

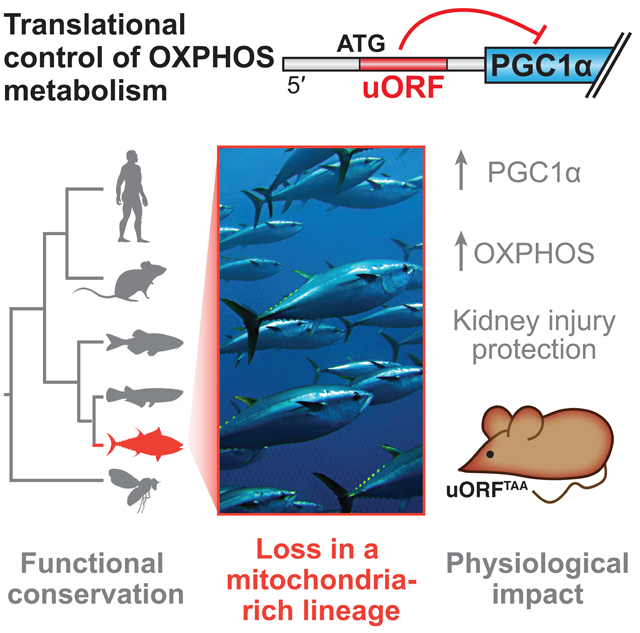
SUMMARY
Mitochondrial abundance and function are tightly controlled during metabolic adaptation, but dysregulated in pathological states such as diabetes, neurodegeneration, cancer, and kidney disease. We show here that translation of PGC1α, a key governor of mitochondrial biogenesis and oxidative metabolism, is negatively regulated by an upstream open reading frame (uORF) in the 5′ untranslated region of its gene (PPARGC1A). We find that uORF–mediated translational repression is a feature of PPARGC1A orthologs from human to fly. Strikingly, whereas multiple inhibitory uORFs are broadly present in fish PPARGC1A orthologs, they are completely absent in the Atlantic bluefin tuna, an animal with exceptionally high mitochondrial content. In mice, an engineered mutation disrupting the PPARGC1A uORF increases PGC1α protein levels and oxidative metabolism, and confers protection from acute kidney injury. These studies identify a translational regulatory element governing oxidative metabolism and highlight its potential contribution to the evolution of organismal mitochondrial function.
Graphical Abstract
INTRODUCTION
The transcriptional coactivator PGC1α was discovered as a brown-fat enriched protein that bound PPARg and other transcription factors, and activated mitochondrial and thermogenic gene expression (Puigserver et al., 1998). PGC1α has since been established as a master regulator of oxidative metabolism and mitochondrial biogenesis not only in brown fat, but also in other tissues such as liver, muscle, brain, and kidney (Fernandez–Marcos and Auwerx, 2011; Lin et al., 2002; 2004; Tran et al., 2011; Yoon et al., 2001). Its functions allow animals to adapt their energy metabolism to environmental and nutritional states.
Post-transcriptional regulation of PGC1α is likely to be crucial for adapting PGC1α levels and function to dynamic metabolic demands on a rapid timescale. Consistent with this idea, PGC1α protein has a short half-life (<30 min) and is heavily post-translationally modified. Furthermore, discrepancies have been noted between the amount of mRNA and the corresponding PGC1α protein (Fisher et al., 2012; Pettersson-Klein et al., 2018; Tsukiyama-Kohara et al., 2001). Given the protective effects of elevated PGC1α function in models of pathology including neurodegenerative disease, muscular dystrophy, muscle atrophy, acute kidney ischemia, and ALS, an understanding of the post-transcriptional modes of quantitative PGC1α modulation is of great interest (Da Cruz et al., 2012; Handschin et al., 2007; Sandri et al., 2006; Tran et al., 2016; Zheng et al., 2010).
Upstream open reading frames (uORFs) reside in the 5′ untranslated regions (UTR) of approximately half of human mRNAs (Calvo et al., 2009; McGillivray et al., 2018), where their function is context-dependent but is generally thought to suppress translation of downstream coding regions (Barbosa et al., 2013; Chew et al., 2016). Although fewer uORFs are present in the human genome than expected by chance, those that are present tend to be evolutionarily conserved, arguing for their adaptive function (Chew et al., 2016; Churbanov et al., 2005). Indeed, uORFs can play specialized roles including regulation of downstream protein output during cell stress (Wethmar et al., 2010b).
We show here that PPARGC1A is under translational control by a uORF. Inhibitory uORF action is functionally conserved in PPARGC1A orthologs from flies to humans. Interestingly, the Atlantic bluefin tuna, an animal with an exceptional oxidative metabolism capacity, completely lacks this regulatory element. A knock-in mutation in mice that prevents uORF translation elevates PGC1α function and confers protection from ischemic kidney injury. These data identify a translational regulatory mechanism controlling PGC1α and mitochondrial function in vivo.
RESULTS
The 5′ UTR of PPARGC1A contains a uORF that negatively regulates PGC1α translation
Given the importance of post-transcriptional mechanisms in determining PGC1α1 (hereafter PGC1α) protein levels, we sought to identify regulatory elements controlling translation of the PPARGC1A mRNA. As a first approach, we analyzed ribosome profiling data to determine the distribution of ribosomes on the mRNA. Using published data from Jurkat cells treated with lactimidomycin (Gawron et al., 2016), an antibiotic that stalls initiating ribosomes, we noticed an accumulation of ribosome footprints at an ATG trinucleotide (AUG in RNA; notations hereafter based on DNA sequence) within the annotated 5′ UTR of PPARGC1A (Figure 1A). In the same cell type, footprints of elongating ribosomes were observed in the 5′ UTR downstream of this ATG. Ribosome profiling datasets collected in HeLa cells similarly demonstrated the presence of elongating ribosome footprints in the PPARGC1A 5′ UTR (Park et al., 2016; Wang et al., 2014; Zur et al., 2016), at levels comparable to those seen in the exon 1 coding region (Figure 1B). These findings suggest ribosomal engagement with an upstream start codon in the PPARGC1A 5′ UTR.
Figure 1. The PPARGC1A 5′ UTR encodes an inhibitory uORF.

(A) Ribosome profiling data indicating location of initiating (blue) or elongating (red) ribosomes on the PPARGC1A transcript in Jurkat cells. Initiating or elongating ribosomes were stalled using lactimidomycin or cycloheximide, respectively. Data adapted from (Gawron et al., 2016). (B) Ribosome profiling data indicating location of elongating ribosomes on the PPARGC1A transcript in HeLa cells. Data adapted from (Park et al., 2016; Wang et al., 2014; Zur et al., 2016). (C) Immunoblot (left) or RT– qPCR (right) of PGC1α protein or mRNA following transfection of each indicated expression vector in 293E cells (n=3). Dash indicates untransfected sample.
We next sought to test whether ribosome engagement with this upstream start codon influences translation of PGC1α. The upstream start codon initiates a 16-codon uORF that does not overlap with the PGC1α coding region. We generated an expression vector in which a CMV promoter drives the PGC1α coding region preceded by a truncated PPARGC1A 5′ UTR lacking the uORF (Short UTR, SU) (Figure 1C). We also generated a construct encoding the entire 139 nt PPARGC1A 5′ UTR (‘WT’), and a construct in which the upstream ATG is mutated to TAA (‘TAA’). Importantly, all constructs contain the same endogenous Kozak sequence surrounding the PGC1α start codon.
Transfection of the SU–PGC1α expression construct into HEK293E cells resulted in greater PGC1α protein levels as compared to WT–PGC1α, suggesting that the PPARGC1A 5′ UTR negatively regulates translation (Figure 1C). The TAA-PGC1α construct also led to greater PGC1α protein levels. To more directly test the idea that ribosome initiation at the upstream ATG is the cause of decreased translation of PGC1α, we also created an expression construct in which the endogenous weak Kozak sequence surrounding the upstream ATG is altered to an optimal Kozak environment (RCCATGG). Indeed, transfection of this construct (‘Strong Kozak’) reduced PGC1α protein levels below those observed with the WT 5′ UTR construct. The PPARGC1A transcript level was similar amongst all constructs (Figure 1C).
To test whether similar regulation could be observed in vitro, we performed translation experiments using a rabbit reticulocyte system. As in intact cells, the full length PPARGC1A 5′ UTR inhibited PGC1α translation as compared to the short 5′ UTR (Figure 2A). The 5′ UTR’s inhibitory effect could be enhanced by strengthening the uORF Kozak sequence and was mostly lost by mutating the uORF start codon to TAA.
Figure 2. The PPARGC1A uORF engages ribosomes to negatively regulate translation of PGC1α.

(A) Immunoblot of PGC1α following translation of each expression construct in reticulocyte lysate. Dash indicates no vector added. (B) Top: schematic of expression constructs. The uORF stop codon and the PGC1α start codon are deleted in the ΔTAA ΔATG2 construct. Bottom: immunoblot of PGC1α following translation of each indicated construct in reticulocyte lysate. (C) Immunoblot of PGC1α following transfection of each indicated expression vector in 293E cells. (D) GFP mRNA level in 293 cells transfected with expression vectors indicated in (C). n=3. (E) Vertebrate phylogeny in which green dots indicate conservation of the human PPARGC1A uORF start codon. White dots indicate species that do not encode an ATG at the genomic position corresponding to the human uORF start codon, but do not rule out the presence of an unrelated upstream ATG.
We next tested whether the PPARGC1A uORF can itself serve as a template for translation. We generated an expression construct in which both the uORF stop codon and the PGC1α start codon were deleted (Figure 2B). Because the uORF is in frame with the PGC1α ORF, translation initiating at the uORF start codon would in this construct be expected to generate an N-terminal extension of PGC1α. Indeed, PGC1α protein produced from this construct migrates at a higher molecular weight upon resolution by SDS–PAGE.
We then asked whether the PPARGC1A 5′ UTR could function outside the context of its specific mRNA. To this end, we first tested whether the inhibitory effect of the 5′ UTR on PGC1α translation could occur in trans. The 5′ UTR did not affect translation of the PPARGC1A coding sequence when each element was encoded on a separate mRNA, nor did addition of a synthetic oligopeptide corresponding to the uORF sequence affect PGC1α translation in vitro (Figure S1A,B). Second, we asked whether the PPARGC1A 5′ UTR could function, in cis, on a novel downstream coding region.
We fused the UTR sequences described above to a green fluorescent protein (GFP) coding sequence. Upon transfection into HEK293E cells, the TAA-GFP and SU-GFP constructs both elevated GFP production relative to the WT–GFP construct, without altering levels of GFP mRNA (Figure 2C,D).
Our finding that the PPARGC1A uORF can engage ribosomes led to a prediction that cellular signals acting to reduce ribosomal preinitiation complex (PIC) scanning fidelity might alter the uORF’s inhibitory potential by allowing the PIC to bypass the uORF in favor of the downstream PGC1α start codon (Barbosa et al., 2013). Consistent with this idea, arsenite treatment, which is expected to cause global suppression of translation and reduce PIC scanning fidelity (Andreev et al., 2015), dampened the PPARGC1A uORF’s quantitative effect on translation, as measured by luciferase assay (Figure S1C).
The PPARGC1A uORF is functionally conserved from human to fly, but absent in Atlantic bluefin tuna
To examine its evolutionary conservation, we assessed the presence of the murine PPARGC1A uORF throughout vertebrates. Since a uORF’s function does not necessarily depend on the polypeptide sequence it encodes, we identified relatives of the murine PPARGC1A uORF by asking which vertebrate genomes, when aligned with the mouse genome, encode a uORF with an ATG start codon at the precise equivalent position (Blanchette et al., 2004). We found that such PPARGC1A uORFs are broadly preserved from humans to lobe-finned fish, the clade of fish that gave rise to terrestrial tetrapods (Sarcopterygii—e.g., coelacanth) (Figure 2E). Supporting the accuracy of the uORF predictions, an alignment of polypeptides encoded by these uORFs revealed related sequences, with particular conservation of a set of serine residues (human positions 2, 5, 7), valines (positions 3, 6), and a cysteine (position 4) (Figure S1D). In contrast to the case of lobe-finned fish, a uORF related to the murine PPARGC1A uORF by predicted amino acid sequence or ATG position is not generally found in the more distantly related ray-finned fish (Actinopterygii), nor is it found in invertebrates.
Importantly, a species that does not contain a direct relative of the murine PPARGC1A uORF may still encode an unrelated uORF in its PPARGC1A gene, possibly preserving uORF functionality. This possibility was assessed in Drosophila and zebrafish owing to the availability of experimental tools for these species. Spargel, the Drosophila PPARGC1A ortholog, encodes an eight-codon uORF. Its primary sequence and start codon location are distinct from those of the murine uORF. To test its function, we generated expression constructs in which the Drosophila Act promoter drives a GFP coding region fused to a 5′ UTR encoding the PPARGC1A or spargel uORF (Figure 3A). We also generated ‘TAA’ versions of these constructs in which their respective uORF start codons are mutated. When assessed in the Drosophila cell line S2R+, disruption of either the mouse or Drosophila uORF similarly elevated GFP levels. These findings suggest that the spargel uORF, though not evolutionarily related by sequence to the murine PPARGC1A uORF, performs a similar inhibitory function.
Figure 3. Inhibitory PPARGC1A uORFs are functionally conserved in fly and zebrafish but not in Atlantic bluefin tuna.

(A) Top: schematic of GFP expression constructs that contain the mouse or fly PPARGC1A/spargel 5′ UTR. Bottom: Constructs were transfected into Drosophila SR2+ cells and GFP was monitored by microscopy. (B) Expression construct mRNA was injected into zebrafish zygotes and analyzed by fluorescence (images) or bioluminescence (graph). mCherry-Luc2 served as control for injection efficiency. N=20; p<0.01. (C) Alignment of conserved uORF within fish ppargc1a 5′ UTRs. The uORF start codon is highlighted in red, whereas the PGC1α start codon is highlighted in green. (D) Expression construct mRNA was injected into zebrafish zygotes and analyzed by fluorescence (images) or bioluminescence (graph). mCherry-Luc2 served as control for injection efficiency. N=20; p<0.01.
The annotated zebrafish ppargc1a 5′ UTR contains two uORFs. Their primary sequences and start codon locations are distinct from the murine and fly uORFs. To test their function, we built DNA constructs in which the zebrafish 5′ UTR (with or without uORF start codon mutations) was fused to the reporter gene ZsGreen::NanoLuc. After in vitro transcription, capped mRNAs were injected into zebrafish zygotes, followed by in vivo fluorescence and bioluminescence measurements 48 hours later. A separate DNA construct encoding the mCherry::Luc2 reporter was used as a control for injection efficiency. uORF mutation in this context significantly increased GFP expression (~3–fold). Therefore, although zebrafish do not encode a ppargc1a uORF directly related to the murine uORF, they have preserved 5′ UTR repressive functionality via uORFs that evolved independently of the murine uORF (Figure 3B).
Using the zebrafish ppargc1a 5′ UTR as a guide, we assessed a diverse set of ray-finned fish: medaka, turquoise killifish, and tilapia. Ppargc1a orthologs in each of these species preserved the second zebrafish uORF, all initiating from an equivalent start codon position (Figure 3C). These findings imply broad conservation of ppargc1a uORF function amongst ray-finned fish.
Another ray-finned fish, the Atlantic bluefin tuna (Thunnus thynnus), is known for its exceptionally high endurance, illustrated by its ability to swim thousands of miles across the Atlantic Ocean (Block et al., 2005). This can be explained, at least in part, by the high abundance of ‘red’ oxidative muscle and mitochondria in this animal (Dalziel et al., 2005). The elevated red muscle content and oxidative capacity in Atlantic bluefin tuna is in stark contrast to what is observed in most fish species (and mammals), which have mainly white glycolytic muscle (Katz, 2002; Korsmeyer and Dewar, 2001; Moyes, 2003). We hypothesized that the red fiber appearance in the Atlantic bluefin tuna, which is similar to the skeletal muscle observed in mice that overexpress PGC1α (Lin et al., 2002), may be due to high activity of PGC1α. This could, in principle, involve alterations in the uORF described here.
The Thunnus thynnus genome has not been sequenced. To investigate its ppargc1a 5′ UTR, we caught a wild Atlantic bluefin tuna in the waters off Cape Cod, MA and performed 5′ RACE. Strikingly, the T. thynnus ppargc1a 5′ UTR is short and lacks any uORFs, including those that we observed in other ray-finned fish species (Figure 3C). To test its ability to support translation, DNA constructs were built encoding the 5′ UTRs of tuna or zebrafish fused to the ZsGreen::NanoLuc reporter gene. When assessed by microinjection, the T. thynnus 5′ UTR directed significantly greater NanoLuc translation than did the zebrafish 5′ UTR (Figure 3D). These findings are consistent with some of the remarkable features of this fish species.
Mutation of the PPARGC1A uORF in mice increases PGC1α function in multiple tissues and protects from acute kidney injury
To determine the functional significance of the mammalian PPARGC1A uORF in vivo, we generated uORF-deficient mice via knock-in mutation of the uORF start codon (Figures 4A and S2A,B). Mutant mice (uORFTAA) were born at expected Mendelian ratios with no gross abnormalities, and their body weight was normal at 8 weeks of age (Figure S2C,D).
Figure 4. uORFTAA mice exhibit elevated PGC1α protein levels and are protected from ischemic kidney injury.

(A) Design of PPARGC1A uORFTAA mouse, in which uORF start codon is mutated. (B) RT–qPCR assessing PPARGC1A mRNA in injured uORFTAA mouse kidney. (C) Immunoblot for PGC1α in injured kidney after immunoprecipitation from kidney lysates. TBP immunoblot of input. (D) Top: Protein lysates of uninjured kidneys were labeled with isobaric tags and subjected to tandem mass spectrometry (n=4). Bottom: Gene set enrichment plot. Proteins detected in MS were ranked based on relative amount in uORFTAA kidney vs wild-type kidney. The first ranked protein is the most up-regulated in uORFTAA kidney (log2 scale). Hash marks represent positions in the ranked list corresponding to members of a given gene set. Normalized enrichment score (NES) indicates whether these members are enriched towards the up-regulated end of this list (positive NES) or down-regulated end of this list (negative NES) as compared to chance expectation. (E) Injured kidney protein lysates blotted for protein components (right) of mitochondrial oxidative phosphorylation complexes I–V (left). (F) LC/MS measurements of serum creatinine from mice 24 hr after bilateral renal ischemia-reperfusion injury (WT n=22; uORFTAA n=15) *p=0.02. (G) Hematoxylin and eosin stain from kidneys in (E). Top: Dotted line surrounds medullary vascular congestion in kidney medulla. Scale bar: 500 μ m. Bottom: Arrows indicate necrotic tubular epithelium (left), whereas arrowheads indicate preserved tubular epithelium (right) in kidney cortex of uORFTAA mice. Scale bar: 50 μ m.
Having obtained homozygous uORFTAA mice, we investigated kidney function, as this tissue has among the highest levels of PGC1α and mitochondria in the body in order to support to the energetic demands of solute transport (Pagliarini et al., 2008; Tran et al., 2011). We were particularly motivated to investigate the pathological setting of acute kidney injury (AKI), a common condition amongst hospitalized patients and major contributor to kidney disease and kidney failure (Chawla et al., 2014). Previous work has established that PPARGC1A is downregulated in the context of chemical, genetic, obstructive, and ischemic kidney injury models (Han et al., 2017; Lynch et al., 2018). Importantly, genetic loss of PGC1α exacerbates experimental AKI, whereas increased expression of PGC1α in the kidney—even to a mild extent relative to endogenous PGC1α—protects from AKI (Tran et al., 2011; 2016). We therefore asked whether quantitative control of PGC1α protein via the PPARGC1A uORF would be sufficient to protect the kidney from ischemia-reperfusion injury. Impairment of renal function can then be quantified by accumulation of serum creatinine, a waste product of endogenous metabolism normally cleared by healthy kidneys.
In the baseline state prior to ischemic injury, uORFTAA mice exhibited normal gross kidney anatomy, urine albumin content, and serum creatinine levels, similar to those of wild-type mice (Figure S3A–F). Levels of PPARGC1A mRNA in kidney were equivalent between the two genotypes, but PGC1α protein was elevated in uORFTAA mice (Figure S3G,H). A separate cohort of mice was subjected to 20 min of kidney ischemia and evaluated after 24 hours of reperfusion, a time point at which serum creatinine elevation is near maximal (Tran et al., 2016). At this time point, PGC1α translation remained derepressed in uORFTAA mice as compared to wild-type mice (Figure 4B,C).
To assess the consequences of uORF ablation on AKI, we first used tandem mass spectrometry after isobaric peptide tagging to compare protein abundance in injured kidneys from uORFTAA versus wild-type mice (Figure 4D, Table S1). Prior work has defined genes that are induced in the context of ischemic kidney injury (‘AKI– correlated’); they are enriched for involvement in functions including leukocyte chemotaxis, cytokine signaling, and inflammatory response (Table S2) (Liu et al., 2017). Gene set enrichment analysis showed that the AKI–correlated gene set was significantly depressed in uORFTAA kidney, implying a reduced extent of kidney injury in uORFTAA mice (Figure 4D). Conversely, proteins that were elevated in uORFTAA kidney included components of multiple OXPHOS complexes, consistent with increased PGC1α function and consequent augmentation of mitochondrial respiratory chain abundance (Figure 4E). Of note, when assessed in the absence of ischemic injury, the uORFTAA mutation’s effect on AKI-associated gene sets was more mild (Figure S3I, Table S1), and the mutation did not affect OXPHOS complex abundance (Figure S3J).
We next assessed a more physiologic readout of kidney injury: serum creatinine levels. Mice carrying the uORFTAA mutation exhibited lower creatinine levels 24 hr after injury, as compared to wild-type littermates, demonstrating relative tolerance to AKI (Figure 4F). Histology confirmed that wild-type mice exhibited a larger zone of vascular congestion in the kidney medulla, a reflection of endothelial swelling in the context of ischemic injury (Figure 4G; dotted line). In the kidney cortex, the site of the most metabolically active tubular epithelium, epithelial damage and necrotic tubules were more pronounced in wild-type mice (Figure 4G; arrows). Together, these results indicate that disruption of the PPARGC1A uORF raises PGC1α levels and function to an extent that alters the course of ischemic kidney injury.
In addition to the kidney, preliminary characterization of skeletal muscle and brown adipose tissue (BAT) in uORFTAA mice further supports a role for the PPARGC1A uORF in regulating PGC1α function and OXPHOS metabolism. In both quadriceps muscle and BAT, gene set enrichment analysis of RNA–seq data comparing uORFTAA versus wild-type mice revealed ‘Oxidative Phosphorylation’ as the most significantly enriched gene set (Figure S4A–E). Skeletal muscle of uORFTAA mice also showed enrichment of a ‘PGC1α signature’ gene set, as defined by genes that are upregulated in the muscle of MCK–PGC1α transgenic mice (Lin et al., 2002). In BAT, the gene expression changes in uORFTAA mice were associated with elevated PGC1α protein (but unchanged PPARGC1A mRNA), as well as increased tissue oxygen consumption (Figure S4F–H). Thus, our data establish the murine PPARGC1A uORF as a functional and physiologically impactful translation regulatory element in vivo.
DISCUSSION
Mitochondrial biogenesis is a crucial process, allowing efficient use of fuels to produce ATP or heat. On the other hand, mitochondria are a source of potentially damaging reactive oxygen species and are energetically costly to produce, requiring assembly of over 1,500 proteins, from two distinct genomes, into specific subcellular structures (Scarpulla, 2011). As a consequence, mitochondrial biogenesis is tightly coupled to environmental cues. While other transcriptional components are also crucial for the expression of mitochondrial genes, it is typically PGC1α that is quantitatively regulated in states of greater mitochondrial demand (Uldry et al., 2006). Our studies show that PGC1α is under regulation by an evolutionarily conserved uORF in the PPARGC1A 5′ UTR.
The extensive conservation of inhibitory uORF function in PPARGC1A orthologs, as well as our observation that multiple PPARGC1A uORFs have arisen independently in vertebrate evolution, highlight the idea that uORF alterations can contribute to the evolution of differences in mitochondrial density and distribution between species. In particular, the Atlantic bluefin tuna stands out as unusual because nearly its entire muscle bed is visibly red, a result of both increased mitochondrial-rich (‘slow–twitch’) muscle as well as an unusually high oxidative capacity within its ‘fast-twitch’ muscle (Korsmeyer and Dewar, 2001; Moyes, 2003). The tuna’s unique metabolic phenotype likely underlies the swimming endurance for which it is famed. Although tuna is not currently experimentally tractable, our findings using the cloned tuna ppargc1a 5′ UTR in zebrafish systems suggest that tuna, by virtue of uORF loss and likely other 5′ UTR alterations, encodes a ppargc1a mRNA that is more robustly translated, perhaps contributing at least in part to its unique physiology.
Cap-dependent translation is a decision point at which regulation can allow complex biological control (Jackson et al., 2010). That uORFs might contribute to such regulation has gained appreciation as ribosome profiling studies have highlighted their prevalence (Ingolia et al., 2014; McGillivray et al., 2018). Indeed, studies in model systems have established roles for uORFs in coordinating dynamic gene expression programs, including in the context of cell differentiation or stress (Brar et al., 2012; Young and Wek, 2016). The conservation of a uORF in PPARGC1A, as well as our finding that its inhibitory effect can be modulated by pharmacological induction of cellular stress, raises the likelihood that this uORF contributes to dynamic regulation of PGC1α. The uORF may also confer regulation by virtue of inclusion or exclusion in different transcript isoforms (Cheng et al., 2018); we note that it is absent in transcripts encoding PGC1α4 and PGC1α–b (Martínez-Redondo et al., 2015).
Given the generally modest effects of uORFs, many insights regarding their function have been obtained by genome-wide analysis. The extent to which an individual uORF can influence mammalian physiology by quantitative modulation of its downstream protein (as opposed to by effects on protein isoform choice) has been unclear (Wethmar et al., 2010a). Our studies demonstrate that preventing ribosome recognition of a single uORF element in the PPARGC1A gene is sufficient to alter PGC1α functional output in multiple metabolically important tissues, and to confer protection from AKI. The setting of AKI may be particularly sensitive to PGC1α levels given that PGC1α function and oxidative phosphorylation systems decrease in the context of renal injury (Tran et al., 2016). Thus, it is possible that the PPARGC1A uORF mutation confers protection from AKI by preventing the AKI–associated drop in PGC1α function. It is also possible that the PGC1α elevation per se, present in baseline conditions, is responsible for the mutation’s therapeutic effect.
Our findings provide a proof of principle that translational control of PGC1α operates within a physiologic window large enough to influence disease processes. Furthermore, our mouse model offers new opportunities to investigate PGC1α function and translational control specifically at times and sites in which the PPARGC1A mRNA is normally expressed, a potential advantage over transgenic models that force heterologous expression.
LIMITATIONS OF STUDY
Our study identifies a translational regulatory element controlling PGC1α expression and investigates its importance through several approaches: evolutionary conservation, functional effects on translation, and a knock-in mouse model. Evolutionary approaches are challenged by the relatively limited genomic annotations in non-model organisms, and by our inability to perform experiments using tuna or their cells. Translational reporter assays lack the natural complexity of the PPARGC1A promoter, which expresses multiple transcript isoforms. For this reason our knock-in mouse model is especially important for demonstrating the uORF’s impact on PPARGC1A translation, as well as on pathophysiology. It will be valuable in the future to examine these mice not only under challenges that cause decreased PGC1α expression (like AKI), but also under challenges that elevate PGC1α (like exercise). Such studies may help reveal physiologic signals that act by modulating uORF function.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce Spiegelman (bruce_spiegelman@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were housed at 23°C under a 12 hr light/dark c ycle with free access to food and water. All experiments used 8–12 week old matched littermates. PPARGC1A uORFTAA mice and littermate uORFWT controls were generated by breeding heterozygous mice in a C57BL/6J background. Animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee (IACUC) of the Beth Israel Deaconess Medical Center.
Generation of uORF deficient animals
uORFTAA heterozygous mice were generated by Applied Stem Cell Inc. mPpargc1a.g9 is 5′-CACACAGCACACACTCATGCAGG–3′. Protospacer Adjacent Motif is underlined. mPpargc1a.g9-ssODN is: 5′ctactttttaatagctttgtcatgtgactggggactgtagtaagacaggtgccttcagttcactctcagtaaggggctggtTg cctgctaaagtgtgtgctgtgtgtcagagtggattggagttgaaaaagcttgactggcgtcattcgggagctggatggcttgg ga-3′. The resultant heterozygous mutant mouse was backcrossed with C57BL/6J for a minimum of three generations before carrying out experimental procedures.
To detect the uORF mutation, PCR was performed with the primers DE144 (5′– ggagtttgtgcagcaagcttg–3′) and DE384 (5′–GAAAGAGTTTCTAACTGCTAC–3′). The PCR product was sequenced using primer DE383 (5′-GACTGGGGACTGTAGTAAG-3′) by Eton Bio.
Zebrafish
AB zebrafish were raised at 28°C under standard housing conditions. All experimental procedures were carried out according to Swiss and EU ethical guidelines and were approved by the animal experimentation ethical committee of Canton of Vaud (permit VD3177).
Cell culture
Human HEK293E cells and monkey COS cells (ATCC) were cultured in DMEM containing 10% fetal bovine serum (HyClone) and penicillin/streptomycin at 37°C in 5% CO2. Drosophila S2R+ cells were cultured in Schneider’s medium (Invitrogen) containing 10% heat-inactivated FBS and penicillin/streptomycin at 25°C.
METHOD DETAILS
Plasmids
The cDNA for mouse PPARGC1A (NM_008904.2) was cloned from a brown fat cDNA library and inserted into pDONR221 using BP clonase (Invitrogen). Site-directed mutagenesis was performed using QuikChange IIXL (Stratagene). For expression, plasmids were then cloned into pcDNA6.2 V5 dest (Invitrogen) using LR reaction.
Gene expression analysis (RT-qPCR)
Total RNA was extracted from frozen tissue or cells using TRIzol (Invitrogen), purified with RNeasy Mini spin columns (Qiagen) and reverse transcribed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). The resultant cDNA was analyzed by RT-qPCR using SYBR green fluorescent dye 2x qPCR master mix (Promega) in a ABI PRISM 7900HT real time PCR system (Applied Biosystems). Relative mRNA levels were normalized to TBP or GAPDH mRNA and calculated using the ΔΔCt method. Primer sequences are shown in Table S3 (Wang et al., 2012).
Immunoblotting
Tissue or cell culture samples were prepared in ice-cold Lysis Buffer (20 mM Tris, pH 7.4, 180 mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X–100, 2.5 mM pyrophosphate, 50mM NaF, 5mM glyerco-phosphoate, 50nM calyculin A, 1mM Na3VO4 and supplemented with Complete EDTA–free protease inhibitor tablet (Roche). Lysates were sonicated, then were centrifuged at 16,000 g for 15 min at 4°C, and the supernatants were used for subsequent analyses. Protein concentration was determined using the bicinchoninic acid assay (Pierce). Protein lysates were denatured in Laemmli buffer (60 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.05% bromophenol blue, 0.7 M β–mercaptoethanol), resolved by 4–12% NuPAGE Bis-Tris SDS–PAGE (Invitrogen) and transferred to a polyvinylidene difluoride (PVDF 0.45 m) membrane. Primary antibodies were diluted in TBS containing 0.05% Tween (TBS–T), 5% BSA, and 0.02% NaN3. Membranes were incubated overnight with primary antibodies at 4°C. For secondary antibody incubation, anti-rabbit or anti-mouse HRP (Promega) was diluted in TBS–T containing 5% dry nonfat milk. Results were visualized with enhanced chemiluminescence (ECL) western blotting substrates (Pierce). Densitometry was performed using ImageJ 1.52k. For each image, signal was normalized such that the average of all control samples equaled 1.0.
Immunoprecipitation
PGC1α detection in mouse tissue required immunoprecipitation prior to immunoblotting (Ruas et al., 2012). For BAT, one lobe of material was lysed by mechanical homogenization in Lysis Buffer (recipe above). Lysates were sonicated, centrifuged at 16,000 g for 15 min at 4°C, and the supernatants removed. Two mg total protein was used for each IP. To pre-clear the lysate, the input protein was incubated 2 hr 4°C with Protein A/G magnetic beads (Thermo) (10 l slurry, washed prior to addition). Beads were removed and then 4 g anti-PGC1α was added (D5, Santa Cruz), followed by overnight rotation at 4°C. The next day, 15 l Protein A/G magnetic bead slurry was washed and added to each IP, followed by rotation at 4°C for 3 hr. Beads were washed 3x in lysis buffer and immunoprecipitated protein was eluted in Laemmli buffer. For kidney, one half kidney was lysed by mechanical homogenization in low-salt buffer (50 mM Tris pH 7.6, 50 mM KCl, 1% NP–40, 20% glycerol, 2 mM EDTA, protease inhibitor). Lysate was centrifuged 12,000 g for 10 min at 4°C, and supernatant was removed. The pellet was extracted with high-salt buffer (50 mM Tris pH 7.6, 400 mM KCl, 1% NP–40, 20% glycerol, 2 mM EDTA, protease inhibitor), sonicated, and centrifuged 16,000 g for 15 min at 4°C. The supernatant was used for protein quantitation and subsequent IP following protocol above, with washes in medium-salt buffer (50 mM Tris pH 7.6, 200 mM KCl, 1% NP–40, 20% glycerol, 2 mM EDTA, protease inhibitor),
DNA transfection
For transient expression of proteins, HEK293E cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen) and lysed for immunoblotting 24 hr later. Drosophila S2R+ cells were transfected with Effectene (Qiagen).
In vitro translation assay
TNT® Quick Coupled Transcription/Translation Systems kit (Promega) was used for in vitro transcription and translation. One microgram of each plasmid was used per reaction, and 1 l of each reaction was used for immunoblotting.
Luciferase translation assay
The full-length PPARGC1A 5′ UTR was cloned upstream of the nanoLuc coding sequence (Promega), with preservation of the endogenous PPARGC1A Kozak sequence. This sequence was then amplified using a forward PCR primer containing a T7 promoter sequence, and the resulting DNA was purified. RNA was transcribed and polyadenylated from this template using the mMESSAGE mMACHINE T7 Ultra kit (Invitrogen), checked for integrity by denaturing gel electrophoresis, purified by phenol-chloroform extraction and precipitation, and quantified by A260 using a NanoDrop (Thermo Fisher).
For translation assays, RNA was transfected into COS cells using Lipofectamine 2000 in a ratio of 2.5 l Lipofectamine 2000 per 1 g RNA. COS cells were allowed to reach 90% confluence in 24-well plates prior to transfection. Immediately prior to transfection, medium was replaced with serum-free DMEM with or without sodium arsenite (Sigma). Cells were harvested 3 hr after transfection and luciferase activity was assessed using Nano-Glo luciferase assay (Promega) and a FLUOstar Omega plate reader (BMG Labtech).
Cloning of full-length PPARGC1A cDNA from bluefin tuna Thunnus thynnus
A wild Atlantic bluefin tuna (Thunnus thynnus) was caught off Cape Cod. Skeletal muscle tissues from the tail were dissected and immediately frozen on dry ice. Total RNA from the skeletal muscle tissue was extracted with TRIzol and purified using RNeasy spin columns (Qiagen). Full-length cDNA library was constructed from purified tuna mRNAs using the SMART cDNA synthesis (TAKARA Bio). A partial cDNA coding tuna ppargc1a was amplified in the tuna cDNA library by using primers (Forward 5’– CACAGAACCATGCAGCCAGCA –3’ and Reverse 5’– CCA GGT GCT CTT GGC TTC AC –3’) that were designed for conserved regions of ppargc1a sequences among vertebrates. Subsequently, amplified cDNAs were cloned into a pCR4–TOPO vector (Invitrogen) for sequencing. 3’ RACE was performed by amplifying tuna PPARGC1A cDNAs using a tuna specific primer (5’–GGC TTC ATA ACT TAC CGC TAC ACC–3’) and adaptor primers for tuna cDNA library. Similarly, 5’ RACE was performed by using a tuna specific primer (5’–TCT CTT GAC TGG CTT TGT GTG G–3’) and adaptor primers specific to the 5’ linker regions. Full-length cDNA sequence was confirmed by reading more than five independent clones both in 3’ RACE and 5’ RACE.
mRNA injections into zebrafish
In vitro transcription of plasmids −0.25ppargc1a:ZsGreen-Nanoluc-pCS2+, −0.25ppargc1a-0.225ATG>TAA,−0.126ATG>TAA:ZsGreen-Nanoluc-pCS2+, −0.12tuna_ppargc1a:ZsGreen-Nanoluc-pCS2, and −0.25ppargc1a:mCherry-Luc2-pCS2 were carried out using an Ambion mMessage Sp6 in vitro transcription kit after linearization with NotI according to the manufacturer’s instructions. One nanoliter of 12.5ng/μl capped mRNA was injected into 1-cell zygotes and visualized 48 hours post fertilization for fluorescence expression as a read-out for translation efficiency using a Leica M165F stereoscope. 0.25ppargc1a:mCherry-Luc2 mRNA was co-injected to control for comparable mRNA levels among experiments. Fluorescence intensity was quantified using FIJI. Quantification of translation efficiency was performed using a Perkin Elmer EnVision plate reader. In brief, zebrafish zygotes were injected with 1nl of 1 ng/μl of the above listed plasmids and luminescence emission values from Nanoluc luciferase were normalized to the Firefly Luciferase values of a co-injected −0.25ppargc1a:mCherry-Luc2 control mRNA using a Promega Dual-Glo assay system according the manufacturer’s instructions.
Mouse ischemia-reperfusion injury experiments
Bilateral renal ischemia-reperfusion injury (IRI) was performed as previously described (Tran et al., 2016). Blood and kidneys were harvested 24 hours after IRI. Serum creatinine measurements were analyzed by LC/MS–MS at the University of Alabama Birmingham O’Brien Core Center for Acute Kidney Injury Research.
Histology
Kidneys were fixed in 4% paraformaldehyde. Paraffin embedding and sectioning were done by the Dana-Farber/Harvard Cancer Center Research Pathology core facility and stained with hematoxylin and eosin.
Tissue respiration
Freshly isolated BAT was rapidly weighed and minced using scissors until homogeneous (3 min). The resulting homogenate was added to 1 ml tissue respiration buffer (10 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, 2% essentially fatty acid free BSA, 1 mM sodium pyruvate, 25 mM glucose, pH 7.4). Oxygen consumption rate was measured in a Clark-type oxygen electrode (Strathkelvin Instruments) at 37°C and normalized to tissue mass.
RNA-seq library preparation and sequencing
Libraries were prepared using Kapa stranded mRNA Hyper Prep sample preparation kits from 500 ng of total RNA isolated from frozen mouse tissue using RNeasy columns (Qiagen). The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR using the Kapa Biosystems library quantification kit according to manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
RNA-seq and gene set enrichment analysis
Sequenced reads were aligned to the UCSC mm9 reference genome assembly and gene counts were quantified using STAR (v2.5.1b) (Dobin et al., 2013). Differential gene expression testing was performed by DESeq2 (v1.10.1) (Love et al., 2014), and resulting fold change values were used as a pre-ranked gene list for gene set enrichment analysis (Mootha et al., 2003; Subramanian et al., 2005). ‘Classic’ mode was used for enrichment statistic calculation. For unbiased discovery of enriched gene sets, the HALLMARK gene sets were used as queries (Liberzon et al., 2015). In a separate query, muscle gene expression data were examined using a custom gene set: ‘PGC1α muscle gene set.’ This gene set contains all genes upregulated >2-fold in the gastrocnemius muscle of MCK–PGC1α transgenic mice (Lin et al., 2002) (Table S2).
Tissue harvest, protein digest, and peptide isobaric labeling
Kidney tissues were lysed with a mechanical homogenizer using lysis buffer composed of 50 mM HEPES pH 8.5, 250 mM NaCl, and EDTA-free protease inhibitor cocktail (Roche), 5 mM TCEP in 2 % SDS. Kidney lysates were centrifuged at 10,000 × g for 10 minutes. Protein content was measured using a BCA assay (Thermo Scientific); disulfide bonds were reduced and cysteine residues alkylated with iodoacetamide (14 mM) essentially as previously described (Huttlin et al., 2010). Protein lysates were purified by methanol-chloroform precipitation and 200 μg was digested overnight with LysC (Wako, Japan) in a ½00 enzyme/protein ratio in 2 M urea and 25 mM HEPES, pH 8.5. Digests were acidified with 10% acetic acid (AA) to a pH of ~ 2 and subjected to C18 solid-phase extraction (50 mg SPE) (Sep–Pak, Waters).
Isobaric labeling of the peptides was performed using 10-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific). Reagents, 5.0 mg, were dissolved in 252 μl acetonitrile (ACN) and 1/10 of the solution were added to 100 μg of peptides dissolved in 100 μl of 200 mM HEPES, pH 8.5. After 1 hour (RT), the reaction was quenched by adding 3 μl of 5% hydroxylamine. Labeled peptides were combined and acidified prior to C18 SPE on Sep-Pak cartridges (50 mg).
Basic pH reversed-phase separation (BPRP)
TMT labeled peptides were solubilized in 500 l solution containing 5% ACN/10 mM ammonium bicarbonate, pH 8.0 and 300 μg of TMT labeled peptides was separated by an Agilent 300 Extend C18 column (3.5 mm particles, 4.6 mm ID and 250 mm in length). An Agilent 1260 binary pump coupled with a photodiode array (PDA) detector (Thermo Scientific) was used to separate the peptides. A 45 minute linear gradient from 10% to 40% acetonitrile in 10 mM ammonium bicarbonate pH 8 (flow rate of 0.6 mL/min) separated the peptide mixtures into a total of 96 fractions (36 seconds). A total of 96 fractions were consolidated into 24 samples in a checkerboard fashion, acidified with 20 μl of 10% formic acid and vacuum dried. Each sample was desalted via StageTips and re-dissolved in 12 μl 5% FA/ 5% ACN, prior to LC–MS/MS analysis.
Liquid chromatography separation and tandem mass spectrometry (LC-MS/MS)
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 75 m inner diameter microcapillary column packed with 35 cm of GP-18 resin (2.6 m, 200 Å, Sepax). For each sample ~1.5 g of peptides were separated using a 3 hr gradient of 6–27% acetonitrile in 0.125% formic acid with a flow rate of 400 nL/min.
Each analysis used an MS3-based TMT method as described previously (McAlister et al., 2014; Ting et al., 2011). The data were acquired using a mass range of m/z 400 – 1400, resolution 120,000, AGC target 5 × 105, maximum injection time 100 ms, dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 1.5 × 105, maximum injection time of 150 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Peptide quantification and gene set enrichment analysis
A compendium of in-house developed software was used to convert mass spectrometric data (Raw file) to the mzXML format, as well as to correct monoisotopic m/z measurements (Elias and Gygi, 2007). All experiments used the Mouse UniProt database (downloaded April 2018) where reversed protein sequences and known contaminants such as human keratins were appended. SEQUEST searches were performed using a 20 ppm precursor ion tolerance, while requiring peptide amino/carboxy (N/C) terminus to have trypsin protease specificity and allowing up to two missed cleavages (Eng et al., 1994). Ten-plex TMT tags on peptide N termini and lysine residues (+ 229.162932 Da) and carbamidomethylation of cysteine residues (+57.02146 Da) were set as static modifications while methionine oxidation (+ 15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target-decoy database search strategy (Elias and Gygi, 2007). Filtering was performed using an in-house linear discrimination analysis(LDA) method to create one combined filter parameter from the following peptide ion and MS2 spectra metrics: SEQUEST parameters XCorr and ΔCn, peptide ion mass accuracy and charge state, in-solution charge of peptide, peptide length and mis-cleavages. Linear discrimination scores were used to assign probabilities to each MS2 spectrum for being assigned correctly and these probabilities were further used to filter the dataset with an MS2 spectra assignment FDR of smaller than a 1% at the protein level (Huttlin et al., 2010).
For quantification, a 0.003 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were further de-normalized based on their ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions (as determined by the manufacturer). The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially uneven TMT labeling and/or sample handling variance.
Fold change values were used to generate a pre-ranked gene list for gene set enrichment analysis (Liberzon et al., 2015; Mootha et al., 2003; Subramanian et al., 2005). ‘Classic’ mode was used for enrichment statistic calculation. To query this gene list, a custom gene set was generated that corresponded to genes up-regulated in the context of ischemic kidney injury: ‘AKI–correlated’ genes. Existing RNA–seq data examining kidney gene expression 24 hr after bilateral ischemic injury (as compared to sham surgery) was used to create this gene set (Liu et al., 2017). AKI–correlated genes were defined as those that were upregulated >4–fold in the context of injury (Table S2).
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental replicates and quantification
Replicate numbers are described in the figure legends. For cellular assays, n corresponds to the number of experimental replicates (e.g., independent transfections). For animal assays or tissue extracted from animals, n corresponds to the number of mice used per genotype or condition. All data are presented as mean and error bars represent standard error.
Statistical analysis
The two-tailed Student’s t test was used for single comparisons. Familywise-error rate (FWER) p values are displayed for gene set enrichment analysis to correct for multiple hypothesis testing.
Supplementary Material
Figure S1, related to Figure 2. PPARGC1A uORF-encoded polypeptide sequences. (A) Immunoblot of PGC1α following in vitro translation of an expression construct encoding PPARGC1A but lacking its uORF. Synthesized uORF–encoded oligopeptide or nonspecific control oligopeptide was added to the rabbit reticulocyte lysate reactions at the indicated concentrations. (B) Top: schematic of the expression construct encoding mouse PPARGC1A 5′ UTR (lacking its uORF) followed by the PGC1α coding sequence. Plasmids added in trans encoded versions of the PPARGC1A 5′ UTR but lacked the PGC1α coding sequence. Bottom: immunoblot of PGC1α following transfection of the indicated constructs in HEK293 cells. (C) Luciferase signal 3 hr after RNA transfection in the presence or absence of sodium arsenite (200 M) (n=4). (D) Relatives of the murine PPARGC1A uORF were identified by asking which vertebrate genomes, when aligned with the mouse genome, encode a uORF with an ATG start codon at the precise equivalent position (Blanchette et al., 2004). Their predicted polypeptide sequences are aligned here.
Figure S2, related to Figure 4. Creation of PPARGC1A uORFTAA mouse with CRISPR/Cas9 (A) Upper: Endogenous mouse PPARGC1A locus highlighting guide RNA used to edit genome. PAM sequence is lower case. Targeted uORF ATG in green. Lower: Resulting uORFTAA edited locus. (B) Confirmation of targeted uORF mutation in homozygous mutant mice by Sanger sequencing. (C) Frequency of genotypes produced from uORFTAA heterozygous mouse intercrosses. Numbers in parentheses indicate the expected number by Mendelian ratio. (D) Body weight of male and female mice at 8 weeks of age.
Figure S3, related to Figure 4. Gross and molecular characterization of kidney in uORFTAA mice. (A) Gross appearance of kidney from wild-type or uORFTAA mice. (B) Gross appearance of left kidney (K), ureter (arrowheads), and bladder (B) in wild-type or uORFTAA mice. Trypan blue dye was injected into renal pelvis to stain ureter and bladder. (C) Kidney length along long axis (n=10–11). (D) Kidney mass normalized to mouse mass (n=10–11). (E) Serum creatinine level (n=19–20). (F) Urine albumin level (n=7–8). (G) RT–qPCR assessing expression of PPARGC1A mRNA in uninjured uORFTAA mouse kidney (n=14). (H) Immunoblot of PGC1α protein in uninjured kidney after immunoprecipitation from kidney lysate. Tubulin immunoblot of input. (I) Top: Protein lysates of uninjured kidneys were labeled with isobaric tags and subjected to tandem mass spectrometry (n=4). Bottom: Gene set enrichment plot. Proteins detected in MS were ranked based on relative amount in uORFTAA kidney vs wild-type kidney. The first ranked protein is the most up-regulated in uORFTAA kidney (log2 scale). Hash marks represent positions in the ranked list corresponding to members of a given gene set. Normalized enrichment score (NES) indicates whether these members are enriched towards the up-regulated end of this list (positive NES) or down-regulated end of this list (negative NES) as compared to chance expectation. (J) Uninjured kidney protein lysates blotted for protein components (right) of mitochondrial oxidative phosphorylation complexes I–V (left).
Figure S4, related to Figure 4. PGC1α levels and function in skeletal muscle and brown adipose tissue of uORFTAA mice. (A) RNA-seq analysis of quadriceps muscle in uORFTAA mice (n=4). Gene set enrichment analysis was used to determine whether any HALLMARK gene sets were enriched amongst genes upregulated in uORFTAA mice. All gene sets enriched with p value <0.01 are shown. (B) Gene set enrichment plots. Genes detected in RNA-seq were ranked based on relative amount in uORFTAA muscle versus wild-type, with the first ranked gene corresponding to the gene most up-regulated in uORFTAA kidney (log2 scale). Hash marks represent positions in the ranked list corresponding to members of a given gene set. Normalized enrichment score (NES) indicates whether these members are enriched towards the up-regulated end of this list (positive NES) or down-regulated end of this list (negative NES) as compared to chance expectation. Top: PGC1α muscle gene set contains genes upregulated >2-fold in MCK–PGC1α transgenic mice. Bottom: HALLMARK OXPHOS gene set. (C) RT–qPCR assessing the 15 genes most upregulated in muscle of MCK-PGC1α transgenic mice (n=8–10). (D) RNA–seq analysis of BAT in uORFTAA mice (n=4). Gene set enrichment analysis was used to determine whether any HALLMARK gene sets were enriched amongst genes upregulated in uORFTAA mice. All gene sets enriched with p value <0.001 are shown. (E) Gene set enrichment plots. HALLMARK OXPHOS and Adipogenesis gene sets are shown. (F) BAT respiration as assessed by oxygen consumption measurement of minced tissue (n=13–14). (G) RT-qPCR assessing PPARGC1A mRNA in uORFTAA mouse BAT (n=6–7). (H) Immunoblot of PGC1α protein in BAT. PGC1α was immunoprecipitated prior to immunoblotting. Tubulin immunoblot of input.
Table S3, Related to STAR Methods. Primers used for RT-qPCR.
Table S1, Related to Figure 4. Quantitative proteomics comparing kidney from wild-type and uORFTAA mutant mice, in the setting of AKI or at baseline.
Table S2, Related to Figure 4. Gene sets used for gene set enrichment analysis.
HIGHLIGHTS (3–4, each 85 characters or fewer).
An upstream open reading frame represses translation of the PPARGC1A mRNA
uORF function is a conserved feature of human, fish, and fly PPARGC1A 5′ UTRs
Atlantic bluefin tuna PPARGC1A 5′ UTR lacks a uORF and supports elevated translation
Mouse PPARGC1A uORF ablation increases PGC1α function and protects from kidney injury
CONTEXT AND SIGNIFICANCE.
Mitochondria are the cell’s powerhouses. Their number and function are tightly controlled so that energy needs are carefully balanced though this can go awry in diseases such as diabetes or kidney disease. PGC1a is key molecular regulator of mitochondria. Researchers at Harvard Medical school identified a naturally occurring conserved genetic element that slows down production of the PGC1α protein. Genetic manipulation of this element in mice increased mitochondrial function and protected them from kidney injury. Although conserved from flies to humans, this PGC1a element is absent in the highly migratory Atlantic bluefin tuna, which has high metabolic demands. This finding is reflective of evolutionary adaptations and opens new avenues to raise PGC1a levels to mitigate metabolic diseases.
ACKNOWLEDGMENTS
This study was aided by the JPB Foundation, the National Institutes of Health (NIH DK061562 to B.M.S; 1F32DK112638–01A1 to D.E.), and the Damon Runyon Cancer Research Foundation (P.A.D.). We thank the Harvard histopathology core for histology. Work in S.M.P.’s laboratory is supported by R01–DK095072 and R35–HL139424. We thank members of the Spiegelman lab for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
BMS is a consultant for Calico, Inc.
REFERENCES
- Andreev DE, O’Connor PBF, Fahey C, Kenny EM, Terenin IM, Dmitriev SE, Cormican P, Morris DW, Shatsky IN, and Baranov PV (2015). Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. Elife 4, e03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Peixeiro I, and Romão L (2013). Gene expression regulation by upstream open reading frames and human disease. PLoS Genet 9, e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Research 14, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block BA, Teo SLH, Walli A, Boustany A, Stokesbury MJW, Farwell CJ, Weng KC, Dewar H, and Williams TD (2005). Electronic tagging and population structure of Atlantic bluefin tuna. Nature 434, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, and Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, and Mootha VK (2009). Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA 106, 7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla LS, Eggers PW, Star RA, and Kimmel PL (2014). Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med 371, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923.e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew G-L, Pauli A, and Schier AF (2016). Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat Commun 7, 11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, Vetto AP, Petrosyan S, Marsala M, Murphy AN, et al. (2012). Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 15, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel AC, Moore SE, and Moyes CD (2005). Mitochondrial enzyme content in the muscles of high-performance fish: evolution and variation among fiber types. Am. J. Physiol. Regul. Integr. Comp. Physiol 288, R163–R172. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, and Auwerx J (2011). Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr 93, 884S–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. (2012). FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawron D, Ndah E, Gevaert K, and Van Damme P (2016). Positional proteomics reveals differences in N-terminal proteoform stability. Mol. Syst. Biol 12, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Wu M-Y, Nam BY, Park JT, Yoo T-H, Kang S-W, Park J, Chinga F, Li S-Y, and Susztak K (2017). PGC-1α Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol 28, 3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, and Spiegelman BM (2007). PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21, 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, Wills MR, and Weissman JS (2014). Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep 8, 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SL (2002). Design of heterothermic muscle in fish. J. Exp. Biol 205, 2251–2266. [DOI] [PubMed] [Google Scholar]
- Korsmeyer KE, and Dewar H (2001). Tuna metabolism and energetics. In Tuna: Physiology, Ecology, and Evolution, (Elsevier; ), pp. 35–78. [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang C-Y, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. (2002). Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418, 797–801. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu P-H, Tarr PT, Lindenberg KS, St-Pierre J, Zhang C-Y, Mootha VK, Jäger S, Vianna CR, Reznick RM, et al. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119, 121–135. [DOI] [PubMed] [Google Scholar]
- Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, et al. (2017). Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MR, Tran MT, and Parikh SM (2018). PGC1α in the kidney. Am. J. Physiol. Renal Physiol 314, F1–F8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Redondo V, Pettersson AT, and Ruas JL (2015). The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 58, 1969–1977. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillivray P, Ault R, Pawashe M, Kitchen R, Balasubramanian S, and Gerstein M (2018). A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Research 46, 3326–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Moyes CD (2003). Controlling muscle mitochondrial content. J. Exp. Biol 206, 4385–4391. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong S-E, Walford GA, Sugiana C, Boneh A, Chen WK, et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-E, Yi H, Kim Y, Chang H, and Kim VN (2016). Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol Cell 62, 462–471. [DOI] [PubMed] [Google Scholar]
- Pettersson-Klein AT, Izadi M, Ferreira DMS, Cervenka I, Correia JC, Martinez-Redondo V, Southern M, Cameron M, Kamenecka T, Agudelo LZ, et al. (2018). Small molecule PGC-1α1 protein stabilizers induce adipocyte Ucp1 expression and uncoupled mitochondrial respiration. Mol Metab 9, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, and Spiegelman BM (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. [DOI] [PubMed] [Google Scholar]
- Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, et al. (2012). A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, and Spiegelman BM (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103, 16260–16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, et al. (2016). PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al. (2011). PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest 121, 4003–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, Gingras AC, Katsume A, Elchebly M, Spiegelman BM, et al. (2001). Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med 7, 1128–1132. [DOI] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, and Spiegelman BM (2006). Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 3, 333–341. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, and Seed B (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Research 40, D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wethmar K, Bégay V, Smink JJ, Zaragoza K, Wiesenthal V, Dörken B, Calkhoven CF, and Leutz A (2010a). C/EBPbetaDeltauORF mice--a genetic model for uORF-mediated translational control in mammals. Genes Dev 24, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wethmar K, Smink JJ, and Leutz A (2010b). Upstream open reading frames: molecular switches in (patho)physiology. Bioessays 32, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. (2001). Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138. [DOI] [PubMed] [Google Scholar]
- Young SK, and Wek RC (2016). Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J Biol Chem 291, 16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, et al. (2010). PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2, 52ra73–52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Aviner R, and Tuller T (2016). Complementary Post Transcriptional Regulatory Information is Detected by PUNCH-P and Ribosome Profiling. Sci Rep 6, 21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 2. PPARGC1A uORF-encoded polypeptide sequences. (A) Immunoblot of PGC1α following in vitro translation of an expression construct encoding PPARGC1A but lacking its uORF. Synthesized uORF–encoded oligopeptide or nonspecific control oligopeptide was added to the rabbit reticulocyte lysate reactions at the indicated concentrations. (B) Top: schematic of the expression construct encoding mouse PPARGC1A 5′ UTR (lacking its uORF) followed by the PGC1α coding sequence. Plasmids added in trans encoded versions of the PPARGC1A 5′ UTR but lacked the PGC1α coding sequence. Bottom: immunoblot of PGC1α following transfection of the indicated constructs in HEK293 cells. (C) Luciferase signal 3 hr after RNA transfection in the presence or absence of sodium arsenite (200 M) (n=4). (D) Relatives of the murine PPARGC1A uORF were identified by asking which vertebrate genomes, when aligned with the mouse genome, encode a uORF with an ATG start codon at the precise equivalent position (Blanchette et al., 2004). Their predicted polypeptide sequences are aligned here.
Figure S2, related to Figure 4. Creation of PPARGC1A uORFTAA mouse with CRISPR/Cas9 (A) Upper: Endogenous mouse PPARGC1A locus highlighting guide RNA used to edit genome. PAM sequence is lower case. Targeted uORF ATG in green. Lower: Resulting uORFTAA edited locus. (B) Confirmation of targeted uORF mutation in homozygous mutant mice by Sanger sequencing. (C) Frequency of genotypes produced from uORFTAA heterozygous mouse intercrosses. Numbers in parentheses indicate the expected number by Mendelian ratio. (D) Body weight of male and female mice at 8 weeks of age.
Figure S3, related to Figure 4. Gross and molecular characterization of kidney in uORFTAA mice. (A) Gross appearance of kidney from wild-type or uORFTAA mice. (B) Gross appearance of left kidney (K), ureter (arrowheads), and bladder (B) in wild-type or uORFTAA mice. Trypan blue dye was injected into renal pelvis to stain ureter and bladder. (C) Kidney length along long axis (n=10–11). (D) Kidney mass normalized to mouse mass (n=10–11). (E) Serum creatinine level (n=19–20). (F) Urine albumin level (n=7–8). (G) RT–qPCR assessing expression of PPARGC1A mRNA in uninjured uORFTAA mouse kidney (n=14). (H) Immunoblot of PGC1α protein in uninjured kidney after immunoprecipitation from kidney lysate. Tubulin immunoblot of input. (I) Top: Protein lysates of uninjured kidneys were labeled with isobaric tags and subjected to tandem mass spectrometry (n=4). Bottom: Gene set enrichment plot. Proteins detected in MS were ranked based on relative amount in uORFTAA kidney vs wild-type kidney. The first ranked protein is the most up-regulated in uORFTAA kidney (log2 scale). Hash marks represent positions in the ranked list corresponding to members of a given gene set. Normalized enrichment score (NES) indicates whether these members are enriched towards the up-regulated end of this list (positive NES) or down-regulated end of this list (negative NES) as compared to chance expectation. (J) Uninjured kidney protein lysates blotted for protein components (right) of mitochondrial oxidative phosphorylation complexes I–V (left).
Figure S4, related to Figure 4. PGC1α levels and function in skeletal muscle and brown adipose tissue of uORFTAA mice. (A) RNA-seq analysis of quadriceps muscle in uORFTAA mice (n=4). Gene set enrichment analysis was used to determine whether any HALLMARK gene sets were enriched amongst genes upregulated in uORFTAA mice. All gene sets enriched with p value <0.01 are shown. (B) Gene set enrichment plots. Genes detected in RNA-seq were ranked based on relative amount in uORFTAA muscle versus wild-type, with the first ranked gene corresponding to the gene most up-regulated in uORFTAA kidney (log2 scale). Hash marks represent positions in the ranked list corresponding to members of a given gene set. Normalized enrichment score (NES) indicates whether these members are enriched towards the up-regulated end of this list (positive NES) or down-regulated end of this list (negative NES) as compared to chance expectation. Top: PGC1α muscle gene set contains genes upregulated >2-fold in MCK–PGC1α transgenic mice. Bottom: HALLMARK OXPHOS gene set. (C) RT–qPCR assessing the 15 genes most upregulated in muscle of MCK-PGC1α transgenic mice (n=8–10). (D) RNA–seq analysis of BAT in uORFTAA mice (n=4). Gene set enrichment analysis was used to determine whether any HALLMARK gene sets were enriched amongst genes upregulated in uORFTAA mice. All gene sets enriched with p value <0.001 are shown. (E) Gene set enrichment plots. HALLMARK OXPHOS and Adipogenesis gene sets are shown. (F) BAT respiration as assessed by oxygen consumption measurement of minced tissue (n=13–14). (G) RT-qPCR assessing PPARGC1A mRNA in uORFTAA mouse BAT (n=6–7). (H) Immunoblot of PGC1α protein in BAT. PGC1α was immunoprecipitated prior to immunoblotting. Tubulin immunoblot of input.
Table S3, Related to STAR Methods. Primers used for RT-qPCR.
Table S1, Related to Figure 4. Quantitative proteomics comparing kidney from wild-type and uORFTAA mutant mice, in the setting of AKI or at baseline.
Table S2, Related to Figure 4. Gene sets used for gene set enrichment analysis.