

Abstract
Pilocarpine injection induces epileptic seizures in rodents, an experimental paradigm extensively used to model temporal lobe epilepsy in humans. It includes conspicuous neuronal death in the forebrain and previous work has demonstrated an involvement of the neurotrophin receptor p75NTR in this process. Following the identification of Galectin-1 (Gal-1) as a downstream effector of p75NTR, we examine here the role of this endogenous lectin in pilocarpine-induced cell death in adult mice. We found that most somatostatin-positive neurons also express Gal-1 and that in mice lacking the corresponding gene Lgals1, pilocarpine-induced neuronal death was essentially abolished in the forebrain. We also found that the related lectin Galectin-3 (Gal-3) was strongly upregulated by pilocarpine in microglial cells. This upregulation was absent in Lgals1 mutants and our results with Lgals3-null animals show that Gal-3 is not required for neuronal death in the hippocampus. These findings provide new insights into the roles and regulation of endogenous lectins in the adult CNS and a surprisingly selective proapoptotic role of Gal-1 for a subpopulation of GABAergic interneurons.
Introduction
Neuronal death is a significant feature of temporal epilepsy in humans, a frequent condition that can be usefully modeled in rodents by injecting the muscarinic receptor agonist pilocarpine (Curia et al., 2008). Like in temporal epilepsy, pilocarpine-induced seizures cause cell death and decrease the number of parvalbumin-positive and somatostatin-positive interneurons in the dentate gyrus of rodents, thus explaining reduced inhibition in granule cells (Kobayashi and Buckmaster, 2003). With regard to mechanistic insights, it has been reported that in the hippocampus, the entorhinal, and the piriform cortex, pilocarpine induces the overexpression of the neurotrophin receptor p75NTR (Roux et al., 1999), a molecule associated with cell death in the nervous system, both during development and in a variety of CNS pathologies in the adult (Dechant and Barde, 2002). Furthermore, cell death was shown to be significantly reduced in animals lacking p75NTR (Troy et al., 2002) by mechanisms involving the p75NTR interactor neurotrophin receptor-interacting factor (NRIF) (Volosin et al., 2008). Using neurons generated from engineered embryonic stem cells (Bibel et al., 2004), we previously showed that Galectin-1 (Gal-1) is a downstream effector of p75NTR, which triggers the disintegration of axons and cell death (Plachta et al., 2007). Gal-1 is a 14 kDa protein belonging to a multimember lectin family with affinity for β-galactoside moieties present in a wide range of cell surface glycoproteins (Barondes et al., 1994). Its reported role in the nervous system is complex and includes both positive as well as negative effects on neurogenesis, cell survival, and axonal elongation (for a recent review, see Sakaguchi and Okano, 2012). Here we assess the role of Gal-1 in pilocarpine-induced seizures using mice lacking Lgals1. We also examine the potential involvement of Galectin-3 (Gal-3) following the realization that pilocarpine also increases the levels of this lectin in the brain.
Materials and Methods
Pilocarpine-induced status epilepticus and brain sections.
All animal experiments were approved by the local veterinarian authorities regulating animal experimentation in Basel. Three-month-old adult mice of either sex were injected with methyl scopolamine (1 mg/kg, i.p.) and 30 min after with pilocarpine hydrochloride (320 mg/kg, i.p.). The mice were wild-type C57BL/6J, Lgals1−/− on a C57BL/6J background, or Lgals3−/− on a C57BL/6J background. Animal behavior was recorded for the next 2 h (Morrison et al., 1996), and then animals were injected with diazepam (10 mg/kg) to stop the seizures. Control mice received the same treatment except that they were injected with saline (0.9% NaCl in H2O, i.p.) instead of pilocarpine. Animals were injected subcutaneously with Hartman's solution (130 mm NaCl, 4 mm KCl, 3 mm CaCl, 28 mm lactate) during the recovery period. Only mice reaching a crisis level of 5–6 (Morrison et al., 1996) were included in the analysis. Fluoro-JadeB (FJB)-positive neurons were detected in all wild-type pilocarpine-injected mice. Twenty-four or 72 h following pilocarpine injection, animals were deeply anesthetized (ketamine 100 mg/kg, Rompun 0.2%, i.p.) and transcardially perfused first with ice-cold PBS and followed by 4% PFA in PBS. Brains were removed and postfixed for 1–2 h in the same fixing solution, cryoprotected with 30% sucrose, embedded in O.C.T. (Optimal Cutting Temperature compound) and dry-ice frozen. Serial coronal sections (30 μm) were collected using a Leica 3050S cryostat mounted on laminin-coated microscope slides and stored at −70°C.
FJB staining.
Consecutive whole-brain coronal sections (30 μm; ∼−2.18 to −2.92 mm posterior to bregma) were processed for FJB staining (Millipore Bioscience Research Reagents) as described previously (Schmued and Hopkins, 2000). Brain sections were visualized under axioplan2 Zeiss epi-fluorescence microscope and posterior analyzed using Photoshop software.
TUNEL.
TUNEL (terminal deoxynucleotidyl transferase-mediated UTP nick end labeling) was performed using the In Situ Cell Death Detection Kit, TMR red (Roche), following provider instructions. Consecutive whole-brain coronal sections (30 μm; ∼−1.82 to −2.18 mm posterior to bregma) were labeled and visualized under an axioplan2 Zeiss epi-fluorescence microscope.
Immunohistochemistry.
Serial coronal sections were processed for immunohistochemistry. The sections were washed in PBS, permeabilized by incubating in PBS containing 0.2% Triton X-100, and blocked with 10% inactivated horse serum, except for p75NTR staining examined under nonpermeabilizing condition by omitting Triton X-100. The slices were then incubated overnight at 4°C with primary antibodies. We used goat anti-Gal-1 at 1:500 (R&D Systems), goat anti-Gal-3 at 1:1000 (R&D Systems), mouse anti-NeuN at 1:200 (Millipore), rabbit anti-p75NTR at 1:500 (Millipore Bioscience Research Reagents), mouse anti-active anti-caspase-3 at 1:100 (Millipore), mouse anti-GFAP at 1:1000 (Sigma-Aldrich), rabbit anti-parvalbumin at 1:200 (Swant), rabbit anti-somatostatin at 1:200 (Immunostar), and rat anti-CD11b (Abcam). After washes, secondary fluorochrome-conjugated antibodies (1:500; Jackson Laboratory) were applied for 1 h at room temperature. Sections were mounted and images collected on either a Zeiss axioplan2 epi-fluorescence microscope or on a Leica SP1 laser scanning confocal microscope and processed and analyzed using Photoshop software.
Data analysis.
All results presented here correspond to the mean of the quantifications established for each experimental condition (3–6 sections per animal and 3 or more animals for each time point). The variation between animals is indicated as SEM and analyzed by unpaired Student's t test. A 95% confidence interval was used and values of p < 0.05 were considered as statistically significant. Means were considered significantly different according to the p value indicated with the following symbols: *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. Calculations and statistical analysis were done with Excel and GraphPad Prism 4.0.
Results
Pilocarpine injection in Lgals1-mutant mice
Mice lacking Lgals1, the gene encoding Gal-1, do not show any overt phenotype (Poirier and Robertson, 1993), and we found no difference between wild-type and mutant animals in the inducibility or severity of seizures triggered by pilocarpine systemic injection (data not shown). Neuronal death in the forebrain was quantified with FJB staining at two different time points following pilocarpine injection. Following saline injections in wild-type animals, we found 1.0 ± 1.0 and 1.4 ± 1.8 FJB-positive neurons after 24 h in the hippocampus and in the parietal and pirifom cortex, respectively, and none after 72 h (Fig. 1A,B). Twenty-four hours following pilocarpine injection, there were 1035 ± 17 and 3228 ± 66 FJB-positive neurons in the hippocampus and the cortex, respectively. After 72 h, the counts were 510 ± 82 and 2977 ± 127 in the same structures, suggesting that many neurons prone to death following pilocarpine injection had already died at early time points (Fig. 1A,B). By contrast, remarkably few FJB-positive neurons were found in the forebrain of Lgals1 mutants after pilocarpine injection (24 ± 6 and 8 ± 2 FJB-positive neurons after 24 h; 0 and 1 ± 1 FJB-positive neurons after 72 h in the hippocampus and the cortex, respectively) (Fig. 1A,B). As the nature of FJB staining is still somewhat unclear with regard to what is currently known about the mechanisms of cell death, we also quantified apoptotic cell death in the cerebral cortex using the reaction involving TUNEL and active caspase-3 staining (Fig. 2). In wild-type mice, pilocarpine injection triggers apoptotic death after 24 h (47.6 ± 4.4 TUNEL-positive cells per mm2 vs 1.5 ± 0.5 for saline-injected animals). As with FJB staining, the number of TUNEL-positive cells detected in Lgals1-mutant mice injected with pilocarpine was only marginally increased (4.4 ± 1) and not significantly different from saline-injected wild-type animals. After pilocarpine injection, the proportion of caspase-3-positive and NeuN-positive neurons was also much lower in Lgals1-mutant mice compared with wild-type animals (5 ± 1% vs 32.6 ± 1.9%, respectively), and not significantly different from wild-type control injected with saline (5.5 ± 0.6%). These two alternative methods confirm the results obtained with FJB staining and indicate that, in absence of Gal-1, pilocarpine injection fails to trigger apoptosis of forebrain neurons. Because our previous work indicated that Gal-1 is upregulated by increased levels of p75NTR (Plachta et al., 2007), we then wondered whether the decreased number of Gal-1-expressing neurons following pilocarpine injection is the result of their elimination by programmed cell death. Twenty-four hours after pilocarpine-induced seizures, we found that caspase-3 was activated in neurons also expressing Gal-1 (Fig. 2). In control animals, no caspase-3-positive neurons could be detected that were also Gal-1-positive. These results suggest that neurons expressing Gal-1, compared with Gal-1-negative neurons, may be more prone to degeneration caused by pilocarpine injections.
Figure 1.
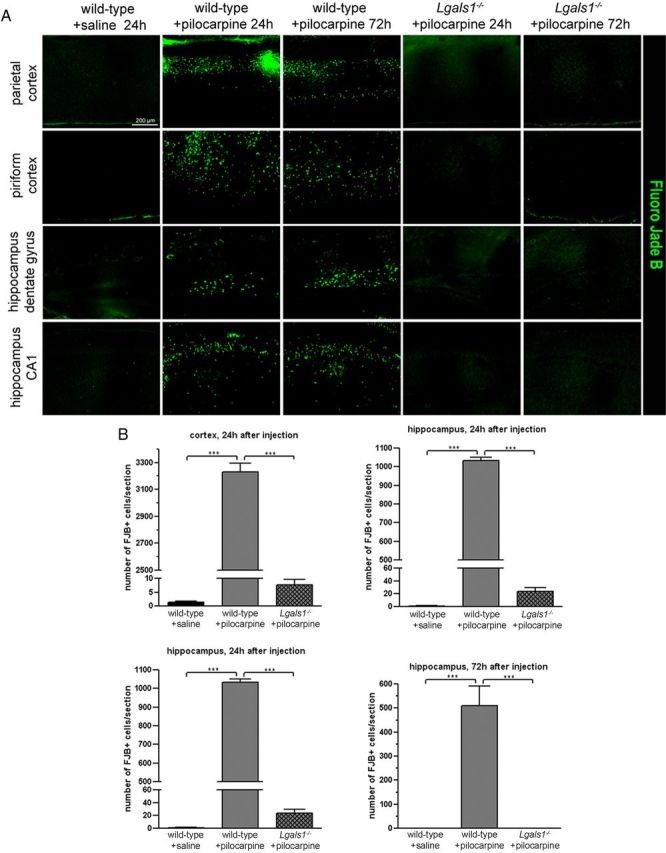
Neuronal death induced by epileptic seizures is blocked in Lgals1 mutants. A, FJB staining showing degenerating neurons in different areas of the cerebral cortex and hippocampus, in wild-type mice injected with saline solution and in wild-type or Lgals1-mutant animals injected with pilocarpine. B, Quantification of the total number of FJB-positive neurons in the cortex and hippocampus of injected animals, per 30 μm section. Scale bar, 200 μm.
Figure 2.
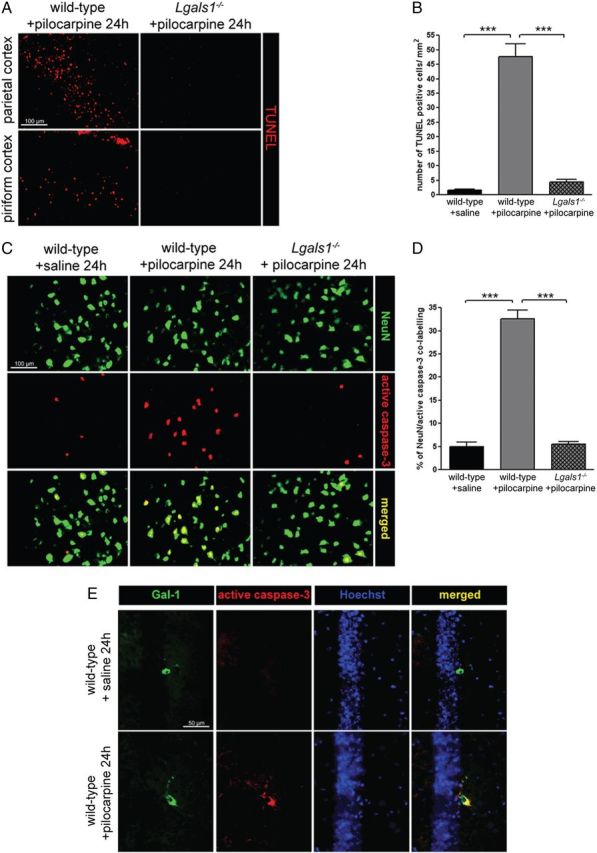
Lack of programmed cell death in Lgals1-mutant mice after pilocarpine-induced seizures. A, TUNEL staining in the parietal cortex, comparing wild-type and Lgals1-mutant mice 24 h after pilocarpine injection. Scale bar, 100 μm. B, Quantification of TUNEL-positive cells in the parietal cortex. C, Coimmunostaining for NeuN and active caspase-3 in the parietal cortex. Scale bar, 100 μm. D, Quantification of the NeuN-positive neuron proportion also positive for active caspase-3 staining. E, Colocalization of active caspase-3 and Gal-1 in the CA1 of the hippocampus following pilocarpine-induced seizures. Scale bar, 50 μm.
Gal-1 expression in the forebrain and pilocarpine injection
In wild-type, untreated animals, scattered cells were found to express Gal-1 mostly in the cerebral cortex and the hippocampus (Fig. 3A) and costaining with NeuN antibodies revealed that essentially all Gal-1-positive cells were neurons (Fig. 3B). This neuron-specific expression of Gal-1 closely matched the pattern of neuronal degeneration in pilocarpine-injected animals (Fig. 3C). Remarkably, the number of neurons labeled with Gal-1 antibodies was markedly reduced, both in the cerebral cortex and in the hippocampus already 24 h after pilocarpine injection (Fig. 3D,E). This number was not further reduced 72 h after injection. In the cerebral cortex, we counted 256 ± 26 Gal-1-positive neurons 24 h after the crisis and 291 ± 26 after 72 h versus 762 ± 38 in saline-injected controls. In the hippocampus, we counted 66 ± 6 (24 h) and 78 ± 10 (72 h) Gal-1-positive neurons in pilocarpine-treated mice versus 272 ± 23 in the saline-injected animals (Fig. 3E). In addition, we observed that many more cells expressed p75NTR after pilocarpine injection compared with control animals (Fig. 3F), in line with previous findings (Roux et al., 1999). As pilocarpine injection in rats has been shown to cause the death of interneurons in the dentate gyrus (Kobayashi and Buckmaster, 2003), we examined the possibility that Gal-1 may colocalize with somatostatin and parvalbumin, two well known markers of subpopulations of interneurons. We found that, in control animals injected with saline, all Gal-1-positive neurons also expressed somatostatin. We found that, conversely, of the total number of somatostatin neurons (N = 487), 95.27 ± 3.8% also expressed Gal-1, while of the total number of parvalbumin cells (N = 628), only 15.6 ± 3.4% coexpressed Gal-1. Following pilocarpine injections, both somatostatin-positive and Gal-1-positive neurons were essentially eliminated after 24 h (saline, N = 42 neurons; pilocarpine, N = 5 neurons; 6 consecutive sections) (Fig. 4A). By contrast, parvalbumin-positive neurons could still be readily detected (saline, N = 115 neurons; pilocarpine, N = 110 neurons; 6 consecutive sections) (Fig. 4B).
Figure 3.
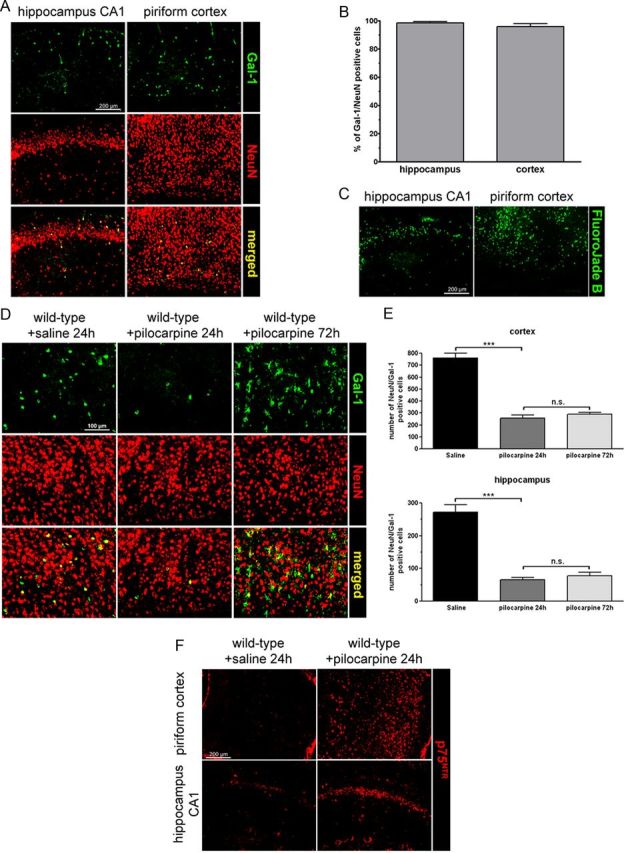
The number of Gal-1-expressing neurons decreases after epileptic seizures. A, Gal-1 immunoreactive cells are almost exclusively neurons. Coimmunostaining in the hippocampus and the cerebral cortex with antibodies against Gal-1 and NeuN. B, The proportion of Gal-1-positive cells identified as neurons is plotted as the percentage of Gal-1-immunoreactive cells also positive for NeuN immunostaining. C, The pattern of neuronal degeneration triggered by pilocarpine injection parallels the pattern of Gal-1 expression in basal conditions. D, In the parietal cortex, the number of neurons expressing Gal-1 decreases 24 and 72 h after pilocarpine-induced seizures. E, Quantification of the total number of Gal-1/NeuN double-positive neurons in the piriform cortex and hippocampus, per 30 μm section. F, p75NTR immunoreactivity in the hippocampus and piriform cortex 24 h after pilocarpine-induced seizures. Scale bars: A, C, F, 200 μm; D, 100 μm. n.s., not statistically significant.
Figure 4.
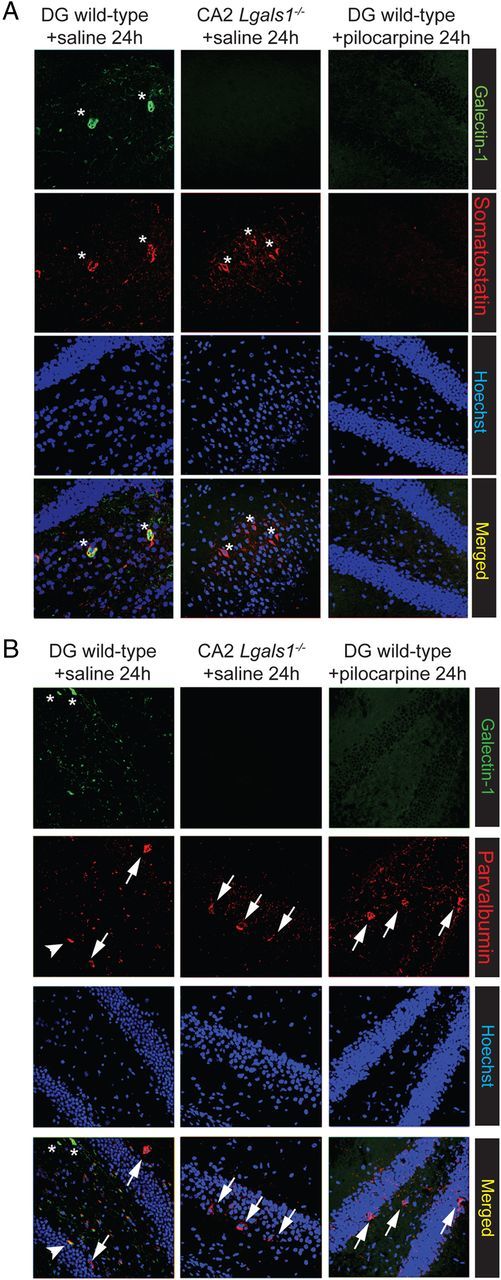
Gal-1-positive neurons are also somatostatin-positive. A, Colocalization of Gal-1-positive and somatostatin-positive neurons (asterisks) in the hippocampus of wild-type mice. Note that somatostatin neurons are still present in the hippocampus of Lgals1−/− mice. B, Most (arrows), but not all (arrowhead), paravalbumin-positive neurons do not express Gal-1 (asterisks).
While very few Gal-1-positive cells are also positive for the astrocytic marker GFAP under control conditions, this proportion increased markedly with time following pilocarpine injection and seizures (Fig. 5A,B). In control animals, only 2.2 and 1.1% Gal-1-positive cells also expressed GFAP in the hippocampus and the cortex, respectively, while 18.3 and 6.3% were double-labeled 24 h after pilocarpine treatment, and as many 78.8 and 74.2% at 72 h. These results are in line with a previous report using injection of kainic acid to trigger neuronal degeneration (Kajitani et al., 2009).
Figure 5.
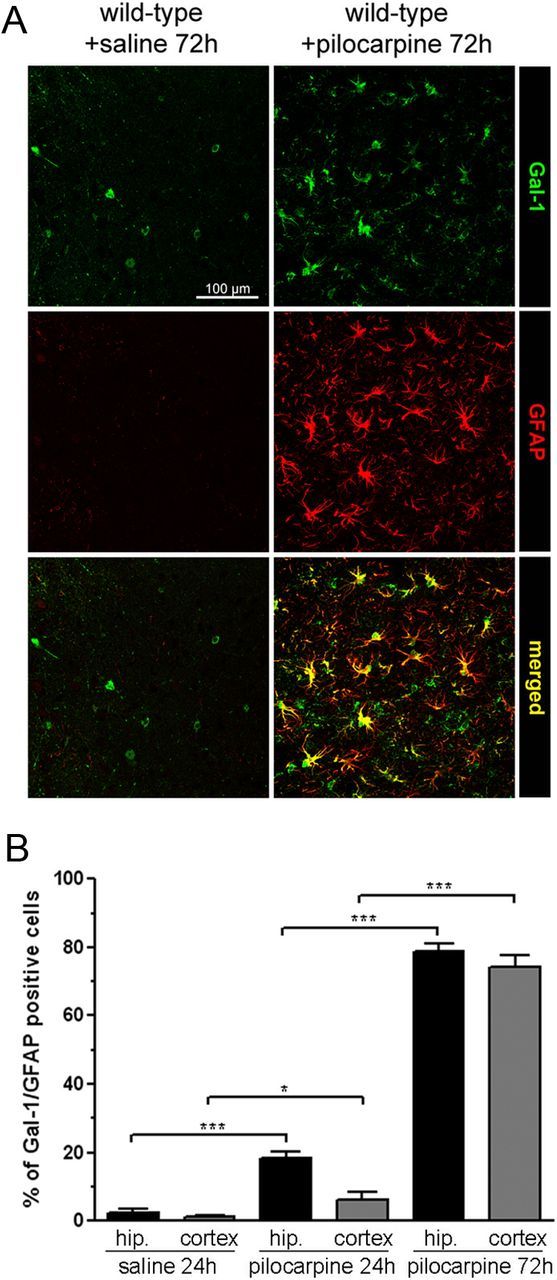
Gal-1 expression increases in astrocytes 72 h after epileptic seizures. A, Coimmunostaining for Gal-1 and GFAP in the piriform cortex. B, Quantification of the number of Gal-1-positive cells also positive for GFAP in the cerebral cortex and the hippocampus. Scale bar, 100 μm.
Glial cells reactivity and p75NTR expression in wild-type and Lgals1-mutant brains after pilocarpine injection
Neuronal degeneration is well known to be accompanied by a strong glial reaction (Barres, 2008) and, as expected, we observed a large increase in the number of GFAP-positive astrocytes and of CD11b-positive microglial cells in the forebrain of wild-type animals treated with pilocarpine (Fig. 6A,B). By contrast, no signs of glial reactivity were observed at 24 or 72 h following pilocarpine injection in Lgals1 animals (Fig. 6A,B). Despite the absence of neuronal death and glial reactivity, we found that p75NTR was still overexpressed in these Lgals1-mutant animals after pilocarpine injection (Fig. 7A). Conversely, all cells expressing Gal-1 24 h after pilocarpine injections were also positive for p75NTR, both in the dentate gyrus and in CA2 (Fig. 7B). Together, these results indicate that p75NTR overexpression per se is not sufficient to cause cell death following pilocarpine injection.
Figure 6.

Glial reactivity is blocked in Lgals1-mutant mice after pilocarpine injection. A, Astrocyte-specific GFAP immunostaining in the cerebral cortex and hippocampus, comparing wild-type and Lgals1-mutant mice, 24 and 72 h after injection. B, Microglia-specific CD11b immunostaining in the cerebral cortex and hippocampus in wild-type versus Lgals1-mutant mice 24 h after injection. Scale bar, 200 μm.
Figure 7.
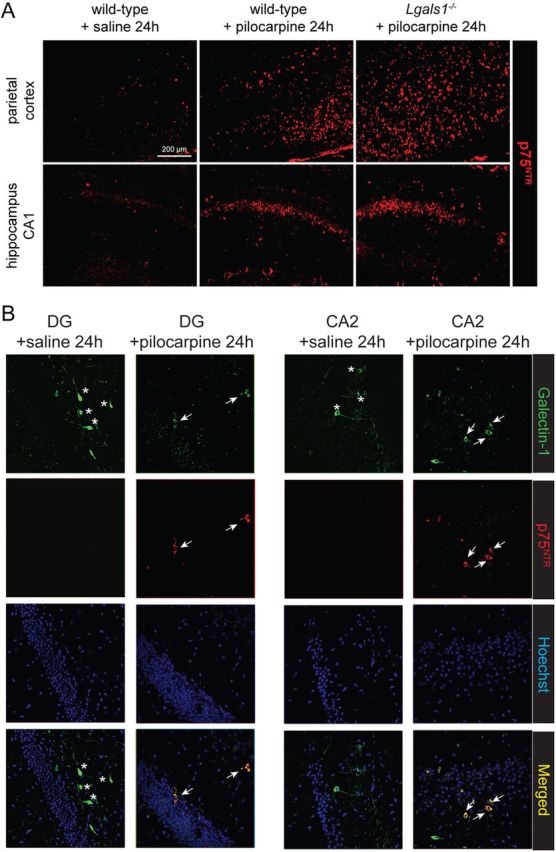
p75NTR is overexpressed in Lgals1 mutant mice after pilocarpine injection. A, p75NTR immunostaining in the parietal cortex and hippocampus, comparing wild-type and Lgals1-mutant mice, 24 h after injection. Scale bar, 200 μm. B, p75NTR is expressed in the Gal-1-positive neurons (asterisks) 24 h after pilocarpine injections in the hippocampus (arrows).
Expression and role of Galectin-3 in the forebrain
Gal-3 is structurally related to Gal-1 and is involved in T-cell apoptosis (Hsu et al., 2009). Also, its role has been recently investigated in the context of cerebral ischemia (Doverhag et al., 2010; Lalancette-Hébert et al., 2012). We found Gal-3 to be strongly upregulated as early as 24 h after pilocarpine-induced seizures (Fig. 8), with an expression pattern closely matching neuronal degeneration revealed by FJB staining in the hippocampus and cortex (Fig. 9). Using the microglial marker CD11b, Gal-3 was found to be exclusively expressed by microglial cells, and not by astrocytes or neurons (Fig. 8). These results are in line with those reported in a recent study indicating that microglial cells express Gal-3 in a mouse model of cerebral ischemia (Lalancette-Hébert et al., 2012). In animals lacking Gal-1, we found that Gal-3 was not upregulated in these or any other cells in the forebrain (Fig. 8), indicating that Gal-1 is required both for the induction of neuronal death and the upregulation of Gal-3 by microglial cells following pilocarpine injection. We next directly examined the possibility that Gal-3 may also be required, like Gal-1, for cell death following pilocarpine-induced seizures. Using mice lacking Lgals3, we found a small, but significant reduction of cell death in the cerebral cortex, but not in the hippocampus (Fig. 9). It thus seems that despite upregulation in microglial cells following pilocarpine injection, Gal-3 plays a marginal role compared with Gal-1 as a cell death effector. Unlike in the model of cerebral ischemia (Lalancette-Hébert et al., 2012), our results do not suggest a protective role for Gal-3 with regard to the prevention of cell death.
Figure 8.

Gal-3 is expressed in microglial cells after epileptic seizures, but not in Lgals1-mutant mice. Coimmunostaining for Gal-3 and CD11b in the parietal cortex. Scale bar, 100 μm.
Figure 9.
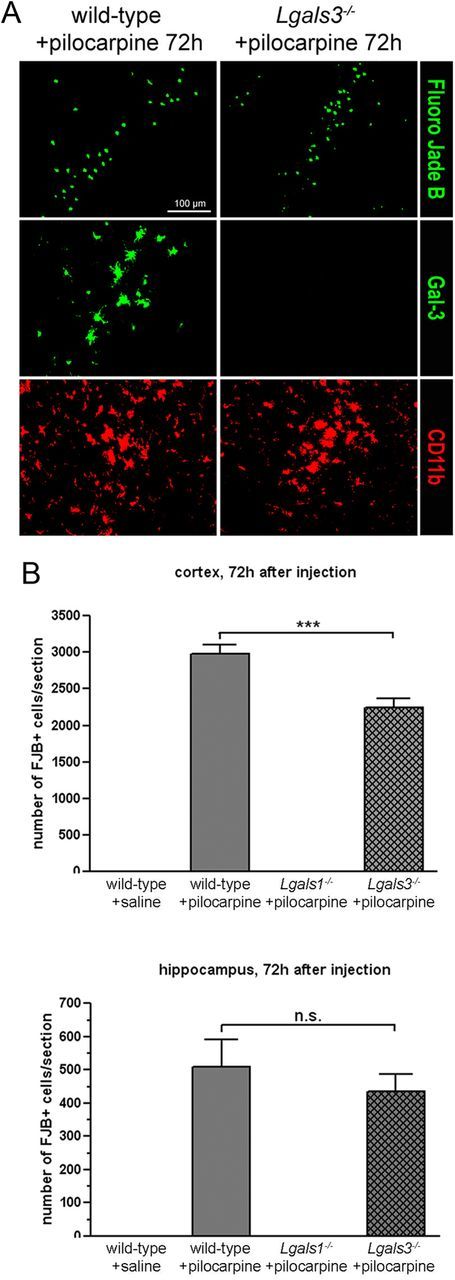
Pilocarpine-induced seizures trigger neuronal degeneration in Lgals3-mutant mice. A, Coimmunostaining for Gal-3 and CD11b in the parietal cortex 72 h after pilocarpine injection. B, Quantification of the total FJB-positive neurons in the cerebral cortex and hippocampus of injected animals. Scale bar, 100 μm. n.s., not statistically significant.
Discussion
Our results reveal that Gal-1 plays an essential role in the neurodegeneration process triggered in the forebrain following systemic pilocarpine administration. Gal-1 is expressed predominantly by somatostatin-positive interneurons and is required for their death during seizures. By contrast, cell death is only marginally reduced in animals lacking Gal-3, and only in the cerebral cortex, not in the hippocampus, despite a strong Gal-3 expression throughout the forebrain by microglial cells following pilocarpine injection.
Galectin-1 and cell death
Gal-1 is known to induce the cell death in the immune system and to regulate T-cell homoeostasis during the development in the thymus as well as after their activation in the periphery (Perillo et al., 1995; Rabinovich et al., 2007). By contrast, still very little is known about the role of galectins in the mouse nervous system, which seems to develop without gross alterations in the absence of both Gal-1 and Gal-3. Also, mice lacking both genes are fertile and do not display any overt phenotype. Upon closer examination, animals constitutively lacking Gal-1 show discrete alterations in the projections of olfactory axons (Puche et al., 1996) and in subsets of sensory afferents (McGraw et al., 2005). Previous reports also noted that Gal-1 expression by GFAP-positive cells promotes basal and kainate-induced proliferation of neuronal progenitors in the hippocampus (Sakaguchi et al., 2007; Sakaguchi and Okano, 2012) and recovery after stroke (Ishibashi et al., 2007). By contrast, our previous results indicated that Gal-1 promotes axonal degeneration caused by p75NTR overexpression and that it participates in excitotoxic cell death following ibotenic acid injections in rats, a treatment that also includes the upregulation of p75NTR (Plachta et al., 2007). It is not easy at this point to reconcile these somewhat disparate findings. They may in part result from the fact that Gal-1 interacts with a number of membrane glycoproteins as well as with molecules of the extracellular matrix (Camby et al., 2006). Also, Gal-1 is only expressed in the forebrain by a small population of neurons largely corresponding to somatostatin-positive interneurons, a subpopulation that is essentially eliminated during the course of seizures triggered by pilocarpine injections. This is functionally interesting as the loss of inhibitory neurons has been previously suggested to be part of the physiopathology of seizure in the rodent dentate gyrus (Kobayashi and Buckmaster, 2003).
The mechanisms by which Gal-1 kills neurons expressing this lectin remain unclear and, given the number of molecules expressing β-galactoside moieties, a deeper understanding of these mechanisms may not be straightforward. In fact, it is not even clear how Gal-1 reaches the cell surface as it does not have the characteristics of a secretory protein (Vasta, 2012). While it is conceivable that Gal-1 may act intracellularly, our previous in vitro observations indicate that it is accessible to extracellular reagents as the killing activity of Gal-1 can be prevented by the addition of antibodies or of excess galactose to cultured cells (Plachta et al., 2007). Our results also indicate that Gal-1 expression is not limited to neurons. Following pilocarpine injection, most astrocytes become strongly immunoreactive for Gal-1, though this is a comparatively late process, reaching completion ∼72 h after seizure initiation, significantly later than the onset of neuronal death. Also, this late increase does not cause the death of reactive astrocytes. In fact, the number of GFAP-positive cells increases markedly at 72 h (Figs. 5, 6A) and, as observed in many experimental models of brain damage (Barres, 2008), this astrocytic reaction may be a consequence, rather than a cause, of neuronal death and it is not observed in Lgals1 mutants after pilocarpine injection (Fig. 6).
Pilocarpine-induced seizure, Gal-3, and p75NTR
Gal-3 is closely related to Gal-1 and has also been shown to trigger the death of activated T-cells, albeit by the activation of cell surface receptors that differ from those used by Gal-1 (Stillman et al., 2006). We found that while there is very little Gal-3 expression in the CNS under control conditions, microglial cells become strongly positive for Gal-3 after pilocarpine. However, this is a process that is strictly Gal-1 dependent (Fig. 8). The conclusion that Gal-3 is expressed by microglial cells after pilocarpine injection is in agreement with results recently published by Lalancette-Hébert and colleagues (2012). Following middle cerebral artery occlusion, Gal-3 was found to be markedly upregulated in Iba-1-positive cells used as microglia markers. In Lgals3 mutants, this lectin was also shown to be required for microglia activation and proliferation after ischemia. However, in this paradigm, the lack of Gal-3 also correlated with increased cell death, which was increased by ∼2-fold after temporary artery occlusion (Lalancette-Hébert et al., 2012). This is very different from what we observed in the pilocarpine-induced seizure paradigm: there is a modest decrease of cell death in the cortex of the Lgals3 mutant with no change in the hippocampus (Fig. 9). Apparently then, the role of Gal-3 in CNS pathologies also depends on the type of lesion inflicted with ischemia likely to generate much higher levels of cell death in the brain then localized seizure episodes of short duration. While the functional role of Gal-3 expressed by microglial cells after seizures remains unclear, it could be related to the phagocytosis of dead neurons or cellular debris resulting from neuronal death. This hypothesis is also supported by the pattern of Gal-3 expression and of neuronal degeneration (Fig. 9), and the observation that some Gal-3-positive microglial cells were seen to engulf dying neurons (data not shown). We note that in the lesioned peripheral nervous system, Gal-3, then known under the name of the MAC-2 antigen, was suggested early on to facilitate the removal of debris of injured axons and thus to promote axonal regeneration (Rotshenker, 2011).
In our experimental seizure paradigm, Gal-1 appears to be a more accurate predictor of cell death than p75NTR as many more cells express this pan-neurotrophin receptor and survive pilocarpine injection than is the case for neurons expressing both p75NTR and Gal-1. Also, pilocarpine-induced neuronal death is essentially abolished in the Lgals1 mutant while a proportion of neurons still die in mutants lacking p75NTR (Troy et al., 2002). In Lgals1 mutants injected with pilocarpine, p75NTR overexpression does not seem to be markedly reduced, suggesting that Gal-1 is not required for the expression of this receptor and that overexpression of p75NTR alone is not a faithful predictor of neuronal death.
In conclusion, our results demonstrate that Gal-1 plays a key role in the death of interneurons in the pilocarpine seizure-inducing model, bringing new insights into the understanding of neurodegeneration caused by excessive neuronal activity. Interfering with the activity of Gal-1 in temporal epilepsy may then represent a new possibility to reduce the loss of neurons in humans. This is of special interest given the difficulties encountered using glutamate receptor antagonist (for review, see Wang and Qin, 2010).
Footnotes
V.B. was supported by European Molecular Biology Organization and Human Frontier Science Program fellowships.
The authors declare no competing financial interests.
References
- Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins. Structure and function of a large family of animal lectins. J Biol Chem. 1994;269:20807–20810. [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Bibel M, Richter J, Schrenk K, Tucker KL, Staiger V, Korte M, Goetz M, Barde YA. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat Neurosci. 2004;7:1003–1009. doi: 10.1038/nn1301. [DOI] [PubMed] [Google Scholar]
- Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–157R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal epilepsy. J Neurosci Meth. 2008;172:143–157. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Doverhag C, Hedtjärn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Sävman K. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2010;38:36–46. doi: 10.1016/j.nbd.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Hsu DK, Chen HY, Liu FT. Galectin-3 regulates T-cell functions. Immunol Rev. 2009;230:114–127. doi: 10.1111/j.1600-065X.2009.00798.x. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Kuroiwa T, Sakaguchi M, Sun L, Kadoya T, Okano H, Mizusawa H. Galectin-1 regulates neurogenesis in the subventricular zone and promotes functional recovery after stroke. Exp Neurol. 2007;207:302–313. doi: 10.1016/j.expneurol.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Kajitani K, Nomaru H, Ifuku M, Yutsudo N, Dan Y, Miura T, Tsuchimoto D, Sakumi K, Kadoya T, Horie H, Poirier F, Noda M, Nakabeppu Y. Galectin-1 promotes basal and kainate-induced proliferation of neural progenitors in the dentate gyrus of adult mouse hippocampus. Cell Death Differ. 2009;16:417–427. doi: 10.1038/cdd.2008.162. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw J, Gaudet AD, Oschipok LW, Steeves JD, Poirier F, Tetzlaff W, Ramer MS. Altered primary afferent anatomy and reduced thermal sensitivity in mice lacking galectin-1. Pain. 2005;114:7–18. doi: 10.1016/j.pain.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- Plachta N, Annaheim C, Bissière S, Lin S, Rüegg M, Hoving S, Müller D, Poirier F, Bibel M, Barde YA. Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nat Neurosci. 2007;10:712–719. doi: 10.1038/nn1897. [DOI] [PubMed] [Google Scholar]
- Poirier F, Robertson EJ. Normal development of mice carrying a null mutation in the gene encoding the L14 S-type lectin. Development. 1993;119:1229–1236. doi: 10.1242/dev.119.4.1229. [DOI] [PubMed] [Google Scholar]
- Puche AC, Poirier F, Hair M, Bartlett PF, Key B. Role of galectin-1 in the developing mouse olfactory system. Dev Biol. 1996;179:274–287. doi: 10.1006/dbio.1996.0257. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Liu FT, Hirashima M, Anderson A. An emerging role for galectins in tuning the immune response: lessons from experimental models of inflammatory disease, autoimmunity and cancer. Scand J Immunol. 2007;66:143–158. doi: 10.1111/j.1365-3083.2007.01986.x. [DOI] [PubMed] [Google Scholar]
- Rotshenker S. Wallerian degeneration: the innate-immune response to traumatic nerve injury. J Neuroinflammation. 2011;8:109. doi: 10.1186/1742-2094-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Colicos MA, Barker PA, Kennedy TE. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci. 1999;19:6887–6896. doi: 10.1523/JNEUROSCI.19-16-06887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Okano H. Neural stem cells, adult neurogenesis, and galectin-1: From bench to bedside. Dev Neurobiol. 2012;72:1059–1067. doi: 10.1002/dneu.22023. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Imaizumi Y, Okano H. Expression and function of galectin-1 in adult neural stem cells. Cell Mol Life Sci. 2007;64:1254–1258. doi: 10.1007/s00018-007-6476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, Baum LG. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem. 2002;277:34295–34302. doi: 10.1074/jbc.M205167200. [DOI] [PubMed] [Google Scholar]
- Vasta GR. Galectins as pattern recognition receptors: structure, function, and evolution. Adv Exp Med Biol. 2012;946:21–36. doi: 10.1007/978-1-4614-0106-3_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volosin M, Trotter C, Cragnolini A, Kenchappa RS, Light M, Hempstead BL, Carter BD, Friedman WJ. Induction of proneurotrophins and activation of p75NTR-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. J Neurosci. 2008;28:9870–9879. doi: 10.1523/JNEUROSCI.2841-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Quin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–1402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]