at and near the injection site (Figs. 9A,B,E,F, 10A,D). Furthermore, inclusions labeled with either pSer129/81A or Syn506 antibodies were frequently found within both of these glial cell types (Figs. 9C,D,G,H, 10B,C,E,F). Quantitative analysis of pSer129/81A-positive inclusions near the injection site of M20 Tg mice revealed that 44 ± 5 and 10 ± 2% were found in GFAP- or IBA-1-labeled cells, respectively. To assess whether this finding was unique to M20 Tg mice, we performed a similar analysis using the tissue from M83 Tg mice that we described recently (Sacino et al., 2014). Similar to the findings in M20 Tg mice, this challenge resulted in significant induction of gliosis, and a significant proportion of the induced αS inclusion pathology was found in the astrocytes and to a lesser extent in microglia (Fig. 11). Furthermore, αS inclusion pathology in the entorhinal cortex at 4 months after injection of Δ71–82 αS was found sparsely in both astrocyte and microglia cells (Fig. 12).
Figure 9.
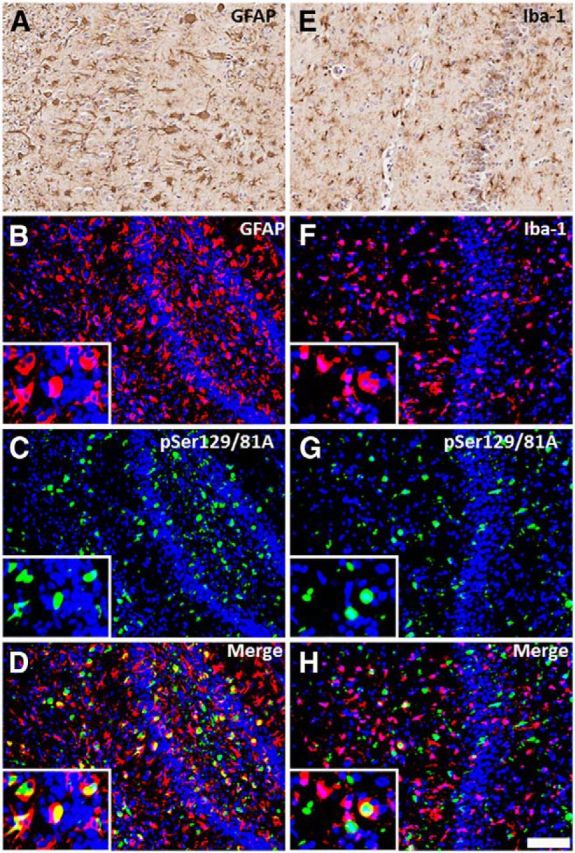
Hippocampal pSer129/81A immunoreactivity induced by 21–140 hfib αS 4 months after injection in M20 Tg mice are abundantly found within astrocytes and microglia. At 4 months after hippocampal injection of hfib αS, immunohistochemistry with anti-GFAP antibody (A) and anti-IBA1 antibody (E) shows robust astrogliosis and microgliosis, respectively. Immunofluorescence analysis of the hippocampus, at the site of injection, shows both astrocytes (B–D) and microglia (F–H) accumulate abundant pSer129/81A-reactive inclusions. Colocalization of pSer129/81A staining (green; C, G) with the astrocyte marker GFAP (red; B) and the microglia marker IBA-1 (red; F) shows that pSer129/81A-reactive inclusions can be found in both cell types. Cell nuclei were stained with DAPI. Scale bar: 250 μm; insets, 62.5 μm.
Figure 10.
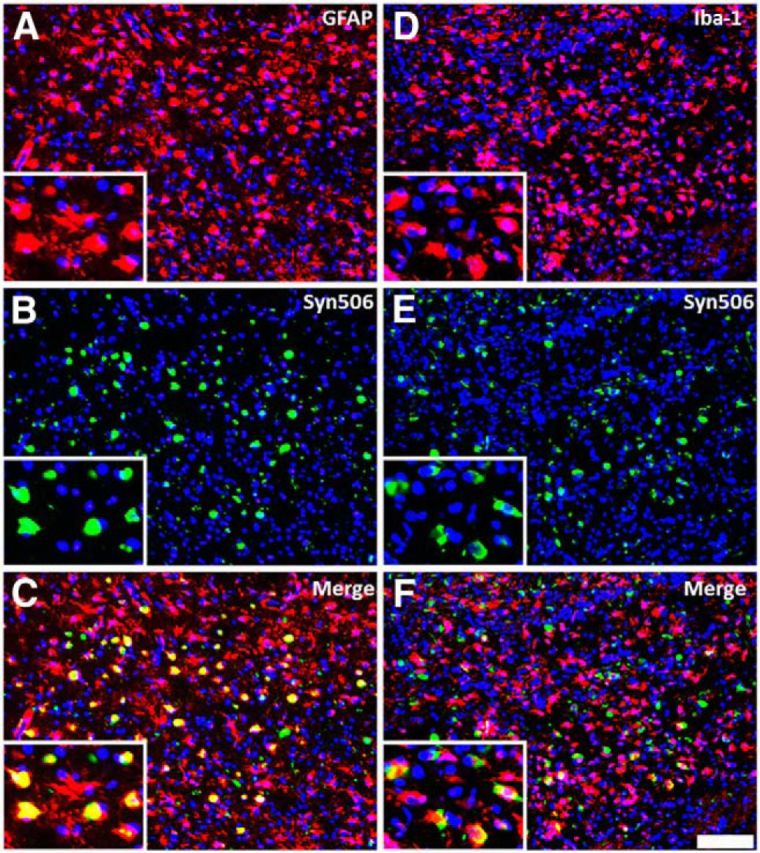
Hippocampal αS inclusions induced by 21–140 hfib αS 4 months after injection in M20 Tg mice are abundantly found within astrocytes and microglia. At 4 months after hippocampal injection of hfib αS, immunofluorescence analysis of the hippocampus, at the site of injection, shows both astrogliosis (A–C) and microgliosis (D–F) containing abundant Syn506-reactive αS inclusions. Colocalization of Syn506 staining (green; B, E) with the astrocyte marker GFAP (red; A) and the microglia marker IBA-1 (red; D) shows that αS inclusions can be found in both cell types. Cell nuclei were stained with DAPI. Scale bar: 250 μm; insets, 62.5 μm.
Figure 11.
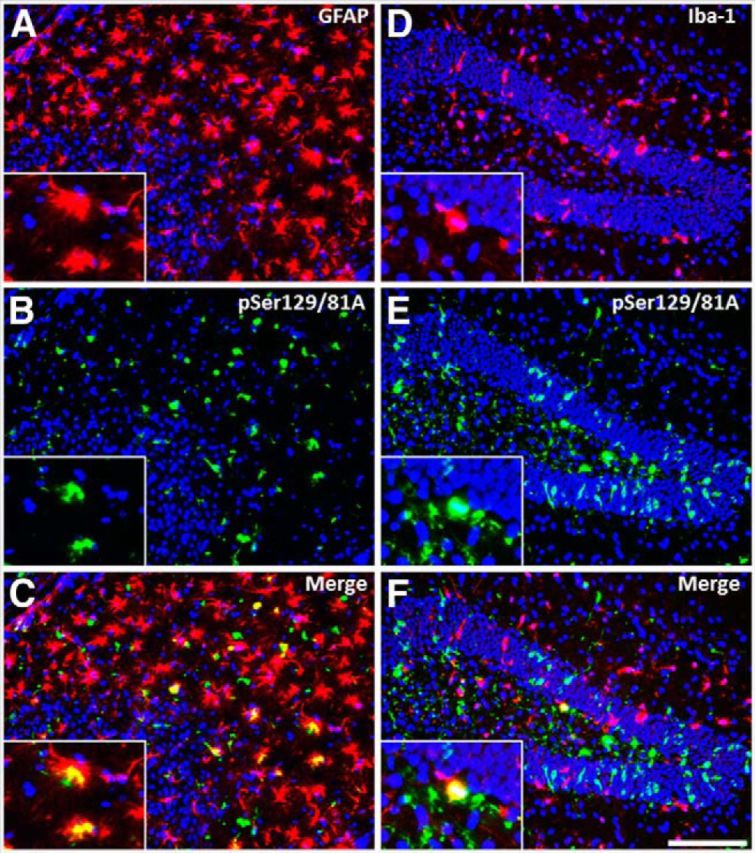
Hippocampal αS inclusions induced by 21–140 hfib αS 2 months after injection in M83 Tg mice are abundantly found within astrocytes. At 2 months after hippocampal injection of hfib αS, immunofluorescence analysis of the hippocampus, at the site of injection, shows both astrogliosis (A–C) and microgliosis (D–F) with pSer129-reactive αS inclusions. Colocalization of pSer129 staining (green; B, E) with the astrocyte marker GFAP (red; A) and the microglia marker IBA-1 (red; D) shows that αS inclusions can be found in both cell types but predominantly in astrocytes. Cell nuclei were stained with DAPI. Scale bar: 250 μm; insets, 62.5 μm.
Figure 12.
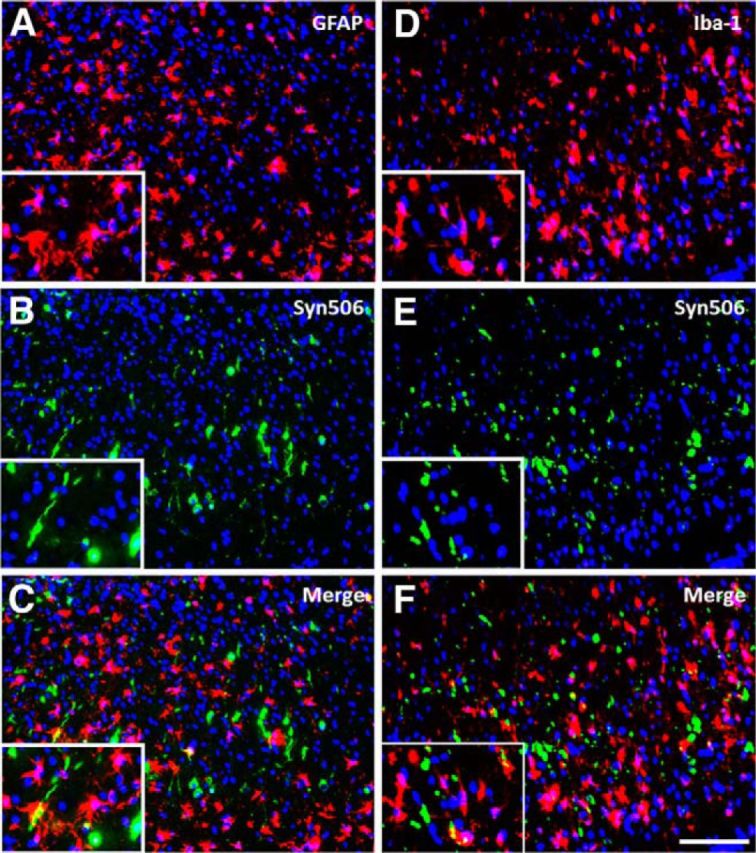
αS inclusions induced by Δ71–82 αS 4 months after injection in M20 Tg mice are rarely found within astrocytes and microglia. At 4 months after hippocampal injection of Δ71–82 αS, immunofluorescence analysis in the entorhinal cortex, shows both astrogliosis (A–C) and microgliosis (D–F). Colocalization of Syn506 staining (green; B, E) with the astrocyte marker GFAP (red; A) and the microglia marker IBA-1 (red; D) shows that αS inclusions are found scantly in both cell types. Cell nuclei were stained with DAPI. Scale bar: 250 μm; insets, 62.5 μm.
Discussion
We show that the intrahippocampal injection of both soluble and aggregated αS in M20 Tg mice leads to the induction of pSer129/81A-reactive inclusion pathology that can progress from the site of injection. These findings are similar to those reported previously by our group after neonatal intracerebral injection of soluble and aggregated αS in M20 Tg mice (Sacino et al., 2013a). Most of these intracellular aggregates near the site of injection appear to be comprised of αS because they are robustly stained with other αS antibodies and these are abundantly present in both neurons and glia. A significant proportion of these inclusions were also reactive for p62, and neuronal inclusions also accumulated NFL.
The composition of the inclusion pathology formed distal to the injection site was heterogonous. A fraction of these inclusions are stained with antibodies to unmodified αS, but a significant proportion of the pSer129/81A-stained inclusions are negative with a battery of αS antibodies. Most of these αS-negative inclusions stain with an antibody to p62, but there are also pSer129/81A-positive inclusions that are p62 negative. In addition, p62-positive inclusions not labeled with pSer129/81A antibody and even more so with antibodies to unmodified αS can readily be observed. These findings are consistent with our recent findings that the antibody pSer129/81A is not completely specific for αS phosphorylated at Ser129 because it cross-reacts with NFL phosphorylated at Ser473 (Sacino et al., 2014). However, at 4 months after hippocampal injection of amyloidogenic αS, most of the pSer129/81A-positive/αS-negative inclusions distal from the injection site are also negative for NFL staining, suggesting the aggregation of an additional unknown protein(s) that can be detected with pSer129/81A antibody. The aggregation of this protein may also account for some of the additional p62-reactive inclusions that are not recognized by antibodies to αS or NFL.
Induction and spread of protein inclusion pathology is greatly accentuated in M20 Tg mice compared with nTg mice in which only local inclusion formation was observed using the same protein to induce intracerebral aggregation (Sacino et al., 2014). Thus, the overexpression of αS in the M20 Tg mice appears to facilitate this process, but the mechanisms involved are likely complex and involve several aberrant processes (Brundin et al., 2008; Golde et al., 2013). Because αS challenge induces multiple inclusion pathologies and gliosis, multiple mechanisms, including prion-like protein conformational templating, neuroimmune activation, injury response and/or general perturbation of proteostatis, or some combination of these could contribute to the phenotype observed.
Unlike previous studies that suggested spread of aggregation of αS along white matter neuroanatomical tracts by staining with pSer129/81A antibody (Luk et al., 2012a,b), we observed only sparse authentic αS inclusion pathology (using antibodies to unmodified αS) in white matter tracts because most of the white matter track staining is attributable to cross-reactivity to NFL phosphorylated at Ser473 (Sacino et al., 2014). As a possible mechanism of inclusion pathology induction and spread, exogenous αS could lead to cellular toxicity, as has been reported in many studies (El-Agnaf et al., 1998; Kayed et al., 2003; Liu et al., 2005; Danzer et al., 2007; Cremades et al., 2012), and/or immune activation as demonstrated by the massive astrogliosis and microgliosis at and near the injection site, with these immune cells containing αS inclusions even at 4 months after injection. These findings are consistent with cell culture studies that showed that both soluble and aggregated αS are potent activators of inflammation (Zhang et al., 2005; Klegeris et al., 2006, 2008; Reynolds et al., 2008; Roodveldt et al., 2008; Su et al., 2008; Lee et al., 2009; Tansey and Goldberg, 2010; Alvarez-Erviti et al., 2011; Béraud et al., 2011; Codolo et al., 2013; Fellner et al., 2013; Kim et al., 2013). A role for immune activation is also suggested by the growing evidence that exogenous amyloidogenic proteins, such as αS, especially when aggregated, act as danger-associated molecular patterns to activate innate immunity (Rubartelli and Lotze, 2007; Golde et al., 2013). Furthermore, extracellular, exogenous αS could lead to a positive-feedback process of cellular damage and inclusion formation, followed by more release of αS aggregates and spread of toxicity. However, we also cannot exclude the possibility that the spread of pathology is attributable to the simple diffusion of the initial injected αS fibrils, resulting in protein inclusion formation by direct seeding or indirectly because of general cellular toxicity.
Collectively, our findings that several types of protein aggregates are induced from the challenge to exogenous αS provide some evidence that impairment to the cellular protein homeostasis mechanism might also be involved in the progressive spread of inclusion pathology. Many lines of evidence suggest that impairment or imbalance of proteostasis contributes to the progression of neurodegenerative diseases (Morimoto and Cuervo, 2009; Kikis et al., 2010; Vekrellis et al., 2011). It is possible that exogenous αS seeds lead to formation of inclusion pathology by overwhelming normal mechanisms that maintain the proteostasis network, such that affected cells can no longer prevent the misfolding and clear misfolded proteins efficiently enough to prevent accumulation. These aggregates of misfolded protein produce an added burden on the proteostasis network by occupying various activities required to dissociate such aggregates and degrade the misfolded proteins, which also result in the aggregation of other proteins or “secondary proteinopathies.” Recent findings indicate that specific proteins that are metastable in nature are more vulnerable as bystanders that can be readily induced to aggregate once the proteostasis network is perturbed (Xu et al., 2012). Furthermore, the presence of misfolded proteins or aggregation-prone protein can disturb the protein homeostasis system causing broad effects on the folding of cellular proteins promoting the misfolding or aggregation of metastable proteins (Gidalevitz et al., 2006; Morimoto and Cuervo, 2009; Kikis et al., 2010; Xu et al., 2012). Future studies will try to address these issues by investigating whether the brain injection of other types of protein aggregates that may perturb protein homeostasis but without the ability to directly induce conformation templating of αS aggregation could also induce the spread of αS inclusion pathology in this model. In addition, direct or indirect failure of the proteasome and/or autophagy that can be inhibited by various forms of aggregated αS could contribute to the spread of αS pathology (Giasson and Lee, 2003; Cookson, 2005; Vekrellis et al., 2011; Ebrahimi-Fakhari et al., 2012, 2013; Manzoni and Lewis, 2013; Xilouri et al., 2013). The notion that impaired proteostasis likely contributes to the spread of aggregate formation is consistent with the induction of inclusion pathology comprising αS, NFL, peripherin, and still other undetermined components. Furthermore, a spread of disease involving a cycle of exogenous secretion/release of aggregated αS and toxicity could be a therapeutic target. However, the prion-like conformational template of αS to form inclusions may also play an important role in the induction and spread of αS inclusion formation. Importantly, all these mechanisms could be involved in the spread of pathological inclusions in human diseases and would be consistent with Braak staging of disease progression (Braak et al., 2006). Future studies will be needed to directly investigate the contribution of various specific mechanisms in the spread of proteinopathy induced by exogenous challenge to αS.
Footnotes
This work was supported by the Ellison Medical Foundation Senior Scholar Award (T.E.G.), the Wilder Family Fellowship (A.N.S.), and funding from the University of Florida.
Dr. Gerry Shaw is the founder, owner, and chief scientific officer at EnCor Biotechnology. The other authors declare no competing financial interests.
References
- Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res. 2011;69:337–342. doi: 10.1016/j.neures.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Gearing M, Bigio EH, Cruz-Sanchez FF, Duyckaerts C, Mackenzie IR, Perry RH, Skullerud K, Yokoo H, Cairns NJ. The spectrum and severity of FUS-immunoreactive inclusions in the frontal and temporal lobes of ten cases of neuronal intermediate filament inclusion disease. Acta Neuropathol. 2011;121:219–228. doi: 10.1007/s00401-010-0753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Kriz J, Julien JP. Induction of peripherin expression in subsets of brain neurons after lesion injury or cerebral ischemia. Brain Res. 2002;946:153–161. doi: 10.1016/S0006-8993(02)02830-5. [DOI] [PubMed] [Google Scholar]
- Béraud D, Twomey M, Bloom B, Mittereder A, Ton V, Neitzke K, Chasovskikh S, Mhyre TR, Maguire-Zeiss KA. α-Synuclein alters toll-like receptor expression. Front Neurosci. 2011;5:80. doi: 10.3389/fnins.2011.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brain and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/S1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Braak H, Bohl JR, Müller CM, Rüb U, de Vos RA, Del Tredici K. Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- Brundin P, Li JY, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci. 2008;9:741–745. doi: 10.1038/nrn2477. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR, Gearing M, Duyckaerts C, Yokoo H, Nakazato Y, Jaros E, Perry RH, Lee VM, Trojanowski JQ. alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004;164:2153–2161. doi: 10.1016/S0002-9440(10)63773-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One. 2013;8:e55375. doi: 10.1371/journal.pone.0055375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Giasson BI, Crowe A, Kung MP, Zhuang ZP, Trojanowski JQ, Lee VM. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J Neurochem. 2003;86:1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VMY, Trojanowski JQ. Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol. 2000;59:830–841. doi: 10.1093/jnen/59.9.830. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VMY, Trojanowski JQ. Novel antibodies to oxidized α-synuclein reveal abundant neuritic pathology in Lewy body disease. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, McLean PJ, Unni VK. Alpha-synuclein's degradation in vivo: opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy. 2012;8:281–283. doi: 10.4161/auto.8.2.18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Saidi LJ, Wahlster L. Molecular chaperones and protein folding as therapeutic targets in Parkinson's disease and other synucleinopathies. Acta Neuropath Commun. 2013;1:79. doi: 10.1186/2051-5960-1-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E, Pessi A, Neill D, Wallace A. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 1998;440:71–75. doi: 10.1016/S0014-5793(98)01418-5. [DOI] [PubMed] [Google Scholar]
- Emmer KL, Waxman EA, Covy JP, Giasson BI. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem. 2011;286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Gwinn-Hardy K, Hutton M, Hardy J. The genetics of disorders with synuclein pathology and parkinsonism. Hum Mol Genet. 1999;8:1901–1905. doi: 10.1093/hmg/8.10.1901. [DOI] [PubMed] [Google Scholar]
- Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Lee VMY. Are ubiquitination pathways central to Parkinson's disease? Cell. 2003;114:1–8. doi: 10.1016/S0092-8674(03)00509-9. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VMY. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Murray IV, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/S0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Goedert M. Familial Parkinson's disease. The awakening of alpha-synuclein. Nature. 1997;388:232–233. doi: 10.1038/40767. [DOI] [PubMed] [Google Scholar]
- Golde TE, Borchelt DR, Giasson BI, Lewis J. Thinking laterally about neurodegenerative proteinopathies. J Clin Invest. 2013;123:1847–1855. doi: 10.1172/JCI66029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha -synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–769. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol. 2010;694:138–159. doi: 10.1007/978-1-4419-7002-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, Giasson BI, Zhang H, Maguire J, Pelech S, McGeer PL. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006;20:2000–2008. doi: 10.1096/fj.06-6183com. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 2008;29:739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Krüger R, Müller T, Riess O. Involvement of alpha-synuclein in Parkinson's disease and other neurodegenerative disorders. J Neural Transm. 2000;107:31–40. doi: 10.1007/s007020050002. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62:1241–1253. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- Lee SB, Park SM, Ahn KJ, Chung KC, Paik SR, Kim J. Identification of the amino acid sequence motif of alpha-synuclein responsible for macrophage activation. Biochem Biophys Res Commun. 2009;381:39–43. doi: 10.1016/j.bbrc.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Widner H, Rehncrona S, Björklund A, Lindvall O, Brundin P. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson's disease. Mov Disord. 2010;25:1091–1096. doi: 10.1002/mds.23012. [DOI] [PubMed] [Google Scholar]
- Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012a;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Lewis PA. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013;27:3424–3429. doi: 10.1096/fj.12-223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI, Cuervo AM. Protein homeostasis and aging: taking care of proteins from the cradle to the grave. J Geront A Biol Sci Med Sci. 2009;64:167–170. doi: 10.1093/gerona/gln071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VMY. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209:889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- Roodveldt C, Christodoulou J, Dobson CM. Immunological features of alpha-synuclein in Parkinson's disease. J Cell Mol Med. 2008;12:1820–1829. doi: 10.1111/j.1582-4934.2008.00450.x. [Erratum 2008 12:2875] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI. Induction of CNS alpha-synuclein pathology by fibrillar and non-amyloidogenic recombinant alpha-synuclein. Acta Neuropathol Commun. 2013a;1:38. doi: 10.1186/2051-5960-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Thomas MA, Ceballos-Diaz C, Cruz PE, Rosario AM, Lewis J, Giasson BI, Golde TE. Conformational templating of alpha-synuclein aggregates in neuronal-glial cultures. Mol Neurodegen. 2013b;8:17. doi: 10.1186/1750-1326-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014;127:645–665. doi: 10.1007/s00401-014-1268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792:616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. A novel, high-efficiency cellular model of fibrillar alpha-synuclein inclusions and the examination of mutations that inhibit amyloid formation. J Neurochem. 2010;113:374–388. doi: 10.1111/j.1471-4159.2010.06592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J Neurosci Res. 2011a;89:231–247. doi: 10.1002/jnr.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011b;31:7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Duda JE, Giasson BI. Characterization of antibodies that selectively detect alpha-synuclein in pathological inclusions. Acta Neuropathol. 2008;116:37–46. doi: 10.1007/s00401-008-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Mazzulli JR, Giasson BI. Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization. Biochemistry. 2009;48:9427–9436. doi: 10.1021/bi900539p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47:537–551. doi: 10.1007/s12035-012-8341-2. [DOI] [PubMed] [Google Scholar]
- Xu G, Stevens SM, Jr, Kobeissy F, Brown H, McClung S, Gold MS, Borchelt DR. Identification of proteins sensitive to thermal stress in human neuroblastoma and glioma cell lines. PLoS One. 2012;7:e49021. doi: 10.1371/journal.pone.0049021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhöfer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]