

Abstract
Chronic inadequate sleep is associated with increased risk of cardiometabolic diseases. The mechanisms involved are poorly understood but involve changes in insulin sensitivity, including within adipose tissue. The aim of this study was to assess the effects of sleep restriction on nonesterified fatty acid (NEFA) suppression profiles in response to an intravenous glucose tolerance test (IVGTT) and to assess whether 2 nights of recovery sleep (a “weekend”) is sufficient to restore metabolic health. We hypothesized that sleep restriction impairs both glucose and lipid metabolism, specifically adipocyte insulin sensitivity, and the dynamic lipemic response of adipocyte NEFA release during an IVGTT. Fifteen healthy men completed an inpatient study of 3 baseline nights (10 h of time in bed/night), followed by 5 nights of 5 h of time in bed/night and 2 recovery nights (10 h of time in bed/night). IVGTTs were performed on the final day of each condition. Reductions in insulin sensitivity without a compensatory change in acute insulin response to glucose were consistent with prior studies (insulin sensitivity P = 0.002; acute insulin response to glucose P = 0.23). The disposition index was suppressed by sleep restriction and did not recover after recovery sleep (P < 0.0001 and P = 0.01, respectively). Fasting NEFAs were not different from baseline in either the restriction or recovery conditions. NEFA rebound was significantly suppressed by sleep restriction (P = 0.01) but returned to baseline values after recovery sleep. Our study indicates that sleep restriction impacts NEFA metabolism and demonstrates that 2 nights of recovery sleep may not be adequate to restore glycemic health.
Keywords: metabolism, NEFA, sleep
INTRODUCTION
Chronic sleep loss increases the risk of the development of type 2 diabetes and obesity (31). Sleep restriction, or chronic partial sleep loss, disrupts glucose metabolism by reducing systemic insulin sensitivity without a compensatory increase in acute insulin secretion (9, 31). Although obesity is characterized by hyperplasia and hypertrophy of adipose tissue (13), sleep-obesity research has focused on glucose metabolism (26). Adipocytes, the lipid storage tissue, are an insulin-sensitive integrator of systemic metabolism, yet the mechanisms connecting sleep restriction to adipocyte function have not been characterized (23, 28).
Adipocyte insulin resistance disrupts nonesterified fatty acid (NEFA) metabolism (10, 15, 20). One of the rapid, primary effects of insulin on the adipocyte is suppression of insulin-responsive intracellular triglyceride lipolysis, which therefore suppresses NEFA release (10, 15). Ex vivo insulin-stimulated subcutaneous adipose biopsies from sleep-restricted subjects have reduced protein kinase B phosphorylation (5), a key step in the insulin-response cascade that is proximal to hormone sensitive lipase suppression (10, 17, 33). Furthermore, sleep restriction causes transiently elevated overnight and early morning NEFA levels that are correlated with the acquired decrease in insulin sensitivity as measured by glucose disposal (4). These findings provide strong preliminary evidence that adipocyte insulin sensitivity is impaired during sleep restriction.
NEFAs are the preferred fuel source for skeletal muscle (16). Aberrantly elevated NEFAs contribute to reductions in whole body insulin sensitivity by inhibiting skeletal muscle glucose uptake via the Randle cycle (16, 24). The observed sleep loss-induced impairments in plasma glucose clearance could be due to either impaired uptake by glucose consuming cells (primarily skeletal muscle) or substrate-level competition driven by inadequate NEFA suppression in the adipose tissue (17, 28). Prior sleep-metabolism work has focused on insulin sensitivity as evidenced by glycemic clearance, omitting the systemic lipemic responses to a glucose load. In particular, changes in the dynamic in vivo suppression of NEFA during sleep restriction have not been assessed, even though subtle changes in NEFA metabolism are present in early insulin resistance and sleep restriction impacts adipocyte-specific insulin sensitivity (2, 5, 6, 29).
The relationship of recent findings of the effects of sleep restriction on metabolism to the development of cardiometabolic diseases remains unclear. Increasing the translatability of sleep studies is a critical step in untangling the various mechanisms linking short sleep and disease. Assessing tissue-specific adaptations to simulated real-world metabolic challenges provides insight into how sleep loss impacts human metabolism in everyday life. Additionally, there is a dearth of studies examining the metabolic recovery process following a bout of sleep restriction. We designed this study to examine the effects of a work week of sleep restriction on dynamic lipemic responses to a glucose challenge and to establish whether any detrimental metabolic changes are restored with a weekend of recovery sleep.
This study addresses several knowledge gaps that are critical for advancing our understanding of the relationship between sleep restriction and chronic disease risk. NEFA kinetics are a dynamic in vivo marker of metabolic health, indicating both adipocyte-specific insulin sensitivity and providing insight into fuel selection processes by skeletal muscle (2). We hypothesized that sleep restriction impairs both glucose and lipid metabolism, specifically adipocyte insulin sensitivity, and the dynamic lipemic response of adipocyte NEFA release during an intravenous glucose tolerance test (IVGTT). Therefore, we quantified the impact of sleep restriction on adipocyte insulin sensitivity by measuring time-dependent changes in NEFA during an IVGTT in healthy human participants. We demonstrate that sleep restriction [5 h of time in bed (TIB) per night for 5 consecutive nights] induces a delayed and incomplete rebound of NEFA, which is restored after 2 nights of recovery sleep. Furthermore, we demonstrate that 2 nights (a “weekend”) of recovery sleep is insufficient to restore the reductions in insulin sensitivity and delayed glucose clearance caused by 5 nights of sleep restriction.
METHODS
See Fig. 1 for a schematic of this study. All procedures were reviewed and approved by the Institutional Review Board at The Pennsylvania State University and conducted according to the principles established in the Declaration of Helsinki. Informed consent for study procedures took place in two phases. First, participants provided written, informed consent for screening procedures. Second, after the screening steps described below, eligible participants met with a senior study investigator to provide written, informed consent for the in-laboratory study portion of the protocol.
Fig. 1.

Study schematic. Black bars represent time in bed (TIB). For at least 1 wk before admission, participants maintained a 10-h TIB routine from 22:00–08:00. After admission, participants were kept on the same 10-h TIB schedule for 3 additional nights (baseline condition; TIB: 22:00–08:00); then sleep was curtailed to 5 h/night, maintaining nocturnal midpoint, for 5 nights (sleep restriction condition; TIB: 00:30–5:30). Finally, participants resumed the 10-h TIB schedule for 2 nights (recovery condition; TIB: 22:00–08:00). An intravenous glucose tolerance test (IVGTT) was performed on the final day of each condition (days 4, 9, and 11).
Participant recruitment and screening.
Participants were recruited through a mix of electronic and posted advertisements and completed a secure online screening questionnaire (Qualtrics, Seattle, WA). Exclusion criteria included recent medication and drug/tobacco use, shift work or recent travel across time zones, and ongoing medical disorders, diseases, and conditions. After providing written, informed consent for screening procedures, eligible participants received a physical examination with a study clinician and were excluded for measures of poor cardiometabolic health [waist circumference > 102 cm, body mass index (BMI) ≤ 18 kg/m2, seated systolic blood pressure > 130 mmHg or diastolic blood pressure > 85 mmHg, HbA1c ≥ 5.7%, HDL cholesterol < 40 mg/dl, LDL cholesterol ≥ 145 mg/dl, fasting plasma triglycerides ≥ 150 mg/dl, fasting glucose > 100 mg/dl]. Instead of excluding participants with overweight/obese BMIs, participants with a waist circumference > 102 cm were excluded to screen for central adiposity/abdominal obesity (a component of the metabolic syndrome that is associated with increased risk of cardiometabolic dysfunction in adults) (1, 21). Participants completed additional screening questionnaires for sleep disorders and medical history and submitted a urine sample for toxicology screen. Participants completed a sleep-wake log and wore an actigraph (Spectrum, Philips-Respironics, Murrysville, PA) for 1 wk to assess habitual sleep. A clinical psychologist interviewed participants to assess suitability to participate in the in-laboratory protocol as well as willingness to comply with study protocol.
Prestudy conditions.
Before admission, participants maintained a 10-h TIB routine each night for at least 1 wk (6 nights) (22:00–08:00), with no more than ± 1 h deviation of TIB. Compliance with the sleep protocol was assessed via three concurrent methods: each night as participants went to bed and each morning as they got out of bed, they called into a time-stamped messaging system; participants recorded their bed times and wake times on a sleep-wake log; and participants wore an actigraph throughout the prestudy conditions, which recorded movement, ambient light exposure, and wear/nonwear status. Participants were instructed to refrain from alcohol, drugs, tobacco, and caffeine (coffee, energy drinks, tea, chocolate, etc.), and a second urine sample for toxicological screening was collected upon admission to the in-laboratory protocol.
In-laboratory study conditions.
This study protocol included 3 conditions (see Fig. 1): 3 baseline nights (days 1–4), during which sleep opportunity (TIB) was 10 h/night; 5 sleep restriction nights (days 5–9) with 5 h TIB/night; and 2 recovery nights (days 10–11) with 10 h TIB/night. Participants were put to bed and awoken by study staff at the designated bed times (22:00 baseline and recovery conditions; 00:30 restriction condition) and wake times (08:00 baseline and recovery conditions; 05:30 restriction condition) to ensure protocol compliance. During the sleep restriction condition, sleep was curtailed evenly from bed time and wake time to maintain the nocturnal midpoint. This minimizes phase shifting of the central circadian pacemaker and minimizes the concomitant effects of circadian misalignment and sleep loss on metabolism (8, 30). Participants lived in a private room at the Clinical Research Center (Pennsylvania State University) throughout the study. Participant wakefulness was monitored at all times (excluding when participants showered or used the bathroom) during scheduled wake periods by study staff. Light exposure was tightly controlled [<100 lux during wake periods, complete darkness (0 lux) during scheduled sleep periods], and ambient temperature was held constant. Participants were not allowed to sit or recline on the bed during scheduled wake times (except during IVGTTs as described below) and were instructed to remain upright (sitting or standing) throughout the day; exercise was limited to light stretching.
Controlled diet.
The study dietician prepared a controlled nutrient diet in the Clinical Research Center’s metabolic kitchen using weighed foods with predetermined macro- and micronutrient content that met the following specifications each day: 55–60% of calories from carbohydrate, 15–17% of calories from protein, 25–30% of calories from fat, 800–1,000 mg calcium/day, 100 ± 2 milliequivalent (meq) K, and 200 ± 2 meq Na. Food volumes were adjusted based on each participant’s estimated total daily energy expenditure. Total daily energy expenditure was calculated as an average of the Harris-Benedict and the Mifflin-St. Jeor equations with low-active activity factors of 1.1 and 1.5, respectively (11, 12). Participants were instructed to eat all foods provided for each meal (breakfast, lunch, and dinner) within 30 min. Participants’ fasted, postvoid body weights were measured each morning. On the day before baseline, sleep restriction, and recovery IVGTT procedures, participants ate exactly the same meals.
Frequently sampled intravenous glucose tolerance test.
Metabolic assessments were performed once during each condition on days 4, 9, and 11 at ~09:30. IVGTT procedures have been previously described (9). After an overnight fast, two intravenous lines were placed at 08:45, one in each of the participant’s forearms. Participants remained in a reclined position for 45 min, during which baseline samples were drawn. After the baseline period, 0.3 g glucose/kg body wt was infused in one arm while frequent blood samples were drawn from the other. Twenty minutes after the glucose infusion, 0.02 U insulin/kg body wt was infused. Blood sampling continued until 3 h after the glucose infusion. Medical staff monitored participant safety and blood glucose values throughout the procedure, particularly after insulin administration, and were prepared to intervene if hypoglycemia occurred with oral glucose gels and tablets, as well as glucagon and D50 glucose for intravenous injection. Blood was collected in 3% EDTA tubes and immediately placed on ice. Samples were spun at 4°C for 10 min at 3,000 relative centrifugal force, then held at 4°C until aliquoted. Aliquoted samples were stored at −80°C until analysis.
Assays.
Glucose and NEFA were quantified using glucose hexokinase-linked and acyl-CoA synthetase-linked colorimetric assays (Fisher Diagnostics, Middletown, VA and FUJIFILM Wako Diagnostics, Mountain View, CA, respectively). Insulin, c-peptide, leptin, glucagon, and glucagon-like peptide 1 (GLP-1; active) were quantified using fluorescent microbead multiplex assays (MilliporeSigma, Burlington, MA). Samples were run in triplicate on colorimetric assays and duplicate on multiplex assays. All plates included a standard curve and two human plasma controls. Multiplex plates also used a validated standard plasma matrix for background wells. Interassay variability was monitored and plates were repeated if > 5%.
Actigraphy.
Habitual, prestudy (10-h TIB), and in-laboratory sleep were assessed via actigraphy using wrist-worn accelerometers (Philips-Respironics, Murrysville, PA). Recordings were reviewed for participant compliance and data quality, then double-scored by trained, condition-blinded scorers. Discrepancies (if any) exceeding 15 min per sleep interval were resolved before unblinding. Actigraphy scoring methods have previously been described (19). Briefly, the beginning of sleep intervals were scored from an epoch with > 10 activity counts followed by 5 consecutive epochs with ≤10 activity counts. The end of sleep intervals was set to the first epoch of >10 activity counts after 5 consecutive epochs with ≤10 activity counts. Device measurements of light levels and nonwear/wear status were used to aid scoring.
Statistical analyses.
Mixed models with random effects for individuals were used for glycemic summary measures (SI, DI, AIRg, etc.), leptin, glucagon-like peptide-1 (GLP-1; active), and fasted analyte means (JMP Pro 14 by SAS, Cary, NC). Glucose, endogenous insulin, c-peptide, NEFA, and glucagon time courses (min 10–180; min 10–20 for endogenous insulin) were analyzed in mixed effects models with random effects for individuals. Minutes 1–8 were not included in the statistical models due to likelihood of errant findings caused by variable mixing of the glucose bolus throughout the body (3). Condition and subject were defined as categorical variables, whereas time (within the procedure) was considered continuous. The fixed effects mean structure was a function of condition, condition × time, and time, with time included up to the third degree polynomial. Residual normality was assessed and transformations were performed if needed. The spatial power covariance structure was used (with time into procedure as distance) for all combinations of subject and condition to account for repeated effects of condition (SAS 9.4M6).
RESULTS
Participants.
Fifteen healthy men (means ± SD: age 22.33 ±2.82 yr; BMI 24.69 ± 2.99 kg/m2) completed this study. Two participants did not complete IVGTT procedures as a result of hypoglycemia and procedural error (reversal of glucose and insulin administration order), respectively. A third participant completed baseline and sleep restriction IVGTTs but withdrew from participation during the recovery IGVTT because of intravenous catheter placement difficulty. As a result, there are thirteen participants in analyses comparing baseline to restriction measures and twelve participants in analyses comparing baseline to recovery condition.
Actigraphic measurements of sleep.
Habitual sleep duration, 10-h TIB prestudy protocol, and in-laboratory sleep were assessed via actigraphy (Fig. 2). Mean ± SD habitual sleep duration was 8.3 ± 1.5 h/night. Sleep duration during the week of 10-h TIB prestudy was 9.2 ± 1.1 h/night, a significant increase from habitual sleep duration (P < 0.0001). Sleep duration during the baseline condition (also 10 h TIB/night) was 9.6 ± 0.4 h/night. Baseline sleep duration was not significantly different than the 10-h TIB prestudy sleep duration (P = 0.16). As expected, sleep duration was significantly shorter during the sleep restriction condition compared with baseline (4.9 ± 0.2 h/night; P < 0.0001). Sleep duration was 9.5 ± 0.6 h/night in the recovery condition and was not significantly different than the baseline condition (P = 0.98).
Fig. 2.
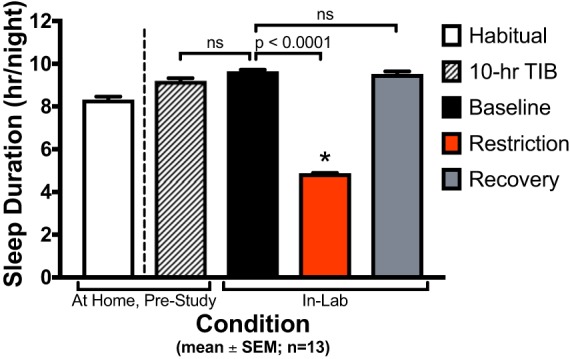
Actigraphy sleep duration by condition. Values are plotted as means ± SE (n = 13 participants). Habitual sleep duration was 8.3 ± 1.5 h/night (mean ± SD). Sleep duration during the prestudy protocol [10-h time in bed (TIB) for ≥ 6 nights] was 9.2 ± 1.1 h/night. Baseline condition sleep duration (10 h TIB/night for 3 nights) was 9.6 ± 0.4 h/night. Sleep duration during sleep restriction condition (5 h TIB/night for 5 nights) was 4.9 ± 0.2 h/night. Recovery condition sleep duration (10 h TIB/night for 2 nights) was 9.5 ± 0.6 h/night. Sleep duration during prestudy 10-h TIB protocol was not significantly different than baseline sleep duration (P = 0.16). Sleep duration during the sleep restriction condition was significantly decreased from baseline sleep duration (P < 0.0001). Recovery sleep duration was not significantly different from baseline sleep duration (P = 0.98).
Acute insulin response and glycemic measurements of insulin sensitivity.
The Bergman minimal model was used to quantify insulin sensitivity using insulin and glucose values (3). All participants (n = 13) had a decrease in insulin sensitivity in response to sleep restriction (3.80 ± 1.12 (mU/l)−1·min−1; P = 0.002) compared with baseline condition (6.13 ±2.45 (mU/l)−1·min−1; Fig. 3A). There was no restoration of insulin sensitivity after 2 nights of recovery sleep (3.75 ± 1.11 (mU/l)−1·min−1; P = 0.003). The acute insulin response to glucose was not significantly affected by sleep restriction (P = 0.23); nor did it change in the recovery condition (P = 0.28; Fig. 3B). Sleep restriction decreased the disposition index from 2,897 ± 1,101 at baseline to 1,996 ± 807 (P < 0.0001; Fig. 3C). The disposition index remained suppressed after 2 nights of recovery sleep (2,103 ± 1,153; P = 0.01).
Fig. 3.
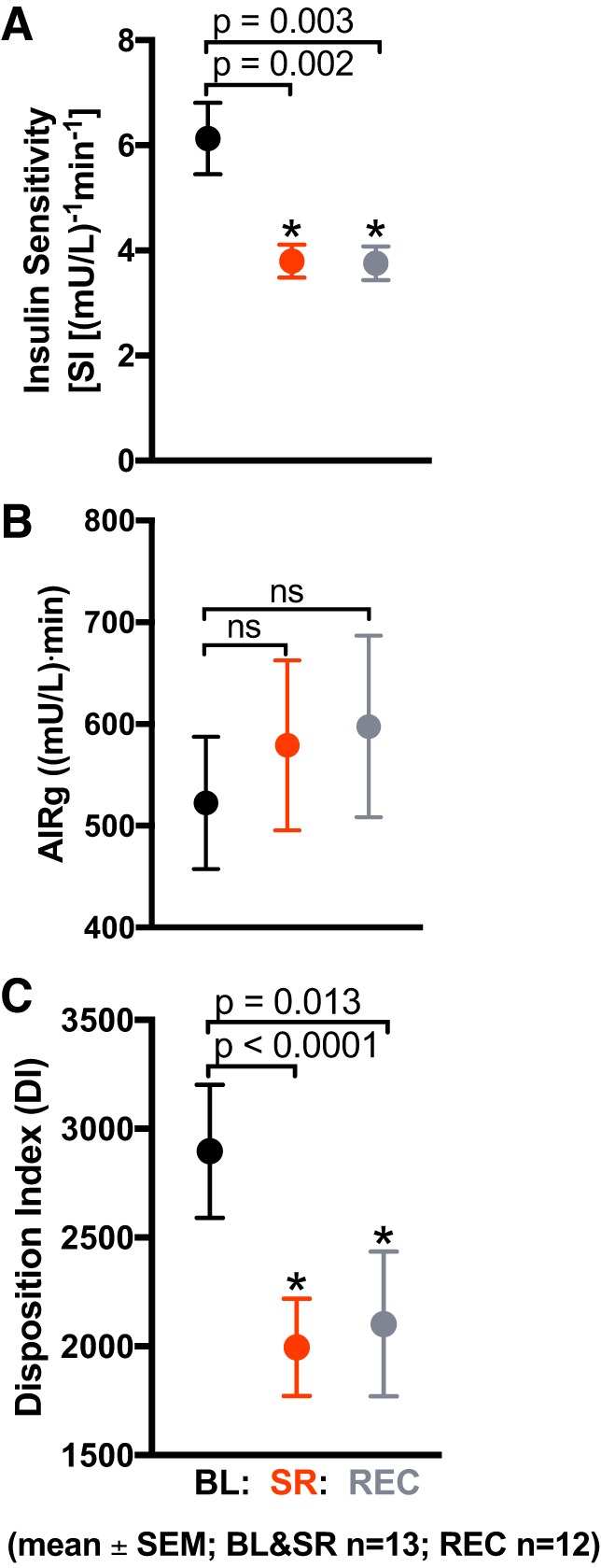
Effects of sleep restriction on insulin sensitivity (SI) and acute insulin response. Values are plotted as means ± SE. Black circles: baseline (BL; n = 13 participants) [10 h/night time in bed (TIB) for 3 nights]; red circles: sleep restriction (SR; n = 13 participants) (5 h/night TIB for 5 nights); gray circles: recovery (REC; n = 12 participants) (10 h/night TIB for 2 nights). A: SI was significantly decreased in the SR condition compared with BL (P = 0.002). SI remained significantly decreased after 2 nights of REC sleep (P = 0.003). B: acute insulin response to glucose (AIRg) did not change from BL after SR or 2 nights of REC sleep (P = 0.23 and P = 0.28, respectively). C: disposition index (DI), a measure of insulin sensitivity adjusted for acute insulin secretion, was significantly decreased by SR (P < 0.0001). DI remained significantly suppressed after 2 nights of recovery sleep (P = 0.01).
Insulin secretion and lipemic measurements of insulin sensitivity.
Glucose, insulin, c-peptide, NEFA, and glucagon profiles are depicted in Fig. 4. IVGTT profiles exhibited the expected postload increase in glucose, insulin, and c-peptide levels, as well as the characteristic insulin-mediated suppression and rebound of NEFA and glucagon. There was no difference by condition in fasting (minutes −15 through 0 of the procedure) glucose (P = 0.07), insulin (P = 0.27), NEFA (P = 0.09), or glucagon (P = 0.55) between baseline and sleep restriction (Fig. 4, A, C, G, and I). There was, however, a marginally significant effect of condition in fasting c-peptide during restriction compared with baseline (P = 0.05; Fig. 4E).
Fig. 4.
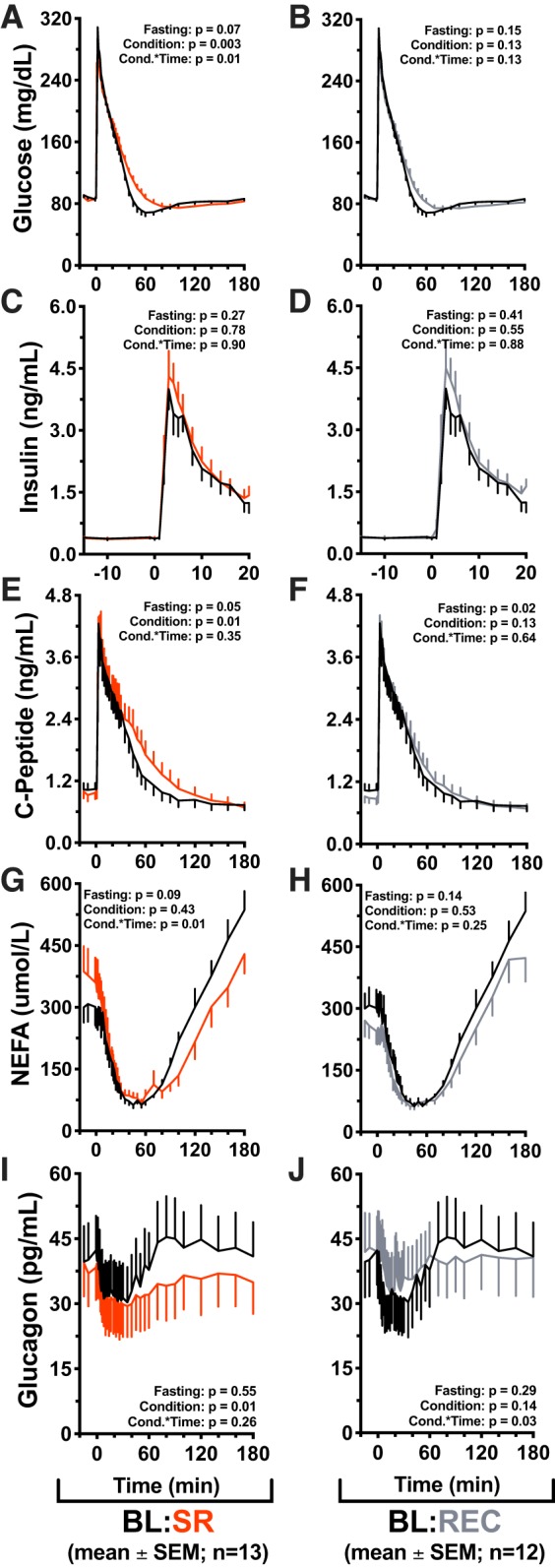
Effects of sleep restriction on intravenous glucose tolerance test (IVGTT) responses. Values are plotted as means ± SE; n, number of participants. Effects of sleep restriction on intravenous glucose tolerance test (IVGTT) responses. Mean values are ± SE, P values are reported on each graph: Fasting = differences during min −15 to 0, Condition = significance of the condition effect, Cond.*Time = significance of the condition × time interaction. Graphs are oriented relative to glucose infusion time (infusion = minute 0). Black lines: baseline (BL; n = 13) (10 h/night time in bed (TIB) for 3 nights); red lines: sleep restriction (SR; n = 13) (5 h/night TIB for 5 nights); gray lines: recovery (REC; n = 12) (10 h/night TIB for 2 nights). A: baseline vs. sleep restriction glucose values during the IVGTT procedure. Significant effect of condition (P = 0.003) and condition × time interaction (P = 0.01). B: baseline vs. recovery IVGTT glucose. C and D: acute phase insulin values (minute 0 through minute 20). E: IVGTT c-peptide values in baseline vs. sleep restriction conditions. Significant effect of condition (P = 0.01), marginally significant difference in fasting values (P = 0.05). F: baseline vs. recovery IVGTT c-peptide. Significant difference in fasting values (P = 0.02). G: baseline vs. sleep restriction IVGTT nonesterified fatty acid (NEFA) values. Significant condition × time interaction (P = 0.01). Post hoc test for differences by condition during NEFA rebound (minutes 70 through 180) found that NEFA rebound was significantly suppressed in the sleep restriction condition (P = 0.01). H: baseline vs. recovery IVGTT NEFA values. I: IVGTT glucagon values in baseline vs. sleep restriction. Significant effect of condition (P = 0.01). J: IVGTT glucagon values in baseline vs. recovery sleep conditions. Significant condition × time interaction (P = 0.03).
Across the IVGTT time course (minutes 10–180), glucose was significantly increased in restriction compared with baseline (P = 0.003), and there was a significant condition × time interaction (P = 0.01; Fig. 4A). There were no differences in endogenous insulin production (minutes 0–20) between baseline and sleep restriction (condition P = 0.78, condition × time P = 0.90; Fig. 4C). C-peptide was significantly increased in the sleep restriction condition (P = 0.01), with no condition × time interaction (P = 0.35; Fig. 4E). There was no significant effect of condition in NEFA between restriction and baseline (P = 0.43); however, there was a significant condition × time interaction in NEFA (P = 0.01; Fig. 4G). A post hoc test for differences by condition in NEFA rebound (minute 70 through minute 180) found that NEFA were significantly suppressed and delayed by sleep restriction (P = 0.01; Fig. 4G) but were not different from baseline IVGTT values after two nights of recovery sleep (P = 0.10; Fig. 4H). Glucagon was decreased in restriction compared with baseline (P = 0.01); there was no condition × time interaction (P = 0.26; Fig. 4I).
Between baseline and recovery conditions, there was no difference in fasting glucose (P = 0.15), insulin (P = 0.41), NEFA (P = 0.14), or glucagon (P = 0.29) (Fig. 4, B, D, H, and J). Fasting c-peptide was decreased from 1,035 ± 407 pg/ml in the baseline condition to 897 ± 354 pg/ml in the recovery condition (P = 0.02; Fig. 4F). There was no effect of condition (P = 0.13) or condition × time (P = 0.13) in glucose between baseline and recovery (Fig. 4B). Endogenous insulin was not different between baseline and recovery (condition P = 0.55, condition × time P = 0.88; Fig. 4D). There was no effect of recovery condition on c-peptide compared with baseline (P = 0.13) nor was there an effect of condition × time (P = 0.64; Fig. 4F). NEFAs were not different by condition (P = 0.53) compared with baseline; condition × time was not significant (P = 0.25; Fig. 4H). There was no effect of recovery condition on glucagon compared with baseline (P = 0.14); however, there was a significant condition × time interaction (P = 0.03; Fig. 4J). GLP-1 (active) and leptin were not different between conditions (P = 0.65 and P = 0.94, respectively) nor were there effects of condition × time (P = 0.57 and P = 0.06, respectively) (Supplementary Figure S1 [DOI: 10.6084/m9.figshare.7568585]). Supplementary Table S2 contains fasting values of measured analytes [DOI: 10.6084/m9.figshare.7568699].
DISCUSSION
We evaluated the metabolic effects of 5 nights of sleep restriction (5 h TIB/night) followed by a weekend of 2 nights of sleep recovery (10 h TIB/night) in healthy, young adult men. The present study extended past findings on the metabolic impact of sleep restriction on lipid metabolism by assessing kinetic changes in NEFA metabolism during an IVGTT. Compared with a 10-h TIB baseline sleep-replete condition, our results replicated prior studies of chronic sleep restriction, showing a significant reduction in insulin sensitivity on an IVGTT without a compensatory increase in acute (first phase) insulin response (9, 32). These impairments of glucose metabolism did not recover with 2 nights of sleep extension, supporting recommendations for consistent adequate sleep duration instead of relying on weekends to recover (27). This study further assessed the characteristic insulin-mediated decline in NEFA levels after a glucose load to quantify the effects of sleep restriction on whole body adipocyte metabolism. We found that the dynamic suppression of NEFA in response to a glucose challenge was not impaired by sleep restriction; rather, the rebound of the NEFA after glucose clearance from the plasma was impaired and suppressed by sleep restriction. It is possible that the delayed NEFA rebound is driven by the delayed glucose clearance from the plasma during sleep restriction. However, after 2 nights of recovery sleep, dynamic NEFA responses recovered to baseline levels, whereas glucose impairments failed to recover. This suggests that there may be complex metabolic shifts occurring that impact NEFA rebound.
A previous study examining the effects of 2 nights of recovery sleep on glucose metabolism in healthy, young men found that 2 nights was sufficient to restore insulin sensitivity to baseline values (7). That study used a sleep restriction model of 4 nights of 4.5 h TIB/night and allowed 12 h of recovery sleep on the first night after sleep restriction and 10 h on the second night. Despite the 2 additional hours of TIB opportunity on the first night of their recovery condition, participants in the Broussard 2016 study (7) averaged 9.7 ± 0.2 h of sleep/night (means ± SE); participants in our study averaged nearly the same during our recovery condition (9.5 ± 0.6 h/night; means ± SD). It is important to note that the participants in our study had a relatively high habitual sleep time (8.3 ± 1.5 h/night). Habitual sleep was not reported in the previous study; however, it is possible that our participants experienced a greater relative sleep restriction, and 2 nights of 10 h TIB was insufficient for their glycemic recovery. The differences in our findings could be explained by differences in degree and length of sleep restriction or because of greater sleep recovery opportunity in the study design by Broussard et. al. Future studies should be designed to evaluate the recovery process and determine what factors are necessary for return to glycemic baseline.
Fasting NEFA levels were low in the baseline condition and elevated by the sleep restriction condition but did not reach statistical significance in this study. Previous studies found elevations in early morning and fasted NEFA values in response to 4 nights of 4.5 h TIB/night and 5 nights of 4 h TIB/night (4, 25). Our fasting sampling began at ~09:15, 15 min before the injection of the glucose bolus (not less than 30 min after successful intravenous placement). Broussard et al. (4) demonstrated that early morning NEFAs are transiently elevated by sleep restriction and are not significantly elevated after 8:45, returning to sleep-replete levels by 09:15. Participants in our study rose at the same time as in the Broussard et al. study (05:30), thus, it is possible that our fasted NEFA samples do not show a significant difference simply because the window of elevated NEFA had already passed by the time we began our metabolic procedures. Regardless, the trend in our data is in agreement with prior published results of elevations in fasting NEFA values.
Plasma NEFA levels in reaction to a glucose load and insulin response reflect a dynamic system balanced by the opposing forces of entry into the plasma via lipolysis and loss from the system through uptake and re-esterification and/or oxidation (14, 22). Therefore, the delayed NEFA rebound during the sleep restriction IVGTT could be due to decreased or delayed lipolysis in the adipose tissue or increased NEFA uptake and utilization in peripheral tissues. A study by Rao et al. (25) in 2015 found that sleep restriction increases whole body lipid oxidation [evidenced by decreased respiratory quotient (RQ)]. Our finding of delayed NEFA rebound, plausibly due to increased peripheral lipid oxidation, is particularly significant in light of the Randle cycle, whereby NEFAs are the preferred fuel source for skeletal muscle and inhibit glucose uptake from the plasma and glucose oxidation in skeletal muscle (16, 24). Increased peripheral NEFA oxidation may at least partially explain sleep restriction-induced insulin resistance.
Limitations.
This study is limited by small sample size and nonrandomized treatment design. The recovery condition must, by definition, follow the sleep restriction condition. Future studies could randomize baseline and sleep restriction conditions on separate visits to retain within-subject primary comparisons. Although we included participants from a range of ethnicities, because of the pilot nature of our study, our sample is limited to young, healthy men. Future studies should include women and older individuals, particularly because the metabolic consequences of chronic sleep restriction are most clinically relevant in older individuals (18). Our methodology was not able to determine cause of the decreased NEFA rebound; future studies including isotope infusions could further characterize the mechanisms underlying these phenomenological changes. There was insufficient blood volume to perform 24-h or overnight blood sampling procedures to ascertain dim-light melatonin onset in each condition and confirm that our metabolic procedures were performed at the same circadian phase. Despite our maintenance of the nocturnal midpoint across conditions, it is possible that our results are compounded by circadian phase differences. Future studies utilizing randomized crossover designs with sufficient blood-volume recovery time between treatments should consider including measurement of dim-light melatonin onset through overnight blood sampling to control for circadian phase of the metabolic procedures. Studies should also be designed to directly evaluate the differences in nocturnal/early morning light exposure that may be part of the mechanisms by which sleep restriction influences physiology in real-world settings.
Perspectives and Significance
We investigated the effects of sleep restriction on lipid metabolism by assessing the dynamic NEFA responses to an intravenous glucose challenge. We found that sleep restriction impairs NEFA rebound after the characteristic insulin-mediated suppression. These results are consistent with previous findings of increased whole body lipid oxidation in response to sleep restriction (25). Our findings, when taken in light of the current literature, indicate that future studies should examine the effects of sleep restriction on skeletal muscle fuel selection and uptake. The effects of sleep restriction on tissue-level changes within skeletal muscle remain unknown; however, whole body findings suggest that the observed decreases in insulin-responsiveness may be partially due to elevated NEFA uptake and metabolism. Furthermore, we assessed the effects of 2 nights of recovery sleep (10 h TIB/night) on glycemic measures of insulin sensitivity after 5 nights of 5 h TIB/night. We found that this weekend of recovery sleep was insufficient to restore glycemic health to baseline values. It is clear that despite the dearth of sleep-recovery literature, future studies will need to account for individual sleep requirements and personalized degree of restriction when assessing metabolic recovery. Our sleep-recovery findings have particular relevance for public health and contribute to the growing body of evidence indicating that regular healthy sleep habits are an important lifestyle choice for optimal metabolic wellbeing.
GRANTS
This project was supported by a pilot grant (PI: A.-M. Chang) from the Pennsylvania State University Clinical and Translational Sciences Institute [funded by the National Center for Advancing Translational Sciences, National Institutes of Health (NIH), through Grant UL1TR002014] and institutional funds from the College of Health and Human Development of the Pennsylvania State University (to A. -M. Chang and O. M. Buxton. K. M. Ness was supported, in part, by NIH Grant T32GM108563 and the College of Health and Human Development of the Pennsylvania State University.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Pennsylvania State University.
DISCLOSURES
G. C. Shearer serves on the Scientific Advisory Board of Amarin Pharmaceuticals. Outside of the current work, O. M. Buxton received two subcontract grants to Pennsylvania State from Mobile Sleep Technologies (National Science Foundation/Small Business Technology Transfer Program no. 1622766 and NIH/National Institute on Aging Small Business Innovation Research Program R43AG056250). None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
K.M.N., A.-M.C., O.M.B., and G.C.S. conceived and designed research; K.M.N. and S.M.S. performed experiments; K.M.N. and G.C.S. analyzed data; K.M.N., O.M.B., and G.C.S. interpreted results of experiments; K.M.N. prepared figures; K.M.N. drafted manuscript; K.M.N., S.M.S., N.G.N., A.-M.C., O.M.B., and G.C.S. edited and revised manuscript; K.M.N., S.M.S., N.G.N., A.-M.C., O.M.B., and G.C.S. approved final version of manuscript.
REFERENCES
- 1.American Heart Association. Metabolic Syndrome (Online). https://www.heart.org/en/health-topics/metabolic-syndrome/about-metabolic-syndrome [23 December 2018].
- 2.Boston RC, Moate PJ. A novel minimal model to describe NEFA kinetics following an intravenous glucose challenge. Am J Physiol Regul Integr Comp Physiol 294: R1140–R1147, 2008. doi: 10.1152/ajpregu.00749.2007. [DOI] [PubMed] [Google Scholar]
- 3.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther 5: 1003–1015, 2003. doi: 10.1089/152091503322641060. [DOI] [PubMed] [Google Scholar]
- 4.Broussard JL, Chapotot F, Abraham V, Day A, Delebecque F, Whitmore HR, Tasali E. Sleep restriction increases free fatty acids in healthy men. Diabetologia 58: 791–798, 2015. doi: 10.1007/s00125-015-3500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann Intern Med 157: 549–557, 2012. doi: 10.7326/0003-4819-157-8-201210160-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broussard JL, Kolka CM, Castro AVB, Asare Bediako I, Paszkiewicz RL, Szczepaniak EW, Szczepaniak LS, Knutson KL, Kim SP, Bergman RN. Elevated nocturnal NEFA are an early signal for hyperinsulinaemic compensation during diet-induced insulin resistance in dogs. Diabetologia 58: 2663–2670, 2015. doi: 10.1007/s00125-015-3721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broussard JL, Wroblewski K, Kilkus JM, Tasali E. Two nights of recovery sleep reverses the effects of short-term sleep restriction on diabetes risk. Diabetes Care 39: e40–e41, 2016. doi: 10.2337/dc15-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buxton OM, Cain SW, O’Connor SP, Porter JH, Duffy JF, Wang W, Czeisler CA, Shea SA. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med 4: 129ra43, 2012. doi: 10.1126/scitranslmed.3003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buxton OM, Pavlova M, Reid EW, Wang W, Simonson DC, Adler GK. Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes 59: 2126–2133, 2010. doi: 10.2337/db09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr 27: 79–101, 2007. doi: 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frankenfield D, Roth-Yousey L, Compher C. Comparison of predictive equations for resting metabolic rate in healthy nonobese and obese adults: a systematic review. J Am Diet Assoc 105: 775–789, 2005. doi: 10.1016/j.jada.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Harris JA, Benedict FG. A biometric study of human basal metabolism. Proc Natl Acad Sci USA 4: 370–373, 1918. doi: 10.1073/pnas.4.12.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc 71: 332–338, 2012. doi: 10.1017/S0029665112000092. [DOI] [PubMed] [Google Scholar]
- 14.Holloway GP, Luiken JJFP, Glatz JFC, Spriet LL, Bonen A. Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: an overview. Acta Physiol (Oxf) 194: 293–309, 2008. doi: 10.1111/j.1748-1716.2008.01878.x. [DOI] [PubMed] [Google Scholar]
- 15.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans 31: 1120–1124, 2003. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 16.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297: E578–E591, 2009. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kraemer FB, Shen W-J. Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J Lipid Res 43: 1585–1594, 2002. doi: 10.1194/jlr.R200009-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Kraja AT, Borecki IB, North K, Tang W, Myers RH, Hopkins PN, Arnett D, Corbett J, Adelman A, Province MA. Longitudinal and age trends of metabolic syndrome and its risk factors: the Family Heart Study. Nutr Metab (Lond) 3: 41, 2006. doi: 10.1186/1743-7075-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marino M, Li Y, Rueschman MN, Winkelman JW, Ellenbogen JM, Solet JM, Dulin H, Berkman LF, Buxton OM. Measuring sleep: accuracy, sensitivity, and specificity of wrist actigraphy compared to polysomnography. Sleep (Basel) 36: 1747–1755, 2013. doi: 10.5665/sleep.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mook S, Halkes CJ, Bilecen S, Cabezas MC. In vivo regulation of plasma free fatty acids in insulin resistance. Metabolism 53: 1197–1201, 2004. doi: 10.1016/j.metabol.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 21.National Institutes of Health (NIH) Heart, Lung, and Blood Institute (NHBLI). Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults—the evidence report. Obes Res 6, Suppl 2: 51S–209S, 1998. [PubMed] [Google Scholar]
- 22.Nielsen TS, Jessen N, Jørgensen JOL, Møller N, Lund S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52: R199–R222, 2014. doi: 10.1530/JME-13-0277. [DOI] [PubMed] [Google Scholar]
- 23.Papackova Z, Cahova M. Fatty acid signaling: the new function of intracellular lipases. Int J Mol Sci 16: 3831–3855, 2015. doi: 10.3390/ijms16023831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281: 785–789, 1963. doi: 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 25.Rao MN, Neylan TC, Grunfeld C, Mulligan K, Schambelan M, Schwarz J-M. Subchronic sleep restriction causes tissue-specific insulin resistance. J Clin Endocrinol Metab 100: 1664–1671, 2015. doi: 10.1210/jc.2014-3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reutrakul S, Van Cauter E. Interactions between sleep, circadian function, and glucose metabolism: implications for risk and severity of diabetes. Ann NY Acad Sci 1311: 151–173, 2014. doi: 10.1111/nyas.12355. [DOI] [PubMed] [Google Scholar]
- 27.Reutrakul S, Van Cauter E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism 84: 56–66, 2018. doi: 10.1016/j.metabol.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444: 847–853, 2006. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sips FLP, Nyman E, Adiels M, Hilbers PAJ, Strålfors P, van Riel NAW, Cedersund G. Model-based quantification of the systemic interplay between glucose and fatty acids in the postprandial state. PLoS One 10: e0135665, 2015. doi: 10.1371/journal.pone.0135665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.St Hilaire MA, Klerman EB, Khalsa SBS, Wright KP Jr, Czeisler CA, Kronauer RE. Addition of a non-photic component to a light-based mathematical model of the human circadian pacemaker. J Theor Biol 247: 583–599, 2007. doi: 10.1016/j.jtbi.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.St-Onge M-P, Grandner MA, Brown D, Conroy MB, Jean-Louis G, Coons M, Bhatt DL; American Heart Association Obesity, Behavior Change, Diabetes, and Nutrition Committees of the Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; and Stroke Council . Sleep duration and quality: impact on lifestyle behaviors and cardiometabolic health: a scientific statement from the American Heart Association. Circulation 134: e367–e386, 2016. doi: 10.1161/CIR.0000000000000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Cauter E, Spiegel K, Tasali E, Leproult R. Metabolic consequences of sleep and sleep loss. Sleep Med 9, Suppl 1: S23–S28, 2008. doi: 10.1016/S1389-9457(08)70013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res 50: 3–21, 2009. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]