

Abstract
A failure in the control of proliferation of cerebellar granule neuron precursor cells (GCPs), located in the external granular layer (EGL) of the cerebellum, gives rise to medulloblastoma. To investigate the process of neoplastic transformation of GCPs, we generated a new medulloblastoma model by crossing Patched1 heterozygous mice, which develop medulloblastomas with low frequency, with mice lacking the Tis21 gene. Overexpression of Tis21 is known to inhibit proliferation and trigger differentiation of GCPs; its expression decreases in human medulloblastomas. Double-knock-out mice show a striking increase in the frequency of medulloblastomas and hyperplastic EGL lesions, formed by preneoplastic GCPs. Tis21 deletion does not affect the proliferation of GCPs but inhibits their differentiation and, chiefly, their intrinsic ability to migrate outside the EGL. This defect of migration may represent an important step in medulloblastoma formation, as GCPs, remaining longer in the EGL proliferative niche, may become more prone to transformation. By genome-wide analysis, we identified the chemokine Cxcl3 as a target of Tis21. Cxcl3 is downregulated in Tis21-null GCPs of EGL and lesions; addition of Cxcl3 to cerebellar slices rescues the defective migration of Tis21-null GCPs and, remarkably, reduces the area of hyperplastic lesions. As Tis21 activates Cxcl3 transcription, our results suggest that Tis21 induces migration of GCPs through Cxcl3, which may represent a novel target for medulloblastoma therapy.
Introduction
Cerebellar granule neuron precursor cells (GCPs) intensely proliferate postnatally within the external granular layer (EGL), at the surface of the developing cerebellum (Hatten, 1999). Proliferation of GCPs is triggered by Sonic Hedgehog (Shh), secreted by Purkinje neurons (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999).
The prolonged mitotic activity of GCPs makes them potential targets of transforming insults (Wang and Zoghbi, 2001). In fact, ∼25% of medulloblastomas, the tumor of cerebellum and among the most common brain tumors in childhood, arises from GCPs (Kadin et al., 1970; Marino, 2005; Schüller et al., 2008; Yang et al., 2008; Gibson et al., 2010). Other medulloblastoma subtypes may originate from neural precursors of the cerebellar embryonic anlage, different from GCPs and dependent on Wnt signaling (Gibson et al., 2010; Hatten and Roussel, 2011).
Inherited or sporadic mutations in the human Patched1 gene—which encodes the Shh receptor and functions as an inhibitor of the Shh pathway in the absence of the ligand—or the lack of one Patched1 allele in mice models, lead to the development of medulloblastomas, indicating that overactivation of the Shh pathway is important in the etiology of the tumor (Hahn et al., 1996, 1998; Goodrich et al., 1997; Pietsch et al., 1997; Raffel et al., 1997; Wolter et al., 1997; Pomeroy et al., 2002; Lee et al., 2003). The Shh-driven proliferation of GCPs in the EGL is maximal 1 week after birth, but after 3 weeks GCPs have differentiated and migrated from the EGL inwardly to the molecular layer (ML) and internal granular layer (IGL), their final destination (Hatten, 1999). In contrast, at the same age, the EGL of Patched1 heterozygous mice presents clusters of highly proliferating GCPs. These anomalous GCPs, which differ from normal GCPs for their expression profile and for the additional loss of the wild-type Patched1 allele, can be considered a preneoplastic intermediate between GCPs and medulloblastoma cells [preneoplastic GCPs (pGCPs)] (Goodrich et al., 1997; Kim et al., 2003; Oliver et al., 2005). Within 3–6 months, in 15–25% of Patched1 heterozygous mice, the pGCPs will develop into larger lesions and then into tumors (Goodrich et al., 1997; Kim et al., 2003; Kessler et al., 2009).
Tis21, also known as PC3 or BTG2 (in mouse, rat, and human, respectively), is a transcriptional cofactor that acts in cerebellar GCPs and in neural progenitor cells of different areas of the brain—such as the hippocampus and the subventricular zone—by inducing them to exit the proliferative state and differentiate (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2008, 2009). Tis21 inhibits the cell cycle in neural progenitor cells through direct repression of the cyclin D1 promoter and activates proneural genes through direct repression of the promoter of Id3, an inhibitor of proneural bHLH genes (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2009). Moreover, upregulation of PC3 (the rat homolog of Tis21) in pGCPs within the EGL of Patched1 heterozygous mice inhibits their proliferation and triggers their differentiation, highly reducing the incidence of medulloblastoma (Farioli-Vecchioli et al., 2007).
Considering that Tis21 is downregulated in human medulloblastomas (Farioli-Vecchioli et al., 2007), we asked whether the transformation of pGCPs is affected by a loss of the PC3-dependent control of proliferation and differentiation.
We found that genetic ablation of Tis21 greatly induced the frequency of medulloblastoma in Patched1 heterozygous mice without affecting the proliferation of GCPs, but reducing their differentiation. Chiefly, however, ablation of Tis21 impaired the process of migration from the EGL and lesions to the IGL. This migration defect appears to be dependent on Cxcl3, as it is rescued by this chemokine, and may play a critical effect on the conversion of pGCPs into tumor cells.
Materials and Methods
Mouse lines and genotyping.
The Tis21 knock-out mice have been generated previously as described previously (Park et al., 2004) in C57BL/6 (B6) strain by replacing with the neomycin resistance cassette exon II of the Tis21 gene. Patched1 heterozygous mice (Ptch1+/−) were generated in a CD1 background through disruption of exons 6 and 7 (Hahn et al., 1998). Ptch1−/− embryos die before E14. The crossing of Patched1 heterozygous with Tis21−/− mice generated Patched1/Tis21 double-mutant mice that were interbred to obtain the genotypes studied; the progeny was made isogenic before starting any analysis, by further interbreeding for six or more generations.
Math1-green fluorescent protein mice (Math1-GFP) express GFP driven by the Math1 enhancer (Lumpkin et al., 2003). Genotyping of Patched1/Tis21 mice was routinely performed by PCR, using genomic DNA from tail tips. To identify mice carrying the Tis21 null or wild-type alleles, three pairs of primers were used, one complementary to the neo cassette and the others complementary either to exon I or to the targeted exon II, and were amplified together in the PCR to obtain different patterns of amplification specific for each allele: neo(−), 5′-GATGCCTGCTTGCCGAATAT-3′; exI(+), 5′-TCTCCAGTCCTCCTGAGGACT-3′; exII(−), 5′-GCCATCACATAGTTCTTCGAG-3′. Patched1 wild-type and Patched1-null alleles were identified by PCR using three pairs of primers complementary to the neo insert and wild-type regions of Patched1 knock-out mice, as described previously (Hahn et al., 1998; Farioli-Vecchioli et al., 2007). Math1-GFP mice were crossed with Patched1/Tis21 double knock-out mice to obtain Ptch1+/−/Tis21+/−/Math1-GFP+/− mice, which were interbred at least four times before further analysis, in this way generating the different genotypes under study. Genotyping of Math1-GFP pups was performed using a “GFP flashlight” (Nightsea) that made the GFP+ pups glow.
Experiments were performed with mice of either sex, and all animal procedures were completed in accordance with the current European (directive 2010/63/EU) Ethical Committee guidelines.
Tumor and lesion quantification and histological analysis.
Mice were observed for symptoms of medulloblastoma daily for a period of 12 months. On the appearance of symptoms of medulloblastoma (head doming, hunched posture, preferential turning to one side, severe weight loss, paralysis, ruffling of fur, or inactivity), they were killed and autopsied. The cerebellar tumor was either snap frozen in liquid nitrogen for mRNA studies, or fixed in 4% paraformaldehyde (PFA) by immersion overnight, and then cryoprotected before sectioning in 30% sucrose in PBS–DEPC. Samples were then embedded in Tissue-Tek OCT (Sakura Finetek), sectioned serially, and stained with hematoxylin/eosin to confirm by histological analysis that the necropsied brain tissue was medulloblastoma. Lesions were identified by visualizing the GFP+ proliferating GCPs in asymptomatic Ptch1+/−/Tis21−/−/Math1-GFP+ mice.
Bromodeoxyuridine treatment of mice.
GCPs entering in S-phase were visualized 1 h after an injection of bromodeoxyuridine (BrdU) (95 mg/kg, i.p.), according to existing protocols (Canzoniere et al., 2004; Qiu et al., 2010).
Immunohistochemistry: sample preparation, BrdU labeling, antibodies, and image analysis.
Histology and immunostaining of sections, stained for multiple labeling and BrdU incorporation using fluorescent methods, was performed as described previously (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2007). Briefly, cerebella of P7 (i.e., 7 d of age) and P14 mice (for EGL or lesion analysis) were dissected out and fixed by immersion overnight in 4% PFA in PBS–DEPC, whereas cerebella of P42 and P84 mice (for lesion analysis) were dissected out after transcardiac perfusion with 4% PFA in PBS–DEPC and kept overnight in PFA. Fixed cerebella were cryoprotected before sectioning in 30% sucrose in PBS–DEPC overnight at 4°C and frozen at −80°C until use. Cerebella were then embedded in Tissue-Tek OCT (Sakura Finetek), and midsagittal sections of 20 μm were cut on a rotary microtome. Lesion studies (proliferation, differentiation, apoptosis, migration, and volume quantifications) were performed on free-floating sagittal sections 40 μm thick. BrdU incorporation was detected following pretreatment of sections to denature the DNA, with 2N HCl 45 min at 37°C and then with 0.1 m sodium borate buffer, pH 8.5, for 10 min. Primary antibodies used were a rat monoclonal antibody against BrdU (AbD Serotec; MCA2060; 1:150), a mouse monoclonal antibody raised against NeuN (Millipore Bioscience Research Reagents; MAB377; 1:100), a goat polyclonal antibody against NeuroD1 (R&D Systems; AF2746; 1:100), or rabbit polyclonal antibodies against cleaved (activated) Caspase-3 (Cell Signaling Technology; 9661; 1:100) or Cxcr2 receptor (Santa Cruz Biotechnology; sc-682; 1:200). Secondary antibodies used to visualize the antigen were either donkey anti-rat IgG tetramethylrhodamine isothiocyanate (TRITC)-conjugated (Jackson ImmunoResearch; BrdU), or donkey anti-rabbit TRITC-conjugated (Jackson ImmunoResearch; Caspase-3), donkey anti-goat Cy2-conjugated (Jackson ImmunoResearch; NeuroD1), or donkey anti-mouse Cy2-conjugated and Alexa 647-conjugated (Invitrogen; NeuN).
Images of the immunostained sections were obtained by laser-scanning confocal microscopy using a TCS SP5 microscope (Leica Microsystems) and were analyzed by the I.A.S. software (Delta Sistemi).
Quantification of cell numbers in EGL and lesions.
Quantifications of proliferating, differentiating, or apoptotic cells in the EGL were performed on five nonadjacent midsagittal sections at the midpoint of the fifth, seventh, and ninth folia of each section, analyzing five sections per mouse and three mice for each genotype. The EGL cell number was expressed as percentage ratio of proliferating (BrdU+), differentiating (NeuroD1+ or NeuN+), or apoptotic (cleaved Caspase-3+) cells, to the total number of cells (labeled by Hoechst 33258; 1 mg/ml in PBS), counted for the entire length of the EGL in each photomicrograph field, from digital images.
BrdU+, NeuroD1+, NeuN+, or cleaved Caspase-3+ cells in lesions were counted in the whole lesion area, defined by the presence of GFP+ cells, on at least five nonadjacent midsagittal sections per lesion. We examined all lesions present in each mouse analyzed, whose number for each genotype and time point is indicated in Figure 2E.
Figure 2.

Ablation of Tis21 in Patched1 heterozygous mice increases the frequency of mice with hyperplasia lesions as well as the number of lesions per cerebellum. A, Representative images of lesions in cerebellar sagittal sections from Patched1 heterozygous mice, either Tis21 wild-type (Ptch1+/−/Tis21WT/GFP) or Tis21-null (Ptch1+/−/Tis21KO/GFP) at 2, 6, and 12 weeks of age. Sections were stained with Hoechst 33258, and lesions were identified by the presence of GCPs expressing GFP, which is driven by the enhancer of Math1 in proliferating, normal and neoplastic, GCPs. Scale bar, 470 μm. B–D, Percentage of mice with lesions (B), frequency of lesions per cerebellum (C), and volume of lesions (D) in Ptch1+/−/Tis21WT/GFP or Ptch1+/−/Tis21KO/GFP mice at 2, 6, and 12 weeks of age. E, Number of mice analyzed in B–D. *p < 0.05 or **p < 0.01, χ2 test in B and Student's t test in C and D. Error bars indicate SEM.
Cell migration assays, layer areas, lesion areas, and volumes.
In experiments of migration from the EGL, the number of BrdU-labeled cells in each defined layer (i.e., EGL, ML, or IGL) was counted as percentage of total BrdU-labeled cells in all three layers, in five nonadjacent midsagittal sections at the midpoint of the fifth, seventh, and ninth folia. Three mice per genotype were analyzed. Similarly, in experiments of migration from lesions, the number of BrdU-labeled cells was counted in each defined layer neighboring the lesion (i.e., ML and IGL) as percentage of total BrdU-labeled cells present in the lesion and layers, in three nonadjacent sagittal sections per lesion.
Planimetric measurements of the EGL, ML, or IGL and lesion area were performed for the whole length of the layer or for the whole extension of the lesion in each (5×) photomicrograph field. The area was obtained by tracing the outline of the whole layer or lesion on a digital picture captured and measured using the I.A.S. software (Delta Sistemi).
The volume of each lesion was calculated multiplying the average lesion area by section thickness and by number of sections in which the lesion was observed.
Isolation of cerebellar granule progenitor cells (from EGL and lesions).
GCPs were isolated from cerebella of P7 mice or of P7 Wistar rats following a described procedure, consisting in the enzymatic digestion of cerebella chopped into small pieces, followed by separation onto a step gradient of 35 and 65% Percoll and then by depletion of adherent cells with two consecutive incubations on tissue culture dishes (Wechsler-Reya and Scott, 1999).
Organotypic slice cultures (BrdU labeling, retroviral infection, and immunostaining).
Slices were taken and cultured following the methods of Stoppini et al. (1991) and Polleux et al. (1998). Briefly, the brains of P7 pups from the different genotypes were isolated under aseptic condition following decapitation, the meninges were removed, and the cerebellum was isolated from the rest of the brain. Cerebella were cut on a McIlwain tissue chopper into 350-μm-thick parasagittal slices. After cutting, slices were washed in calcium- and magnesium-free HBSS with 36 mm glucose and 15 mm HEPES, pH 7.4, for 30 min at 4°C, carefully separated, and transferred to 23-mm-diameter semiporous membrane culture inserts with a 0.4 μm pore size (Falcon cell culture inserts; BD Biosciences Discovery Labware) with culture medium containing DMEM/Ham's F-12 with N2 supplement (Invitrogen), 20 mm KCl, 36 mm glucose, and penicillin–streptomycin.
For migration experiments, slices were cultured for 120 h with 100 ng/ml Cxcl3 (R&D Systems) or with vehicle (PBS, 0.1% BSA) alone, and the medium was replaced every 48 h. To label and subsequently track migrating cells from EGL, cultures were pulse-labeled with BrdU (10 μg/ml; Sigma-Aldrich) added at t0 for 18 h. In experiments of retroviral infection with the cDNA sequence of Tis21, organotypic slices were infected by adding directly onto the slice the volume of viral stock (pCAG-Tomato-Tis21 retrovirus) corresponding to 1.4 × 106 TU (Boukhtouche et al., 2006), pulsed the day after with 10 μg/ml BrdU for 6 h, and cultured for an additional 72 h. For each animal, one-half of the cerebellar slices served as control and were infected with pCAG-Tomato-empty virus and the other one-half with pCAG-Tomato-Tis21. At the end of each experiment, slices were fixed in 4% PFA and immunostained. For proliferation and differentiation experiments, slices were cultured for 48 h with 100 ng/ml Cxcl3 (R&D Systems) or with vehicle alone (PBS, 0.1% BSA); after a 2 h pulse with BrdU, slices were fixed and analyzed for incorporation of BrdU and expression of NeuroD1 or NeuN. For retroviral experiments, a goat polyclonal DsRed antibody (Santa Cruz) was used at 1:100 in 5% normal donkey serum overnight at 4°C.
In vitro migration assay.
The migration of GFP+ GCPs purified from GFP P7 mice was measured using poly-d-lysine-coated membranes (0.1 mg/ml; Sigma-Aldrich) with 8 μm pore size (0.3 cm2) in modified Boyden chambers as previously described (Lu et al., 2001). To obtain GCPs, cerebella were removed, chopped into small pieces, and resuspended in HBSS Ca–Mg-free with 0.5 mm EDTA through Pasteur pipettes of two sequentially decreasing bore size (10 times each). The cell resuspension was then passed through a 30 μm cell strainer (BD Biosciences). The filtrate was centrifuged at 200 × g to sediment the dissociated cells, resuspended in 200 μl of PBS with 5 mm EDTA, and separated by fluorescence-activated cell sorting (FACS) on a MoFlo high-speed cell sorter (Beckman Coulter). Isolated GCPs were resuspended in 200 μl of DMEM/Ham's F-12 with 1% fetal bovine serum and directly seeded (1 × 105) onto the membrane filter in the upper chamber (BD Biosciences; 353182). In the lower chamber, medium with 5% fetal bovine serum was added in order to establish a 1–5% serum gradient. After 16 h at 37°C in 5% CO2, cells were removed from the upper membrane using cotton swab and washing twice with PBS. The cells on the lower surface were fixed in 4% PFA, stained with Hoechst 33258, and counted by confocal microscopy. Twenty randomly chosen high-magnification fields were counted in each well; three experiments were performed.
Design of small interference RNAs and transfection of GCPs.
Several 19 nt small interference RNAs (siRNAs) specific to Cxcl3 were designed by the on-line Design Tool software (MWG); two of them (siRNA7-Cxcl3 and siRNA6-Cxcl3, the latter specific to both mouse and rat sequences) were selected for their ability to inhibit Cxcl3 expression. GCPs were transfected immediately after the preparation of the cell suspension from P7 rat cerebella, according to a described protocol (Zhokhov et al., 2008). For this procedure, we used an electroporation-based Amaxa Nucleofector technology (rat neuron nucleofection kit; Amaxa Biosystems). The siRNA7-Cxcl3 sequence was 5′-ACAAGGTCGTCTCAGTGTA-3′; the siRNA6-Cxcl3 sequence was 5′-CAACTCCTGAGA GTTCATA-3′; the control siRNA was from the luciferase gene, 5′-CTTACGCTGAGTACTTCGA-3′. For each transfection procedure, 6 × 106 GCPs were centrifuged for 2 min at 100 × g and then resuspended in 100 μl of transfection buffer; 15 μg of either siRNA7-Cxcl3 or siRNA6-Cxcl3, or of siRNA-control, previously dissolved in the same buffer, was then added. Transfection was performed in cuvettes part of the kit. Transfected GCPs were suspended in DMEM with 10% fetal bovine serum and plated in uncoated 35 mm dishes for 3 h under gentle agitation, then resuspended in DMEM/Ham's F-12 with 1% fetal bovine serum and seeded onto the membrane filter of the modified Boyden chamber for migration analysis for 16 h, as described above (see In vitro migration assay). Alternatively, transfected GCPs were plated in coated dishes for 4 h, and then the medium was replaced with serum-free DMEM/Ham's F-12 with N2 supplement, and after 16 h, GCPs were analyzed for differentiation or collected for RNA analysis.
Transfection of cerebellar slices.
Cerebellar slices were isolated from P7 mice and incubated for 4 h onto semiporous membrane, as described previously [see above, Organotypic slice cultures (BrdU labeling, retroviral infection, and immunostaining)]. siRNA targeting Cxcl3 (siRNA6-Cxcl3) or siRNA-control was incubated for 20 min with 8 μl of Lipofectamine 2000. The medium of cerebellar slices was then substituted with the transfection mixture added to serum-free N2 medium with 20 nm final concentration of each siRNA (1.5 ml total volume, 1 ml at bottom of cultures, 0.5 ml at top of slices). After 16 h, the siRNA-containing medium was replaced with regular serum-free N2-supplemented medium containing BrdU, replaced again after 6 h with medium without BrdU, and left for 72 h. Then the percentage of GCPs migrated out of EGL (BrdU+) was determined.
DNA constructs and retrovirus production.
The retroviral vector pCAG-IRES-tdTomato, kindly provided by Dr. Mu-ming Poo (University of California, Berkeley, Berkeley, CA) (Cancedda et al., 2007), was used to express the cDNA of Tis21 (i.e., the murine sequence) only in dividing GCPs. The full open reading frame of Tis21 cDNA was cloned in the sites XhoI-5′/SmaI-3′ of pCAG-IRES-tdTomato, obtaining pCAG-IRES-tdTomato-Tis21. The construct was checked by DNA sequencing.
Retroviruses were propagated as previously described (Farioli-Vecchioli et al., 2008).
RNA extraction, real-time RT-PCR.
Total RNA, extracted from GCPs and reverse-transcribed as described previously (Guardavaccaro et al., 2000), was analyzed by real-time RT-PCR amplification, using TaqMan probe-based fluorogenic 5-nuclease chemistry in duplicate samples and a 7900HT System (Applied Biosystems). The mRNA relative expression values, obtained by the comparative cycle threshold method (Livak and Schmittgen, 2001), were normalized to endogenous controls TATA-binding protein and GAPDH. Statistical analysis of mRNA expression values was performed by Student's t test on data normalized to the endogenous controls but not relativized in fold expression of the calibrator sample. Specific real-time RT-PCR primers were designed by the software Beacon Designer 7.1 (Premier Biosoft International); their sequence is available on request.
Microarray analysis.
Genome-wide expression analysis of GCPs isolated from the EGL of P7 mice was performed with Whole Mouse Genome Microarrays (Agilent Technologies). Significant changes in gene expression were identified when comparing Tis21-null and Tis21 wild-type mice, either in Patched1 wild-type background (Ptch1+/+/Tis21−/− vs Ptch1+/+/Tis21+/+) or in Patched1 heterozygous background (Ptch1+/−/Tis21−/− vs Ptch1+/−/Tis21+/+). Following extraction of GPCs from Ptch1+/+/Tis21+/+ (n = 3), Ptch1+/+/Tis21−/− (n = 3), Ptch1+/−/Tis21+/+ (n = 4), and Ptch1+/−/Tis21−/− (n = 4) mice with Trizol (Invitrogen), RNA integrity was confirmed by using a RNA chip and a 2100 Bioanalyzer (Agilent Technologies) with the protocol outlined by the manufacturer. cRNAs labeled with Cy3-CTP were synthesized from 1 μg of total RNA of each sample using the Low Input Quick Amp Labeling Kit (Agilent Technologies) following the manufacturer's protocol. A reference cRNA, labeled with Cy5-CTP was synthesized from 1 μg of RNA from adult mouse brain. Aliquots (750 ng) of Cy3- and Cy5-labeled cRNA targets were cohybridized on Whole Mouse Genome Microarrays (Design ID 014868; Agilent Technologies). Microarray hybridization and washing were performed using reagents and instruments (hybridization chambers and rotating oven) as indicated by the manufacturer (Agilent Technologies). Microarrays were scanned at 5 μm resolution using a GenePix Personal 4100A microarray scanner and the GenePix Pro 6.0 acquisition and data extraction software (Molecular Devices). Raw data were processed and analyzed by GeneSpring 11.5.1 (Agilent Technologies). Raw signal values were thresholded to 1, log2 transformed, normalized to the 50th percentile, and baselined to the median of all samples. Genes with a corrected p value of <0.05 (one-way ANOVA followed by the Benjamini and Hochberg false discovery rate and the Tukey's post hoc test) were considered differentially expressed.
Chromatin immunoprecipitation.
Primary cultures of cerebellar postmitotic granule neurons from Wistar P7 rats were prepared and infected with adenoviruses expressing Tis21 (rat sequence) or β-galactosidase (β-Gal) as previously described (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2007). Chromatin was released from nuclear preparations of cerebellar granule neurons infected with adeno-Tis21 or adeno-β-Gal by digestion at 37°C with micrococcal nuclease (to obtain fragments of predominantly one to five nucleosomes) and immunoprecipitated with anti-Tis21 antibody as previously described (O'Neill and Turner, 1995; Farioli-Vecchioli et al., 2009). Approximately 4 × 107 cerebellar granule neurons were used for each immunoprecipitation, either with anti-Tis21 A3H antibody or with normal rabbit serum as control. Alternatively, an equivalent number of rat GCPs were used for chromatin immunoprecipitation (ChIP) assays, isolated as described above. The immunoprecipitated DNA and 1/200 dilution of the input DNA were quantified in triplicate samples by real-time PCR using the 2−Ct formula. The DNA immunoprecipitated by immune serum and by normal rabbit serum was calculated as input percentage for each cell treatment. Input percentage is the ratio of the average value of the DNA detected in immunoprecipitated samples to the average value of the DNA present in input lysates (Heard et al., 2001). The amount of DNA promoter immunoprecipitated was finally expressed as fold enrichment (ratio of input percentage of DNA immunoprecipitated by A3H antibody to input percentage of DNA immunoprecipitated by normal rabbit serum). PCR primers used to amplify were as follows: (1) Cxcl3 promoter region 170 nt before transcription start, 5′-TTCTTCTGACACAGGGAC-3′, 5′-ATACATGATGGCTGGAAGA-3′; (2) muscle creatine kinase promoter region 340 nt before transcription start, 5′-GGCTGAGGGCAGGCTGTAAC-3′, 5′-GGGTCAGTAATACTCTGGGTGTCC-3′.
In situ hybridization.
Preparation of sections and hybridization were performed as previously described (Canzoniere et al., 2004). Antisense riboprobes detecting mouse Cxcl3 and Tis21 mRNAs were synthesized by T7 polymerase from the 3′-UTR region of mouse Cxcl3 and Tis21 cDNAs (a 300-bp-long sequence part of the fourth exon of the Cxcl3 gene and a 350-bp-long sequence within the second exon of the Tis21 gene, chosen for the absence of any cross-homology). The Cxcl3 and Tis21 cDNA probes had been previously cloned into the pcDNA3 vector in the NotI site and were checked by sequencing. Riboprobes were labeled with digoxigenin-UTP (transcription kit; Roche), according to the protocol of the manufacturer. No signal was detected by sense probes.
Cxcl3 promoter activity.
Postmitotic cerebellar granule neurons, or the tet-off PC12 cell line expressing a doxycycline-regulated Tis21 cDNA (rat sequence) (Farioli-Vecchioli et al., 2009), were seeded in 35 mm poly-lysine-coated dishes (2 × 106 or 5 × 105 cells per dish, respectively). The following day, cerebellar granule neurons were transfected with a vector expressing Tis21 (rat sequence, pSCT-Tis21) (Guardavaccaro et al., 2000) and with the pGL3-Cxcl3-prom/-628 reporter construct using the Lipofectamine reagent and were harvested after 48 h. PC12 cells were transfected only with the pGL3-Cxcl3-prom/-628 reporter, and Tis21 expression was activated by removing doxycycline from the medium. Luciferase assays were performed by the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions and as previously described (Canzoniere et al., 2004). Luciferase activities were measured in luciferase units per microgram of protein normalized to the activity of the coreporter pRL-TK present in each extract, as a measure of the efficiency of transfection. The pGL3-Cxcl3-prom/-628 construct had been generated by cloning the PCR-amplified region of the rat Cxcl3 promoter (628 nt before transcription start), using genomic rat DNA as template, in the 5′-SacI-3′-BglII site of pGL3-basic. The construct was checked by sequencing.
Results
Tis21 ablation facilitates tumorigenesis
Patched1 heterozygous mice (Ptch1+/−), with low frequency, develop spontaneous medulloblastoma from pGCPs clustering in the EGL (Kessler et al., 2009). To test whether the tumorigenicity of pGCPs is affected by ablation of Tis21, we analyzed the frequency of medulloblastomas developed during 1 year of life in Ptch1+/− mice crossed with Tis21 knock-out mice. We observed that, while Ptch1+/−/Tis21WT mice developed medulloblastoma with a frequency of 25.3%, double-mutant mice, either Ptch1+/−/Tis21KO or Ptch1+/−/Tis21+/−, showed a significantly higher incidence of medulloblastoma [i.e., 80% (p < 0.0001) and 75.9% (p < 0.0001), respectively] (Fig. 1, Table 1). Unexpectedly, however, the average tumor latency was significantly longer in double-mutant than in Ptch1+/−/Tis21WT mice (Table 1). No tumors developed in Ptch1+/+/Tis21−/− or Ptch1+/+/Tis21+/+ mice (hereafter referred to as Tis21KO and Tis21WT; Table 1). Therefore, the ablation of one or both alleles of Tis21 appears to strongly favor tumorigenesis in Ptch1+/− pGCPs.
Figure 1.

Enhancement of medulloblastoma incidence in Patched1 heterozygous mice deprived of Tis21. Ptch1+/−/Tis21WT, Ptch1+/−/Tis21+/− and Ptch1+/−/Tis21KO mice, obtained by interbreeding for at least six generations the progeny of Patched1 heterozygous and Tis21 knock-out mice, were then monitored for the onset of medulloblastoma; p < 0.0001 vs Ptch1+/−/Tis21WT, χ2 test (Table 1).
Table 1.
Statistical analysis of medulloblastoma incidence and latencya
| Genotype | Tis21WT | Tis21KO | Ptch1+/−/Tis21WT | Ptch1+/−/Tis21+/− | Ptch1+/−/Tis21KO |
|---|---|---|---|---|---|
| Medulloblastoma (%) | 0 (0.0) | 0 (0.0) | 118 (25.35) | 41* (75.93) | 60* (80.00) |
| Average latency (weeks) | — | — | 20.09 ± 1.7 | 25.46 ± 1.5† | 25.57 ± 1.3† |
| No. of mice analyzed | 40 | 42 | 71 | 54 | 75 |
aThe five genotypes refer to the progeny of breedings between Patched1 heterozygotes and Tis21 knock-out mice; all mouse genotypes are in the same genetic background, having been cross-mated for at least six generations.
*p < 0.0001 versus Ptch1+/−/Tis21WTgroup (χ2 test).
†p < 0.05 versus Ptch1+/−/Tis21WT group (Student's t test).
To investigate such a Tis21-dependent effect, we analyzed key cellular parameters of the pGCPs resident within the lesions (i.e., proliferation, differentiation, viability, and migration).
Patched1 heterozygous mice at P14 present lesions, characterized by thicker EGL regions, expanded as nodular formations inside the cerebellar lobules and containing highly proliferating pGCPs. These lesions are defined hyperplastic EGL remnants or focal hyperplasias if comprising >30 pGCPs, or diffuse hyperplasias when including >5000 cells (Kim et al., 2003; Thomas et al., 2009). Focal hyperplasias are frequent at P14, but starting from P21 they begin to disappear, being substituted by larger diffuse hyperplasias, which indicates the occurrence of a selection of pGCPs into tumor cells (Kim et al., 2003; Thomas et al., 2009).
Thus, we analyzed the early hyperplasia lesions at P14, when normal GCPs in the EGL are two to three layers thick, having almost completed their migration within the IGL, and the remaining clusters of GCPs can be considered preneoplastic. We also analyzed lesions at 6 and 12 weeks, when the EGL has disappeared and the external cerebellum contains foci of ectopic pGCPs (Oliver et al., 2005).
To identify pGCPs in focal and diffuse hyperplasia lesions, double-mutant Ptch1+/−/Tis21KO mice were crossed with Math1-GFP transgenic mice (hereafter referred to as GFP) (Lumpkin et al., 2003), carrying the GFP transgene under control of the enhancer of Math1, a transcription factor expressed in proliferating, normal, preneoplastic, and tumor GCPs (Ben-Arie et al., 2000; Kim et al., 2003; Lee et al., 2003; Oliver et al., 2005). We verified that the progeny of Ptch1+/−/Tis21KO double mutants crossed with GFP mice presented frequency and volumes of lesions and proliferative parameters of pGCPs equivalent to those of double mutants not crossed with GFP mice (data not shown).
We observed that the percentage of Ptch1+/−/Tis21KO/GFP mice with focal or diffused hyperplasia lesions ranged between 100 and 80% at 2, 6, and 12 weeks after birth, whereas that of Ptch1+/−/Tis21WT/GFP mice was significantly lower at 2 weeks (60%) and decreased to 20% within 12 postnatal weeks (Fig. 2A,B; Ptch1+/−/Tis21KO/GFP vs Ptch1+/−/Tis21WT/GFP; p = 0.02 and p = 0.03 in 2- and 12-week-old mice, respectively). Interestingly, the percentage of mice with lesions at 12 weeks matched exactly in both genotypes the percentage of mice developing medulloblastoma (Fig. 2B, Table 1), indicating that lesions present at 12 postnatal weeks are irreversible. Furthermore, we observed that the number of hyperplasia lesions per mice was significantly higher in Ptch1+/−/Tis21KO/GFP mice at 2 and 6 weeks of age, although this number progressively decreased, matching, at 12 weeks, that observed in Ptch1+/−/Tis21WT/GFP mice (Fig. 2A,C; Ptch1+/−/Tis21KO/GFP vs Ptch1+/−/Tis21WT/GFP; p = 0.006 and p = 0.02 in 2- and 6-week-old mice, respectively). In both genotypes, we observed a progressive increase of lesion volume through successive ages; at 2 weeks of age, however, lesion volumes were significantly greater in double mutants than in Patched1 heterozygous mice (Fig. 2A,D; Ptch1+/−/Tis21KO/GFP vs Ptch1+/−/Tis21WT/GFP; p = 0.007; the number of mice analyzed in Fig. 2B–D is shown in Fig. 2E).
As a whole, these results suggested that, in Ptch1+/−/Tis21KO double-mutant mice, pGCPs were more prone to cluster in lesions and expand into tumors, although with a longer latency than in Patched1 heterozygous mice.
We reasoned that the higher incidence of tumors observed in double mutants could depend on the greater frequency and persistence of lesions. To better understand this aspect, we analyzed pGCPs inside lesions, whose area was visualized by the presence of GFP+ cells. We measured the proliferation rate of pGCPs through BrdU incorporation after a pulse of 1 h, their differentiation, by labeling pGCPs with the neural markers NeuroD1 and NeuN, and their survival. No significant differences were detected between Ptch1+/−/Tis21KO/GFP and Ptch1+/−/Tis21WT/GFP mice in the number of either BrdU+, NeuroD1+, or NeuN+ cells present within the GFP+ lesion area, at 2, 6, or 12 weeks of age (data not shown). Conversely, we observed a significant increase of apoptosis at 6 weeks of age in Ptch1+/−/Tis21KO/GFP versus Ptch1+/−/Tis21WT/GFP mice, as determined by counting the number of cleaved Caspase-3+ cells per lesion area (Fig. 3A,B; p = 0.01). Then, we measured the percentage of pGCPs migrated outside lesions by labeling proliferating pGCPs in mice of 2 and 6 weeks of age with a pulse of BrdU, and following their migration to the ML and IGL over the next 5 d. An evident reduction (>40%) in the percentage of BrdU+ pGCPs migrated from the lesion to both the ML and IGL was observed in Ptch1+/−/Tis21KO/GFP mice at either 2 or 6 weeks of age (Fig. 3C–E; Ptch1+/−/Tis21KO/GFP vs Ptch1+/−/Tis21WT/GFP mice, ML: p = 0.007 at 2 weeks, p = 0.03 at 6 weeks of age; IGL: p = 0.007 at 2 weeks, p = 0.02 at 6 weeks of age). As a whole, these data indicate that ablation of Tis21 impairs the migration of pGCPs outside lesions and suggest that this defect, by delaying the disappearance of lesions from the proliferative area at the surface of cerebellum, favors their conversion into medulloblastoma.
Figure 3.
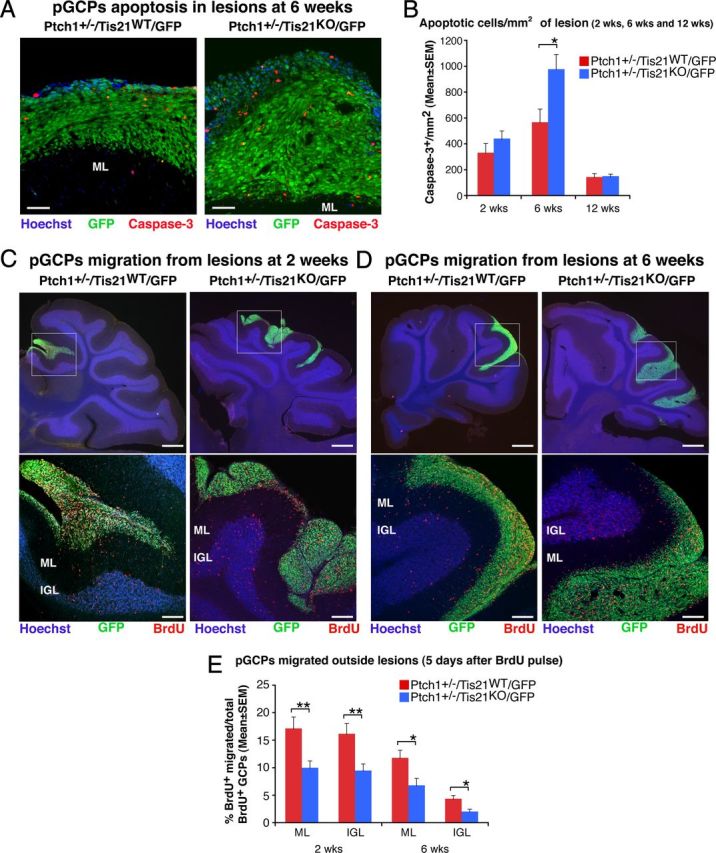
Ablation of Tis21 in Patched1 heterozygous mice increases the apoptosis and impairs the migration from lesions of preneoplastic GCPs. A, Representative confocal images of apoptotic GCPs, identified as cleaved Caspase-3-positive cells, in a diffuse hyperplasia lesion in Patched1 heterozygous mice, either Tis21 wild-type (Ptch1+/−/Tis21WT/GFP) or Tis21-null (Ptch1+/−/Tis21KO/GFP) at 6 weeks of age. Preneoplastic GCPs in the lesion are GFP+; the ML and its boundaries are visualized by Hoechst 33258 staining. Scale bar, 50 μm. B, Apoptotic GCPs per lesion area were quantified as mean ± SEM/mm2 of cleaved Caspase-3+ cells in the whole area of the lesion (defined by the presence of GFP+ cells), on at least five nonadjacent sagittal sections for each lesion. The number of mice analyzed is indicated in Figure 2E. C, D, Representative confocal images of GCPs migrating outside the lesions, identified as BrdU+ cells (red), in Patched1 heterozygous mice either Tis21 wild-type (Ptch1+/−/Tis21WT/GFP) or Tis21-null (Ptch1+/−/Tis21KO/GFP) at 2 or 6 weeks of age. Sections are counterstained with Hoechst 33258 to visualize the ML or the IGL. Lesions in boxed area of top panels (scale bars, 410 μm) are shown at higher magnification in panels below (scale bars, 100 μm). E, GCPs migrating from lesions were quantified as mean ± SEM percentage ratio of BrdU+ cells present within the ML or the IGL area neighboring each lesion 5 d after the BrdU pulse, to the total number of BrdU+ cells in lesion, ML and IGL. Three 2-week-old mice per genotype and at least four 6-week-old mice per genotype were analyzed. In B and E were analyzed all lesions present in each mouse cerebellum. *p < 0.05 or **p < 0.01, Student's t test.
Tis21 ablation inhibits differentiation and migration of GCPs
Having analyzed the pGCPs inside hyperplastic lesions, we sought to define whether the genetic ablation of Tis21, alone (Tis21KO mice) or combined with that of one Patched1 allele (Ptch1+/−/Tis21KO mice), affected the proliferation and differentiation of GCPs in lesion-free areas of the EGL at P7 and P14. These stages precede or correspond to the initial formation of focal hyperplasia lesions, respectively.
At P7—the age of highest expansion of GCPs—the percentage of proliferating GCPs, identified by incorporation of BrdU after 1 h pulse, did not change significantly in Tis21KO or in Ptch1+/−/Tis21KO mice, compared with their respective controls (i.e., Tis21WT and Ptch1+/−/Tis21WT) (Fig. 4A,B). Similar results were obtained at P14 (Fig. 4B). This indicates that the absence of Tis21 does not affect the proliferation rate of GCPs. Consistently, in GCPs isolated at P7, the absence of Tis21 did not affect cyclin D1 mRNA levels (Fig. 4C). This was unexpected, considering that Tis21 exerts an antiproliferative action through inhibition of cyclin D1 when overexpressed in GCPs (Farioli-Vecchioli et al., 2007).
Figure 4.
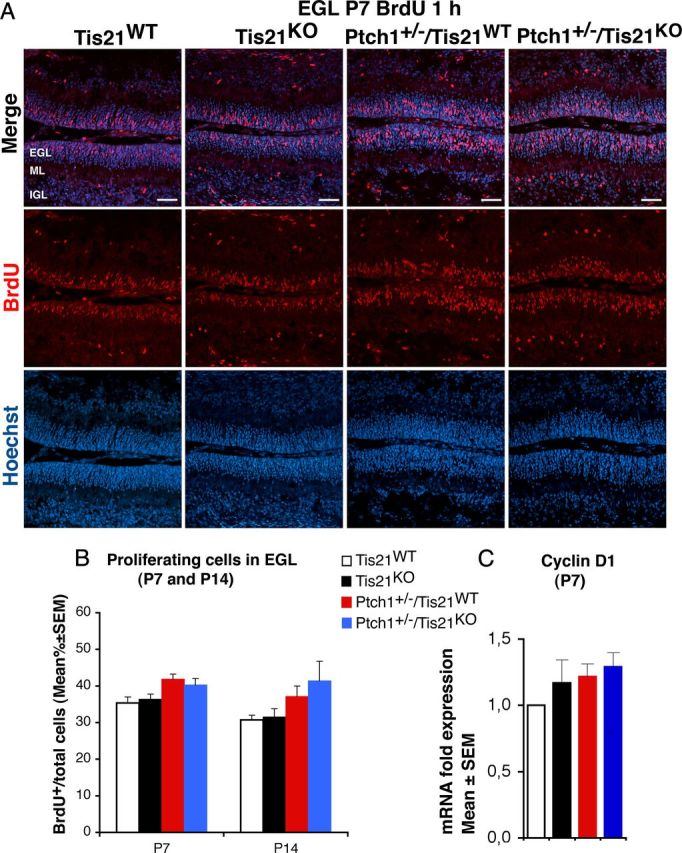
Ablation of Tis21 in Patched1 heterozygous or wild-type mice does not influence the proliferation of GCPs in the EGL. A, Representative confocal images of GCPs in the EGL of P7 mice of the four genotypes indicated, entering in the cell cycle S-phase, identified as BrdU-positive cells after a short pulse of BrdU (1 h). Sections are counterstained with Hoechst 33258 to visualize the EGL and the granule neurons and precursors contained. Scale bar, 50 μm. B, Quantitative analysis in the EGL of P7 or P14 mice with the indicated genotype, of GCPs that have entered the S-phase of cell cycle, measured as mean ± SEM percentage ratio between the number of BrdU+ cells and the total number of cells (labeled by Hoechst 33258; BrdU labeling index). Three mice for each genotype were analyzed. C, Real-time PCR analysis in isolated GCPs from P7 mice of mRNA fold expression relative to wild-type mice (set to unity). Mean ± SEM fold increases are from three independent experiments. TBP and GAPDH were used to normalize data.
Next, we analyzed the differentiation of GCPs in the EGL by labeling GCPs with the neural markers NeuroD1 and NeuN at P7 and P14, respectively. NeuroD1 labels recently differentiated postmitotic GCPs of the inner EGL and is required for their differentiation (Miyata et al., 1999), while NeuN marks postmitotic differentiated granule neurons (Weyer and Schilling, 2003). At P7, the percentage of NeuroD1-positive GCPs in Tis21KO mice was significantly lower (25% decrease) than in Tis21WT mice; Ptch1+/−/Tis21KO and Ptch1+/−/Tis21WT mice presented equivalent percentages (Fig. 5A,C; Tis21KO vs Tis21WT, p < 0.0001; Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p = 0.1). No differences between groups were observed in NeuroD1 expression at P14 (Fig. 5C). However, the ablation of Tis21 induced a decrease of GCPs positive for the late differentiation marker NeuN at P14, in both Tis21KO and Ptch1+/−/Tis21KO mice, compared with their respective controls (i.e., Tis21WT and Ptch1+/−/Tis21WT) (Fig. 5B,D; Tis21KO vs Tis21WT, and Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p = 0.003 and p = 0.002, respectively).
Figure 5.

Ablation of Tis21 in Patched1 heterozygous or wild-type mice reduces the differentiation of GCPs in the EGL. A, B, Representative confocal images of GCPs in the EGL of the four genotypes indicated; NeuroD1 expression in P7 mice (A) and NeuN expression in P14 mice (B). Sections are counterstained with Hoechst 33258 to visualize the EGL and the ML and IGL. Scale bar: 50 μm. C, D, Quantitative analysis of GCPs expressing NeuroD1 in the EGL of P7 and P14 mice (C) or expressing NeuN (D), represented as mean ± SEM percentage ratio between the number of NeuroD1+ or NeuN+ cells, respectively, and the total number of cells (labeled by Hoechst 33258) (n = 3 for each genotype). **p < 0.01 or ***p < 0.001, Student's t test.
Together, these findings indicate that the ablation of Tis21 can impair both the early and late differentiation.
Next, we measured the migration of GCPs from lesion-free areas of the EGL, by labeling them at P7 with a pulse of BrdU and analyzing their migration after 42 h or after 5 d. As shown in Figure 6, A and C, in Tis21-null mice (Tis21KO or Ptch1+/−/Tis21KO), a significantly lower percentage of BrdU-labeled GCPs migrated in the ML and in the IGL after 42 h (Fig. 6A,C; Tis21KO vs Tis21WT, p < 0.003; or Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p < 0.0001; both in ML and IGL). Consistent with the reduced migration outside the EGL, an increase up to 1.5-fold of BrdU-labeled GCPs was observed in the EGL of Tis21KO mice, either Patched1 wild-type or heterozygous (Fig. 6A,C; Tis21KO vs Tis21WT, p < 0.0001; or Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p < 0.0001). The decreased migration of GCPs in the ML and IGL was specifically associated to the ablation of Tis21, as Ptch1+/−/Tis21WT mice did not show any decrease in migration of GCPs when compared with Tis21WT (Fig. 6C; p > 0.05 in ML and IGL). Moreover, 5 d after labeling (at P12), the majority of BrdU-labeled GCPs had migrated outside the EGL in all groups, to reach their final destination, the IGL (Fig. 6B,D). However, in Ptch1+/−/Tis21KO mice, a twofold higher percentage of GCPs was still present in the EGL and a lower percentage had migrated to the ML (Fig. 6B,D; Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p = 0.02 in ML), suggesting that the defect in migration persisted longer if the mutation of Tis21 was associated with that of Patched1. Furthermore, we found no defect in Tis21-null mice, either Patched1 wild-type or heterozygous, in the organization of the Bergmann radial glia, detected by GFAP, which guides granule cell migration (Hatten, 1999) (data not shown).
Figure 6.

Ablation of Tis21 in Patched1 heterozygous or wild-type mice impairs the migration of GCPs from EGL to molecular and internal granular layers. A, B, Representative confocal images of GCPs migrating outside the EGL, identified as BrdU+ cells in Tis21-null and wild-type mice, in either Patched1 heterozygous or wild-type background, as indicated. P7 mice were injected with BrdU and analyzed after 42 h (A) or after 5 d (B). Sections are counterstained with Hoechst 33258 to visualize the ML and the IGL. Scale bars, 50 μm. C, D, Quantification of GCPs migrating from the EGL 42 h after BrdU injection in P7 mice (C), or after 5 d (D), represented as mean ± SEM percentage ratio of BrdU+ cells in the EGL, ML, or IGL to the total BrdU+ cells. Three mice per genotype were analyzed. *p < 0.05, or **p < 0.01, or ***p < 0.001, Student's t test.
We also evaluated the survival of GCPs in the EGL at P7 and P14, and found no significant changes in the percentage of GCPs undergoing apoptosis between the four different genotypes studied, as detected by positivity to cleaved Caspase-3 (data not shown).
As the ablation of Tis21 caused a defective migration of GCPs evident not only in EGL but also in hyperplastic lesions, while differentiation in lesions was normal, we decided to further investigate the migration of GCPs by analyzing organotypic cerebellar slice cultures.
Indeed, a defective migration out of the EGL was observed in BrdU-positive GCPs of organotypic slices from Tis21-null mice, either Patched1 wild-type or heterozygous, compared with Tis21 wild-type mice (Fig. 7A,B; Tis21KO vs Tis21WT, or Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p < 0.0001).
Figure 7.
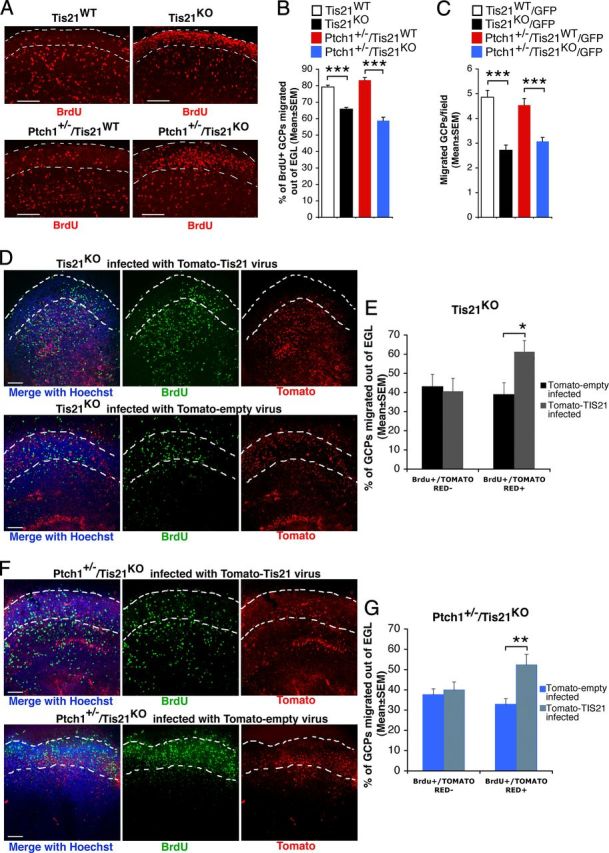
The impairment of migration of GCPs outside the EGL in Tis21 knock-out mice is rescued in organotypic slices by Tis21. A, Representative confocal images of cerebellar organotypic slices, showing BrdU+ GCPs inside or outside the EGL (whose boundaries are indicated by a white broken line). Slices, obtained from P7 mice of the indicated genotype, were pulse-labeled with BrdU for 6 h, cultured for additional 72 h, fixed, and immunostained with anti-BrdU antibody. Scale bars, 100 μm. B, Percentage ratio between BrdU-positive cells localized outside the EGL, and the total number of BrdU-positive cells. Data are mean ± SEM from three experiments (3 mice per genotype were analyzed). ***p < 0.001, Student's t test. C, Test by a modified Boyden chamber of the intrinsic ability of purified GCPs to migrate. GFP+ GCPs isolated by FACS from P7 cerebellum of GFP mice (of the indicated genotype) were placed in the upper chamber, and cells migrating to the lower side of the membrane were counted. Mean cell number per field ± SEM were obtained from three separate experiments, counting 20 fields per well (at least 2 wells per experiment). The number of migrated Tis21-null GCPs, either Patched1 wild-type or heterozygous, was significantly reduced with respect to Tis21 wild-type GCPs. ***p < 0.01, Student's t test. D–G, In cerebellar slices, Tis21-retrovirus rescues the defective migration of Tis21-null GCPs. D, F, Representative confocal images of cerebellar slices, showing BrdU+ and Tomato-Tis21+ or Tomato-empty+ GCPs inside or outside the EGL. Slices, obtained from P7 Tis21 knock-out mice in Patched1 background, either Tis21KO (D) or Ptch1+/−/Tis21KO (F), were infected with Tomato-Tis21 or Tomato-empty retrovirus, pulse-labeled with BrdU for 6 h, cultured for additional 72 h, fixed, and immunostained with anti-BrdU antibody. Scale bars, 100 μm. E, G, The percentage ratio between BrdU+/Tomato+ or BrdU+/Tomato− cells outside the EGL, and the total number of BrdU+ cells was analyzed in slices from either Tis21KO (E) or Ptch1+/−/Tis21KO mice (G), infected with Tomato-Tis21 or Tomato-empty retrovirus. The absence of effect in BrdU+/Tomato− cells is a control of the specificity of infection. Data are mean ± SEM from three experiments (3 mice per genotype were analyzed). *p < 0.05, or **p < 0.01, Student's t test.
We further sought to test whether, either in the presence or in the absence of activated Shh signaling, GCPs lacking Tis21 presented a migration defect per se. To this aim, we analyzed the ability of purified GCPs—isolated at P7 as GFP+ cells by FACS—to migrate in a modified Boyden chamber, as previously described (Lu et al., 2001). Briefly, a membrane filter with uniform pore size divides upper and lower chambers. GCPs are seeded in the upper chamber, and the number of those that have migrated to the lower surface of the filter is determined. The number of Ptch1+/−/Tis21KO/GFP double-mutant GCPs that migrated to the lower chamber was significantly lower than that of Ptch1+/−/Tis21WT/GFP GCPs (Fig. 7C; 32% decrease; p < 0.0001). A quantitatively similar decrease of migration was observed also in GCPs of Tis21-null Patched1 wild-type mice (Fig. 7C; Tis21KO/GFP vs Tis21WT/GFP; p < 0.0001). This indicates that the ablation of Tis21 has an effect on the intrinsic ability of GCPs to migrate, both in the presence or absence of activated Shh signaling.
Moreover, when we infected organotypic cerebellar slices from Tis21-null mice, either Patched1 wild-type or heterozygous, with a retrovirus expressing Tis21 and the Tomato red fluorescent protein, the defective migration of GCPs was significantly rescued (Fig. 7D–G; Tomato-Tis21+ vs Tomato-empty+ in Tis21KO mice, p = 0.02; Tomato-Tis21+ vs Tomato-empty+ in Ptch1+/−/Tis21KO mice, p = 0.003). This indicates that the defect of migration is reversible and specifically dependent on the loss of Tis21.
Tis21 knock-out/medulloblastoma-specific gene expression
To start investigating the molecular mechanisms underlying the complex phenotype observed in Tis21-null cerebellum, by real-time PCR we analyzed the expression levels of components of three main pathways known to regulate migration of cerebellar neurons, namely BDNF and its receptor TrkB (Borghesani et al., 2002; Kokubo et al., 2009), astrotactin1/astrotactin2 (Wilson et al., 2010), and neuregulin and its receptor Erb4 (Rio et al., 1997). However, we could not detect significant changes of these mRNAs in GCPs isolated from P7 mice, either double mutant or Patched1 wild-type/Tis21-null, compared with the respective Tis21 wild-type controls (data not shown). Therefore, we performed a genome-wide mRNA analysis by microarray in GCPs isolated from the EGL of P7 mice. We compared gene expression between Tis21-null and Tis21 wild-type mice, either in Patched1 wild-type background (Tis21KO vs Tis21WT) or in Patched1 heterozygous background (Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT). We assumed that genes whose expression was significantly modified only in the first comparison may be related to Tis21 knock-out non-tumor-specific cerebellar phenotypes, while genes significantly modified in the second comparison (or in both comparisons) may be specifically involved in the Tis21 knock-out-dependent enhancement of medulloblastoma formation. In this way, we identified 344 differentially expressed genes, of which 179 were Tis21 knock-out-specific and 165 medulloblastoma-specific (Fig. 8A,B). A comprehensive picture of these transcriptional changes is shown with a hierarchical clustering method (Fig. 8B), in which the 344 genes are grouped on the basis of similarity in their expression patterns. Among these, we identified 11 Tis21 knock-out-specific and 22 medulloblastoma-specific genes known to be involved in the processes of migration, neurogenesis, differentiation, or cell adhesion and motility, as defined by gene ontology annotations (Fig. 8C,D). Among the 22 medulloblastoma-specific genes, 5 of them showed concordant expression changes also in Patched1 wild-type background (Tis21KO vs Tis21WT) and have been previously associated to the neuronal phenotype. These genes are the chemokines Cxcl3 and Cxcl12 (de Haas et al., 2007), Pag1 (Feng et al., 2009; Georgakopoulos et al., 2011), Efna4 (Wilkinson, 2001), and Jmy (Coutts et al., 2009; Zuchero et al., 2009). Their expression changes were validated by real-time PCR in GCPs from the EGL of P7 mice (Fig. 8E). All these genes showed significantly different expression in double-mutant mice compared with Patched1 heterozygous mice, thus matching the microarray results, with Cxcl3 showing the highest decrease (∼45%; Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT, p = 0.003). Remarkably, in pGCPs isolated from diffused lesions in 6-week-old double-mutant mice as GFP+ cells by FACS, Cxcl3 expression almost disappeared (95% decrease; p = 0.0007; Fig. 8E), while the other genes did not differ (data not shown).
Figure 8.
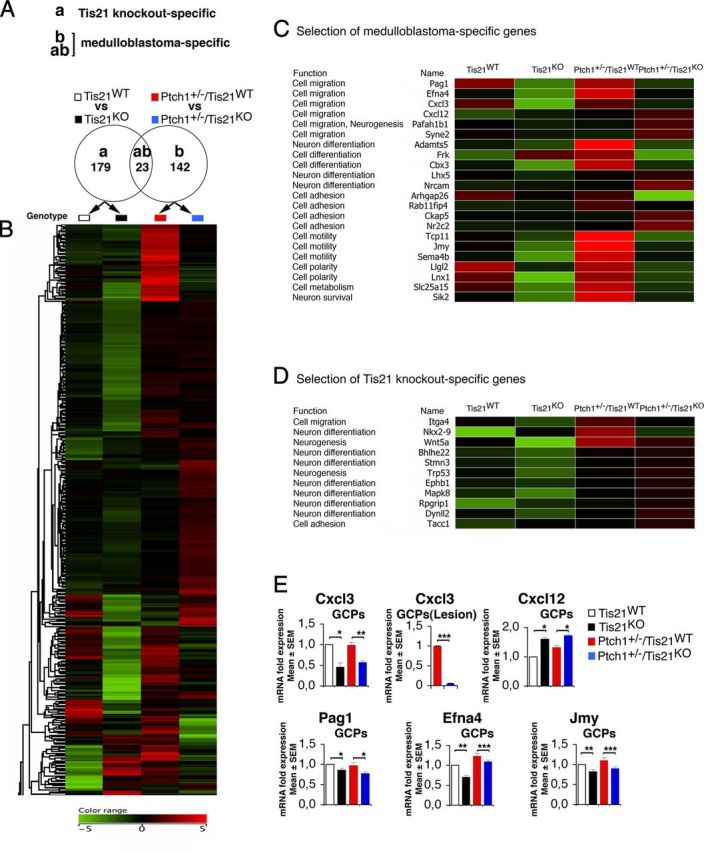
Medulloblastoma-specific gene expression in Patched1 heterozygous/Tis21-null model. A, As shown in the Venn diagram, microarray analysis of GCPs in P7 mice identified 179 Tis21 knock-out-specific genes, whose expression significantly differed only in pairwise comparison (a): Patched1 wild-type/Tis21-null versus wild type (Tis21KO vs Tis21WT). Likewise, 165 medulloblastoma-specific genes were identified whose expression significantly differed in pairwise comparison (b): Patched1 heterozygous/Tis21-null versus Patched1 heterozygous/Tis21 wild-type (Ptch1+/−/Tis21KO vs Ptch1+/−/Tis21WT). Twenty-three genes significantly differed in both comparisons (a) and (b). B, A hierarchical clustering algorithm (similarity measure, euclidean; linkage rule, centroid) was used to order the 344 genes differentially expressed in both pairwise comparisons (a) and (b). Colors indicate high expression (red) to low expression (green), corresponding to changes of normalized values relative to the median value. C, Twenty-two medulloblastoma-specific genes are involved in cell migration, differentiation, or adhesion processes. D, Eleven Tis21 knock-out-specific genes are involved in cell migration or differentiation processes. E, The differential expression of five neuronal medulloblastoma-specific genes in GCPs from the EGL at P7 or from lesions (GFP+) at 6 weeks was confirmed by real-time PCR, analyzing mRNA fold expression relative to wild-type mice (set to unity). Mean ± SEM fold increases are from three independent experiments. TBP and GAPDH were used to normalize data. *p < 0.05, **p < 0.01, or ***p < 0.001, Student's t test.
Cxcl3 counteracts the GCPs migration defect and reduces the lesion area
To investigate the functional implications of Cxcl3 downregulation, we tested the ability of Cxcl3 to revert the defective migration of Tis21-null GCPs. To this end, we treated organotypic cerebellar slices from Tis21-null mice at P7, either Patched1 wild-type or heterozygous, with the Cxcl3 recombinant protein for 5 d. We observed that Cxcl3 significantly neutralized the defect of migration of GCPs from the EGL, as indicated by the percentage of BrdU+ GCPs migrated out of EGL (Fig. 9A,B; with Cxcl3 vs without: in Tis21KO mice, p = 0.001, or in Ptch1+/−/Tis21KO mice, p < 0.0001). In particular, in double-mutant mice, Cxcl3 induced ∼75% of GCPs to migrate outside the EGL (i.e., nearly attaining the basal level of migration observed in wild-type mice) (Fig. 7B). Additionally, we checked whether Cxcl3 treatment affected the proliferation or differentiation of GCPs, by analyzing them in the EGL of cerebellar slices obtained from P7 mice of the four genotypes under study, exposed to the Cxcl3 protein for 48 h. GCPs treated with Cxcl3 did not present significant differences of proliferation, as defined by the percentage of BrdU+ cells after a 2 h pulse, or differentiation (NeuroD1+ and NeuN+ cells), relative to untreated control slices (Fig. 9C). We further sought to test whether the positive effect of Cxcl3 on the migration of GCPs was cell autonomous or whether it was indirect. To this aim, we silenced Cxcl3 in preparations of purified GCPs from P7 wild-type rats, by transfecting two specific siRNAs interacting with Cxcl3 mRNA, siRNA7-Cxcl3 or siRNA6-Cxcl3. These were selected between several 19 nt candidate sequences targeting Cxcl3 designed by the MWG Design Tool software (MWG), as sequences capable to reduce the levels of Cxcl3 mRNA, relative to control GCPs transfected with a control siRNA targeting the luciferase gene (Fig. 9D, right; siRNA7-Cxcl3, p = 0.0001; siRNA6-Cxcl3, p = 0.002). A significantly lower number of GCPs transfected with either siRNA7-Cxcl3 or siRNA6-Cxcl3 were able to migrate to the lower part of a modified Boyden chamber, compared with GCPs transfected with siRNA-control (Fig. 9D, left; p = 0.002 or p = 0.006, respectively). A parallel analysis of the state of differentiation of the same preparations of GCPs, either silenced for Cxcl3 or control, did not show differences in the percentage of expression of NeuN (63.8 ± 2.2 for GCPs transfected by siRNA7-Cxcl3, 61.9 ± 1.1 for siRNA6-Cxcl3, and 62.9 ± 3.2 for siRNA-control; three independent experiments of isolation and transfection of GCPs). Similarly, cerebellar slices from P7 wild-type mouse transfected with siRNA6-Cxcl3 presented a lower number of GCPs migrated out of EGL, relative to GCPs transfected with siRNA-control (data not shown). Therefore, these data indicate that Cxcl3 plays a cell-autonomous role required in the migration of the GCPs out of the EGL.
Figure 9.
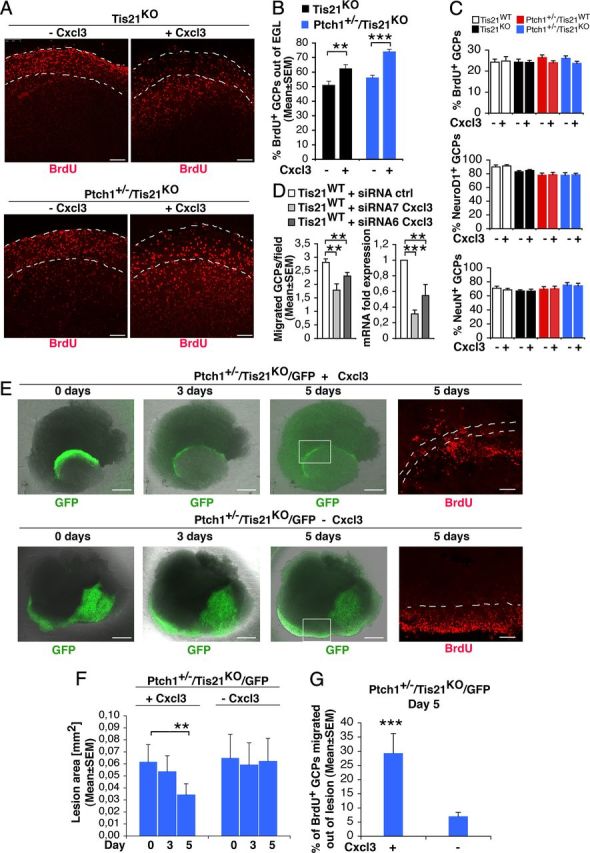
Cxcl3 rescues the defective migration of GCPs outside the EGL or outside lesions, and reduces the area of lesions in Patched1 heterozygous/Tis21 knock-out mice. A, Representative confocal images of cerebellar organotypic slices from Tis21-null/Patched1 wild-type or heterozygous mice treated with or without Cxcl3, showing BrdU+ GCPs inside or outside the EGL (indicated by a white broken line). Scale bars, 100 μm. B, Quantification of BrdU+ GCPs migrated outside the EGL. Slices from P7 mice were cultured 5 d with or without Cxcl3 (adding BrdU at t0 for 18 h), fixed, and immunostained with anti-BrdU antibody. Shown is the mean ± SEM percentage ratio between BrdU-positive cells outside EGL and total number of BrdU-positive cells (from 3 experiments; 3 mice per genotype were analyzed). **p < 0.01 or ***p < 0.001, Student's t test. C, Quantification of proliferating (BrdU+) or differentiating (NeuroD1+ or NeuN+) GCPs in the EGL after treatment with Cxcl3. Slices from P7 mice were cultured 48 h with or without Cxcl3 (adding BrdU 2 h before analysis), fixed, and immunostained with antibodies against BrdU, NeuroD1, or NeuN (mean ± SEM percentage ratio between BrdU+, NeuroD1+, or NeuN+ cells in the EGL and total number of cells, from 3 experiments; 3 mice per genotype were analyzed). D, RNAi knockdown of Cxcl3 in GCPs leads to inhibition of their ability to migrate (graph on the left). Purified GCPs from wild-type P7 rat cerebella were transfected by electroporation with siRNAs targeting Cxcl3 (either siRNA7-Cxcl3 or siRNA6-Cxcl3) or with a control siRNA; immediately thereafter, GCPs were seeded (6 × 106) onto 35 mm dishes in recovery medium (with high serum) for 3 h, and then transferred in the upper part of a modified Boyden chamber. After 16 h, the GCPs migrating to the lower side of the membrane were counted. Mean cell number per field ± SEM were obtained from three separate experiments, counting 20 fields per well (at least 2 wells per experiment). The graph on the right shows the levels of Cxcl3 mRNA in the same preparations of purified GCPs transfected for migration tests (mean ± SEM fold increases; TBP was used to normalize data). **p < 0.01, ***p < 0.001, Student's t test. E, Representative confocal images of diffuse hyperplasia lesions in cerebellar organotypic slices treated with or without Cxcl3, from Patched1 heterozygous/Tis21-null mice (Ptch1+/−/Tis21KO/GFP) at 4 weeks of age, showing GFP+ GCPs inside the lesion (green), and a magnification of BrdU+ GCPs (red) migrating outside lesions (bordered by white broken line) in the presence or absence of Cxcl3. Scale bars: 500 or 100 μm for GFP or BrdU panels, respectively. F, G, Quantification of lesion area (F) and percentage of BrdU+ GCPs migrated outside the lesion (G). Slices obtained from P42 mice were treated with Cxcl3 for 5 d and pulse-labeled with BrdU added at t0 for 18 h, fixed, and immunostained with anti-BrdU antibody. Data are mean ± SEM from three experiments (3 mice per genotype were analyzed). **p < 0.01 or ***p < 0.001, Student's t test.
Having ascertained this, we further analyzed the effect of Cxcl3 on GCPs within lesions. Remarkably, at P30, 5 d of treatment with Cxcl3 significantly reduced the area of lesions in slices (Fig. 9E,F; Cxcl3 day 0 vs day 5, in Ptch1+/−/Tis21KO/GFP mice, p = 0.003) and, correspondingly, significantly reversed the defect of migration of pGCPs (labeled by BrdU+) outside lesions (Fig. 9E,G; with Cxcl3 vs without, in Ptch1+/−/Tis21KO/GFP mice, p = 0.0006). This indicates that the addition of exogenous Cxcl3 can rescue the Tis21-dependent defect of migration of GCPs also at a preneoplastic stage within lesions and, in consequence of this, reduces the area of lesions.
Tis21 is recruited to the Cxcl3 promoter and induces its activity
The above data indicate that Cxcl3 plays a central role in the migration of normal and preneoplastic GCPs and that its expression is controlled by Tis21. Given that Tis21 is a transcriptional cofactor known to be recruited to the promoter of cyclin D1, Id3, and RAR-β (Passeri et al., 2006; Farioli-Vecchioli et al., 2007, 2009), we sought to define whether Tis21 directly controls the transcription of Cxcl3. First, we determined the effect of Tis21 overexpression on Cxcl3 mRNA levels in primary cultures of postmitotic cerebellar granule neurons from P7 wild-type rats, infected with adenoviruses expressing either Tis21 (rat sequence) or β-Gal. As shown in Figure 10, A and B, the expression of exogenous Tis21 in cerebellar granule neurons was associated with a corresponding increase of the levels of Cxcl3 mRNA, relative to control (p = 0.012; Fig. 10B).
Figure 10.
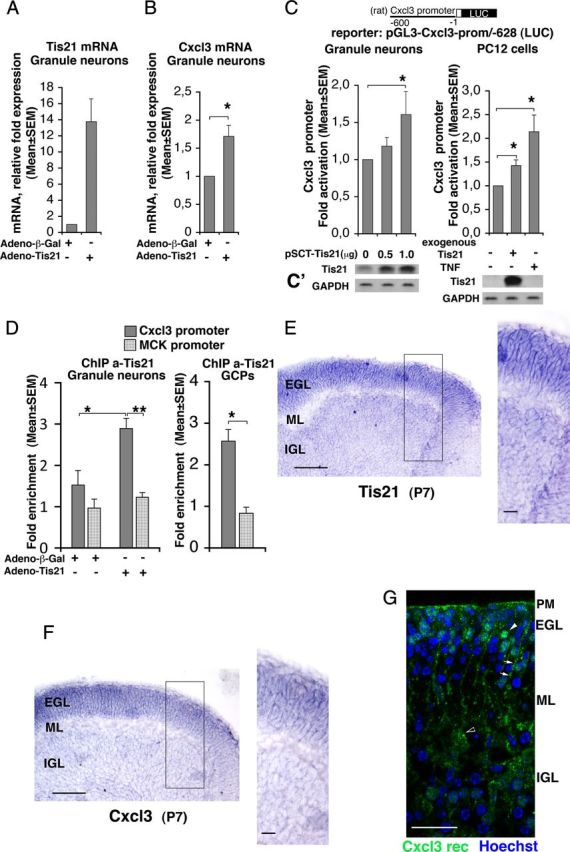
Tis21 associates to the Cxcl3 promoter and induces Cxcl3 transcription. A, B, Real-time RT-PCR analysis of Tis21 and Cxcl3 mRNAs in primary cultures of cerebellar granule neurons. Cells, obtained from P7 rats, were plated and infected with recombinant adenoviruses adeno-Tis21 or control adeno-β-Gal the same day of plating (DIV 0), and harvested after 24 h. The primers amplifying Tis21 (rat sequence) detected both the endogenous and exogenous mRNA. Shown are mean ± SEM values are from five separate experiments. TATA-binding protein mRNA was used as endogenous control for normalization. *p < 0.05 versus control, Student's t test (performed on data normalized to the endogenous controls but not yet relativized as fold expression). C, Cxcl3 promoter activity in cerebellar granule neurons from P7 wild-type rats transfected with an expression vector for Tis21 (pSCT-Tis21) and in PC12 cells expressing inducible Tis21 (rat sequence) increased significantly, relative to the corresponding controls expressing endogenous levels of Tis21. The Cxcl3 promoter construct comprises 628 nt 5′ to the putative transcription start, placed upstream of a luciferase reporter (construct pGL3-Cxcl3-prom/-628). TNF-α, known inducer of Cxcl3 promoter activity, is used in PC12 cells as positive control. Luciferase activity is represented as mean ± SEM fold increase from five separate experiments. *p < 0.05 versus the corresponding control without exogenous or ectopic Tis21, Student's t test. C′, Parallel cultures were analyzed for Tis21 protein expression by Western blot. D, ChIP analysis of Tis21 binding to the Cxcl3 promoter and to the muscle creatine kinase promoter (MCK) (negative control) either in cerebellar granule neurons (graph on the left), infected with adeno-Tis21 or adeno-β-Gal as in A and B, or in GCPs (graph on the right), both isolated from P7 wild-type rats. The amount of Cxcl3 or MCK promoters present in immunoprecipitates, obtained using anti-Tis21 antibody A3H, is quantified by real-time PCR and is expressed as fold enrichment (ratio of the percentage of the Tis21-immunoprecipitated amount of Cxcl3 or MCK promoter detected in the input cell lysates to the percentage of the normal serum-immunoprecipitated amount detected in the input cell lysates). The binding of Tis21 protein to the Cxcl3 promoter (darker gray columns) significantly increases above basal levels (i.e., above the binding levels to the MCK negative control promoter) in correlation with the overexpression of Tis21 mRNA (compare also A). Shown are mean ± SEM values are from five separate experiments performed with cerebellar granule neurons and from three separate experiments with GCPs. *p < 0.05 versus control, Student's t test. E, F, Expression of Tis21 and Cxcl3 mRNAs, respectively, as detected by in situ hybridization in cerebellar midsagittal sections from P7 wild-type mice; their expression colocalizes and is very high in GCPs, throughout the whole EGL width. Black box, Area at higher magnification. Scale bars: 100 μm; enlargement, 20 μm. G, Expression of Cxcr2, receptor of Cxcl3, immunostained by a specific antibody in a representative confocal image of a cerebellar midsagittal section from P7 wild-type mice. Nuclei are visualized by Hoechst 33258 staining. Clusters of receptors are clearly detectable in GCPs (white arrowhead in EGL) and to a lower extent in the pial membrane (PM) and in Purkinje cells (black-white arrowhead); staining of putative Bergmann's glia is indicated by white arrows. Scale bar, 25 μm.
We further sought to check whether the upregulation of Cxcl3 mRNA elicited in cerebellar granule neurons by overexpression of Tis21 corresponded to an induction of Cxcl3 promoter activity. Therefore, we generated the construct pGL3-Cxcl3-prom/-628, carrying the Cxcl3 promoter region upstream of the luciferase reporter gene. This construct was either cotransfected into cerebellar granule neurons together with a Tis21-expressing vector (pSCT-Tis21), or transfected in PC12 cells expressing an inducible Tis21 gene (rat sequence) under control of the tetracycline-regulatable tTA transactivator (PC12/tetoff) (Farioli-Vecchioli et al., 2009). Cxcl3 promoter activity was significantly induced in both cerebellar granule neurons and PC12 cells expressing Tis21, relative to the corresponding controls (Fig. 10C,C′; p = 0.020 and p = 0.021, respectively). The extent of induction of the Cxcl3 promoter by Tis21 in PC12 cells was comparable with that exerted by TNF-α, which is a known inducer of the Cxcl3 promoter (Anisowicz et al., 1991) (p = 0.040 vs control; Fig. 10C,C′).
Next, we asked whether the Tis21-dependent increase of Cxcl3 promoter activity was related to recruitment of Tis21 to the Cxcl3 promoter. We performed ChIP experiments on primary cultures of cerebellar granule neurons and on purified GCPs, obtained from P7 wild-type rats, using the anti-Tis21 antibody A3H, as described previously (Farioli-Vecchioli et al., 2009). The recruitment of Tis21 to the Cxcl3 promoter was significantly induced in cerebellar granule neurons infected with Tis21 virus, relative to control neurons infected with β-Gal virus (Fig. 10D; p = 0.02 vs column on the left). The increased association of exogenous Tis21 with the Cxcl3 promoter sequences was specific, as it was not detected with the muscle creatine kinase promoter (negative control, inactive in cerebellar granule neurons; Fig. 10D; p = 0.002 vs column on the right). We also observed a recruitment of endogenous Tis21, although below statistical significance (Fig. 10D; column on the left). To further test whether endogenous Tis21 is recruited to the Cxcl3 promoter, we analyzed GCPs isolated from rat cerebellum; the association of Tis21 with the Cxcl3 promoter resulted significantly above the background association with the negative control promoter (threefold increase; Fig. 10D, graph on the right; p = 0.02) and was higher than that observed for endogenous Tis21 in cerebellar granule neurons. Therefore, the endogenous Tis21 protein specifically interacts with the Cxcl3 promoter.
Together, this indicates that Tis21 may induce the transcription of Cxcl3 following recruitment to its promoter.
To extend and confirm our real-time PCR data indicating that Cxcl3 is expressed like Tis21 in purified GCPs [Fig. 8E and our microarray data (see Notes)], we sought to assess in cerebellum the cellular localization of Cxcl3 and Tis21 as well as that of the Cxcl3 receptor. Therefore, by in situ hybridization, we analyzed the expression of Cxcl3 and Tis21 mRNAs in the cerebellar layers of P7 Tis21WT mice. We observed that Tis21 and Cxcl3 mRNAs colocalize and are very strongly expressed in GCPs within the whole EGL (Fig. 10E,F). A weaker signal appears detectable also in cerebellar granule neurons of the IGL. Moreover, Cxcr2 (i.e., the receptor of Cxcl3) (Rossi and Zlotnik, 2000) is detectable in clusters at the surface of the plasma membrane of GCPs and, to a lower extent, also in Purkinje cells and in the pial membrane (Fig. 10G). As Cxcl3 is a secreted chemokine, this suggests that the migration of GCPs to the ML and IGL may be controlled by Cxcl3 through an autocrine loop, regulated by Tis21 expression.
Discussion
We have generated a new murine medulloblastoma model that enabled us to identify new mechanisms underlying the plasticity and neoplastic transformation of GCPs. In this model, the ablation of Tis21 strikingly enhances, from 25 to 80%, the incidence of medulloblastoma spontaneously occurring in Patched1 heterozygous mice. At the origin of the higher medulloblastoma incidence is the higher frequency with which Tis21-null GCPs become preneoplastic in early postnatal life and cluster in the EGL, to form focal and diffused hyperplastic lesions that will develop after 12 weeks of age into medulloblastomas. In GCPs lacking Tis21, we observe a remarkable delay in their migration from the EGL and also from lesions, to the neighboring molecular and internal granular layers. Such an effect is intrinsic to the GCP and is peculiar to the null mutation of Tis21—given that Patched1 heterozygosity by itself does not inhibit migration of GCPs. The migration defect of Tis21-null GCPs is quantitatively comparable with that observed in mice deprived of genes known to regulate the migration of GCPs, such as BDNF (Borghesani et al., 2002; Kokubo et al., 2009). Moreover, Tis21-null GCPs also show a delay of differentiation in the EGL but not in lesions. Surprisingly, no change is observed in the proliferation rate of GCPs lacking Tis21, either in the EGL or in lesions. Thus, our data indicate that Tis21 is required for migration and terminal differentiation of GCPs, while its known action as inhibitor of proliferation of GCPs, emerging upon its upregulation (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2007), appears to be redundant. In fact, it is worth noting that, in other neural and non-neural progenitor cell types, such as those of the hippocampus, subventricular zone, and muscle, or in embryo fibroblasts, the knock-out of Tis21 elicits an increase of proliferation, consistently with its physiological antiproliferative activity (Boiko et al., 2006; Evangelisti et al., 2009; Farioli-Vecchioli et al., 2009). This suggests that, in cerebellar progenitor cells, other genes might contribute redundantly with Tis21 to maintain the tightly regulated control of proliferation.
It has been shown that GCPs proliferate when they are exposed to the microenvironment of the EGL proliferative niche and exit the cell cycle as a result of migrating away from this environment (Choi et al., 2005). Thus, differentiation and migration are correlated as differentiation can occur only after GCPs have migrated outside the proliferative EGL region, under control of Shh, a strong mitogen produced by Purkinje cells (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999; Lewis et al., 2004; Choi et al., 2005). The delay in migration, detectable in Tis21-null GCPs, either Patched1 wild-type or heterozygous, causes their prolonged permanence in the EGL and thus exposure to the proliferative action of Shh. We hypothesize that this may result in the higher frequency of lesions and higher incidence of medulloblastomas observed in the double-mutant genotype (Ptch1+/−/Tis21KO). It appears that, in Ptch1+/−/Tis21KO double-mutant mice, tumorigenesis follows two steps: (1) at 2 weeks, when the EGL is still present (i.e., since the initial postnatal stages), the percentage of double-mutant mice with lesions as well as the number of lesions per cerebellum increases significantly, with respect to Ptch1+/−/Tis21WT; (2) at 12 weeks of age, the percentage of Ptch1+/−/Tis21WT mice with lesions has decreased, while that of double-mutant mice remains high. In addition, the percentage of mice with lesions at 12 weeks of age exactly matches that of mice developing medulloblastoma. Overall, this suggests that the ablation of Tis21 in Patched1 heterozygous mice, by causing from the early postnatal period a lengthened exposure of GCPs in the EGL area to the transforming effect exerted by Shh, may favor their preneoplastic transformation and an additional burst of tumorigenesis after 12–15 weeks of age. This latter would result in a delayed attainment of the plateau in the medulloblastoma incidence curve, which may also explain the longer average latency of medulloblastoma onset in double mice. We cannot exclude, however, that a component of the longer latency may be a reduced diffusion of tumor GCPs and thus a reduced tumor expansion.
Our data indicated that, in Tis21-null mice, no change occurs in the expression of genes of known pathways regulating migration of cerebellar neurons, such as BDNF/TrkB (Borghesani et al., 2002; Kokubo et al., 2009), astrotactin1/astrotactin2 (Wilson et al., 2010), and neuregulin/Erb4 (Rio et al., 1997). However, a genome-wide expression analysis by microarray of GCPs isolated from mice at P7, followed by real-time PCR expression analysis, showed that in double-mutant mice several other genes involved in neuronal cell migration presented a highly decreased mRNA expression, namely, the chemokine Cxcl3, ephrin4A (Wilkinson, 2001), and Jmy, a transcriptional coactivator of p53 that regulates cell motility (Coutts et al., 2009; Zuchero et al., 2009). In particular, we noticed Cxcl3, as it presented a strong decrease of expression in GCPs, and even more strikingly in pGCPs of lesions in double-mutant Patched1 heterozygous/Tis21-null mice, compared with Patched1 heterozygous/Tis21 wild-type mice. Chemokines have been described to regulate cell migration in leukocytes and in several other cell types (de Haas et al., 2007). More recently, a number of chemokines have been shown to be expressed in neurons, where they may play a role in development, synaptic transmission, and neuroinflammation (Bertollini et al., 2006; Charo and Ransohoff, 2006; Ubogu et al., 2006). Cxcl2, a chemokine closely related to Cxcl3 as it shares the same sequence of four conserved cysteines in the N-terminal region (Rossi and Zlotnik, 2000), is expressed in cerebellar granule neurons (Yabe et al., 2004). Interestingly, the genetic ablation of CXCR4, the receptor of the Cxcl12 chemokine, has been shown to cause a defect of migration of cerebellar granule cells (Ma et al., 1998). Our demonstration that Cxcl3 rescues the defect of migration of GCPs of double-mutant mice, without affecting their differentiation or proliferation, points to Cxcl3 as responsible for this defect. Indeed, the reduced migration observed in isolated GCPs silenced for Cxcl3 expression, suggests a cell-autonomous role of Cxcl3 in this process. Furthermore, a causal role in tumorigenesis for Cxcl3 is strongly suggested for the first time by our observation that exogenous Cxcl3 reduces the extension of diffused lesions, which are formed by pGCPs and represent a stage contiguous to the full-blown tumor. As Cxcl3 reduces the area of lesions by inducing the migration of pGCPs outside lesions, we can speculate that this migration-promoting action of Cxcl3 may induce pGCP to exit from the neoplastic program. Moreover, we observe that the transcription of Cxcl3 is directly controlled by Tis21 in GCPs, where, after secretion, Cxcl3 can activate its receptor.
Together, these findings support our hypothesis of a migration defect as the cause of the enhancement of medulloblastoma tumorigenesis following Tis21 ablation. By microarray analysis, we also identified a chemokine, Cxcl12, whose levels increase in the EGL of Tis21-null mutant mice. As Cxcl12 seems to exert an action opposite to Cxcl3, since it promotes the localization of GCPs to the EGL by chemoattraction (Klein et al., 2001), it is also possible that the decrease of Cxcl3 in GCPs prevents their migration outside the EGL in synergy with Cxcl12.
Interestingly, in line with our data, another example of a genetic defect of GCPs migration leading to increase of medulloblastoma frequency has been very recently reported, represented by Patched1 heterozygous mice knock-out for nitric oxide synthase (Haag et al., 2012).
Thus, Tis21, by controlling chemokine levels in GCPs, can play a key role in the control of their migration (Fig. 11, model). Importantly, the expression of Tis21 has been demonstrated to decrease in human medulloblastomas, in particular those of the desmoplastic subtype, and also in medulloblastomas of Patched1 heterozygous mice (Farioli-Vecchioli et al., 2007). Hence, this new Tis21/Patched1 medulloblastoma model displaying defective GCPs migration, as well as altered expression of several genes identified here, may highlight novel features of the physiopathological conditions occurring in human medulloblastoma. While it is possible that other genes identified here may concur to the defective migration of GCPs and increased lesion formation caused by ablation of Tis21, Cxcl3, being able to rescue this complex Tis21-dependent defective phenotype, might be investigated as a new target for medulloblastoma therapy.
Figure 11.

A working model, in conditions of gain of function of Tis21 (A), or loss of function of Tis21 in either Ptch1+/+ (B) or Ptch1+/− context (C). Proliferation, differentiation, and migration out of EGL are referred to GCPs. The gray lines in A refer to data from previous reports (Canzoniere et al., 2004; Farioli-Vecchioli et al., 2007). B, C, In Tis21KO and in Ptch1+/−/Tis21KO mice, the expression of Cxcl3 in GCPs and the migration of GCPs out of EGL decrease. C, In Ptch1+/−/Tis21KO mice, this decrease correlates (dotted line) with the enhancement of medulloblastoma frequency. The defect of migration is rescued in both genotypes by exogenous Cxcl3, which, in Ptch1+/−/Tis21KO mice, reduces the area of cerebellar lesions (C). The dashed line on the left indicates no effect on proliferation relative to controls.
Notes
Supplemental material for this article is available at http://www.inmm.cnr.it/tirone/JNsuppl.zip. The supplemental material includes supplemental Figure S1, Bergmann glia.tif, Bergmann's glia analysis in Tis21 WT and knock-out mice, either in Patched1 heterozygous or wild-type background; supplemental Figure S2, small.tif, which shows that RNAi knockdown of Cxcl3 in cerebellar slices inhibits the ability of GCPs to migrate out of EGL, indicating a cell-autonomous action of Cxcl3; and Farioli-Vecchioli_Table S1_.xls, the original microarray data. Table S1 has not been peer reviewed.
Footnotes
This work was supported by Associazione Italiana Ricerca sul Cancro Grant 9251 (F.T.) and the Italian Ministry of Economy and Finance to Consiglio Nazionale delle Ricerche (Project FaReBio). L.M. was supported by Finanziaria Laziale di Sviluppo. We thank Jane Johnson for the gift of Math1-GFP mice and Richard Butler and Maurizia Caruso for critical reading.
The authors declare no competing financial interests.
References
- Anisowicz A, Messineo M, Lee SW, Sager R. An NF-κB-like transcription factor mediates IL-1/TNF-α induction of gro in human fibroblasts. J Immunol. 1991;147:520–527. [PubMed] [Google Scholar]
- Ben-Arie N, Hassan BA, Bermingham NA, Malicki DM, Armstrong D, Matzuk M, Bellen HJ, Zoghbi HY. Functional conservation of atonal and Math1 in the CNS and PNS. Development. 2000;127:1039–1048. doi: 10.1242/dev.127.5.1039. [DOI] [PubMed] [Google Scholar]
- Bertollini C, Ragozzino D, Gross C, Limatola C, Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology. 2006;51:816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Boiko AD, Porteous S, Razorenova OV, Krivokrysenko VI, Williams BR, Gudkov AV. A systematic search for downstream mediators of tumor suppressor function of p53 reveals a major role of BTG2 in suppression of Ras-induced transformation. Genes Dev. 2006;20:236–252. doi: 10.1101/gad.1372606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghesani PR, Peyrin JM, Klein R, Rubin J, Carter AR, Schwartz PM, Luster A, Corfas G, Segal RA. BDNF stimulates migration of cerebellar granule cells. Development. 2002;129:1435–1442. doi: 10.1242/dev.129.6.1435. [DOI] [PubMed] [Google Scholar]
- Boukhtouche F, Janmaat S, Vodjdani G, Gautheron V, Mallet J, Dusart I, Mariani J. Retinoid-related orphan receptor a controls the early steps of Purkinje cell dendritic differentiation. J Neurosci. 2006;26:1531–1538. doi: 10.1523/JNEUROSCI.4636-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzoniere D, Farioli-Vecchioli S, Conti F, Ciotti MT, Tata AM, Augusti-Tocco G, Mattei E, Lakshmana MK, Krizhanovsky V, Reeves SA, Giovannoni R, Castano F, Servadio A, Ben-Arie N, Tirone F. Dual control of neurogenesis by PC3 through cell cycle inhibition and induction of Math1. J Neurosci. 2004;24:3355–3369. doi: 10.1523/JNEUROSCI.3860-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Choi Y, Borghesani PR, Chan JA, Segal RA. Migration from a mitogenic niche promotes cell-cycle exit. J Neurosci. 2005;25:10437–10445. doi: 10.1523/JNEUROSCI.1559-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AS, Weston L, La Thangue NB. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc Natl Acad Sci U S A. 2009;106:19872–19877. doi: 10.1073/pnas.0906785106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36:137–151. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti C, Astolfi A, Gaboardi GC, Tazzari P, Pession A, Goto K, Martelli AM. TIS21/BTG2/PC3 and cyclin D1 are key determinants of nuclear diacylglycerol kinase-zeta-dependent cell cycle arrest. Cell Signal. 2009;21:801–809. doi: 10.1016/j.cellsig.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Tanori M, Micheli L, Mancuso M, Leonardi L, Saran A, Ciotti MT, Ferretti E, Gulino A, Pazzaglia S, Tirone F. Inhibition of medulloblastoma tumorigenesis by the antiproliferative and prodifferentiative gene PC3. FASEB J. 2007;21:2215–2225. doi: 10.1096/fj.06-7548com. [DOI] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Saraulli D, Costanzi M, Pacioni S, Cinà I, Aceti M, Micheli L, Bacci A, Cestari V, Tirone F. The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol. 2008;6:e246. doi: 10.1371/journal.pbio.0060246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Saraulli D, Costanzi M, Leonardi L, Cinà I, Micheli L, Nutini M, Longone P, Oh SP, Cestari V, Tirone F. Impaired terminal differentiation of hippocampal granule neurons and defective contextual memory in PC3/Tis21 knockout mice. PLoS One. 2009;4:e8339. doi: 10.1371/journal.pone.0008339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Lu X, Man X, Zhou W, Jiang LQ, Knyazev P, Lei L, Huang Q, Ullrich A, Zhang Z, Chen Z. Overexpression of Csk-binding protein contributes to renal cell carcinogenesis. Oncogene. 2009;28:3320–3331. doi: 10.1038/onc.2009.185. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Xu J, Xu C, Mauger G, Barthet G, Robakis NK. Presenilin1/gamma-secretase promotes the EphB2-induced phosphorylation of ephrinB2 by regulating phosphoprotein associated with glycosphingolipid-enriched microdomains/Csk binding protein. FASEB J. 2011;25:3594–3604. doi: 10.1096/fj.11-187856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, Patay Z, Scoggins M, Ogg R, Pei Y, Yang ZJ, Brun S, Lee Y, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, Corrente G, Covone F, Micheli L, D'Agnano I, Starace G, Caruso M, Tirone F. Arrest of G1-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol Cell Biol. 2000;20:1797–1815. doi: 10.1128/mcb.20.5.1797-1815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag D, Zipper P, Westrich V, Karra D, Pfleger K, Toedt G, Blond F, Delhomme N, Hahn M, Reifenberger J, Reifenberger G, Lichter P. Nos2 inactivation promotes the development of medulloblastoma in Ptch1+/− mice by deregulation of Gap43-dependent granule cell precursor migration. PLoS Genet. 2012;8:e1002572. doi: 10.1371/journal.pgen.1002572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Roussel MF. Development and cancer of the cerebellum. Trends Neurosci. 2011;34:134–142. doi: 10.1016/j.tins.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell. 2001;107:727–738. doi: 10.1016/s0092-8674(01)00598-0. [DOI] [PubMed] [Google Scholar]
- Kadin ME, Rubinstein LJ, Nelson JS. Neonatal cerebellar medulloblastoma originating from the fetal external granular layer. J Neuropathol Exp Neurol. 1970;29:583–600. doi: 10.1097/00005072-197010000-00005. [DOI] [PubMed] [Google Scholar]
- Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, Wang F, Wechsler-Reya RJ. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Nelson AL, Algon SA, Graves O, Sturla LM, Goumnerova LC, Rowitch DH, Segal RA, Pomeroy SL. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol. 2003;263:50–66. doi: 10.1016/s0012-1606(03)00434-2. [DOI] [PubMed] [Google Scholar]
- Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, Luster AD. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–1981. doi: 10.1242/dev.128.11.1971. [DOI] [PubMed] [Google Scholar]
- Kokubo M, Nishio M, Ribar TJ, Anderson KA, West AE, Means AR. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J Neurosci. 2009;29:8901–8913. doi: 10.1523/JNEUROSCI.0040-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, Curran T, Gilbertson RJ, McKinnon PJ. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428–5437. [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell. 2001;105:69–79. doi: 10.1016/s0092-8674(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, Johnson JE. Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns. 2003;3:389–395. doi: 10.1016/s1567-133x(03)00089-9. [DOI] [PubMed] [Google Scholar]
- Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A. 1998;95:9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med. 2005;11:17–22. doi: 10.1016/j.molmed.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- O'Neill LP, Turner BM. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 1995;14:3946–3957. doi: 10.1002/j.1460-2075.1995.tb00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Lee YJ, Lee HJ, Seki T, Hong KH, Park J, Beppu H, Lim IK, Yoon JW, Li E, Kim SJ, Oh SP. B-cell translocation gene 2 (Btg2) regulates vertebral patterning by modulating bone morphogenetic protein/smad signaling. Mol Cell Biol. 2004;24:10256–10262. doi: 10.1128/MCB.24.23.10256-10262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeri D, Marcucci A, Rizzo G, Billi M, Panigada M, Leonardi L, Tirone F, Grignani F. Btg2 enhances retinoic acid-induced differentiation by modulating histone H4 methylation and acetylation. Mol Cell Biol. 2006;26:5023–5032. doi: 10.1128/MCB.01360-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sörensen N, Berthold F, Henk B, Schmandt N, Wolf HK, von Deimling A, Wainwright B, Chenevix-Trench G, Wiestler OD, Wicking C. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085–2088. [PubMed] [Google Scholar]
- Polleux F, Giger RJ, Ginty DD, Kolodkin AL, Ghosh A. Patterning of cortical efferent projections by semaphorine-neuropilin. Science. 1998;282:1904–1906. doi: 10.1126/science.282.5395.1904. [DOI] [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Cang Y, Goff SP. Abl family tyrosine kinases are essential for basement membrane integrity and cortical lamination in the cerebellum. J Neurosci. 2010;30:14430–14439. doi: 10.1523/JNEUROSCI.2861-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–845. [PubMed] [Google Scholar]
- Rio C, Rieff HI, Qi P, Khurana TS, Corfas G. Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron. 1997;19:39–50. doi: 10.1016/s0896-6273(00)80346-3. [DOI] [PubMed] [Google Scholar]
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E, Norris MD, Haber M, Ellis T, Wainwright B, Marshall GM. Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene. 2009;28:1605–1615. doi: 10.1038/onc.2009.3. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27:48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Weyer A, Schilling K. Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res. 2003;73:400–409. doi: 10.1002/jnr.10655. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nat Rev Neurosci. 2001;2:155–164. doi: 10.1038/35058515. [DOI] [PubMed] [Google Scholar]
- Wilson PM, Fryer RH, Fang Y, Hatten ME. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci. 2010;30:8529–8540. doi: 10.1523/JNEUROSCI.0032-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57:2581–2585. [PubMed] [Google Scholar]
- Yabe T, Herbert JT, Takanohashi A, Schwartz JP. Treatment of cerebellar granule cell neurons with the neurotrophic factor pigment epithelium-derived factor in vitro enhances expression of other neurotrophic factors as well as cytokines and chemokines. J Neurosci Res. 2004;77:642–652. doi: 10.1002/jnr.20196. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schüller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhokhov SS, Desfeux A, Aubert N, Falluel-Morel A, Fournier A, Laudenbach V, Vaudry H, Gonzalez BJ. Bax siRNA promotes survival of cultured and allografted granule cell precursors through blockade of caspase-3 cleavage. Cell Death Differ. 2008;15:1042–1053. doi: 10.1038/cdd.2008.29. [DOI] [PubMed] [Google Scholar]
- Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, Mullins RD. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009;11:451–459. doi: 10.1038/ncb1852. [DOI] [PMC free article] [PubMed] [Google Scholar]