

Abstract
Intra-acinar trypsinogen activation occurs in the earliest stages of pancreatitis and is believed to play important roles in pancreatitis pathogenesis. However, the exact role of intra-acinar trypsin activity in pancreatitis remains elusive. Here, we aimed to examine the specific effects of intra-acinar trypsin activity on the development of pancreatitis using a transgenic mouse model. This transgenic mouse model allowed for the conditional expression of a mutant trypsinogen that can be activated specifically inside pancreatic acinar cells. We found that expression of this active mutated trypsin had no significant effect on triggering spontaneous pancreatitis. Instead, several protective compensatory mechanisms, including SPINK1 and heat shock proteins, were upregulated. Notably, these transgenic mice developed much more severe acute pancreatitis, compared with control mice, when challenged with caerulein. Elevated tissue edema, serum amylase, inflammatory cell infiltration and acinar cell apoptosis were dramatically associated with increased trypsin activity. Furthermore, chronic pathological changes were observed in the pancreas of all transgenic mice, including inflammatory cell infiltration, parenchymal atrophy and cell loss, fibrosis, and fatty replacement. These changes were not observed in control mice treated with caerulein. The alterations in pancreata from transgenic mice mimicked the histological changes common to human chronic pancreatitis. Taken together, we provided in vivo evidence that increased intra-acinar activation of trypsinogen plays an important role in the initiation and progression of both acute and chronic pancreatitis.
NEW & NOTEWORTHY Trypsinogen is activated early in pancreatitis. However, the roles of trypsin in the development of pancreatitis have not been fully addressed. Using a genetic approach, we showed trypsin activity is critical for the severity of both acute and chronic pancreatitis.
Keywords: cell death, digestive enzyme, inflammation, mouse models, pancreatic disorders
INTRODUCTION
Acute pancreatitis is an inflammatory condition of the pancreas associated with upper abdominal pain, bloating, and nausea and vomiting; it usually requires emergency medical treatment. Acute pancreatitis is associated with significant morbidity and mortality; however, the pathogenic mechanisms of both acute and chronic pancreatitis are still not well understood. Thus there are currently no targeted preventative medications or therapies available for the clinic (2). More than 100 years ago, Chiari (4) proposed that acute pancreatitis is a result of autodigestion. Intracellular trypsinogen activation has consistently been observed in the early phases of pancreatitis in several developed animal models as well as in clinical patient specimens (23, 26, 41). Given the character of trypsin as a digestive enzyme and its capability of activating other zymogens, pathological intra-acinar trypsinogen activation was hypothesized to be the central mechanism of pancreatitis initiation. This notion is strongly supported by the observation that human hereditary pancreatitis is closely associated with gain-of-function trypsinogen mutations or loss-of-function of endogenous trypsin inhibitors [e.g., serine protease inhibitor Kazal type 1 (SPINK)] (7, 31, 40). In addition, direct intra-acinar activation of trypsinogen causes pancreatic acinar cell damage and initiates the development of acute pancreatitis in mice (8, 16).
Recently, it has also been reported that a genetic mouse model was developed in which trypsinogen isoform-7, the most prominent trypsinogen in mice, was deleted (5). The deficiency of pathological activation of trypsinogen prevents acinar cells from death in vitro and decreases acinar cell necrosis in vivo. However, local and systemic inflammations are not different between wild-type mice and mice bearing trypsinogen isoform-7 knockout (5). Furthermore, these mice also had similar levels of atrophy, histomorphological features of chronic pancreatitis, and chronic inflammation (34). These findings necessitate a paradigm shift in our understanding of acute pancreatitis. The role of trypsin in the initiation of acute pancreatitis and its progression to chronic pancreatitis needs to be further clarified.
Herein, using a transgenic mouse model in which trypsinogen was engineered to be activated by paired basic amino acid-cleaving enzyme (PACE)/furin in the secretory pathway of the pancreatic acinar cells (8, 16), we demonstrated that the elevated trypsin activity in the secretory pathway causes severe pancreatitis under caerulein stimulation. In our study, the level of trypsin activity was positively correlated with the severity of acute pancreatitis. Additionally, increased trypsin activity promoted the development of chronic pancreatitis. Altogether, our data illustrate that early trypsin activation in pancreatic acinar cells may be the driving force that aggravates the severity of acute pancreatitis and results in the progression to chronic pancreatitis.
METHODS AND EXPERIMENTAL PROCEDURES
Transgenic mice.
All research has been reviewed and approved by our Institutional Animal Care and Use Committee. The generation of the PACE-trypsinogen transgenic mouse model has been described previously (8). LGL (loxP-GFP-stop-loxP)-PACE-PRSS2 transgenic mice were bred with transgenic mice expressing CreERT from a full-length elastase promoter in a bacteria artificial chromosome (BAC) for specific targeted expression of PACE-trypsinogen in pancreatic acinar cells. Because of high levels of CreERT expression in differentiated acinar cells, tamoxifen-independent recombination is in approximately ~50% of adult acinar cells at 2 mo of age, and the recombination rate increases with age (18).
Genotyping.
Mice tails (0.2 cm) were snipped at 2 wk old. DNA was prepared for PCR by incubating the tail with 250 μl of 50 mM sodium hydroxide at 95°C for 15 min followed by neutralization with 25 μl of 1 M Tris (pH 8.0) containing 10 mM EDTA. Primers designed were as follows: Cre primer forward, gcctgcattaccggtcga; Cre primer reverse, tatcctggcagcgatcgc; and PRSS2 primer forward, ggacttgtagcagtggcctgc; PRSS2 primer reverse, tggagttcgtgaccgccgccg.
Induction of pancreatitis with caerulein.
Control mice expressing CreERT alone (BAC) and transgenic mice expressing PRSS2 (Try/BAC) were challenged with caerulein or l-arginine. For caerulein-induced pancreatitis, caerulein was solubilized in PBS at a final concentration of 15 µg/ml and injected intraperitoneally (50 µg/kg every hour). For acute pancreatitis, 8 hourly injections were utilized, and pancreatitis was assessed 12–24 h after the final caerulein treatment. For chronic pancreatitis, mice were treated with eight hourly injections 1 day/week for 2 wk, and pancreatitis was assessed 6 wk after the first injection. For arginine-induced pancreatitis, mice were treated with l-arginine (8% in normal saline, 4 g/kg) twice with an hour interval between injections. The pancreatic tissues were collected 72 h after initial l-arginine injection (6).
Isolation of pancreatic acini: trypsin activity assay.
Pancreatic acinar cells were isolated from the Try, BAC, or Try/BAC mice. The detailed protocol is described in the previous study (17). Briefly, the isolated pancreas was digested by 1KU CLSPA collagenase (Worthington, Freehold, NJ) in the presence of 0.1 mg/ml trypsin inhibitor (Sigma). The green fluorescent protein (GFP) signal in acinar cells was observed by fluorescence microscopy. CBZ-Ile-Pro-Arg-R110, a fluorescent trypsin substrate (Invitrogen), was used to measure trypsinogen activation by fluorescence microscopy. Ethidium bromide was used for detecting dead acinar cells.
RNA isolation and quantitative PCR.
Total RNA was prepared from mouse pancreatic tissues with TRIzol reagent (Invitrogen) and was further purified with the RNeasy Plus Mini Kit (Qiagen). cDNA was synthesized using a reverse transcription kit (Promega, Madison, WI). Quantitative (q) PCR was performed using the Quant Studio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). Primers were as follows: mouse SPINK forward, CCTATATGACCACTCCTCAGTTTC; mouse SPINK reverse, GTCCCACACACAGGATCATAAA; mouse heat shock protein 70 (HSP70) forward, CCATCGAGGAGGTGGATTAGA; mouse HSP70 reverse, AGTGCTGCTCCCAACATTAC; mouse HSP90aA forward, GGACAGCAAACATGGAGAGA; mouse HSP90aA reverse, CCAGACTGAAGCCAGAAGATAG; mouse HSP90aB forward, CGGATTCTACTCGGCCTATCTA; mouse HSP90aB reverse, CTTTCTCTTCCTCTGCCTCATC. Results were normalized to GAPDH and shown as fold-change to control.
Measurement of edema, serum amylase, and trypsin activity.
To determine the level of edema, we used the formula whole pancreata weight/body weight × 100. Serum amylase activity was measured using the Phadebas test as recommended (Pharmacia Diagnostics). Trypsin enzymatic activity was measured in homogenates of the pancreas using a trypsin activity kit (Abcam).
Histology.
Standard hematoxylin and eosin was used for histological examination of the pathology of the pancreas. For quantification, the histology was examined by a pathologist specialized in pancreatic diseases in a blinded manner. Ten random fields (×100) per animal were evaluated as previously described (34, 43). The representative images and results were reported.
Masson staining and immunohistochemistry.
Masson staining of collagen in paraffin-embedded pancreatic tissue sections was performed to detect collagen deposits. Samples were stained in the core laboratory at Mayo Clinic. Immunohistochemical staining was performed with the Leica Bond Max automated system (Leica Biosystems, Bannockburn, IL) using a Leica-Refine detection kit. Immunostaining was preceded by Leica high-pH epitope retrieval for 25 min. The primary antibodies [CD11b, Abcam AB-133357, 1:2,000; F4–80 ebioscience 14–4801, 1:250; a-smooth muscle actin (SMA), Abcam AB-5694, 1:600; and HSP70, CST 4872, 1:200] were incubated for 60 min at room temperature followed by the Leica polymer and post-polymer (each 15 min), DAB for 10 min, and a hematoxylin counterstain for 5 min.
Terminal transferase-mediated dUTP nick end-labeling assay.
Terminal transferase-mediated dUTP nick end-labeling (TUNEL) staining was conducted using an in situ cell death detection kit (POD; Roche, Germany) according to the manufacturer’s instructions. Briefly, following deparaffinization and rehydration, the sections were treated with 10 mM protease K for 15 min. The slides were immersed in a TUNEL reaction mixture for 60 min at 37°C in a humidified atmosphere in the dark. The slides were incubated in Converter-POD for 30 min to show blue nuclear staining and then analyzed via optical microscopy. The TUNEL index (%) was computed by dividing the ratio of the number of TUNEL-positive cells by the total number of cells; this index was considered in evaluating the apoptosis index of TUNEL-stained pancreas tissues.
Statistical analysis.
Data are presented as means ± SE from at least three independent experiments. P values were analyzed by Student’s t-test or ANOVA using IBM SPSS 20.0 software. A P value <0.05 was considered statistically significant.
RESULTS
Transgenic expression of active trypsin in the secretory pathway did not cause spontaneous pancreatitis.
To study the effects of trypsin activity on the pancreatic acinar cells, we used a conditional PACE-trypsinogen (Try) transgenic mouse line which we developed previously (8, 16). Before Cre recombination, GFP was expressed in pancreatic acinar cells (Fig. 1, A and B). Upon Cre recombination, the floxed GFP-stop cassette was removed, resulting in PACE-trypsinogen being expressed in the Try/BAC double transgenic mice. PACE-trypsinogen is subsequently activated by intracellular PACE (also called furin) cleavage of a PACE sequence (Fig. 1A). To obtain mice with pancreatic-specific expression of PACE-trypsinogen (Try/BAC), Try mice were crossed with pancreatic acinar-specific elastase promoter-driven CreERT mice (BAC) (18). BAC mice express high levels of CreERT, which possesses tamoxifen-independent Cre recombination that increases with age (18). Therefore, in this study the double transgenic mice were not induced with tamoxifen. High recombination efficiency (~90%) was observed in the pancreata of these mice (3 mo old), as indicated by the loss of GFP and expression of hemagglutinin-tagged trypsinogen (Fig. 1, B and C). In contrast to previous findings that tamoxifen rapidly induced PACE-trypsinogen expression and caused the development of acute pancreatitis, we found here that there were no differences in pancreas weight and histology between BAC mice and Try/BAC mice (Fig. 1, C and D). These data led us to suppose whether compensatory mechanisms in the pancreas may have neutralized the effects of active trypsin. Indeed, higher mRNA levels of SPINK1, which can inhibit prematurely activated trypsin, were observed in Try/BAC mice compared with BAC mice (Fig. 1E). Other pancreatic protection mechanisms, such as HSP70 and HSP90α isoform A were also enhanced by active trypsin (Fig. 1E). Despite expression of active trypsin in mouse pancreatic acinar cells, no pathological phenotype was noted in the animals up to 450 days (data not shown). Taken together, active trypsin expression in pancreatic acinar cells has no effect on a spontaneous pancreatitis phenotype, which might be explained by increased compensatory mechanisms.
Fig. 1.

Expression of active trypsin in the secretory pathway activated compensatory protective mechanisms. A: strategy to generate hemagglutinin (HA)-paired basic amino acid-cleaving enzyme (PACE)-trypsin/BAC-Ela-CreErT (Try/BAC) double transgenic mice. B: pancreatic acinar green fluorescent protein (GFP) expression was lost after Cre recombination. C: Western blot showed the expression of HA-tagged PACE-trypsinogen. No pathological phenotype was noted in Try/BAC mice. D: comparison of relative pancreas weight-to-body weight ratio (mg/g). E: expressions of serine protease inhibitor Kazal type 1 (SPINK1), heat shock protein-70 (HSP70), HSP90aA, and HSP90aB in the pancreas were detected by q-RTPCR.
Trypsin induced cell death in pancreatic acinar cells from Try/BAC mice.
In our study, trypsin activity was slightly elevated in the transgenic Try/BAC mice under stable conditions, which may be explained by compensatory protective mechanisms. However, with the treatment of caerulein, Try/BAC mice showed a marked increase in trypsin activity compared with BAC mice (Fig. 2A). Additionally, we confirmed increased trypsin activity by CCK-8 stimulation, and the result showed that pancreatic acinar cells of Try/BAC mice presented elevated fluorescent R110 substrate levels contrasted with those of BAC mice (Fig. 2B), suggesting that trypsin activity was increased in Try/BAC mice. We further isolated pancreatic acini from mice to examine the role of trypsin in pancreatic acinar damage. The data showed that trypsin activation induced more acinar cell blebbing and promoted cell death as revealed by ethidium bromide staining (Fig. 2C). Therefore, our data demonstrated that increased trypsin activity induced more severe pancreatic acinar cell damage.
Fig. 2.

Trypsin activation induced cell death in pancreatic acinar cells. A: trypsin activity was measured 30 min after caerulein injection (50 μg/kg ip). Significant activation of trypsin was seen in Try/BAC mice [(*P < 0.01 vs. BAC. #P < 0.05 vs. BAC+Caerulein (Cer); n = 3)]. B: trypsin activity was visualized in green by R110 (CBZ-Ile-Pro-Arg-R110) in CCK-simulated pancreatic acini from BAC and Try/BAC mice. C: damage of acinar cells from BAC and Try/BAC mice after CCK-8 stimulation (2 h) was compared by morphological observation and ethidium bromide (EB) staining in red.
Trypsin activation exacerbated caerulein-induced acute pancreatitis in mice.
To determine whether the expression of active trypsin would affect the severity of caerulein-induced acute pancreatitis, we challenged the Try/BAC transgenic mice and control BAC littermates with 8 hourly caerulein (50 µg/kg) injections (Fig. 3A). Pancreatitis was examined 16 h after the last injection. Mice with PACE-trypsinogen expression developed more severe acute pancreatitis, as indicated by increased edema and serum amylase, compared with BAC mice (Fig. 3, B–D). We also detected histological changes, including characteristic edema, acinar cell damage, and inflammatory cell infiltration. The histology results suggested trypsin activation significantly increased the severity of pancreatitis (Fig. 3E). Similar to what we observed previously (8), elevated levels of trypsin induced more acinar cell apoptosis, as indicated by TUNEL staining (Fig. 3F). Therefore, the data demonstrated that increased levels of trypsin activation aggravated the severity of caerulein-induced acute pancreatitis.
Fig. 3.

Trypsin activation exacerbated caerulein-induced acute pancreatitis in mice. A: acute pancreatitis was induced by administration of caerulein (50 μg/kg ip hourly for 8 h). B: representative pictures showing pancreas swelling in BAC and Try/BAC mice. C: pancreas edema was measured by pancreas-to-body weight ratio (*P < 0.01 vs. BAC+Cer; n = 5). D: serum amylase comparison. (*P < 0.01 vs. BAC+Cer; n = 5). E: histology comparisons of caerulein-treated BAC and Try/BAC mice. F: Try/BAC mice displayed increased number of terminal transferase-mediated dUTP nick end-labeling-positive cells compared with BAC mice.
Increased trypsin promoted the progression of acute pancreatitis to chronic pancreatitis.
Caerulein-induced mild edematous acute pancreatitis in wild-type mice does not develop into chronic pancreatitis unless the stimulation is repeated for many weeks (43). We hypothesized that elevated trypsin-induced severe acute pancreatitis would result in the progression of acute pancreatitis to chronic pancreatitis, as observed in human hereditary pancreatitis caused by gain-of-function trypsinogen mutations (40). To test this hypothesis, animals were given 8 hourly injections of caerulein (50 μg/kg), and groups of mice were euthanized at different times for evaluation of pancreatitis (Fig. 4A). With this schedule of injections, caerulein induced mild pancreatitis in control BAC mice, and the pancreatic damage was resolved within a week. In contrast, the caerulein-induced pancreatitis in Try/BAC mice gradually progressed with greater inflammatory cell infiltration and pancreatic acinar cell death (Fig. 4B). Immunohistochemical studies revealed markedly increased CD11b leukocyte and F4/80 macrophage infiltration in Try/BAC mice (Fig. 4, C and D). Compared with control BAC mice, HSP70 expression was lower in Try/BAC mice, suggesting that increased trypsin activity downregulated the protective mechanism during the development of pancreatitis (Fig. 4E). The severe pancreatic damage observed in Try/BAC mice was accompanied by upregulation of SMA expression, an early feature of developing chronic pancreatitis (Fig. 4F).
Fig. 4.

Progressive pancreatic damage of caerulein-induced pancreatitis was prominent in Try/BAC mice. A: control and Try/BAC mice were treated with repeated caerulein injection (50 μg/kg for each injection) on day 1. Groups of mice were euthanized at the end of day 1 (d1; 24 h), d3, or d7. B: pathology of the pancreas was examined by hematoxylin and eosin staining. C: immunohistochemistry showed marked increase in CD11b leukocytes in the pancreas of Try/BAC mice. D: F4/80-positive macrophages were predominant in the pancreas of Try/BAC mice. E: heat shock protein-70 (HSP70) expression was downregulated in Try/BAC mice. F: increased smooth muscle actin (SMA) expression, a marker of stellate activation during the early phase of chronic pancreatitis, was evident in Try/BAC mice.
To further examine whether typical chronic pancreatitis will develop in these mice, pancreatitis was evaluated at the sixth week after injections of caerulein (Fig. 5A). As expected, caerulein did not cause obvious chronic changes in pancreatic histology in control BAC animals. In contrast, all the pancreata of Try/BAC mice shrank (Fig. 5B). Histologically, features of chronic pancreatitis with increased fibrosis, fat replacement, acinar cell loss and atrophy, and inflammatory cell infiltration were evident in Try/BAC mice but not in BAC mice (Fig. 5C). Additionally, the results from immunohistochemistry suggested that the infiltrating inflammatory cell population included neutrophils and macrophages (Fig. 6A). We further performed Masson staining to identify collagen deposition and found that transgenic Try/BAC mice presented higher levels of collagen deposition compared with BAC mice (Fig. 6B). Acinar cell apoptosis was still observed several weeks after caerulein treatment (Fig. 6C). Altogether, these data suggested that trypsin activation caused prolonged pancreatic damage, which promoted the development of chronic pancreatitis (Fig. 6D).
Fig. 5.
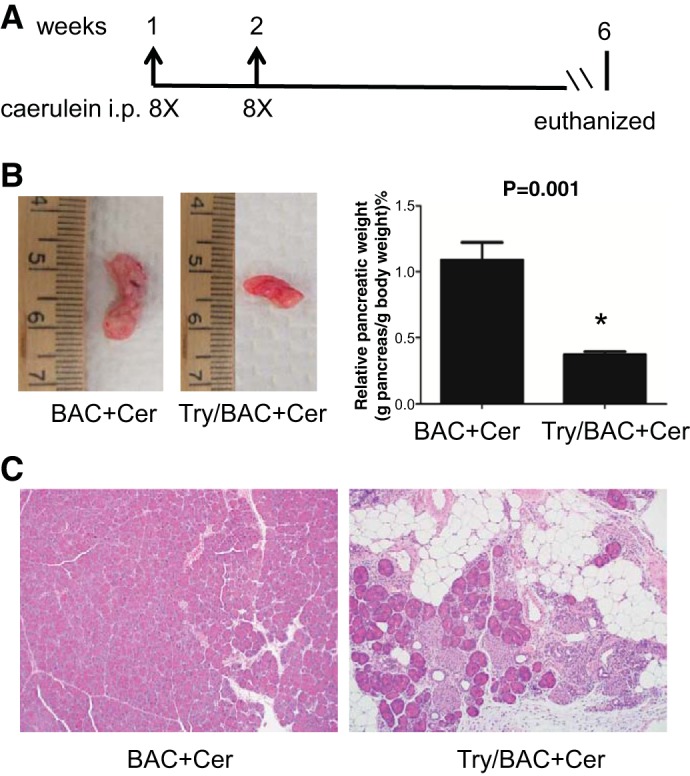
Elevated trypsin promoted the development of chronic pancreatitis. A: chronic pancreatitis was induced by repeated episodes (once a week for 2 wk) of acute pancreatitis with caerulein hyperstimulation (50 μg/kg ip hourly for 8 h). B: whole mount pancreas size (left) and relative pancreas weight (pancreas weight vs. body weight; right) comparisons. (*P < 0.001; n = 4 mice/group). C: representative pancreas histology of chronic pancreatitis in BAC and Try/BAC mice.
Fig. 6.

Characterizing the parameters of chronic pancreatitis. A: inflammatory cells (indicated by CD11b, F4/80, and GR1 stain) in chronic pancreatitis of BAC and Try/BAC mice. There were more CD11b-, F4/80-, and GR1-positive stained cells in Try/BAC mice (IHC stain). B: Try/BAC mice displayed increased fibrosis (in blue) compared with BAC mice. C: caspase-3-positive cells increased in chronic pancreatitis of Try/BAC mice compared with BAC mice. D: comparison of chronic pancreatitis histological scores between BAC and Try/BAC mice (*P < 0.001; n = 4 mice/group).
Expression of PACE-trypsinogen-sensitized mice to arginine-induced pancreatitis.
To further study that the adverse effects of trypsin activation on the severity of pancreatitis are not dependent on a disease model, we utilized l-arginine-induced acute pancreatitis, which has been widely used for causing necrotizing pancreatitis in rats and mice (6, 43). The mice were treated twice with l-arginine (4 g/kg) by intraperitoneal injections, and pancreatic damage was measured after 72 h (Fig. 7A). As expected, the BAC mice showed a slight increase in edema, elevated serum amylase levels, mild inflammation and necrosis of the pancreas (Fig. 7, B and C). However, the transgenic Try/BAC mice exhibited much more severe pancreatic edema, higher serum amylase levels, and more widespread necrosis after l-arginine treatment compared with BAC mice undergoing the same l-arginine treatment (Fig. 7, B and C). The overwhelming inflammatory cell infiltration was also diffusely distributed between the glandular cells in Try/BAC mice (Fig. 7D). Therefore, elevated trypsin can cause severe pancreatitis in multiple animal models of experimental pancreatitis.
Fig. 7.
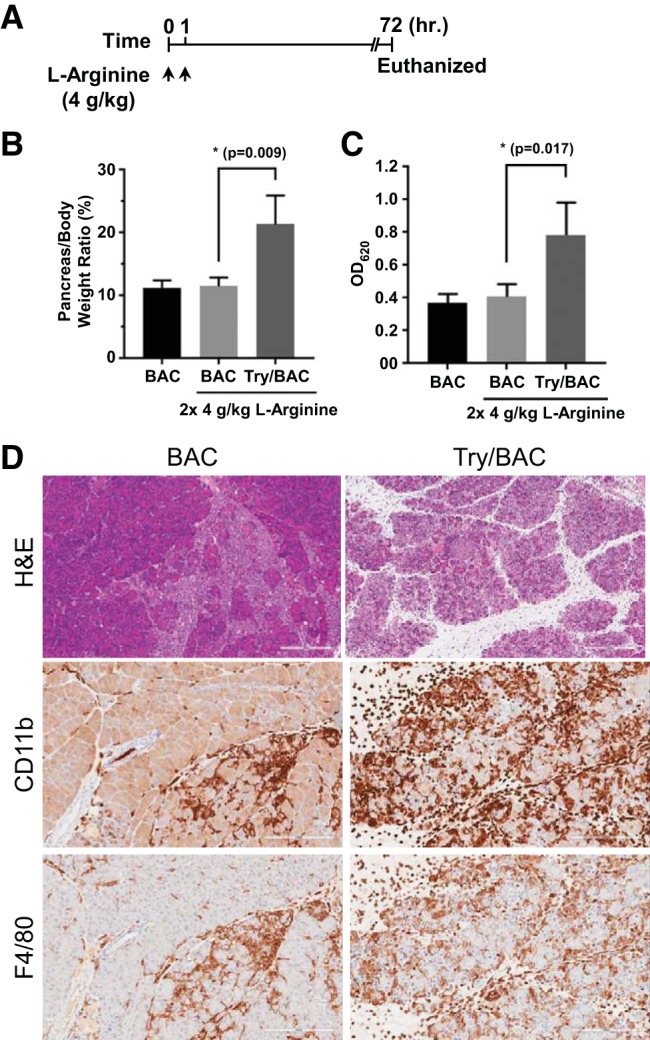
Trypsin activity was complicated in the severity of pancreatitis induced by l-arginine. A: l-arginine-inducing pancreatitis was induced by administration of l-arginine (4 g/kg ip hourly for 2 h). Mice were euthanized in 72 h. B and C: comparisons of relative pancreas weight and serum amylase levels. D: histology and inflammatory cells (indicated by CD11b and F4/80 IHC staining) are shown in BAC and Try/BAC mice (×200).
DISCUSSION
In this study, we investigated the effects of intracellular trypsin activity on the severity of pancreatitis using genetically engineered mouse models. We confirmed that transgenic mice that express acinar-specific PACE-trypsinogen exhibited a higher level of trypsin activity. Elevated trypsin is associated with severe acute pancreatitis, which was demonstrated by dramatically increased levels of edema, serum amylase, inflammatory cell infiltration, and acinar cell damage. Furthermore, high intra-acinar trypsinogen activation resulted in a significant increase in the severity of chronic pancreatitis.
It has been proposed that trypsinogen activation plays an important role in the pathogenesis of acute pancreatitis, which was supported by several lines of evidence: first, intracellular trypsinogen activation has consistently been observed during the course of pancreatitis in experimental models as well as in clinical pancreatitis (23, 36, 41); second, it has been identified in hereditary pancreatitis that a series of gain-of-function mutations in trypsinogen or loss-of-function mutations in trypsin inhibitors resulted in increased intra-acinar trypsin activity (38); third, studies using inhibition or knockout of cathepsin B, phosphatidylinositol 3-kinase knockout, or expression of protease inhibitors to suppress the trypsinogen activation demonstrated that inhibition of trypsinogen activation leads to a reduction in the severity of pancreatitis (12, 33, 37). Our data in this study indicated that increasing intra-acinar trypsinogen activation augments the severity for both acute and chronic pancreatitis. This notion is also in agreement with previous studies which showed that intra-acinar expression of active trypsin in vivo induced typical acute pancreatitis with widespread pancreatic cell death and inflammatory cell infiltration (8, 10).
In our previous study, initiation of acute pancreatitis was observed with high levels of PACE-trypon expression by maximal-rapid induction (8). However, in this study mice showed normal pancreas histology and pancreatic size when the expression of PACE-trypsinogen was not induced by tamoxifen. Instead the recombination process relied on the tamoxifen-independent Cre recombination, which is a slow process related to the gradual increase in endogenous elastase promoter activity (18). This progressive recombination process induces a number of protective mechanisms, including SPINK1, which inhibits the activity of trypsin (30); HSP70, which interferes with colocalization of zymogens with lysosomes (15); and HSP90aA, which interacts with proteases (27). Therefore, the pancreas has a capacity to prevent damage from lower levels of trypsin activity and has the ability to upregulate several protective mechanisms. However, once the threshold of protection is overwhelmed, trypsin promotes the development of both acute and chronic pancreatitis.
Cell death and inflammation are key pathological responses of acute pancreatitis. The severity of experimental acute pancreatitis positively correlates with the extent and the type of cell death. Although multiple forms of cell death occur in physiological and pathological conditions (9, 20, 25), apoptosis and necrosis play a major role and are most widely studied in clinical and experimental acute pancreatitis (1). Previous studies showed that intracellular trypsinogen activation can induce acinar cell apoptosis in in vitro and in vivo models (1, 8). Consistent with previous findings, we observed much higher levels of TUNEL-positive and caspase 3-positive cells in PACE-trypon mice compared with wild-type mice (8). Although apoptosis is usually considered immunologically silent, since the cytoplasmic content is packaged in apoptotic bodies and then rapidly taken up by phagocytes or autophagy (25), excessive apoptotic cells can activate the inflammatory and immune responses by releasing nuclear damage-associated molecular pattern molecules (11, 21). Consistent with this notion, researchers have reported that pancreatic acinar cells undergoing apoptosis can release histones, DNA, and high mobility group box 1 (13, 22), which facilitate pancreatic injury and the inflammatory response. Similarly, we have previously found that diphtheria toxin-induced apoptotic acinar cell death in diphtheria toxin receptor transgenic mice caused widespread damage and an acute phase inflammation of the pancreas, which is similar to the outcome of genetically upregulated intra-acinar trypsinogen activation (8). Thus massive apoptosis may be a potential mechanism that intracellular trypsinogen activation enhances in the pathogenesis of pancreatitis. Chronic pancreatitis is characterized by progressive pancreatic damage that eventually results in significant impairment of both exocrine and endocrine functions of the gland (42). The pathophysiological mechanisms of chronic pancreatitis are not well understood. A popular notion is that intrapancreatic trypsinogen activation could be responsible for the pathogenesis of chronic pancreatitis. The strongest support of this idea is the identification of trypsinogen mutations in hereditary pancreatitis, an uncommon form of chronic pancreatitis (38, 40). These gain-of-function mutations result in the production of trypsin that is resistant to degradation or autolysis, thus causing autodigestion of the pancreatic tissue, resulting in pancreatitis (35).
Our current study and many other previous studies have all indicated that active trypsin is a key initiating factor for both acute and chronic pancreatitis (24, 28, 39). However, this conclusion is not fully supported from a more recent genetic mouse model in which trypsinogen isoform-7, the most prominent trypsinogen in mice, was deleted (5). These mice lacked pathological activation of trypsinogen after caerulein treatment that led to near complete inhibition of acinar cell death in vitro and a 50% reduction in acinar necrosis in vivo. However, these mice showed similar degrees of local and systemic inflammation (5). Furthermore, compared with pancreatic tissues from wild-type mice, those trypsinogen knockout mice had similar levels of atrophy, histological features of chronic pancreatitis, and chronic inflammation (34). On one hand, this model challenged the “trypsin central” paradigm. In fact, several previous studies have shown that NFAT, NF-κB, and Ras signaling pathways can mediate pancreatitis (3, 14, 19, 29). On the other hand, these knockout studies also support the finding that human and mouse trypsinogens have different characteristics and a mouse model with human trypsinogen (e.g., PRSS1R122H) expression is needed to study the role of trypsin (32).
In conclusion, we have demonstrated that increased intra-acinar trypsinogen activation led to more severe acute and chronic pancreatitis in experimental pancreatitis mouse models. These findings will improve our understanding of the pathogenesis of pancreatitis and assist the development of therapies, which are still lacking in the clinic.
GRANTS
This work was supported by Grants 5K12 CA090628-18, P50 CA102701, and DoD W81XWH-15-1-0257 (to B. Ji). This work was also partly supported by Natural Science Foundation of China Grant 81370571 (to X. Zhan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.Z. and B.J. conceived and designed research; X.Z., J.W., G.Z., F.G., Y.Z., Y.L., J.G., and R.K.D. performed experiments; X.Z., J.W., G.Z., L.Z., and B.J. analyzed data; X.Z., A.K.S., A.N.H., Y.B., and B.J. interpreted results of experiments; X.Z., J.W., G.Z., and L.S. prepared figures; X.Z., J.W., G.Z., L.S., A.N.H., and B.J. drafted manuscript; X.Z., A.N.H., Y.B., and B.J. edited and revised manuscript; B.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We sincerely thank Brandy H. Edenfield at the Mayo Clinic for histology and immunohistochemistry assistance.
REFERENCES
- 1.Bhatia M. Apoptosis versus necrosis in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 286: G189–G196, 2004. doi: 10.1152/ajpgi.00304.2003. [DOI] [PubMed] [Google Scholar]
- 2.Bi Y, Atwal T, Vege SS. Drug therapy for acute pancreatitis. Curr Treat Options Gastroenterol 13: 354–368, 2015. doi: 10.1007/s11938-015-0058-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 122: 448–457, 2002. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 4.Chiari H. Über die Selbstverdauung des menschlichen Pankreas. Zeitschrift für Heilkunde 17: 69–96, 1896. [Google Scholar]
- 5.Dawra R, Sah RP, Dudeja V, Rishi L, Talukdar R, Garg P, Saluja AK. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141: 2210–2217.e2, 2011. doi: 10.1053/j.gastro.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawra R, Sharif R, Phillips P, Dudeja V, Dhaulakhandi D, Saluja AK. Development of a new mouse model of acute pancreatitis induced by administration of l-arginine. Am J Physiol Gastrointest Liver Physiol 292: G1009–G1018, 2007. doi: 10.1152/ajpgi.00167.2006. [DOI] [PubMed] [Google Scholar]
- 7.Drenth JP, te Morsche R, Jansen JB. Mutations in serine protease inhibitor Kazal type 1 are strongly associated with chronic pancreatitis. Gut 50: 687–692, 2002. doi: 10.1136/gut.50.5.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaiser S, Daniluk J, Liu Y, Tsou L, Chu J, Lee W, Longnecker DS, Logsdon CD, Ji B. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60: 1379–1388, 2011. doi: 10.1136/gut.2010.226175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: 107–120, 2012. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geisz A, Sahin-Tóth M. A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation. Nat Commun 9: 5033, 2018. doi: 10.1038/s41467-018-07347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol 31: 395–399, 2015. doi: 10.1097/MOG.0000000000000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 106: 773–781, 2000. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141: 358–369, 2011. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang H, Liu Y, Daniluk J, Gaiser S, Chu J, Wang H, Li ZS, Logsdon CD, Ji B. Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 144: 202–210, 2013. doi: 10.1053/j.gastro.2012.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang JH, Ryu JK, Yoon YB, Lee KH, Park YS, Kim JW, Kim N, Lee DH, Jeong JB, Seo JS, Kim YT. Spontaneous activation of pancreas trypsinogen in heat shock protein 70.1 knock-out mice. Pancreas 31: 332–336, 2005. doi: 10.1097/01.mpa.0000183377.04295.c3. [DOI] [PubMed] [Google Scholar]
- 16.Ji B, Gaiser S, Chen X, Ernst SA, Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J Biol Chem 284: 17488–17498, 2009. doi: 10.1074/jbc.M109.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji B, Kopin AS, Logsdon CD. Species differences between rat and mouse CCKA receptors determine the divergent acinar cell response to the cholecystokinin analog JMV-180. J Biol Chem 275: 19115–19120, 2000. doi: 10.1074/jbc.M001685200. [DOI] [PubMed] [Google Scholar]
- 18.Ji B, Song J, Tsou L, Bi Y, Gaiser S, Mortensen R, Logsdon C. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis 46: 390–395, 2008. doi: 10.1002/dvg.20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J, Bi Y, Grote T, Longnecker DS, Logsdon CD. Ras activity levels control the development of pancreatic diseases. Gastroenterology 137: 1072–1082.e6, 2009. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol 269: C1295–C1304, 1995. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- 21.Kang R, Lotze MT, Zeh HJ, Billiar TR, Tang D. Cell death and DAMPs in acute pancreatitis. Mol Med 20: 466–477, 2014. doi: 10.2119/molmed.2014.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, Bartlett DL, Whitcomb DC, Chang EB, Zhu X, Wang H, Lu B, Tracey KJ, Cao L, Fan XG, Lotze MT, Zeh HJ III, Tang D. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 146: 1097–1107.e8, 2014. doi: 10.1053/j.gastro.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klöppel G, Dreyer T, Willemer S, Kern HF, Adler G. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol 409: 791–803, 1986. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 24.Logsdon CD, Ji B. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat Rev Gastroenterol Hepatol 10: 362–370, 2013. doi: 10.1038/nrgastro.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mareninova OA, Hermann K, French SW, O’Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119: 3340–3355, 2009. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Messenger SW, Jones EK, Holthaus CL, Thomas DDH, Cooley MM, Byrne JA, Mareninova OA, Gukovskaya AS, Groblewski GE. Acute acinar pancreatitis blocks vesicle-associated membrane protein 8 (VAMP8)-dependent secretion, resulting in intracellular trypsin accumulation. J Biol Chem 292: 7828–7839, 2017. doi: 10.1074/jbc.M117.781815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moretti AI, Rios EC, Soriano FG, de Souza HP, Abatepaulo F, Barbeiro DF, Velasco IT. Acute pancreatitis: hypertonic saline increases heat shock proteins 70 and 90 and reduces neutrophil infiltration in lung injury. Pancreas 38: 507–514, 2009. doi: 10.1097/MPA.0b013e31819fef75. [DOI] [PubMed] [Google Scholar]
- 28.Mounzer R, Whitcomb DC. Genetics of acute and chronic pancreatitis. Curr Opin Gastroenterol 29: 544–551, 2013. doi: 10.1097/MOG.0b013e3283639383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muili KA, Wang D, Orabi AI, Sarwar S, Luo Y, Javed TA, Eisses JF, Mahmood SM, Jin S, Singh VP, Ananthanaravanan M, Perides G, Williams JA, Molkentin JD, Husain SZ. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J Biol Chem 288: 570–580, 2013. doi: 10.1074/jbc.M112.428896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathan JD, Romac J, Peng RY, Peyton M, Macdonald RJ, Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology 128: 717–727, 2005. doi: 10.1053/j.gastro.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 31.Németh BC, Sahin-Tóth M. Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 306: G466–G473, 2014. doi: 10.1152/ajpgi.00419.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Németh BC, Wartmann T, Halangk W, Sahin-Tóth M. Autoactivation of mouse trypsinogens is regulated by chymotrypsin C via cleavage of the autolysis loop. J Biol Chem 288: 24049–24062, 2013. doi: 10.1074/jbc.M113.478800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romac JM, Ohmuraya M, Bittner C, Majeed MF, Vigna SR, Que J, Fee BE, Wartmann T, Yamamura K, Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-1 rescues SPINK3-deficient mice and restores a normal pancreatic phenotype. Am J Physiol Gastrointest Liver Physiol 298: G518–G524, 2010. doi: 10.1152/ajpgi.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sah RP, Dudeja V, Dawra RK, Saluja AK. Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 144: 1076–1085.e2, 2013. doi: 10.1053/j.gastro.2013.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahin-Tóth M. Biochemical models of hereditary pancreatitis. Endocrinol Metab Clin North Am 35: 303–312, 2006. doi: 10.1016/j.ecl.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saluja A, Saluja M, Villa A, Leli U, Rutledge P, Meldolesi J, Steer M. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest 84: 1260–1266, 1989. doi: 10.1172/JCI114293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest 108: 1387–1395, 2001. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teich N, Rosendahl J, Tóth M, Mössner J, Sahin-Tóth M. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat 27: 721–730, 2006. doi: 10.1002/humu.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thrower EC, Gorelick FS, Husain SZ. Molecular and cellular mechanisms of pancreatic injury. Curr Opin Gastroenterol 26: 484–489, 2010. doi: 10.1097/MOG.0b013e32833d119e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14: 141–145, 1996. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 41.Willemer S, Klöppel G, Kern HF, Adler G. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch A Pathol Anat Histopathol 415: 115–123, 1989. doi: 10.1007/BF00784348. [DOI] [PubMed] [Google Scholar]
- 42.Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 132: 1557–1573, 2007. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Zhan X, Wang F, Bi Y, Ji B. Animal models of gastrointestinal and liver diseases. Animal models of acute and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 311: G343–G355, 2016. doi: 10.1152/ajpgi.00372.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]