

Abstract
PTI-125 is a novel compound demonstrating a promising new approach to treating Alzheimer's disease (AD), characterized by neurodegeneration and amyloid plaque and neurofibrillary pathologies. We show that the toxic signaling of amyloid-β42 (Aβ42) by the α7-nicotinic acetylcholine receptor (α7nAChR), which results in tau phosphorylation and formation of neurofibrillary tangles, requires the recruitment of the scaffolding protein filamin A (FLNA). By binding FLNA with high affinity, PTI-125 prevents Aβ42's toxic cascade, decreasing phospho-tau and Aβ aggregates and reducing the dysfunction of α7nAChRs, NMDARs, and insulin receptors. PTI-125 prevents Aβ42 signaling by drastically reducing its affinity for α7nAChRs and can even dissociate existing Aβ42–α7nAChR complexes. Additionally, PTI-125 prevents Aβ-induced inflammatory cytokine release by blocking FLNA recruitment to toll-like receptor 4, illustrating an anti-inflammatory effect. PTI-125's broad spectrum of beneficial effects is demonstrated here in an intracerebroventricular Aβ42 infusion mouse model of AD and in human postmortem AD brain tissue.
Introduction
Alzheimer's disease (AD) represents one of the greatest health care burdens, affecting 35 million persons worldwide and an estimated 115 million persons by 2050 (Wimo and Prince, 2010). AD is a devastating dementia that first presents as progressive memory loss and later can include neuropsychiatric symptoms. Diagnosis is confirmed at autopsy by amyloid deposits and neurofibrillary tangles (NFTs) containing the microtubule-associated protein tau. This neuropathology is estimated to precede symptoms by 10 years (Trojanowski et al., 2010). Current AD treatment is limited to acetylcholinesterase inhibitors, with mild and short-lived cognitive enhancement, and memantine, an NMDAR antagonist that may delay some later stage symptoms.
Amyloid-β (Aβ), in particular Aβ42, is commonly recognized as the principal causative agent in AD, though its mechanism is debated. Cognitive impairment and the magnitude of synaptic deficit in the AD brain are more highly correlated with soluble Aβ than with the abundance of amyloid plaques, suggesting that soluble Aβ inflicts synaptic impairment (Naslund et al., 2000). Robust evidence demonstrates that soluble Aβ elicits a toxic signaling cascade by the α7-nicotinic acetylcholine receptor (α7nAChR), leading to impaired synaptic activities, intraneuronal Aβ42 aggregates, and cognitive deficits (Liu et al., 2001; Pettit et al., 2001; Chen et al., 2006; Dziewczapolski et al., 2009; Wang et al., 2009). This aberrant signaling activates the kinases ERK2 and JNK1, which phosphorylate tau, leading to the formation of NFTs (Wang et al., 2003).
Aβ42 binds α7nAChR with femtomolar affinity (Wang et al., 2000a,b); therefore, to prevent Aβ42-induced α7nAChR toxic signaling, anti-Aβ therapies must either compete with this extremely high-affinity interaction or essentially eliminate all Aβ42. After the initial femtomolar interaction, numerous Aβ42 molecules bind the receptor, eventually causing internalization and amyloid plaques. These additional Aβ42 molecules appear to bind at a lower (picomolar) affinity (Wang et al., 2009); hence, it is possible to remove sufficient amounts of Aβ42 to prevent plaque formation without any impact on the toxic signaling by the high-affinity Aβ42–α7nAChR interaction. Furthermore, pharmacotherapies aiming to prevent Aβ42 binding to α7nAChRs by directly targeting the receptor could, unfortunately, alter α7nAChR sensitivity or cell surface expression, especially if they surpass subpicomolar affinity. Alternative strategies to reduce Aβ42 signaling by α7nAChR are needed.
We show here that Aβ42 toxic signaling requires the recruitment of the scaffolding protein filamin A (FLNA), which otherwise has very low levels of association with α7nAChR. Filamins are large cytoplasmic proteins best known for crosslinking actin but increasingly implicated in cell signaling (Stossel et al., 2001). PTI-125 binds FLNA with 200 femtomolar affinity to disrupt the Aβ-induced FLNA recruitment to α7nAChRs and to reduce Aβ42 signaling. PTI-125 decreases Aβ42 affinity for α7nAChR, dissociating bound Aβ42. Additionally, PTI-125 provides an anti-inflammatory effect by similarly reducing FLNA association with toll-like receptor 4 (TLR4) and preventing cytokine release. Aβ42 activates TLR4 through CD14 (Reed-Geaghan et al., 2009), and we show this activation requires FLNA. Here we use an intracerebroventricular (ICV) Aβ42 infusion mouse model and human postmortem AD brain tissue to demonstrate PTI-125's promising potential as a novel therapeutic for AD. We also examine FLNA–α7nAChR/TLR4 associations and Aβ42–α7nAChR complexes in lymphocytes from AD patients as a potential AD diagnostic and biomarker.
Materials and Methods
Materials and chemicals.
Leupeptin, aprotinin, phenylmethylsulfonyl fluoride (PMSF), pepstatin A, soybean trypsin inhibitor, NaF, sodium vanadate, β-glycerophosphate, 2-mercaptoethanol, glycine, Histopaque-1077, Tween-20, NP-40, anti-α7nAChR (M-220), ATP, choline kinase, physostigmine sulfate, acetylcholine chloride, AgNO3, tetraphenylboran, n-butyronitrile, and digitonin were all purchased from Sigma. 45Ca2+ (10 Ci/mmol) and [methyl-3H] choline chloride (85.1 Ci/mmol) were purchased from PerkinElmer. PNU282987 and NMDA were purchased from Tocris Bioscience. Anti-pY1150/1151ΙRβ/pY1135/1136IGF-1Rβ (44–804G), -pY960ΙRβ (44–800G), and Aβ1–42 were obtained from Invitrogen. Anti-PSD-95 (05494) and -Aβ42 (AB5739) and -NFTs (AB1518) were from Millipore Bioscience Research Reagents. Anti-phosphotyrosine (SC-508), -α7nAChR (SC-5544, SC-65844), -FLNA (SC-28284, SC-271440), -IRS-1 (SC-8038), -TLR4 (SC-293072), -IRβ (SC-20739, SC-81465), -pY1150/1151ΙRβ (SC-81500), -nNOS (SC-5302), -phospholipase C-γ1 (SC-7290), -NR1 (SC-9058), -NR2A (SC-9056), -NR2B (SC-9057), -tau (SC-1995 [IP], SC-58860[WB]), -actin (SC-7210), and -β-actin (SC-47778) were all purchased from Santa Cruz Biotechnology. Anti-γPKC (P82820) was from BD Transduction Laboratories, and anti-pY402PyK2 (A00498) and -pY416Src (A00396) were from GenScript. Anti-pSer202tau (AT-8), anti-pThr231tau (AT-180), anti-pThr181 (AT-270), Reacti-Bind NeutrAvidin High Binding Capacity Coated 96-well plates, covalently conjugated protein A-agarose beads, antigen elution buffer, and chemiluminescent reagents were purchased from Pierce-Endogen. SK-N-MC human neuroblastoma cells were purchased from American Type Culture Collection. All Aβ-derived peptides were dissolved in 50 mm Tris, pH 9.0, containing 10% DMSO and were stored at −80°C. All test agents were freshly made according to the manufacturer's recommendation. If DMSO was used as the solvent, the highest DMSO concentration in the incubation medium was 1%.
ICV Aβ42 infusion mouse study.
Eight-week-old male and female E129 mice (30–35 g) from Taconic were maintained on a 12 h light/dark cycle with food and water. All animal procedures comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the City College of New York Animal Care and Use Committee.
Mice anesthetized with 30 mg/kg sodium pentobarbital intraperitoneally were placed in a mouse stereotaxic surgery apparatus, as described previously (Wang et al., 2010). Mice receiving 7-day continuous ICV Aβ42 infusion were implanted with a minipump (Alzet) that delivers 0.1 μl/h through a surgical glue-secured cannula placed in the left ventricle at the following coordinates: anteroposterior from bregma, 3.0 mm; lateral, 1.0 mm; horizontal, 3.0 mm. The Aβ42 (0.2 nmol/μl) was dissolved in 10% DMSO containing 50 mm Tris, pH 9.0, to prevent aggregation. Each mouse received 4.8 nmol of Aβ42 daily for 7 d. Each daily dose into the 35 μl of CSF volume of a mouse would reach a concentration of 137 μm, assuming no clearance. Control mice received a 7-day ICV infusion of vehicle.
To assess the effect of in vivo PTI-125 on Aβ42-elicited effects, mice received 10 mg/kg PTI-125 by intraperitoneal injection daily for 2 weeks, starting on the day of surgery (day 1: 2 h after recovery from surgery; days 2–14, twice daily between 10 and 11 A.M. and between 3 and 4 P.M.). Twenty-four hours after the last injection, mice were killed by decapitation. Frontal cortex (FCX) and hippocampus from one half brain were homogenized and processed immediately after harvesting to obtain synaptosomes (P2 fraction), as described previously (Wang et al., 2003), for neuropharmacological assessments. Synaptosomes were washed twice and suspended in 2 ml of ice-cold oxygenated Krebs-Ringer (K-R), containing (in mm): 25 HEPES, pH 7.4, 118 NaCl, 4.8 KCl, 25 NaHCO3, 1.3 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 glucose, 100 μm ascorbic acid, mixture of protease and protein phosphatase inhibitors (Roche Diagnostics) that had been aerated for 10 min with 95% O2/5% CO2. The protein concentration was determined using the Bradford method (Bio-Rad). The other brain halves were immersion fixed in cold 0.15 m phosphate-buffered 10% formalin, pH 7.4, and processed for immunohistochemical determinations of intraneuronal Aβ42 aggregates/plaques and NFTs as well as morphological integrity.
Postmortem human brain tissue study.
This study protocol conformed to the tenets of the Declaration of Helsinki as reflected in a previous approval by the City College of New York and City University of New York Medical School human research committee. Each participant underwent a uniform clinical evaluation that included a medical history, complete neurological examination, cognitive testing including Mini-Mental state examination and other cognitive tests on episodic memory, semantic memory and language, working memory, perceptual speed, and visuospatial ability as well as psychiatric rating. Based on this information, subjects received AD diagnoses based on NINCDS-ADRDA criteria (McKhann et al., 1984). Postmortem brain tissue FCX from patients with clinically diagnosed sporadic AD and control tissues from healthy, age-matched, neurologically normal persons were obtained from the Harvard Brain Tissue Resource Center (HBTRC, Belmont, MA) and the UCLA Brain Tissue Resource Center (UBTRC, Los Angeles, CA). Both HBTRC and UBTRC are supported in part by the National Institutes of Health. The postmortem time intervals for collecting these brains were ≤13 h (mean postmortem intervals for collection of AD and control brain samples were 6.0 ± 0.9 h and 5.8 ± 0.8 h, respectively). Diagnostic neuropathological examination was conducted on fixed sections stained with hematoxylin and eosin and with modified Bielschowsky silver staining (Yamamoto and Hirano, 1986) to establish any disease diagnosis according to defined criteria (Hyman and Trojanowski, 1997), and brain tissue from age-matched controls was similarly screened. The presence of both neuritic (amyloid) plaques and neurofibrillary tangles in all AD brains was confirmed by Nissl and Bielschowsky staining and characterized immunohistochemically with anti-Aβ42 and -NFT staining in frontal and entorhinal cortex as well as hippocampus, as described previously (Wang et al., 2000a). Control tissues exhibited only minimal, localized microscopic neuropathology of AD (0–3 neuritic plaques/10× field and 0–6 NFTs/10× field in hippocampus). One gram blocks from Brodmann areas 10 and/or 46 of FCX were dissected using a band saw from fresh frozen coronal brain sections maintained at −80°C. All postmortem tissues were identified by an anonymous identification number, and experiments were performed as a best-matched pair without knowledge of clinical information.
For in vitro assessments, postmortem tissues were gradually thawed (from −80°C to −20°C), sliced using a chilled McIlwain tissue chopper (200 μm × 200 μm × 3 mm) and suspended in ice-cold oxygenated K-R, containing (in mm): 25 HEPES, pH 7.4, 118 NaCl, 4.8 KCl, 1.3 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 glucose, 100 μm ascorbic acid, 50 μg/ml of leupeptin, 0.2 PMSF, 25 μg/ml pepstatin A, and 0.01 U/ml soybean trypsin inhibitor (∼20 mg/1 ml of K-R). After centrifugation and two additional washes with 1 ml of ice-cold K-R, brain slices were suspended in 1 ml of K-R.
To examine the effects of exogenous Aβ42, ∼20 mg of FCX from control subjects was incubated with 0.1 μm Aβ42 at 37°C for 70 min. To test the effects of PTI-125 on Aβ42-incubated control and native AD tissues, PTI-125 (0.1 and 1 nm) was added 10 min after 0.1 μm Aβ42. Incubation continued for 1 h in the dark. The incubation mixture in a total incubation volume of 0.5 ml was aerated for 1 min every 15 min with 95% O2/5% CO2. Reaction was terminated by the addition of 1.5 ml of ice-cold Ca2+-free K-R, and slices were collected by a brief centrifugation.
To assess the effects of various α7nAChR agents on α7nAChR-FLNA linkages, ∼20 mg of FCX from control subjects was incubated with 1 μm nicotine, PNU282987, α-bungarotoxin, methyllycaconitine, galantamine, memantine, and Aβ40 along with 0.1 μm Aβ42. Incubation continued for 1 h in the dark. The incubation mixture in a total incubation volume of 0.5 ml was aerated for 1 min every 15 min with 95% O2/5% CO2. Reaction was terminated by the addition of 1.5 ml of ice-cold Ca2+-free K-R, and slices were collected by a brief centrifugation.
Assessment of FLNA–α7nAChR, FLNA–TLR4, and Aβ42–α7nAChR associations by coimmunoprecipitation.
These assessments used previously described coimmunoprecipitation methods (Wang et al., 2009). Two hundred micrograms of synaptosomes from either postmortem brain slices or prefrontal cortex or hippocampus of treated mice were pelleted by centrifugation, solubilized by brief sonication in 250 μl of immunoprecipitation buffer (containing, in mm: 25 HEPES, pH 7.5, 200 NaCl, 1 EDTA, 50 μg/ml leupeptin, 10 μg/ml aprotinin, 2 μg/ml soybean trypsin inhibitor, 0.04 PMSF, 5 NaF, 1 sodium vanadate, 0.5 β-glycerophosphate, and 0.1% 2-mercaptoethanol containing 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40) and incubated at 4°C with end-to-end shaking for 1 h. After dilution with 750 μl of ice-cold immunoprecipitation buffer and centrifugation (4°C) to remove insoluble debris, the FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR complexes in the lysate were isolated by immunoprecipitation with 16 h incubation at 4°C with respective rabbit anti-FLNA (1 μg) and anti-Aβ42 antibodies (1 μg) immobilized on protein A-conjugated agarose beads. The resultant immunocomplexes were pelleted by centrifugation at 4°C. After three washes with 1 ml of ice-cold PBS, pH 7.2, and centrifugation, the isolated FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR complexes were solubilized by boiling for 5 min in 100 μl of SDS-PAGE sample preparation buffer (62.5 mm Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.1% bromophenol blue). The content of α7nAChRs/TLR4s in 50% of the anti-FLNA and α7nAChRs in 50% of the anti-Aβ42 immunoprecipitate was determined by Western blotting with monoclonal anti-α7nAChR or -TLR4 antibodies. To affirm the effect of PTI-125 on α7nAChR-/TLR4-FLNA linkages, lysates from 200 μg of solubilized synaptosomes derived from control FCX slices incubated with vehicle and 0.1 μm Aβ42 as well as AD FCX slices incubated with vehicle and 1 nm PTI-125 were assessed by a two-step immunoprecipitation. Lysates were immunoprecipitated first with 1 μg of anti-α7nAChR to obtain the α7nAChR-FLNA complexes. The resultant supernatants were then immunoprecipitated with 1 μg of anti-TLR4 to obtain TLR4-FLNA complexes. In addition, lysates from α7nAChR agent-incubated control tissues were immunoprecipitated with anti-α7nAChR to obtain α7nAChR-FLNA complexes. The FLNA–α7nAChR/TLR4 complex blots were stripped and reprobed with monoclonal anti-FLNA, -α7nAChR, or -TLR4 to assess immunoprecipitation efficiency and loading. To determine Aβ42–α7nAChR complex levels, immobilized rabbit anti-actin (0.5 μg)–protein A-conjugated agarose was added together with anti-Aβ42 in the coimmunoprecipitation process. The content of β-actin in resultant immunoprecipitates was analyzed by immunoblotting using monoclonal anti–β-actin to illustrate even immunoprecipitation efficiency and loading.
Tau phosphorylation.
Using an established method (Wang et al., 2003, 2010), tau proteins in synaptosomes from ICV Aβ42-infused mice were immunoprecipitated with immobilized anti-tau (SC-65865), which does not discriminate between phosphorylation states. The levels of phosphorylated tau (pSer202tau, pThr231tau and pThr181tau) and total tau precipitated (loading controls) were assessed by Western blotting using antibodies specific to each individual phosphoepitope and the anti-tau, respectively.
Assessment of Ca2+ influx as indices of α7nAChR and NMDAR function and cell death.
The effect of PTI-125 on Ca2+ influx after stimulation of α7nAChR or NMDAR was assessed in treated mice or postmortem FCX. Because the level of voltage-gated Ca2+ channel activity indicates the integrity of the cells, K+-depolarization mediated Ca2+ influx was used to assess the effect of PTI-125 on Aβ42-induced cell death, dysfunction, or both in treated mice and in postmortem tissues. As described previously (Wang et al., 2010), brain synaptosomes (50–100 μg) were incubated at 37°C for 5 min in oxygenated 1.2 mm (α7nAChR and K+) or 0.3 mm Mg2+ containing K-R (low Mg2+ K-R [LMKR] for NMDAR) containing 5 μΜ 45Ca2+ (10 Ci/mmol) followed by incubation with vehicle, 0.1–10 μm PNU282987, a specific α7nAChR agonist, or 0.1–10 μm NMDA + 1 μm glycine for 5 min or 65 mm K+ (made with isomolar replacement of Na+) for 1 min in a total incubation volume of 0.5 ml. The reaction was terminated by the addition of 0.5 ml of ice-cold Ca2+-free K-R containing 0.5 mm EGTA and centrifugation at 4°C. After two additional washes, 45Ca2+ contents in synaptosomes were assessed using scintillation spectrometry (Beckman). Background 45Ca2+ was estimated using hypotonically lysed synaptosomes. Absolute Ca2+ influx was calculated by subtracting background 45Ca2+ count. The percentage increase in Ca2+ influx was calculated as percentage [(drug-treated − vehicle)/vehicle].
NMDAR and insulin receptor signaling assessments.
NMDAR and insulin receptor (IR) signaling were assessed in LMKR (K-R) and PTI-125 (1 nm)-exposed postmortem FCX and in prefrontal cortex of treated mice. NMDAR activation and signaling were initiated by incubation of ∼10 mg of in vitro treated brain slices with either LMKR (basal) or LMKR containing 10 μm NMDA and 1 μm glycine at 37°C for 30 min. For initiation of IR activation and signaling, ∼10 mg slices were incubated with either K-R (basal) or K-R containing 1 nm insulin at 37°C for 30 min. The incubation mixtures were aerated with 95% O2/5% CO2 every 10 min for 1 min. Ligand stimulation was terminated by the addition of 1 ml of ice-cold Ca2+-free K-R containing 0.5 mm EGTA and 0.1 mm EDTA. Slices were harvested by brief centrifugation and homogenized in 0.25 ml of ice-cold immunoprecipitation buffer. Homogenates were centrifuged at 1000 × g for 5 min (4°C), and the supernatant (postmitochondrial fraction) was sonicated for 10 s on ice. Proteins were solubilized in 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40 for 60 min at 4°C with end-over-end rotation. The resultant lysates were cleared by centrifugation at 50,000 × g for 5 min and diluted with 0.75 ml of immunoprecipitation buffer. Protein concentrations were measured by the Bradford method (Bio-Rad).
To determine NMDAR signaling and the NMDAR association with PSD-95, the levels of NMDAR subunits, PSD-95, and NMDAR-associated signaling molecules were measured in anti-NR1 immunoprecipitates. Brain slice lysates (200 μg) were immunoprecipitated overnight at 4°C with 2 μg of immobilized anti-NR1 onto covalently conjugated protein A-agarose beads (Pierce-Endogen). Anti-NR1 immunoprecipitates were incubated with 75 μl of antigen elution buffer (Pierce-Endogen) and 2% SDS for 2 min on ice, centrifuged to remove antibody–protein A–agarose complexes, and neutralized immediately with 10 μl of 1.5 M Tris buffer, pH 8.8, followed by the addition of 65 μl of 2 × PAGE sample buffer and boiling for 5 min. Seventy-five microliters of the obtained eluates (50%) were then size fractionated on 7.5% SDS-PAGE. Proteins were transferred to nitrocellulose membrane, and the levels of various NMDAR subunits, PSD-95, and signaling proteins were measured using Western blotting with antibodies for NR1, PSD-95, nNOS, phospholipase C-γ1, γPKC, pY402PyK2, pY416Src, or phosphotyrosine. Blots were stripped and reprobed with anti-NR1 to assess loading.
To determine IR activation and signaling, the levels of pY1150/1151- and pY960-IRs as well as insulin receptor substrate (IRS)-1 recruited to IR were measured in anti-IRβ immunoprecipitates after incubation of brain slices with 1 nm insulin in K-R. These experiments followed the above procedures for assessing NMDAR signaling but were immunoprecipitated with immobilized anti-IRβ–protein A–agarose beads and detected levels of activated IR (pY1150/1151 and pY960) and recruited IRS-1 using Western blotting with antibodies for pY1150/1151-IRβ, pY972-IRβ, or IRS-1. Blots were stripped and reprobed with anti-IRβ to assess the loading.
Assessment of cytokine levels in ICV Aβ42-infused mice.
Parietal cortices (∼10 mg) from vehicle-treated sham, PTI-125-treated sham, vehicle-treated ICV Aβ42, and PTI-125-treated ICV Aβ42 mice were first thawed slowly (−80°C to −20°C to −4°C), homogenized in 100 μl of ice-cold homogenization medium (25 mm HEPES, pH 7.5, 50 mm NaCl, mixture of protease and protein phosphatase inhibitors) by sonication, and then solubilized with 0.5% NP-40, 0.2% Na cholate, and 0.5% digitonin at 4°C for 1 h with end-over-end shaking. After centrifugation, the lysate was diluted with 500 μl (total volume, 600 μl) and was used as the source of cytokines.
To determine the levels of cytokines, 0.5 μg/well biotinylated mouse monoclonal anti-TNF-α, -IL-6 and -IL-1β were coated onto streptavidin-coated plates (Reacti-Bind NeutrAvidin High Binding Capacity Coated 96-well plate; Pierce-Endogen). Plates were washed three times with ice-cold 50 mm Tris HCl, pH 7.4, and incubated at 30°C with 100 μl of lysate derived as described above for 1 h. Plates were washed three times with ice-cold 50 mm Tris HCl, pH 7.4, and incubated at 30°C with 0.5 μg/well unconjugated rabbit anti-TNF-α, anti-IL-6, and anti-IL-1β for 1 h. After two washes with 50 mm Tris HCl, pH 7.4, each well was incubated in 0.5 μg/well FITC-conjugated anti-rabbit IgG (human and mouse absorbed) for 1 h at 30°C. Plates were washed twice with 200 μl of ice-cold Tris HCl, and the residual FITC signals were determined by a multimode plate reader (DTX880; Beckman). Each lysate was surveyed twice.
Immunohistochemical studies of ICV Aβ42-infused mice.
Quantitative immunohistochemistry on consecutive 5 μm sections containing prefrontal cortex and entorhinal cortex/hippocampus was used to determine the levels of Aβ42 aggregates/plaques and neurofibrillary pathology (NFT and paired helical filament [PHF] immunoreactivity) using single-labeling immunohistochemistry, as described previously (D'Andrea et al., 2001; Nagele et al., 2002; Wang et al., 2010). One section was immunostained with anti-NFT or anti-PHF. The next (consecutive) section (often containing the same neuron) was immunostained with anti-Aβ42 antibodies to measure relative levels of accumulated Aβ42 peptide in neurons. The relative Aβ42 accumulation was compared among different cell types using computer-assisted image analysis, as described previously (Wang et al., 2000b). Brain tissues were fixed at 4°C in 0.15 M phosphate-buffered 10% formalin, pH 7.4, for 2 weeks, paraffin embedded, serially sectioned at 5 μm, and processed for bright-field microscopy. Aβ42 immunoreactivity was absent when preabsorbing anti-Aβ42 with Aβ42 but not Aβ42–1. Specimens were examined using a Nikon FXA microscope with a Princeton Instruments CCD camera and were recorded digitally. Relative intensities of the NFT/PHF and Aβ42 immunoreactivity were measured and compared among similar and different cell types using Image Pro Plus (Media Cybernetics) and MetaMorph (Molecular Devices) software. The correlations between the amount of NFT/PHF immunoreactivity and Aβ42-positive accumulation within mature neurons were also determined.
Assessment of PTI-125 effects on Aβ42 affinity for α7nAChRs.
To determine the effect of PTI-125 on Aβ42 affinity for the surface α7nAChRs, synaptosomes (200 μg) prepared from postmortem FCX from control subjects or SK-N-MC cells (human neuroblastoma cell line from American Type Culture Collection) were biotinylated using a biotinylation kit (ThermoFisher Scientific). The biotinylated synaptosomes/cells were lysed by brief sonication in hypertonic solution and were used as the tissue source to determine Aβ42 affinity for the α7nAChRs in the presence and absence of 1 nm PTI-125.
Blocking PTI-125 effects with FLNA pentapeptide VAKGL.
Synaptosomes prepared from postmortem FCX of control subjects were used to elucidate whether the pentapeptide FLNA fragment VAKGL functions as an FLNA decoy by binding to PTI-125, thereby blocking the PTI-125 effects and confirming this pentapeptide binding site on FLNA. Briefly, FCX synaptosomes (200 μg) were incubated with oxygenated K-R containing 1 nm PTI-125 and 10 μm VAKGL or alanine-substituted VAAGL (a control pentapeptide that does not bind PTI-125) alone or together with 0.1 μm Aβ42 at 37°C for 30 min. Synaptosomes were harvested by centrifugation and solubilized, and lysates were immunoprecipitated with anti-FLNA or anti-tau. The α7nAChR and TLR4 contents in the anti-FLNA immunoprecipitates, as well as pS202, pT181, and pT231 tau levels in the anti-tau immunoprecipitates, were determined by Western blotting. Blots were stripped and reprobed with anti-FLNA and anti-tau, respectively, to ascertain even immunoprecipitation and loading.
Lymphocyte treatment and assessments of FLNA–α7nAChR/TLR4 and α7nAChR–Aβ42 complex levels.
To prepare lymphocytes, 7 ml of venous blood was collected into an EDTA-containing collecting tube or S-monovette (Sarstedt). Collected blood (6 ml) was layered onto 6 ml of Histopaque-1077 (Sigma) at 25°C, and the anticoagulated blood was centrifuged at 400 × g for 30 min (25°C) to yield plasma (top layer) and lymphocytes (opaque interface). The lymphocytes were washed twice by mixing with 6 ml of oxygenated K-R followed by centrifugation at 250 × g for 10 min and resuspension. The final pellet was resuspended in 250 μl of oxygenated K-R before protein content determination by the Bradford method and assessment of the levels of FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR complexes. To store lymphocytes, the lymphocyte pellet was resuspended in 250 μl of ice-cold oxygenated K-R containing 10% glycerol and was stored at −20°C for 1 h before storage at −80°C.
To determine the levels of FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR complexes in the lymphocytes, lymphocytes (100 μg) from control subjects were incubated for 30 min at 37°C in 0.25 ml of K-R without or with 100 nm Aβ42, and lymphocytes (50 μg) from AD subjects were incubated for 30 min at 37°C in 0.25 ml of K-R. Lymphocytes from control and AD subjects were also simultaneously incubated with or without PTI-125 (1 nm). The incubation mixture was aerated with 95% O2/5% CO2 every 10 min for 1 min during the incubation. If frozen lymphocyte samples were used, lymphocyte suspension was gradually thawed by storing at −20°C for 1 h after removal from −80°C and then thawed at 4°C.
Incubation was terminated by adding 1 ml of ice-cold Ca2+-free K-R containing 0.5 mm EGTA/0.1 mm EDTA, protease, and phosphatase inhibitors followed by centrifugation. The resultant lymphocyte pellet was homogenized in 0.25 ml of ice-cold immunoprecipitation buffer. Homogenates were centrifuged at 1000 × g for 5 min (4°C), and the supernatant (post-mitochondrial fraction) was sonicated for 10 s on ice and solubilized in 0.5% digitonin/0.2% sodium cholate/0.5% NP-40 for 60 min (4°C) with end-to-end rotation. Resultant lysates were cleared by centrifugation at 50,000 × g for 5 min and diluted with 0.75 ml of immunoprecipitation buffer before coimmunoprecipitation to assess FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR complexes.
Western blot analysis.
Solubilized immunoprecipitates derived from coimmunoprecipitation assays were separated by either 7.5 or 10% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Membranes were washed with PBS and blocked overnight at 4°C with 10% milk in PBS with 0.1% Tween-20 (PBST). After three 5 min washes with 0.1% PBST, the membranes were incubated at room temperature for 2 h with the appropriate antibody at 1:500–1:1000 dilutions. After three 2 min washes in 0.1% PBST, membranes were incubated for 1 h with anti-species IgG-HRP (1:5000 dilution) and were washed with 0.1% PBST three times for 2 min each time. Immunoreactivity was visualized by reacting with chemiluminescent reagent (Pierce-Endogen) for exactly 5 min and immediate exposure to x-ray film. Specific bands were quantified by densitometric scanning (GS-800 calibrated densitometer; Bio-Rad).
Statistical analysis.
All data are presented as mean ± SEM. Treatment/group effects were evaluated by one-way ANOVA followed by Newman–Keul's multiple comparison. Two-tailed Student's t test was also used as the post hoc test for between-group differences. The threshold for significance was p < 0.05.
Results
FLNA recruitment required for α7nAChR and tau phosphorylation
In an ICV Aβ42 infusion mouse model of AD, Aβ42 dramatically increased FLNA association with both α7nAChR and TLR4, caused tau phosphorylation at three phosphorylation sites of tau that are found in neurofibrillary tangles and impaired signaling of α7nAChR, NMDARs, and IRs. Twice-daily treatment with 10 mg/kg of PTI-125 markedly reduced these deleterious effects of the infused Aβ42. PTI-125 reduced Aβ42-induced increases in FLNA associations with both α7nAChR and TLR4 (Fig. 1A,B), suggesting a reduction in Aβ42-mediated signaling of both these receptors with corresponding suppression of the tau phosphorylation at all three phosphorylation sites (Fig. 1C). Essentially identical results were found by detecting FLNA in anti-α7nAChR or anti-TLR4 immunoprecipitates (data not shown). FLNA association with α7nAChR and TLR4 was also elevated in postmortem AD compared with control tissue, as evidenced by Western blot detection of α7nAChR or TLR4 in anti-FLNA immunoprecipitates as above (Fig. 2A,B, respectively) or by Western detection of FLNA in anti-α7nAChR or anti-TLR4 immunoprecipitates (Fig. 2C,D). The fact that we can immunoprecipitate with anti-α7nAChR and separately with anti-TLR4 in the same lysate and subsequently detect robust levels of FLNA in each suggests that these receptors are not sharing the same FLNA molecules. The addition of exogenous Aβ42 to control tissue elevated these associations, and PTI-125 incubation for 1 h reduced these associations in both AD and Aβ42-treated control tissue in this ex vivo setting.
Figure 1.

A, Continuous ICV Aβ42 infusion for 1 week increased FLNA association with α7nAChR, and twice-daily intraperitoneal injections of PTI-125 for 2 weeks significantly reduced this interaction. Synaptosomes prepared from prefrontal cortex and hippocampus of treated mice were immunoprecipitated with immobilized anti-FLNA, and α7nAChR levels in the immunoprecipitates were detected by Western blot (WB) using an α7nAChR-specific antibody. B, ICV Aβ42 infusion also increased FLNA association with TLR4, detected in the same anti-FLNA immunoprecipitates with a TLR4-specific antibody. C, Aβ42 strongly promoted tau phosphorylation at Ser202, Thr231, and Thr181, and PTI-125 significantly reduced phosphorylation at all three sites. Levels of tau protein phosphorylated at each site were measured in the immunoprecipitates of an anti-tau antibody that did not distinguish its phosphorylation state by Western blotting with specific antibodies separately recognizing each tau phosphoepitope. Blots (inset) were analyzed by densitometric quantitation. n = 7 or 8. *p < 0.01 vs sham, vehicle; #p < 0.01 vs Aβ42, vehicle.
Figure 2.

A–D, Incubation for 1 h with 1 nm PTI-125 reduced FLNA–α7nAChR/TLR4 associations shown by Western blot detection of α7nAChR (A) or TLR4 (B) in anti-FLNA immunoprecipitates from AD and Aβ42-treated control FCX slices or by Western detection of FLNA in anti-α7nAChR (C) or anti-TLR4 (D) immunoprecipitates. Western blots (inset) were analyzed by densitometric quantitation. n = 11. *p < 0.01 vs vehicle-treated control; #p < 0.01 vs Aβ42-treated control or vehicle-treated AD.
Aβ42-induced FLNA recruitment to α7nAChRs was mimicked by 10-fold higher concentrations of Aβ40 or of the α7nAChR agonists nicotine and PNU282987 with significantly lesser magnitudes. The α7nAChR antagonists α-bungarotoxin and methyllycaconitine and the cholinesterase inhibitor/positive allosteric nicotinic receptor modulator galantamine showed no effect (Table 1). Interestingly, memantine, an antagonist of both NMDAR and α7nAChR, elicited the recruitment of FLNA to α7nAChRs with a magnitude not far from that of Aβ40.
Table 1.
Effects of α7nAChR agents on FLNA recruitment to α7nAChR
| Agent | FLNA/α7nAChR ratio (stimulation vs vehicle, %) | Statistics vs vehicle, p | Aβ42 effect, % | Statistics vs Aβ42, p |
|---|---|---|---|---|
| Vehicle | 0.07 ± 0.01 | |||
| 0.1 μm Aβ42 | 0.73 ± 0.05 (1012.3 ± 104.9) | <0.0001 | 100 | |
| 1 μm Aβ40 | 0.28 ± 0.03 (341.9 ± 65.1) | 0.001 | 34.8 ± 6.6 | <0.001 |
| 1 μm Nicotine | 0.13 ± 0.02 (107.7 ± 31.4) | <0.001 | 11.0 ± 3.2 | 0.001 |
| 1 μm PNU282987 | 0.12 ± 0.02 (87.1 ± 37.9) | 0.01 | 8.9 ± 3.9 | 0.001 |
| 1 μm α-Bungarotoxin | 0.07 ± 0.01 (7.5 ± 6.6) | 0.65 | 0.8 ± 0.7 | <0.001 |
| 1 μm Methyllycaconitine | 0.08 ± 0.02 (19.8 ± 12.2) | 0.21 | 2.0 ± 1.2 | <0.001 |
| 1 μm Galantamine | 0.08 ± 0.02 (17.1 ± 14.6) | 0.37 | 1.7 ± 1.5 | <0.001 |
| 1 μm Memantine | 0.21 ± 0.03 (225.9 ± 38.5) | <0.00001 | 23.0 ± 3.9 | 0.001 |
PTI-125 normalizes α7nAChR, NMDAR, and IR dysfunction
The impact of PTI-125 treatment on Aβ42-induced dysfunction was evaluated by agonist-evoked calcium influx through α7nAChRs and NMDARs. In the sham-treated mice, PNU282987, an α7nAChR full agonist, and NMDA/glycine elicited a dose-dependent increase in Ca2+ influx through α7nAChRs and NMDARs, respectively. Agonist-induced α7nAChR and NMDAR channel activation was greatly reduced in Aβ42-infused animals, and this impairment was normalized by PTI-125 (Fig. 3A,B). Similarly, α7nAChR and NMDAR channel activation was impaired in AD tissue compared with that in age-matched control tissue, and PTI-125 significantly restored these deficits (Fig. 3C,D).
Figure 3.

PTI-125 treatment of ICV Aβ42-infused mice normalized the Aβ42-induced α7nAChR (A) and NMDAR (B) dysfunction, realized by reduced calcium influx in response to varying doses of the full α7nAChR agonist PNU282987 and by the NMDAR co-agonists NMDA and glycine. n = 7 or 8. *p < 0.01 vs sham, vehicle; #p < 0.01 vs Aβ42, vehicle; +p < 0.01 vs vehicle-treated and PTI-125-treated sham groups. Similarly, 1 h incubation with PTI-125 of postmortem AD or Aβ42-treated control tissue restored α7nAChR (C) and NMDAR (D) function. n = 11. *p < 0.01 vs vehicle-treated control; #p < 0.01 vs vehicle-treated AD group; +p < 0.01 vs vehicle-treated and PTI-125-treated control groups.
Calcium influx after depolarization by K+, indicative of healthy cells, was used to assess overall cellular dysfunction or unhealthy cells. This K+-evoked calcium influx was reduced in both ICV Aβ42-infused mice and in AD compared with age-matched control tissue, demonstrating an elevated number of nonfunctioning cells. This depolarization-induced calcium influx was partially restored by PTI-125 incubation or PTI-125 treatment (Fig. 4).
Figure 4.

A, Aβ42 reduced K+-evoked Ca+2 influx, indicating dead, dying, or dysfunctional cells, and PTI-125 attenuated this Aβ42-induced deleterious effect. n = 7 or 8. *p < 0.01 vs sham, vehicle; #p < 0.01 vs Aβ42, vehicle; ++p < 0.05 vs vehicle-treated and PTI-125-treated sham groups. B, Similarly, PTI-125 incubation partially normalized the drastic reduction in depolarization-induced calcium influx in postmortem AD or Aβ42-treated control tissue. n = 11. *p < 0.01 vs vehicle-treated control; #p < 0.01 vs vehicle-treated AD group; +p < 0.01 vs vehicle-treated and PTI-125-treated control groups.
That PTI-125 treatment can reverse NMDAR dysfunction is also evidenced by measuring NMDAR signaling. Both Aβ42-infused mice and AD postmortem tissue showed reductions in NMDA/glycine-induced activation (phosphorylation) and in recruitment to NMDAR of six signaling components. Remarkably, 1 h incubation of AD brain slices with 1 nm PTI-125 normalized all six measures of this NMDAR signaling impairment without affecting the NMDAR subunit assemblies (Fig. 5). PTI-125 treatment of ICV Aβ42-infused mice produced virtually identical results (data not shown).
Figure 5.

Incubation for 1 h with PTI-125 normalized NMDAR signaling impairment in postmortem AD brain slices and Aβ42-treated control brain slices, evidenced by reductions in linkages of several signaling molecules to NR1, the obligatory NMDAR subunit. Western blots (inset) were analyzed by densitometric quantitation. n = 11. *p < 0.01 vs vehicle-treated control group; #p < 0.01 vs vehicle-treated AD group; +p < 0.01 compared with PTI-125-treated control group.
The effect of Aβ42 infusion on IR function was similarly assessed by the magnitude of insulin-induced tyrosine phosphorylation of ΙRβ and the level of insulin-induced IRS-1 recruited to IRs. Both ΙRβ phosphorylation and IRS-1 recruitment were reduced by Aβ42 infusion and in AD tissue versus control tissue, and PTI-125 treatment or incubation significantly restored this signaling impairment (Fig. 6).
Figure 6.

A, PTI-125 treatment ameliorated the Aβ42-induced impairment in IR signaling in ICV Aβ42-infused mice, as measured by the phosphorylation of ΙRβ and its association with the signaling adaptor molecule IRS-1. n = 7 or 8. *p < 0.01 vs sham, vehicle; #p < 0.01 vs Aβ42, vehicle. B, Similarly, the deficit in IR signaling seen in postmortem AD and Aβ42-treated control brain slices was lessened by 1 h PTI-125 incubation. n = 11. *p < 0.01 vs vehicle-treated control group; #p < 0.01 vs vehicle-treated AD group.
PTI-125 reduces inflammatory cytokines
Given that FLNA association with TLR4 is increased by Aβ42 and normalized by PTI-125, we assessed whether the levels of inflammatory cytokines are higher in ICV Aβ42-infused brains and assessed the effect of PTI-125 treatment. ICV Aβ42 infusion increased IL-6, TNF-α, and IL-1β production. PTI-125 treatment completely abolished Aβ42-induced IL-6 production and suppressed TNF-α and IL-1β levels by 86 and 80%, respectively, illustrating a strong anti-inflammatory effect (Fig. 7).
Figure 7.

PTI-125 treatment of ICV Aβ42-infused mice nearly abolished Aβ42-induced production of inflammatory cytokines IL-6, TNF-α, and IL-1β in parietal cortex, measured by a fluorescence ELISA assay. n = 7 or 8. *p < 0.01 vs respective cytokine level in sham, vehicle group; #p < 0.01 vs respective cytokine level in ICV Aβ42, vehicle group.
PTI-125 reduces Aβ42 and pTau immunoreactivity
Given that α7nAChRs can mediate intraneuronal Aβ42 loading and Aβ42-induced tau phosphorylation, we assessed the levels of Aβ deposits and tau phosphorylation indicative of neurofibrillary lesions and NFTs in both FCX and hippocampus after ICV Aβ42 infusion and the effect of PTI-125 treatment by immunohistochemical method. Aβ42 infusion caused substantial pTau immunoreactivity that was markedly reduced by PTI-125 treatment (Fig. 8A). Perhaps more striking was the abolition of amyloid deposits (Aβ42 immunoreactivity) by PTI-125 (Fig. 8B), further supporting the theory that Aβ42 signaling by α7nAChR also underlies the formation of Aβ-containing plaques (Wang et al., 2009).
Figure 8.

A, Representative sections immunostained with an anti-NFT (phospho-tau) antibody show that PTI-125 treatment dramatically reduced NFT immunoreactivity in both prefrontal cortex and hippocampus of mice receiving ICV Aβ42 infusions. B, Representative sections immunostained for Aβ42 aggregates show that PTI-125 treatment greatly reduced Aβ42 deposits in both brain regions of Aβ42-infused mice. Immunostaining was quantified by image analysis software. n = 7 or 8. *p < 0.01 vs sham, vehicle; #p < 0.01 vs Aβ42, vehicle.
PTI-125 reduces Aβ42 affinity for α7nAChR
In reducing the Aβ42-induced FLNA–α7nAChR association, PTI-125 reduced the level of Aβ42 bound to α7nAChRs. In ICV Aβ42-infused mice, systemic PTI-125 treatment reduced the levels of Aβ42–α7nAChR complexes by 60% in hippocampus and 75% in prefrontal cortex (data not shown). PTI-125 also significantly reduced the levels of Aβ42–α7nAChR complexes in AD tissue, illustrating that substantial amounts of Aβ42 already bound to α7nAChRs in late-stage AD tissue can be removed by PTI-125 binding to FLNA (Fig. 9A). Aβ42–α7nAChR complexes induced by Aβ42 treatment of age-matched control tissue were also reduced by PTI-125 (Fig. 9A). This removal of Aβ42 from α7nAChR by PTI-125 appears to occur because its binding to FLNA reduces the affinity of Aβ42 for α7nAChR by 1000-fold in postmortem control tissue (Fig. 9B) and by 10,000-fold in fresh SK-N-MC cells (Fig. 9C).
Figure 9.

A, Aβ42–α7nAChR complexes were elevated in both Aβ42-treated control tissue and AD tissue, and PTI-125 incubation reduced this interaction in each. Blots (inset) of anti-Aβ42 immunoprecipitates were probed with anti-α7nAChR and analyzed by densitometric quantitation. B, PTI-125 incubation reduced Aβ42 binding affinity for α7nAChR 1000-fold from 100 fm to 16 nm (dotted line) in biotinylated synaptic membranes from postmortem frontal cortices of controls without dementia. In the K-R control condition (solid line), there were two binding sites, a high-affinity (EC50_1) and a lower-affinity (EC50_2) binding site, which were also reduced by PTI-125 incubation. C, In fresh SK-N-MC cells, PTI-125 reduced this binding affinity 10,000-fold from 770 fm, the high-affinity binding site in the K-R control condition (solid line), to 1 nm (dotted line). n = 11 for postmortem control tissue; n = 6 for SK-N-MC cells. *p < 0.01 vs vehicle-treated control; #p < 0.01 vs Aβ42-treated control or vehicle-treated AD.
Confirmation of FLNA binding as mechanism of action
To affirm that the beneficial effect of PTI-125 rests on its specific binding to FLNA, we used a pentapeptide that is the sequence of the FLNA binding site (VAKGL) as a decoy. This pentapeptide prevents PTI-125 from binding FLNA in tissue and thereby prevents PTI-125 effects. In control postmortem tissue, the Aβ42-induced association of FLNA with both α7nAChR and TLR4 was blocked by PTI-125, but coincubation with the FLNA pentapeptide prevented PTI-125 blockade (Fig. 10). Similarly, the pentapeptide blocked PTI-125 suppression of tau phosphorylation in Aβ42-treated postmortem control tissue (Fig. 11). We used a pentapeptide with a midpoint alanine for lysine substitution (VAAGL) as an inactive control pentapeptide because it does not bind PTI-125.
Figure 10.

A, Acting as a decoy for the FLNA protein, the pentapeptide binding site of PTI-125 on FLNA (VAKGL) blocked the reduction by PTI-125 in Aβ42-induced FLNA–α7nAChR/TLR4 association in postmortem frontal cortical synaptosomes. The inactive control pentapeptide (VAAGL) did not disrupt PTI-125 reductions of these Aβ42-induced FLNA associations. Western blots (A) were analyzed by densitometric quantification (B). n = 3. *p < 0.01 vs respective basal level; #p < 0.01 vs Aβ42-exposed tissues.
Figure 11.

The VAKGL pentapeptide also blocked PTI-125 reduction in Aβ42-induced tau phosphorylation at three phosphorylation sites in postmortem frontal cortical synaptosomes. Again, the inactive control pentapeptide (VAAGL) did not disrupt PTI-125 reduction of Aβ42-induced tau phosphorylation. Western blots (A) were analyzed by densitometric quantification (B). n = 3. *p < 0.01 vs respective basal level; #p < 0.01 vs Aβ42-exposed tissues.
FLNA–α7nAChR/TLR4 and Aβ42–α7nAChR associations are increased in AD lymphocytes
Because lymphocytes contain α7nAChR and TLR4, we assessed whether the associations of these receptors with FLNA are increased in the lymphocytes of AD patients and whether PTI-125 treatment ex vivo could disrupt these associations. We also tested the effect of PTI-125 on Aβ42–α7nAChR complexes in AD lymphocytes and Aβ42-treated lymphocytes from age-matched control subjects. FLNA associations with α7nAChR and with TLR4 were dramatically increased in AD and Aβ42-treated control lymphocytes compared with vehicle-treated control lymphocytes. Additionally, the level of Aβ42–α7nAChRcomplexes was elevated in AD or Aβ42-treated control lymphocytes. Incubation with 1 nm PTI-125 for 30 min significantly reduced the increased associations with FLNA and the level of Aβ42–α7nAChR complexes (Fig. 12).
Figure 12.

FLNA associations with α7nAChR and TLR4 were markedly increased in AD and Aβ42-treated lymphocytes, and 30 min incubation with PTI-125 reduced these associations. Western blots (A) were analyzed by densitometric quantification (B). n = 2. **p < 0.05, *p < 0.01 vs vehicle-treated control; ##p < 0.05, #p < 0.01 vs Aβ42-treated control or vehicle-treated AD.
Discussion
PTI-125 is a new chemical entity representing a novel approach to the prevention and treatment of AD. A molecule of <300 Da, PTI-125 appears to be orally available and well tolerated because it produces notable plasma and CNS levels but negligible side effects at 2 g/kg administered orally in rats. Although much AD research has focused on Aβ42 signaling through α7nAChR, the present study is the first to demonstrate that Aβ42 signaling requires the recruitment of FLNA to α7nAChRs: there is a profound increase in FLNA coupling to α7nAChR in AD postmortem tissue or after Aβ42 exposure. Basal α7nAChR-associated FLNA levels are negligible. The model we propose is that the initial binding of Aβ42 to α7nAChR recruits FLNA, which causes a conformational change in α7nAChR that both enables the toxic signaling and increases Aβ42 affinity to α7nAChR. The fact that Aβ42 binding blocks Ca2+ influx by α7nAChRs (Wang et al., 2009, 2010) suggests that one conformational change in α7nAChRs may occur in the interface between extracellular and transmembrane domains, the area governing channel opening/desensitization (Bouzat et al., 2008). This conformational change likely exposes a positive charge-rich transmembrane region close to the Aβ42 binding site. FLNA binds this positive charge to stabilize the bound Aβ42 and additional binding of Aβ42 peptides, leading to eventual internalization of Aβ42–α7nAChR complexes (Nagele et al., 2002). PTI-125 disruption of the FLNA–α7nAChR interaction stops the pathological signaling and stops Aβ42 high-affinity anchoring to the receptor (Fig. 13). By binding FLNA instead of the receptor, PTI-125 blocks Aβ42 toxic signaling without disrupting α7nAChR physiological activation by acetylcholine or altering its sensitivity or cell surface expression. PTI-125 efficacy in a mouse model and in human postmortem AD and Aβ42-treated control tissue on numerous AD-related neuropathologies both validates the FLNA pentapeptide target and reinforces Aβ42 aberrant signaling by α7nAChR as a predominant mechanism of AD pathology.
Figure 13.
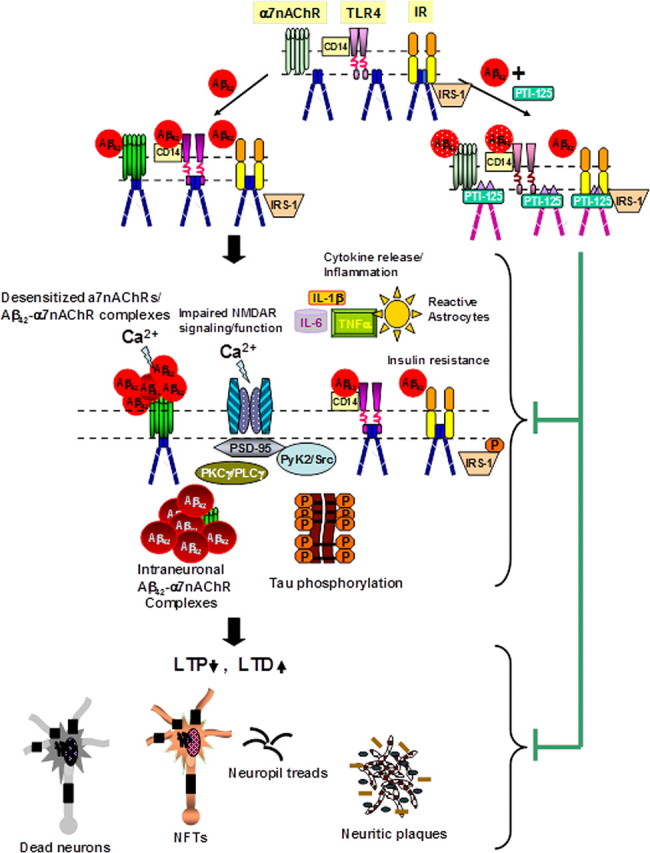
Proposed model of PTI-125 normalizing Aβ42-induced synaptic dysfunction. Soluble Aβ42 or Aβ42 oligomers bind to α7nAChRs in AD brains to induce synaptic dysfunction by first activating α7nAChRs (Dougherty et al., 2003) and TLR4s (Lotz et al., 2005) (by CD14) to recruit FLNA, which enables a high-affinity interaction of Aβ42 with these receptors. The result of FLNA recruitment to TLR4 is enhanced signaling and cytokine release. With FLNA recruited to α7nAChR, Aβ42 binding activates kinases that leads to rapid tau phosphorylation and neurofibrillary lesions (Wang et al., 2003) and causes desensitization, restricting Ca2+ influx through α7nAChRs and impairing downstream NMDAR activity and signaling. In addition, Aβ42 oligomers acting through multiple pathways impair IR function, leading to insulin resistance in neuronal cells (Zhao et al., 2008). Similar to a partial pharmacological blockade of NMDARs, Aβ42-induced hypofunctioning NMDARs are expected to reduce NMDAR-dependent LTP and to increase LTD, leading to dendritic spine shrinkage and retraction (Shankar et al., 2007) that further dampens excitatory neurotransmission. By binding to a defined pentapeptide binding domain with high affinity, PTI-125 may elicit conformational changes in FLNA to prevent its recruitment to α7nAChRs and TLR4s, thereby reducing Aβ42 affinity and facilitating the dissociation of Aβ42 from these receptors. The normalized α7nAChR and TLR4 activities improve NMDAR activation and reduce cytokine release (inflammation). Although neither Aβ42 nor PTI-125 alters FLNA recruitment to IRs, the PTI-125-induced conformational change in FLNA restores Aβ42-suppressed IR function and lessens insulin resistance. The augmented NMDAR activation favors LTP induction and healthier dendritic spines, resulting in more normal excitatory neurotransmission and cognitive processing. The improved IR function leads to healthier cells. Together, PTI-125 treatment lessens neurodegeneration and reduces the burdens of neuritic plaques and neurofibrillary tangles. Changes in color and shape of receptors and FLNA represent conformational or affinity changes.
To assess the specificity of the Aβ42-induced FLNA–α7nAChR association, we examined Aβ40, α7nAChR agonists and antagonists, and a cholinesterase/allosteric nicotinic receptor modulator by incubating postmortem control FCX slices with these agents at 10-fold higher concentrations. Aβ40, known to be less toxic than Aβ42 and to bind α7nAChR with substantially lower affinity (Lee and Wang, 2003), also induced a notable FLNA–α7nAChR association, though, at one-third the level induced by Aβ42, it was less robust. Agonists, but not antagonists, of α7nAChR induced an even milder FLNA–α7nAChR association—∼10% of Aβ42 levels—implying that chronic activation by such agonist drugs could be detrimental. Surprisingly, memantine, an NMDAR and α7nAChR antagonist, was nearly as potent as Aβ40 in inducing an FLNA–α7nAChR association, suggesting that this commonly prescribed AD drug may potentially contribute to disease progression.
AD postmortem tissue or Aβ42 treatment also shows a dramatic increase in FLNA association with TLR4, the innate immune receptor responsible for inflammatory cytokine release. FLNA recruitment may similarly aid in the signaling of TLR4 because PTI-125 also blocks FLNA–TLR4 association and Aβ42-induced inflammatory cytokine release. Although Aβ42 does not itself interact with TLR4, it binds to CD14, which, in turn, binds TLR4 to produce the inflammation noted in AD (Reed-Geaghan et al., 2009). A common feature of neurodegenerative diseases is chronic immune activation, particularly of microglia (Amor, 2010). Hence, a second therapeutic mechanism of action likely to be highly beneficial in AD is an anti-inflammatory effect, particularly against Aβ-induced inflammation.
In an ICV Aβ42 infusion mouse model of AD, Aβ42 induced robust FLNA associations with both α7nAChR and TLR4 that were almost completely blocked by 2-week PTI-125 treatment starting shortly after Aβ42 administration. Remarkably, PTI-125 also reversed this FLNA–α7nAChR association in postmortem AD brain slices and in age-matched control brain tissue incubated with Aβ42. The enabling of Aβ42 signaling through α7nAChR by the FLNA–α7nAChR association is evidenced by a prominent phosphorylation of tau protein at three sites known to be phosphorylated in tau found in NFTs. PTI-125 blockade of FLNA–α7nAChR association dramatically decreased this tau phosphorylation. PTI-125 prevention of Aβ42 signaling and tau phosphorylation resulted in a profound reduction of both NFT (phospho-tau) immunoreactivity and Aβ42 aggregates in the FCX and hippocampus of ICV Aβ42-infused mice. The reduction in Aβ42 aggregates in parallel with the reduced NFT formation supports the theory that tau phosphorylation and NFTs are associated with Aβ42 internalization/deposition and eventual plaque formation (Wang et al., 2009). Evidence that the FLNA–TLR4 association is critical to Aβ42-induced TLR4 signaling is the profound reduction in IL-6, TNF-α, and IL-1β by PTI-125 in ICV Aβ42-infused mice.
Importantly, PTI-125 preserves or restores the signaling functions of α7nAChR, NMDAR, which is regulated by α7nAChR (Snyder et al., 2005; Wang et al., 2009), and IR. The signaling capacity of all three receptors is notably impaired in postmortem AD tissue, Aβ42-treated postmortem control tissue, and Aβ42-infused mice, and PTI-125 normalized function in each case. Calcium influx after receptor stimulation was used to assess the function of α7nAChR and NMDARs, and signaling was also assessed in NMDARs and IRs by the association of each receptor with several signaling molecules, activated (phosphorylated) or recruited as appropriate. PTI-125 does not alter these receptor functions in the absence of Aβ42 because the very low basal levels of FLNA association with α7nAChR or TLR4 are not altered by PTI-125 (e.g., PTI-125 treatment of ICV vehicle-infused mice). Clearly, it is important to preserve NMDAR function in AD because these receptors mediate long-term potentiation (LTP), critical to learning and memory and disrupted by Aβ (Yamin, 2009). α7nAChR, though a subtype of acetylcholine receptors, also plays an important role in cognition and LTP (Timofeeva and Levin, 2011; Wallace and Porter, 2011), and its dysfunction undoubtedly contributes to cognitive decline in AD.
The loss of IR signaling in AD is a recent focus in AD research, underscoring the comorbidity of AD and type 2 diabetes mellitus. Type 2 diabetes mellitus appears to be a significant risk factor for AD (Biessels et al., 2006; Profenno et al., 2010; Schrijvers et al., 2010; Bosco et al., 2011), likely because insulin signaling in the CNS regulates cell survival. Decreased insulin signaling leaves cells vulnerable to oxidative stress and other metabolic insults and can increase inflammation and even tau phosphorylation (Bosco et al., 2011). Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of AD (Ke et al., 2009). The diabetes drug liraglutide has been shown to decrease neuropathology in a transgenic mouse model of AD (McClean et al., 2011), suggesting possible usefulness in AD. PTI-125 does not directly interact with IRs, and though FLNA interacts with IR, this interaction is unchanged by Aβ42 or PTI-125. However, Aβ42 and PTI-125 may both alter the conformation of FLNA to respectively impair and restore IR signaling. It is also possible that Aβ42-induced IR signaling impairments are a result of Aβ42-induced inflammation because inflammation and inflammatory cytokines such as TNF-α and IL-6 are causally linked to insulin resistance (Wellen and Hotamisligil, 2005; de Luca and Olefsky, 2008). The restoration of IR signaling function by PTI-125 could be the result of decreased inflammatory cytokines through the suppression of TLR4 signaling. The restoration of IR signaling is another important therapeutic aspect of PTI-125.
Finally, the femtomolar-to-nanomolar affinity shift in Aβ42 interaction with α7nAChR induced by PTI-125 represents a novel mechanism of action, occurring indirectly by FLNA. The effects of PTI-125 are clearly dependent on its binding to FLNA because both the FLNA–α7nAChR/TLR4 association and tau phosphorylation can be blocked in Aβ42-treated control postmortem tissue by the addition of a decoy pentapeptide that is the compound's binding site on FLNA. Thus far, the best attempt to directly displace Aβ42 from α7nAChR has yielded more modest effects on tau phosphorylation and in vitro requires incubation 10 min before exposure to Aβ42 to compete with its femtomolar affinity for α7nAChR (Wang et al., 2010). PTI-125 dramatically reduces this extraordinarily high affinity of Aβ42 binding indirectly by binding FLNA, which apparently is required to stabilize Aβ42 binding. Hence, PTI-125 can help release Aβ42 already bound to α7nAChR, as evidenced by the reduction in Aβ42–α7nAChR complexes in postmortem AD tissue. This attribute represents a distinct advantage among AD therapeutic candidates because it implies the potential for some cognitive recovery in addition to slowing or halting disease progression.
The fact that dramatic increases in FLNA–α7nAChR and TLR4 associations and in Aβ42–α7nAChR complexes were found not just in brain but also in lymphocytes of AD patients suggests that some high threshold level of these FLNA associations could potentially serve as a noninvasive, blood-based AD diagnostic. Furthermore, PTI-125 reduction of these associations after ex vivo incubation of lymphocytes suggests the usefulness of FLNA–α7nAChR/TLR4 as a biomarker during treatment. Current thinking points to the need for several different simultaneous approaches to treat AD, clearly a disease with multifaceted pathologies. PTI-125's novel and effective mechanism of disrupting Aβ42-induced α7nAChR and TLR4 signaling produces a multitude of therapeutic effects, re-emphasizing the critical roles of Aβ42–α7nAChR signaling and inflammation in AD. With its attenuation of Aβ-driven tau phosphorylation, normalization of multiple receptor functions, and powerful anti-inflammatory activity, PTI-125 shows great potential as a disease-modifying therapeutic for AD.
Footnotes
This work was supported by Pain Therapeutics, Inc. We thank Drs. Nan-Horng (Stan) Lin and Yongmei Xu of Shanghai Medicilon, Inc., for the medicinal chemistry effort that led to PTI-125, a proprietary compound of Pain Therapeutics, Inc.
L.H.B. is an employee of and H.-Y.W. is a consultant for Pain Therapeutics, Inc. The rest of the authors declare no competing financial interests.
References
- Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med. 2011;15:1807–1821. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Bartos M, Corradi J, Sine SM. The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci. 2008;28:7808–7819. doi: 10.1523/JNEUROSCI.0448-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yamada K, Nabeshima T, Sokabe M. alpha7 Nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in beta-amyloid infused rats. Neuropharmacology. 2006;50:254–268. doi: 10.1016/j.neuropharm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF. Deletion of the α7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer's disease. J Neurosci. 2009;29:8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease: the National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Ke YD, Delerue F, Gladbach A, Götz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer's disease. PLoS One. 2009;4:e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Wang HY. Differential physiologic responses of alpha7 nicotinic acetylcholine receptors to beta-amyloid1–40 and beta-amyloid1–42. J Neurobiol. 2003;55:25–30. doi: 10.1002/neu.10203. [DOI] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: Report of the NINCDSADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Accumulation of beta-amyloid1–42 in neurons is facilitated by the alpha7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. β-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120–RC125. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology. 2010;75:1982–1987. doi: 10.1212/WNL.0b013e3181ffe4f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synaptic loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Stossel T, Condeelis J, Cooley L, Hartwig J, Noegel A, Schleicher M, Shapiro S. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- Timofeeva OA, Levin ED. Glutamate and nicotinic receptor interactions in working memory: importance for the cognitive impairment of schizophrenia. Neuroscience. 2011;195:21–36. doi: 10.1016/j.neuroscience.2011.08.038. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, Kaj Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter WZ, Weiner MW, Jack CR, Jr, Jagust W, Toga AW, Lee VM, Shaw LM. Update on the Biomarker Core of the Alzheimer's Disease Neuroimaging Initiative Subjects. Alzheimers Dement. 2010;6:230–238. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Porter RH. Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem Pharmacol. 2011;82:891–903. doi: 10.1016/j.bcp.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davie CB, Shank RP. Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem. 2000a;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer's disease pathology. J Biol Chem. 2000b;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang HY, Li W, Benedetti NJ, Lee DH. α7 Nicotinic acetylcholine receptors mediate β-amyloid peptide-induced tau protein phosphorylation. J Biol Chem. 2003;278:31547–31553. doi: 10.1074/jbc.M212532200. [DOI] [PubMed] [Google Scholar]
- Wang HY, Stucky A, Liu J, Shen C, Trocme-Thibierge C, Morain P. Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer's disease brain. J Neurosci. 2009;35:10961–10973. doi: 10.1523/JNEUROSCI.6088-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Bakshi K, Shen C, Frankfurt M, Trocmé-Thibierge C, Morain P. S 24795 limits β-amyloid–α7 nicotinic receptor interaction and reduces Alzheimer's disease-like pathologies. Biol Psychiatry. 2010;67:522–530. doi: 10.1016/j.biopsych.2009.09.031. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimo A, Prince M. World Alzheimer report 2010: the global economic impact of dementia. London: Alzheimer's Disease International; 2010. [Google Scholar]
- Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer's neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- Yamin G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J Neurosci Res. 2009;87:1729–1736. doi: 10.1002/jnr.21998. [DOI] [PubMed] [Google Scholar]
- Zhao W, De Felice F, Fernandez S, Chen H, Lambert M, Quon M, Krafft G, Klein W. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]