

Abstract
It was demonstrated previously that a positive feedback loop, including protein kinase C (PKC) and mitogen-activated protein kinase (MAPK), is required for the gradual expression of cerebellar long-term depression (LTD). PKC and MAPK are mutually activated in this loop. MAPK-dependent PKC activation is likely to be mediated by phospholipase A2. On the other hand, it is not clear how PKC activates MAPK. Therefore, the entire picture of this loop was not fully understood. We here test the hypothesis that this loop is completed by the PKC substrate, Raf kinase inhibitory protein (RKIP). To test this hypothesis, we used a mutant form of RKIP that is not phosphorylated by PKC and thus constitutively inhibits Raf-1 and MEK, upstream kinases of MAPK. When this RKIP mutant was introduced into Purkinje cells of mouse cerebellar slices through patch-clamp electrodes, LTD was blocked, while wild-type (WT) RKIP had no effect on LTD. Physiological epistasis experiments demonstrated that RKIP works downstream of PKC and upstream of MAPK during LTD induction. Furthermore, biochemical analyses demonstrated that endogenous RKIP dissociates from Raf-1 and MEK during LTD induction in a PKC-dependent manner, suggesting that RKIP binding-dependent inhibition of Raf-1 and MEK is removed upon LTD induction. We therefore conclude that PKC-dependent regulation of RKIP leads to MAPK activation, with RKIP completing the positive feedback loop that is required for LTD.
Introduction
Long-term synaptic depression (LTD) elicited at synapses of cerebellar Purkinje cells receiving inputs from parallel fibers (PFs) is traditionally induced by a few minutes of repeatedly applied conjunctive stimulation of the PFs and the climbing fiber (CF) (Linden, 1994; Ito, 2001). Such short-lasting synaptic activity triggers a transient increase in calcium concentration that is required for the induction of LTD (Ohtsuki et al., 2009; Finch et al., 2012). We found previously that a positive feedback loop (see Fig. 1A) that includes protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) is responsible for prolonged kinase activation that underlies the long-lasting expression of LTD (Tanaka and Augustine, 2008). Considering that long-term synaptic plasticity is generally induced by short-lasting synaptic activity, regenerative mechanisms such as the positive feedback loop may be a common way to fill in the temporal gap between transient synaptic signaling and long-lasting kinase activities.
Figure 1.
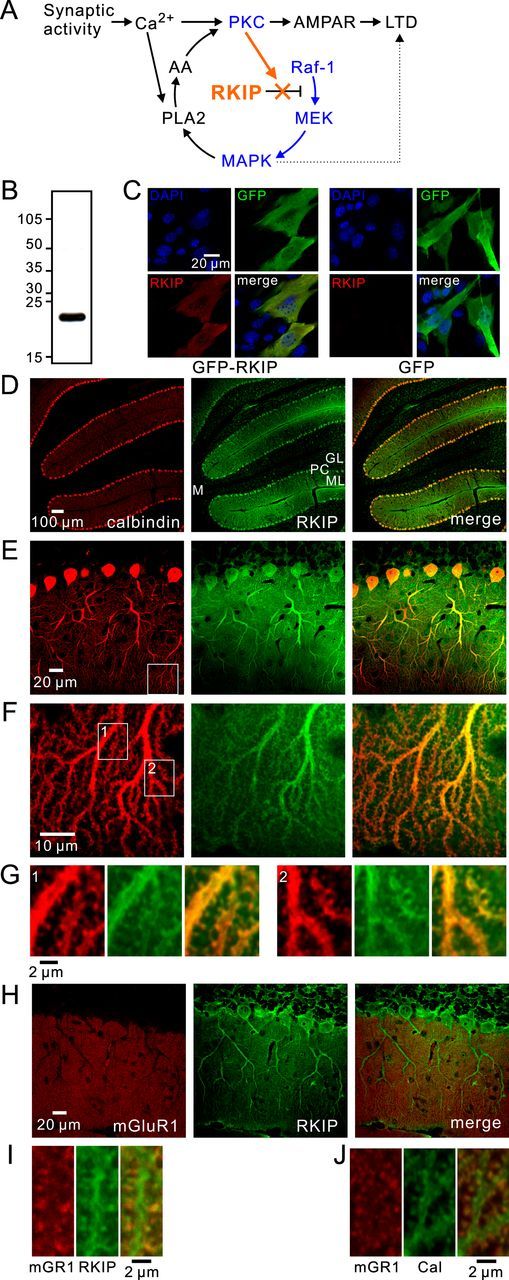
Expression of RKIP in Purkinje cells. A, Proposed model of the involvement of RKIP in the positive feedback loop for LTD (orange). PKC and MAPK pathways are shown in blue. B, Immunoblotting using an antibody against RKIP in cerebellar slices. C, Images of NIH3T3 cells transfected with GFP or GFP-RKIP (green) and stained with a RKIP antibody (red). D–G, Confocal images of a cerebellar slice double stained with antibodies against calbindin (red) and RKIP (green). GL, Granule cell layer; PC, Purkinje cell layer; ML, molecular layer; M, cerebellar medulla. The areas within the white squares shown in E and F are magnified in F and G, respectively. H–J, Confocal images of cerebellar slices double stained with antibodies against mGluR1 (red) and RKIP (green, H, I), or with antibodies against mGluR1 (red) and calbindin (green, J). Magnified images of distal dendrites are shown in I and J.
This positive feedback loop was first proposed by computational studies of signal transduction (Bhalla and Iyengar, 1999). While PKC-dependent activation of MAPK has been experimentally observed in many types of cells (Marais et al., 1998; Corbit et al., 2003; Zheng et al., 2005; Wen-Sheng, 2006), including Purkinje cells (Endo and Launey, 2003; Ito-Ishida et al., 2006; Tanaka and Augustine, 2008), the molecular mechanisms underlying this interaction are extremely varied and depend on cell type and stimulus conditions (Kolch et al., 1993; Marais et al., 1998; Schönwasser et al., 1998; Verin et al., 2000; Corbit et al., 2003; Zheng et al., 2005; Wen-Sheng, 2006). Thus, it remains unknown as to how PKC activates MAPK during cerebellar LTD, and therefore this critical reaction in the positive feedback loop of Purkinje cells is unidentified.
Raf kinase inhibitory protein (RKIP), also known as phosphatidylethanolamine binding protein, was identified in a yeast two-hybrid screening as a protein that interacts with Raf-1, which is a kinase that is upstream of the MAPK kinase, MEK. RKIP also interacts with MEK and competitively disrupts the interaction between Raf-1 and MEK, so that RKIP prevents Raf-1 from activating MEK (Yeung et al., 1999, 2000). In contrast, when RKIP is phosphorylated by PKC, RKIP dissociates from Raf-1, allowing PKC-dependent phosphorylation of RKIP to activate kinases in the MAPK pathway (Corbit et al., 2003; Lorenz et al., 2003). An immunohistochemical study (Frayne et al., 1999) and in situ hybridization analyses (Allen Brain Atlas, http://www.brainatlas.org) have demonstrated strong RKIP expression in cerebellar Purkinje cells. Although the function of RKIP in Purkinje cells has not been described, these findings raise the possibility that RKIP may have an important role in Purkinje cells by regulating MAPK following PKC activation.
In the present study, we tested whether RKIP mediates PKC-dependent MAPK activation to induce LTD (see Fig. 1A). We found that PKC-dependent regulation of RKIP is required upstream of MAPK during LTD. Furthermore, the RKIP-mediated MAPK activation seems to arise at least in part from dissociation of RKIP from Raf-1 and MEK. Thus, our results indicate that RKIP is the molecule that completes the positive feedback loop required for cerebellar LTD.
Materials and Methods
Patch-clamp recording.
Chemicals were obtained from Sigma or Wako Pure Chemical Industries unless specified otherwise. Whole-cell patch-clamp recordings were made from Purkinje cells as described previously (Miyata et al., 2000; Wang et al., 2000). Sagittal slices (200 μm) of cerebella from 14- to 21-d-old mice of either sex were bathed in extracellular solution [artificial CSF (ACSF)] containing the following (in mm): 125 NaCl, 2.5 KCl, 1.3 Mg2Cl, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, and 0.01 bicuculline methochloride (Tocris Bioscience). 12-O-tetradecanoylphorbol-13-acetate (TPA; Calbiochem) was added to the extracellular solution as indicated. Patch pipettes (resistance 5–6 MΩ) were filled with the following (in mm): 130 potassium gluconate, 2 NaCl, 4 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 20 HEPES, pH 7.2, and 0.25 EGTA. For the experiments using RKIP protein or caged, constitutively active MEK (MEK-CA) protein (Tanaka and Augustine, 2008), these proteins (1–2 mg/ml) were added into the internal solution along with dextran-conjugated fluorescein (Invitrogen) to monitor the dialysis of intracellular solutions into Purkinje cells. When GFP-conjugated protein was virally expressed, whole-cell patch-clamp recordings were made from Purkinje cells expressing GFP, which were identified by GFP fluorescence under a microscope (Zeiss Axioskop 2FS or Olympus BX61WI).
EPSCs were evoked in Purkinje cells (holding potential, −70 mV) by activating PFs with a glass stimulating electrode on the surface of the molecular layer (PF-EPSCs). PF-EPSCs were acquired and analyzed using pClamp software (Molecular Devices). To evoke LTD by electrical stimulation (PF&ΔV), PF stimuli were paired with Purkinje cell depolarization (0 mV, 200 ms) 300 times at 1 Hz. Data were accepted if the series resistance changed <20%, the input resistance was >80 MΩ, and the holding current changed <10%. Uncaging MEK proteins did not change the input resistance or holding current, and UV illumination in the absence of caged compound or in the presence of control caged protein (kinase-negative mutant of MEK) did not affect PF synaptic transmission (Tanaka et al., 2007; Tanaka and Augustine, 2008).
For recording miniature EPSC (mEPSC), patch pipettes were filled with the following (in mm): 60 CsCl, 10 Cs d-gluconate, 20 tetraethylammonium-Cl, 20 BAPTA, 2 CaCl2, 4 MgCl2, 4 ATP, 0.4 GTP, and 30 HEPES, pH 7.2, adjusted with CsOH. The extracellular solution included tetrodotoxin (1 μm; Tocris Bioscience). For chemical LTD stimulation, slices were treated with extracellular solution containing 50 mm K+ and 10 μm glutamate (K-glu) for 5 min, and then kept in normal ACSF for 20 min before being transferred to the recording chamber. mEPSC was recorded 30–60 min after K-glu treatment. Miniature events were analyzed using Mini Analysis Program (Synaptosoft).
Live cell imaging and photolysis of caged compounds.
The imaging of fluorescent dyes was performed by a confocal microscope (Zeiss LSM 510 or Olympus FV1000). A similar setup to that described previously (Wang and Augustine, 1995) was used to photolyze caged MEK-CA. Briefly, an N2 pulsed laser and Micropoint photostimulation system (Photonic Instruments) were connected to a Zeiss Axioskop 2FS microscope, and UV light (365 nm) was delivered to the slice via a 60× water-immersion objective to make a light spot with a half-width of ∼5 μm that was focused on the primary or secondary dendrites of a Purkinje cell. Photolysis was accomplished with a train (1 Hz, for 5 min) of two UV light flashes (<0.9 ns flashes at 10 Hz). To photolyze Dendra-fused MEK-CA (Dendra-MEK-CA), violet light (405 nm) was delivered to Purkinje cell dendrites by using the simultaneous (SIM) scanner of an Olympus FV1000 confocal microscope. Photolysis was accomplished with a train of 100 ms light pulses (1 Hz for 5 min). Images of green and red Dendra were taken every 30 s before, during, and after the photolysis.
Purification of recombinant protein.
Recombinant glutathione S-transferase (GST) and GST-fused RKIP were purified from Escherichia coli using glutathione Sepharose (GE Healthcare). RKIP-TV and RKIP-TA possess a single mutation of threonine residue 153 to valine (Corbit et al., 2003) and to alanine (Lorenz et al., 2003), respectively. RKIP-TV-delBD possesses a deletion mutation of amino acids 27–108, which are responsible for its association with MEK and Raf-1 (Yeung et al., 2000), in addition to the mutation in RKIP-TV. Purified proteins were dialyzed in HEPES-buffered solution (20 mm HEPES), pH 7.2, concentrated by centrifugal filter devices (Millipore), and stored at −80°C.
Immunoprecipitation, immunoblotting, and immunohistochemistry.
Primary antibodies used were mouse anti-Raf-1 (BD Biosciences), rabbit anti-MEK (Cell Signaling Technology), rabbit anti-RKIP (Millipore Biotechnology), mouse anti-metabotropic glutamate receptor 1 (mGluR1; BD Biosciences), mouse anti-GFP (Roche Applied Science), rabbit anti-MAPK (Cell Signaling Technology), rabbit anti-phospho-MAPK (Cell Signaling Technology), and mouse (Sigma) or guinea pig (Synaptic Systems) anti-calbindin antibodies. Secondary or tertiary antibodies used were horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (GE Healthcare), conformation-specific anti-rabbit IgG (Cell Signaling Technology), Clean-Blot IP Detection Reagents (Thermo Scientific), DyLight 649-conjugated anti-mouse IgG (Jackson ImmunoResearch), Alexa 647-conjugated anti-rabbit IgG (Invitrogen), and Alexa 488-conjugated anti-rabbit, anti-mouse, or anti-guinea pig IgG (Invitrogen) antibodies.
For immunoprecipitation, freshly prepared cerebellar slices were incubated for 30 min in normal ACSF and transferred to K-glu solution or to solution containing 0.5 μm TPA. In the case of experiments with chelerythrine (Calbiochem) and Go6976 (5,6,7,13-Tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile; Tocris Bioscience), they were added for the entire duration of the experiment. Slices were then lysed in lysis buffer containing the following: 150 mm NaCl, 50 mm Tris, pH 8, 1 mm EDTA, 1% Triton X-100, 10% glycerol, 1 μm staurosporine (Calbiochem), 1× proteinase inhibitor cocktail (Nacalai Tesque), and 1× phosphatase inhibitor cocktail (Nacalai Tesque). After homogenization, samples were centrifuged to remove cell debris. A portion of the supernatant was directly subjected to SDS-PAGE analysis to detect the total amounts of Raf-1, MEK, and RKIP. Polyclonal anti-RKIP was added to the supernatant (∼500 μg protein) to immunoprecipitate RKIP and rotated at 4°C followed by addition of 20 μl protein A-Sepharose (GE Healthcare). The immunoprecipitate was suspended in SDS-PAGE sample buffer, subjected to SDS-PAGE analysis, and transferred to blotting membranes. The membranes were cut into three portions, and Raf-1, MEK, and RKIP were detected by immunoblotting. To quantify the binding of Raf-1 or MEK to RKIP, band intensities were measured by MetaMorph software (Molecular Devices), and the ratios of normalized band intensities of Raf-1 or MEK to those of RKIP were then calculated.
For immunohistochemistry of cerebellar slices, mice were anesthetized and perfused transcardially with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4. The cerebellum was postfixed for 3 h at 4°C followed by sectioning (40 μm) using a vibrating microtome (Leica VT 1200S). The sections were blocked in 5% normal goat serum in PBS, incubated in primary antibodies overnight at 4°C, washed several times, and then incubated in secondary antibodies for 3 h at room temperature. Single optical sections of confocal images were acquired by an A1R laser-scanning confocal microscope (Nikon).
To confirm that the RKIP antibody used in this study is applicable for immunohistochemistry, NIH3T3 cells were transfected with pEGFP or pEGFP-RKIP-WT, the latter of which expresses GFP-fused wild-type RKIP. Approximately 48 h after the transfection, cells were fixed with 2% paraformaldehyde, permeabilized in 0.2% Triton X-100, blocked in 1% bovine serum albumin, and incubated with anti-RKIP and anti-GFP antibodies. Subsequently, cells were incubated with secondary antibodies and mounted in anti-fade solution with DAPI (Olink Bioscience).
To test the effects of nonphosphorylatable RKIP mutants on PKC-dependent MAPK phosphorylation, human embryonic kidney 293T (HEK293T) cells were transfected with pEGFP-RKIP-WT, pEGFP-RKIP-TV, or pEGFP-RKIP-TA. Serum was removed from the culture medium at ∼18 h after transfection, and cells were incubated for another 6 h in serum-free medium. Cells were then treated with or without TPA (0.1 μm) for 5 min and lysed in lysis buffer containing the following: 150 mm NaCl, 50 mm Tris, pH 8, 1 mm EDTA, 0.2% NP-40 alternative (Calbiochem), 10% glycerol, 1× proteinase inhibitor cocktail, and 1× phosphatase inhibitor cocktail. Cell lysates were centrifuged to remove cell debris, and the supernatant was subjected to SDS-PAGE analysis to detect phosphorylated MAPK, MAPK, and GFP by immunoblotting.
In vivo lentiviral injection.
Constructs for vesicular stomatitis virus G (VSV-G) pseudotyped lentiviral vectors were provided by St. Jude Children's Research Hospital (Hanawa et al., 2002). Virus vectors, which express GFP, GFP-RKIP-WT, GFP-RKIP-TV, or Dendra-MEK-CA under the control of the murine stem cell virus (MSCV) promoter, were produced as described previously (Torashima et al., 2006) with slight modifications. In brief, HEK293T cells were cotransfected with a mixture of four plasmids, pCAGkGP1R, pCAG4RTR2, pCAG-VSV-G, and either pCL20c MSCV-GFP, pCL20c MSCV-GFP-RKIP-WT, pCL20c MSCV-GFP-RKIP-TV, or pCL20c MSCV-Dendra-MEK-CA, using Lipofectamine 2000 (Invitrogen). At ∼46 h after transfection, virus vectors were harvested from the culture medium by ultracentrifugation at 25,000 rpm for 90 min, suspended in HBSS, and stored at −80°C.
Virus vectors were injected into the cerebellum of 8- to 13-d-old ICR mice by the procedures described previously (Torashima et al., 2006). Briefly, mice were anesthetized by sodium pentobarbital (40 mg/kg body weight) or Avertin (250 mg/kg body weight) and mounted on a stereotaxic stage. The cranium over the cerebellar vermis was exposed by a midline sagittal incision. A hole was made ∼3 mm caudal from lambda by a tip of forceps, and a glass needle was placed in the molecular layer of the cerebellar vermis. The virus solution (2–3 μl total) was injected at a rate of 200 nl/min using an UltraMicroPump II and Micro4 controller (World Precision Instruments). After the surgery, mice were kept on a heating pad until they recovered from the anesthesia and then were returned to their mother.
Construction and characterization of caged, constitutively active MEK.
Caged MEK-CA was produced by labeling with 4,5-dimethoxy-2-nitrobenzyl chloroformate (NVOC-Cl; Sigma), as described previously (Tanaka and Augustine, 2008). Final concentrations of MEK-CA and NVOC were determined by a commercial protein assay (Bio-Rad) and OD360 measurements (OD360 = 5000 m−1 cm−1), respectively. The labeling ratio typically ranged from five to seven NVOC-Cl per MEK molecule.
To examine the activity of caged or uncaged MEK-CA, in vitro assays of MAPK phosphorylation were performed. To obtain uncaged MEK-CA, caged MEK-CA was photolyzed by 360 nm UV light from a transilluminator for 3 min. Caged or uncaged MEK-CA was mixed with recombinant MAPK and ATP in kinase buffer (Cell Signaling Technology) and incubated for 30 min at 30°C. The kinase reaction was terminated by adding SDS-PAGE sample buffer. Samples were subjected to SDS-PAGE analysis, and phosphorylated MAPK and MEK were detected by immunoblotting.
Statistical analysis.
Statistical differences were determined by the Student's t test (for two group comparisons) and ANOVA followed by the Fisher's PLSD test (for more than two group comparisons). Analysis was performed with OriginPro 8.1 software.
Results
Localization of RKIP in cerebellar Purkinje cells
We first examined the localization of endogenous RKIP in the cerebellum by confocal immunofluorescence analysis. We considered the anti-RKIP antibody used in this study to be applicable for immunofluorescence staining of cerebellar sections because of the following two results. First, we found that in cerebellar slice lysates, the anti-RKIP antibody detected a molecule of ∼21 kDa (Fig. 1B), which is consistent with the published molecular weight of RKIP. Second, our control experiments using NIH3T3 cells demonstrated specific staining in cells expressing RKIP. NIH3T3 cells were reported to show no or very weak endogenous expression of RKIP (Wang et al., 2010), and cells expressing GFP only or nontransfected cells were indeed not clearly stained by the anti-RKIP antibody (Fig. 1C). In contrast, NIH3T3 cells expressing RKIP fused with GFP were positively stained by this antibody (Fig. 1C). These results validate the use of this antibody for immunofluorescence staining of cerebellar sections.
In the cerebellum, RKIP is strongly expressed in both the Purkinje cell layer and molecular layer, and is also expressed in a part of the granule cell layer to a lesser extent, while it is not expressed in the cerebellar medulla (Fig. 1D). As can be seen in the magnified images, RKIP immunoreactivity was clearly detected in both the somata and dendrites of Purkinje cells, which were readily distinguished from other cells by their staining for calbindin (Fig. 1E), consistent with a previous study (Frayne et al., 1999). RKIP was also observed in distal dendrites (Fig. 1F) as well as protrusions attached along dendrites (Fig. 1G), which were also stained by calbindin. We confirmed that these protrusions are dendritic spines by their overlap with mGluR1 (Fig. 1H–J), which is concentrated on dendritic spines (Takács et al., 1997; Yoshida et al., 2006). Although a lesser extent of RKIP immunoreactivity was also detected in other types of cells, presumably glial cells and some interneurons, because RKIP is clearly expressed not only in the soma and proximal dendrites but also in the distal dendrites of Purkinje cells, it is possible that RKIP has synaptic functions in Purkinje cells.
Involvement of RKIP in LTD induction
We next asked whether RKIP is involved in LTD. It was reported previously that RKIP-dependent inhibition of Raf-1 was cancelled by PKC-mediated phosphorylation of human RKIP at serine 153 (Ser-153), which causes the dissociation of RKIP from Raf-1 (Corbit et al., 2003; Lorenz et al., 2003), so that the phosphorylation of RKIP leads to activation of kinases in the MAPK pathway. When Ser-153 is mutated to the nonphosphorylatable residue valine, binding of this mutated RKIP with Raf-1 is not reversed by PKC (Corbit et al., 2003). Thus, the inhibitory effects of this RKIP mutant are maintained even in the presence of PKC activation, demonstrating that this is a dominant-negative mutation (Corbit et al., 2003). Another RKIP mutant, in which Ser-153 was replaced with alanine, was also used as a phosphorylation-deficient RKIP (Lorenz et al., 2003). In the case of mouse RKIP, the phosphorylation site is threonine 153 (Thr-153). Thus we used mouse RKIP in which Thr-153 was mutated to valine (RKIP-TV) or alanine (RKIP-TA), to test whether RKIP phosphorylation by PKC is involved in LTD. We confirmed in experiments using HEK293T cells that these mutants are capable of inhibiting PKC-dependent MAPK activation: while a PKC activator, TPA, phosphorylated MAPK in cells expressing RKIP-WT, this TPA-induced MAPK phosphorylation was severely reduced in cells expressing RKIP-TV or RKIP-TA (Fig. 2A).
Figure 2.

Basic electrophysiological properties of PF-EPSCs in the presence of intracellular RKIP protein. A, Immunoblot analysis of MAPK phosphorylation in HEK293T cells expressing GFP-fused RKIP-WT, RKIP-TV, or RKIP-TA. TPA was extracellularly applied for 5 min. B, CBB staining of purified GST, GST-fused RKIP-WT, RKIP-TV, and RKIP-TA. C, Images of a Purkinje cell containing fluorescein dextran that was introduced with the RKIP protein. Top, A single focal plane image taken at 60 min after whole-cell configuration. Bottom, A z-stack image taken at the end of the recording. D, Amplitudes of PF-EPSCs elicited by PF stimuli of increasing intensity (8, 17, 27, 36, 45, and 54 μA). E, PPF ratios of PF-EPSCs elicited by a pair of PF stimuli with different intervals (50–250 ms). PF-EPSCs included in the results of D and E were recorded from Purkinje cells without protein (n = 11; black symbols), with GST (n = 5; brown symbols), with RKIP-WT (n = 7; blue symbols), with RKIP-TV (n = 11; red symbols), or with RKIP-TA (n = 4; magenta symbols). Error bars represent SEM.
For the electrophysiological experiments in Purkinje cells of cerebellar slices, we purified GST, GST-tagged RKIP-WT, RKIP-TV, and RKIP-TA, and introduced these proteins into Purkinje cells through a patch pipette. These protein samples were >80% pure, according to Coomassie brilliant blue (CBB) staining of an SDS-PAGE gel (Fig. 2B). To allow the proteins to diffuse into Purkinje cells, we waited for 60–90 min and verified cell dialysis by monitoring the delivery of a fluorescent tracer dye, fluorescein, into Purkinje cells (Fig. 2C). The basal level of synaptic transmission was unaffected by the introduction of the proteins, because the relationship between stimulus intensity and the amplitude of PF-EPSC was unchanged (Fig. 2D). Two-way ANOVA (p = 0.192) confirmed that there is no significant difference in the relationship with or without proteins. The decay time constant of PF-EPSC was also unaltered (control, 13.6 ± 0.7 ms, n = 7; GST, 13.9 ± 1.4 ms, n = 6; RKIP-WT, 13.1 ± 1.1 ms, n = 6; RKIP-TV, 14.3 ± 0.6 ms, n = 7; RKIP-TA, 15.3 ± 1.3 ms, n = 7; ANOVA, p = 0.636). It is unlikely that the introduction of RKIP-WT, RKIP-TV, or RKIP-TA into postsynaptic Purkinje cells would affect presynaptic glutamate release, and consistent with this, the paired-pulse facilitation (PPF) ratio was unaltered by the introduction of these proteins (Fig. 2E; ANOVA, p = 0.205).
Pairing electrical stimulation of PFs together with Purkinje cell depolarization at 1 Hz for 5 min induced LTD when GST or RKIP-WT was introduced into Purkinje cells (Fig. 3A,B). The amplitude and time course of LTD were similar to those observed in control Purkinje cells, in which no proteins were introduced (Fig. 3B,C). In contrast, introducing RKIP-TV or RKIP-TA into Purkinje cells caused a significant reduction in the amount of LTD (Fig. 3B,C). Because LTD was blocked by the nonphosphorylatable RKIP mutants but not by RKIP-WT, these results indicate that phosphorylation of RKIP is involved in LTD. Furthermore, deletion of the binding site for Raf-1 and MEK in RKIP-TV (RKIP-TV-delBD) (Yeung et al., 2000) reversed this effect (Fig. 3B,C). This is consistent with the idea that the inhibition of LTD by nonphosphorylatable RKIP mutants is due to its continuous association with Raf-1 or MEK. Thus, dissociation of RKIP from Raf-1 or MEK following RKIP phosphorylation by PKC appears to be required for LTD.
Figure 3.

Effects of RKIP protein introduction into Purkinje cells on LTD. A, Superimposed PF-EPSCs recorded before and 20 min after PF&ΔV in the absence (control) and presence of intracellular GST, RKIP-WT, RKIP-TV, RKIP-TA, and RKIP-TV-delBD. Calibration: 100 pA, 20 ms. B, Time courses of LTD induced by PF&ΔV in the absence of protein (n = 6; black circles) and in the presence of GST (n = 6; brown circles), RKIP-WT (n = 6; blue circles), RKIP-TV (n = 7; red circles), RKIP-TA (n = 7; magenta circles), and RKIP-TV-delBD (n = 5; green circles). PF-EPSC amplitudes are normalized to their mean prestimulation level. C, Average amounts of LTD calculated 20–40 min after the end of PF&ΔV. *p < 0.05, ANOVA followed by the Fisher's PLSD test. Error bars represent SEM.
We also manipulated RKIP function in a second way, to confirm the inhibition of LTD by RKIP-TV. Lentiviral vectors were injected in vivo to express GFP, GFP-RKIP-WT, or GFP-RKIP-TV in Purkinje cells, and PF-EPSCs were recorded from the GFP-positive Purkinje cells of cerebellar slices. The procedures we used for the in vivo injection of lentivirus (Torashima et al., 2006) allowed us to express the proteins in Purkinje cells in a slice (Fig. 4A). Consistent with the results obtained with acute introduction of proteins into Purkinje cells, LTD was blocked in Purkinje cells virally expressing RKIP-TV (Fig. 4B; −0.92 ± 5.9%, n = 6). In contrast, LTD was intact in cells expressing RKIP-WT (Fig. 4B; 34.4 ± 8.5%, n = 6), and the amplitude and time course of LTD were similar to those in control cells expressing only GFP (Fig. 4B; 30.9 ± 3.5%, n = 4). ANOVA confirmed the significance of the difference (p < 0.01), and multiple comparison by Fisher's PLSD test confirmed that LTD in cells expressing RKIP-TV was significantly smaller compared to LTD in cells expressing RKIP-WT or GFP (p < 0.01).
Figure 4.

Effects of virally expressed RKIP-TV on LTD. A, Bright-field (left) and z-stack confocal images (center and right) of live cerebellar slices obtained from a mouse injected with lentiviral vector expressing GFP-RKIP-TV. B, Time courses of LTD recorded from Purkinje cells expressing GFP (n = 4; black circles), GFP-RKIP-WT (n = 6; blue circles), and GFP-RKIP-TV (n = 6; red circles). Error bars represent SEM.
RKIP mediates PKC-dependent MAPK activation
We then examined whether RKIP is involved in the positive feedback loop required for LTD induction. We addressed this hypothesis by asking whether RKIP works downstream of PKC and upstream of MAPK during cerebellar LTD. For this purpose, we used experiments analogous to genetic epistasis analysis (Lev-Ram et al., 1997; Tanaka and Augustine, 2008). Specifically, we asked whether the LTD produced by direct activation of PKC or MAPK could be blocked by RKIP-TV. If RKIP is interposed in a loop between PKC and MAPK, then RKIP-TV will prevent a PKC activator from producing LTD, while RKIP-TV will not prevent a MAPK activator from producing LTD.
A pseudophosphorylated, constitutively active form of MEK1 that directly phosphorylates and activates MAPK has been used to activate MAPK in several types of cells (Cobb and Goldsmith, 1995). To provide temporal control over the activity of MEK-CA, we previously developed a caged version of MEK-CA by covalently attaching a NVOC-Cl caging group (Tanaka and Augustine, 2008). Previous results demonstrated that LTD can be induced by uncaging MEK-CA in Purkinje cells, but not by the uncaging of a control caged protein, kinase-negative mutant of MEK (Tanaka and Augustine, 2008). Thus, caged MEK-CA enables us to observe LTD induced specifically by the activation of MAPK, and not by UV illumination or the by-products of the photolysis reaction. We note that the start of MAPK activation could be temporally controlled by photolyzing caged MEK-CA, but the end of MAPK activation appeared to rely on the diffusion of uncaged MEK-CA. In fact, the experiments using MEK-CA fused to Dendra, a green-to-red photoswitchable fluorescent protein, showed that photoconverted red Dendra-MEK-CA remained around the dendrites of Purkinje cells, where the photolysis was applied, for at least 20 min after 5 min of photolysis (Fig. 5A). This suggests that MAPK activation may be sustained for a while even after the photolysis.
Figure 5.
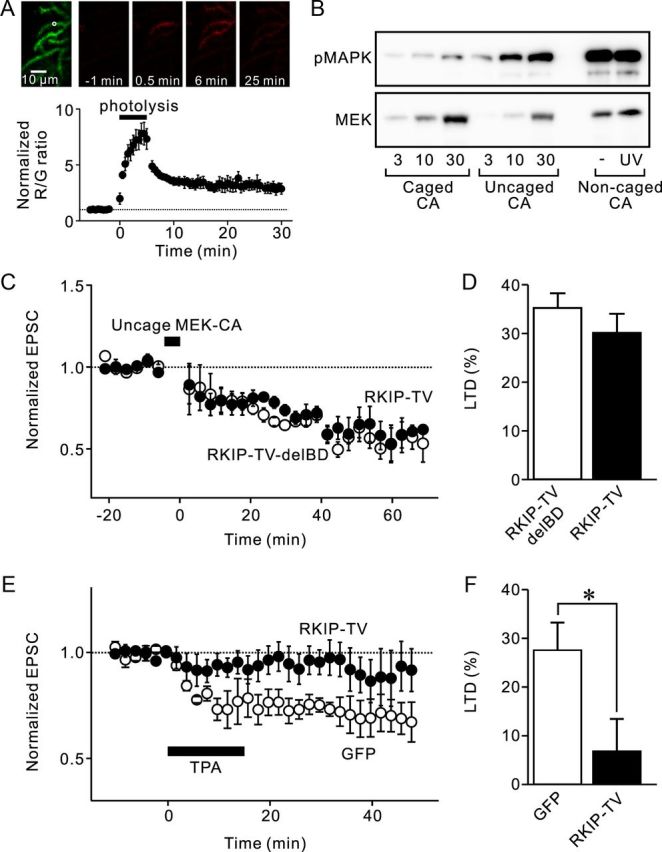
RKIP-TV inhibits LTD induced by PKC activation but not MAPK activation. A, Dynamics of photoconverted red Dendra-MEK-CA in Purkinje cell dendrites after the photolysis. Top images show green Dendra in Purkinje cell dendrites before the photolysis (left) and photoconverted red Dendra at times indicated. The white circle shows the area where photolysis was applied. The bottom graph shows average change in red-to-green ratio with time around the dendrites where photolysis was applied (n = 4). The ratio was normalized to their mean prephotolysis level. Photolysis was applied from 0 to 5 min. B, Phosphorylation of purified MAPK protein by MEK-CA in vitro. Concentrations of MEK-CA in reaction mixtures were 3–30 ng/ml (caged or uncaged MEK-CA) or 20 ng/ml (noncaged MEK-CA). C, The LTD induced by the uncaging of MEK-CA is not blocked by intracellular application of either RKIP-TV (closed circles; n = 5) or RKIP-TV-delBD (open circles; n = 5). D, Summarized data of the averaged reduction of PF-EPSC by the uncaging of MEK-CA. E, The LTD induced by TPA (0.5 μm) is blocked in Purkinje cells expressing GFP-RKIP-TV (closed circles; n = 7), whereas LTD is triggered in cells expressing GFP (open circles; n = 6). F, Summarized data of the averaged reduction of PF-EPSC by TPA. *p < 0.05 (Student's t test). Error bars represent SEM.
We used an in vitro kinase assay to confirm that the covalent addition of the caging group reduced the ability of MEK-CA to phosphorylate MAPK, whereas removing the cage via UV (360 nm) photolysis increased the ability of MEK-CA (Fig. 5B). We then introduced both RKIP and the caged MEK-CA through a patch pipette, waited for ∼90 min to allow these proteins to diffuse into Purkinje cells, and applied UV light to dendrites where PFs were electrically stimulated. When the control RKIP protein, RKIP-TV-delBD, was introduced together with caged MEK-CA, LTD of PF-EPSC was induced upon uncaging of MEK-CA (Fig. 5C). Furthermore, even when RKIP-TV was introduced with caged MEK-CA, LTD was still clearly observed (Fig. 5C). The degree of depression of PF-EPSCs in the presence of RKIP-TV-delBD and RKIP-TV were not significantly different (Fig. 5D; p = 0.32). These results indicate that RKIP is not required for LTD once MAPK is activated. Thus, we conclude that RKIP works upstream of MAPK.
To activate PKC, we used TPA, which directly activates PKC and can induce LTD in cerebellar slices (Kondo et al., 2005; Tanaka and Augustine, 2008), as well as in cultured Purkinje cells (Linden and Connor, 1991; Endo and Launey, 2003). In these experiments, we used lentiviral vectors to express GFP-RKIP-TV or GFP in Purkinje cells and recorded their PF-EPSCs, as was done in the experiments shown in Figure 4. This paradigm allowed us to start recordings immediately after the whole-cell configuration was made. When TPA (0.5 μm) was applied extracellularly for 15 min, LTD was induced in control cells expressing GFP (Fig. 5E). In contrast, LTD was not observed when TPA was applied to cells expressing RKIP-TV (Fig. 5E). The depression of PF-EPSCs in cells expressing RKIP-TV was significantly smaller than that in cells expressing GFP (Fig. 5F; p < 0.05). These results indicate that RKIP works downstream of PKC. Together, our results indicate that RKIP is required for LTD and works downstream of PKC and upstream of MAPK. Thus, RKIP appears to link PKC and MAPK in the positive feedback loop.
Association between endogenous RKIP and Raf-1 or MEK in cerebellar slices
Our electrophysiological analyses demonstrated that phosphorylation of RKIP is involved in the pathway of PKC activating MAPK that is required for LTD. These results, together with previous biochemical reports showing the PKC-dependent regulation of the interaction between RKIP and Raf-1, raise a possibility that at least one of the mechanisms of RKIP working for LTD may also be related to its interaction with Raf-1. Therefore, we tested by immunoprecipitation analysis whether endogenous RKIP is normally associated with Raf-1 under basal conditions in the cerebellum. In cerebellar slices that were maintained in normal ACSF, RKIP was immunoprecipitated by our RKIP antibody (Fig. 6A). We then found that Raf-1 was coimmunoprecipitated by this antibody in control slices, whereas it was much less immunoprecipitated by normal IgG (Fig. 6A). Since it was shown by in vitro assays that RKIP also binds to MEK, and this interaction is sufficient to prevent Raf-1 from activating MEK (Yeung et al., 2000), we tested whether RKIP also interacts with MEK in the cerebellum. Indeed, MEK was also coimmunoprecipitated by the RKIP antibody (Fig. 6A). These results indicate that RKIP is associated with Raf-1 and MEK in cerebellar slices under basal conditions.
Figure 6.
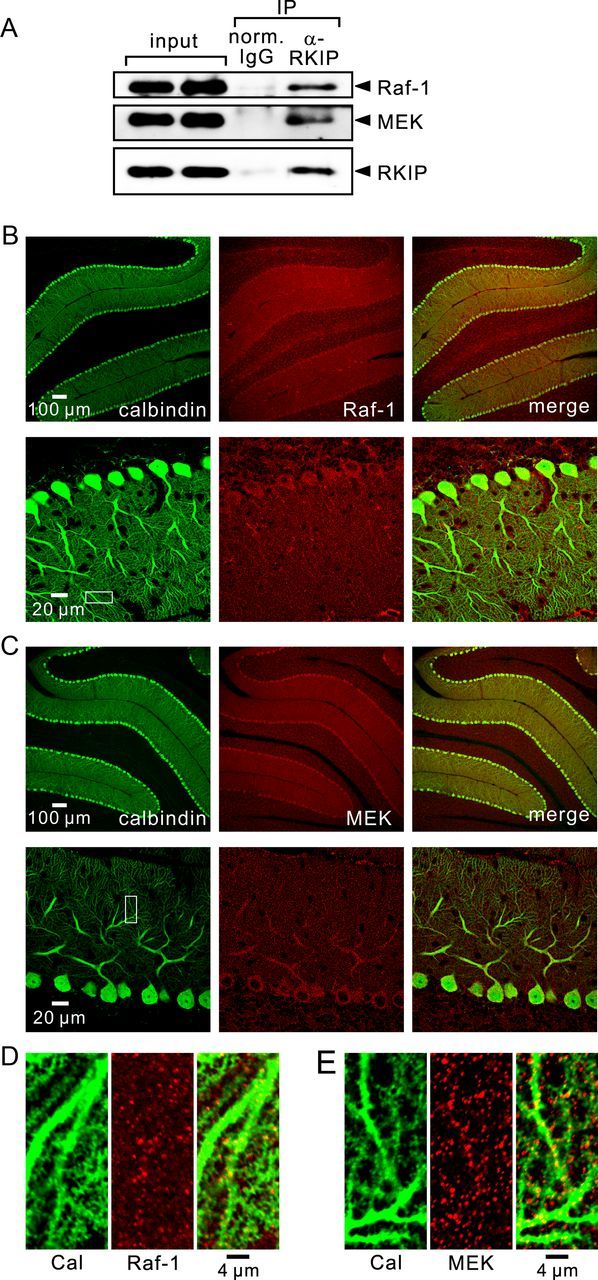
Interaction of Raf-1 and MEK with RKIP, and localization of Raf-1 and MEK in cerebellar slices. A, Immunoblot analysis of coimmunoprecipitated Raf-1, coimmunoprecipitated MEK, and precipitated RKIP in lysates from control cerebellar slices. B–E, Confocal images of cerebellar slices double stained with antibodies against calbindin (green) and Raf-1 (red; B, D), or with antibodies against calbindin (green) and MEK (red; C, E). Areas within the white squares shown in B and C are magnified in D and E, respectively.
As shown in Figure 1, RKIP is predominantly expressed in Purkinje cells within the cerebellum. We next examined the localization of endogenous Raf-1 and MEK in the cerebellum by confocal immunofluorescence analysis. Consistent with a previous report (Morice et al., 1999), our results demonstrated the expression of Raf-1 in Purkinje cells (Fig. 6B). Although Raf-1 expression in Purkinje cells within the cerebellum was not as predominant as RKIP expression, Raf-1 immunoreactivity was detected both in the soma and in the dendrites of Purkinje cells. Similarly, MEK is expressed in Purkinje cells, as well as other types of cells (Fig. 6C). MEK immunoreactivity was relatively strong in the soma and in the proximal dendrites of Purkinje cells compared with other types of cells. Furthermore, both Raf-1 (Fig. 6D) and MEK (Fig. 6E) immunoreactivites were present in distal dendrites of Purkinje cells that were visualized with a calbindin antibody. Considering the expression of RKIP throughout Purkinje cells and the existence of Raf-1 and MEK in Purkinje cells, it is possible that at least a part of RKIP interacts with Raf-1 and MEK in Purkinje cells.
Dissociation of endogenous RKIP from Raf-1 or MEK by LTD stimulation
We next used immunoprecipitation to ask whether the association of RKIP with Raf-1 or MEK is altered by LTD-inducing stimuli. Because such biochemical analyses require a sufficient amount of tissue, we used a chemical LTD induction protocol. This involves treating cerebellar slices with 50 mm K+ and 10 μm glutamate. This protocol was shown previously to cause LTD at the synapses between PF and Purkinje cells in cerebellar slices (Tanaka and Augustine, 2008). In the present study, we confirmed by recording mEPSC that LTD induced by K-glu could also be blocked by RKIP-TV: K-glu treatment significantly (p < 0.05) decreased the amplitude of mEPSC recorded from Purkinje cells expressing GFP, whereas it did not alter the amplitude of mEPSC recorded from Purkinje cells expressing GFP-RKIP-TV (Fig. 7A; p = 0.45).
Figure 7.
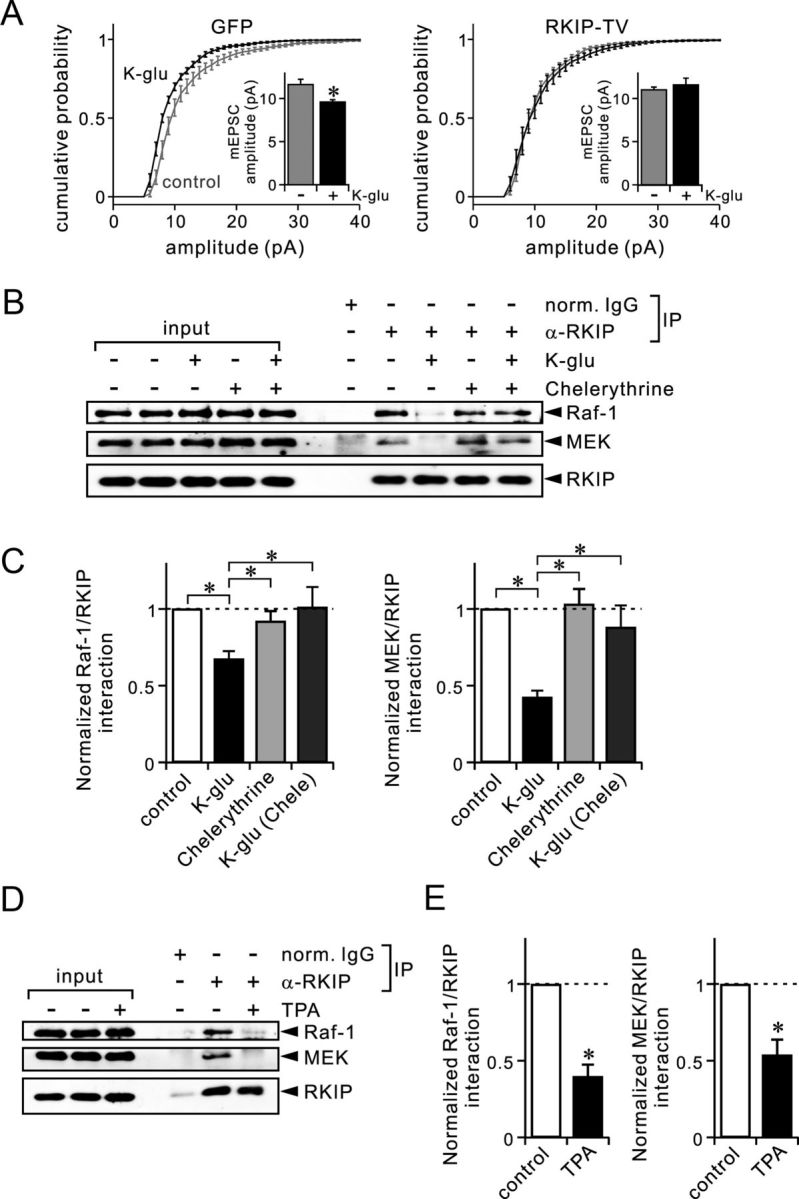
Dissociation of RKIP from Raf-1 and MEK during chemical LTD stimulation depends on PKC. A, Summary of mEPSC amplitudes in Purkinje cells, shown as cumulative distributions and bar graphs (inset). Results were obtained by recording from 10 (control) or 8 (K-glu) cells expressing GFP (left), and by recording from 12 (control) or 8 (K-glu) cells expressing GFP-RKIP-TV (right). B, Immunoblot analysis of coimmunoprecipitated Raf-1, coimmunoprecipitated MEK, and precipitated RKIP in cerebellar slices, which were treated with or without K-glu in the absence or presence of chelerythrine (5 μm). C, Quantification of the interaction between RKIP and Raf-1 (left) or between RKIP and MEK (right) in cerebellar slices. Control slices were incubated only in normal ACSF. Other slices were treated with K-glu or chelerythrine as indicated. Results were obtained from 21 (K-glu only) or 11 (chelerythrine) experiments for Raf-1, and from 4 experiments for MEK. D, Immunoblot analysis of coimmunoprecipitated Raf-1, coimmunoprecipitated MEK, and precipitated RKIP in cerebellar slices, which were treated with or without TPA (0.5 μm). E, Quantification of the interaction of RKIP with Raf-1 (left) or MEK (right) in cerebellar slices, which were treated with or without TPA. Results were obtained from five experiments. *p < 0.05 (A); *p < 0.01 (C, E) [Student's t test (A, E); ANOVA followed by the Fisher's PLSD test (C)]. Ratios of coimmunoprecipitated Raf-1 or MEK to precipitated RKIP shown in C and E were normalized with the ratio of the control condition. Error bars represent SEM.
Compared with the lysate of control slices not treated with K-glu, the amount of Raf-1 and MEK that coimmunoprecipitated with the RKIP antibody was reduced following treatment with K-glu (Fig. 7B). The ratio of intensities of the Raf-1 and RKIP bands was smaller in the K-glu-treated samples compared to the control samples (Fig. 7C). Likewise, the ratio of intensities of the MEK and RKIP bands was also smaller in the K-glu-treated samples compared to the control samples (Fig. 7C). Thus our biochemical analyses demonstrated that endogenous RKIP dissociates from Raf-1 and MEK in response to LTD stimulation of cerebellar slices. This is in line with our electrophysiological results showing that RKIP-TV and RKIP-TA, which presumably do not dissociate from Raf-1, blocked LTD (Fig. 3).
We next asked whether the dissociation of RKIP from Raf-1 or MEK observed during LTD depends on PKC. For this purpose, we used a PKC inhibitor, chelerythrine, which is an inhibitor of all PKC subtypes. Treatment with chelerythrine prevented dissociation of RKIP from Raf-1 or MEK in response to K-glu (Fig. 7B). In the presence of chelerythrine with or without K-glu treatment, the degree of association between RKIP and Raf-1 or MEK was not different from the interaction in control conditions (Fig. 7C). ANOVA confirmed the significance of the difference (p < 0.01) in the association of Raf-1 or MEK with RKIP, and multiple comparison by Fisher's PLSD test confirmed that the association of Raf-1 or MEK with RKIP during K-glu treatment was significantly (p < 0.01) smaller than that in control conditions or in the presence of chelerythrine (Fig. 7C). In addition, Go6976, which is a specific inhibitor of Ca2+-dependent PKC, also prevented the dissociation of RKIP from Raf-1 (Go6976 only, 94.4 ± 2.3% of control, n = 3; K-glu with Go6976, 92.4 ± 8.2% of control, n = 3; p = 0.91 with Fisher's PLSD test). We also confirmed that PKC activation by TPA was capable of causing dissociation of RKIP from Raf-1 or MEK in cerebellar slices (Fig. 7D,E). These results indicate that the dissociation of RKIP from Raf-1 and MEK during LTD induction depends on PKC.
Discussion
Although previous computational and experimental studies have shown that MAPK is activated via PKC during LTD (Endo and Launey, 2003; Ito-Ishida et al., 2006; Tanaka and Augustine, 2008) and that this pathway is an important part of the positive feedback loop required for LTD (Kuroda et al., 2001; Tanaka and Augustine, 2008), the reaction that allows PKC to activate MAPK has not been identified. The present study provides several results indicating that RKIP serves this role: (1) RKIP exists in distal dendrites of Purkinje cells, (2) phosphorylation of RKIP is required for LTD, (3) RKIP works downstream of PKC and upstream of MAPK during LTD, (4) interaction between RKIP and Raf-1 or MEK occurs under basal conditions and is reduced during LTD in the cerebellum, and (5) PKC activation is responsible for the dissociation of RKIP from Raf-1 or MEK during LTD. These results suggest that RKIP, which binds to and inhibits Raf-1 and MEK in the steady state, is phosphorylated by PKC and then dissociates from these proteins after LTD induction, so that Raf-1 can activate MEK and thereby MAPK, which is the downstream kinase. Thus our study reveals one mechanism by which PKC activates MAPK in Purkinje cells during LTD, and further emphasizes the central role of the positive feedback loop in LTD induction.
Signaling molecules involved in the positive feedback loop
The positive feedback loop, including PKC and MAPK, was first postulated to produce a bistable system that prolongs kinase activities in time (Bhalla and Iyengar, 1999). Another computational study used the positive feedback loop model to simulate several features of LTD (Kuroda et al., 2001). The loop includes three signaling pathways: PKC activates Raf-1/MEK/MAPK, MAPK phosphorylates and activates PLA2, and arachidonic acid (AA) produced by PLA2 binds and activates PKC (Fig. 1A). The individual molecules involved in these pathways have been experimentally implicated in cerebellar LTD (Linden and Connor, 1991; Hartell, 1994; Linden, 1995; De Zeeuw et al., 1998; Kawasaki et al., 1999; Reynolds and Hartell, 2001; Leitges et al., 2004), and our previous studies further demonstrated that the positive feedback loop composed with these molecules is indeed required for LTD (Tanaka et al., 2007; Tanaka and Augustine, 2008). Our present study adds RKIP to this loop. We postulate that upon PKC activation, RKIP switches on Raf-1 activity during LTD, although it is still possible that other mechanisms may also contribute to the activation of Raf-1 by PKC, such as direct phosphorylation of Raf-1 by PKC (Kolch et al., 1993). It was proposed previously that cyclooxygenase-2 and prostaglandin D2 or E2 intervene in the pathway between AA and PKC (Le et al., 2010). Because many molecules have been implicated in LTD, it is possible that other signals may be involved in the positive feedback loop.
In our experiments using caged MEK-CA, LTD induced by the activation of MAPK was unaffected by the presence of RKIP-TV. This indicates that RKIP works upstream of MAPK. However, if RKIP-TV disrupts the positive feedback loop that is required for the first 20–30 min of LTD (Tanaka and Augustine, 2008), one might expect that RKIP-TV would block LTD. This result may be due to the persistent activation of uncaged MEK-CA. Indeed, our observation of the photoconverted red Dendra-MEK-CA suggests that uncaged MEK-CA remains for ∼20 min around the dendrites, where photolysis was applied. In this case, even though Raf-1 is inhibited by RKIP-TV, uncaged MEK-CA would persistently activate MAPK, PLA2, and PKC, leading to LTD induction. Therefore our observation that uncaging of MEK-CA triggers LTD in the presence of RKIP-TV suggests that the positive feedback loop is required to activate these enzymes during LTD, but not to sustain this loop itself.
Since the expression of LTD is known to result from the internalization of AMPA-type glutamate receptors (AMPARs), mediated by their PKC-dependent phosphorylation (Chung et al., 2003; Steinberg et al., 2006), PKC has been thought to be the only output signal of the positive feedback loop (Kuroda et al., 2001). Our previous observation that inhibition of MAPK activation completely blocked the LTD induced by TPA (Tanaka and Augustine, 2008) suggests that MAPK has an additional role beyond activating PKC as a component working in the positive feedback loop (Fig. 1A, dashed line). Our present observation that RKIP-TV blocked the LTD induced by TPA supports this idea. Although further work is required to clarify the targets of MAPK, both the MAPK-dependent pathway and PKC-dependent phosphorylation of AMPARs seem to be required for LTD expression.
Role of RKIP interactions in cerebellar LTD
The present study provides the first evidence of the involvement of RKIP in cerebellar LTD. In addition, our results demonstrate that RKIP works downstream of PKC and upstream of MAPK, and that RKIP dissociates from Raf-1 and MEK in a PKC-dependent manner in the cerebellum. Therefore, RKIP seems to dissociate from, and thereby activate, Raf-1 and MEK after being phosphorylated by PKC during LTD. On the other hand, several studies described below, as well as our results showing the involvement of PKC-dependent phosphorylation of RKIP in LTD, raise the possibility that RKIP may also contribute to LTD via another signaling pathway. It was reported that PKC switches RKIP binding from Raf-1 to G-protein-coupled receptor (GPCR) kinase 2 (GRK2) (Lorenz et al., 2003), which inhibits the activation of GPCRs, including mGluR1 (Carman and Benovic, 1998; Dhami et al., 2005). Activation of mGluR1 is well known to be important for the induction of cerebellar LTD (Kano et al., 2008). Binding of phosphorylated RKIP inhibits the activity of GRK2, leading to the enhancement of GPCR-dependent signals (Lorenz et al., 2003). Thus, although it is not yet known whether inhibition of GRK2 by phosphorylated RKIP is involved in LTD, it is possible that RKIP has dual roles in LTD, mediating the activation of MAPK by PKC and enhancing mGluR1 activity. Furthermore, considering the strong expression of RKIP in whole Purkinje cells, other possible mechanisms working for LTD cannot be completely denied. Nevertheless, the pathway of PKC activating MAPK likely relies at least in part on PKC-dependent dissociation of RKIP from Raf-1 and MEK that initiates the activation of Raf-1/MEK/MAPK during LTD.
Our immunohistochemical results show the localization of Raf-1 and MEK mainly in the soma and proximal dendrites, which raises the possibility that Raf-1 and MEK in proximal dendrites are activated via PKC-dependent dissociation of RKIP and are involved in LTD. This is possible specifically when LTD is triggered by K-glu treatment, which stimulates whole Purkinje cells. Consequently, experiments using K-glu treatment would have allowed us to detect the dissociation of RKIP from Raf-1 and MEK by coimmunoprecipitation analysis. In contrast, when LTD is locally triggered in synapses of distal dendrites by synaptic stimulation, it is hard to explain how Raf-1 and MEK activated in proximal dendrites affect synapses undergoing LTD. Although one possibility is MAPK transportation from proximal to distal dendrites, it is unlikely, because a study using hippocampal culture neurons demonstrated that passive diffusion, but not active transportation, underlies dendritic movement of MAPK (Wiegert et al., 2007). Alternatively, it may be possible that Raf-1 and MEK are locally activated via PKC-dependent dissociation of RKIP in distal dendrites around synapses undergoing LTD, because our results showed that small amounts of Raf-1 and MEK also exist in distal dendrites. Spatial information of signals responsible for LTD, including PKC and MAPK, is largely unknown, so further comprehensive studies are required to understand the region where PKC-dependent dissociation of RKIP from Raf-1 and MEK takes place.
Immunoprecipitation has been used previously to demonstrate the association of RKIP with target molecules (Yeung et al., 2001; Corbit et al., 2003; Lorenz et al., 2003). Although the degree of association of Raf-1 or MEK with RKIP in control slices looked small, this could be due to their low binding affinity. A previous report described that the association of Raf-1 with RKIP is sensitive to lysis conditions (Corbit et al., 2003). In the present study, we needed to use somewhat strong conditions to adequately lyse the slices. Even under such conditions, we detected that RKIP bound with Raf-1 and MEK in control slices, and that RKIP dissociated from them after chemical LTD stimulation. Thus, the immunoprecipitation experiments can be used for investigating the interaction between RKIP and Raf-1 or MEK in brain slices.
Significance of RKIP in neural functions
The expression of RKIP has been observed in many brain areas, as well as in other tissues. Within the brain, Purkinje cells are one of the areas exhibiting strong expression of RKIP (Frayne et al., 1999). Nevertheless, the function of RKIP in Purkinje cells has not been described previously. In the hippocampus, RKIP is thought to be a precursor of hippocampal cholinergic neurostimulating peptide (HCNP), which promotes the differentiation of hippocampal cholinergic neurons (Ojika et al., 2000). RKIP in Purkinje cells is unlikely to serve as a precursor of HCNP, because there are few cholinergic neurons in the cerebellum; most cholinergic input is provided by mossy fibers (Jaarsma et al., 1997), which innervate granule cells, but not Purkinje cells. Instead, we found that RKIP participates in synaptic depression in Purkinje cells by mediating PKC-induced activation of MAPK. In general, RKIP has been considered as a suppressor of various diseases, such as cancer and Alzheimer's disease, by maintaining the balance of intracellular signaling (Keller et al., 2005; Granovsky and Rosner, 2008; Klysik et al., 2008). Considering the ability of RKIP to be regulated by phosphorylation, such regulation of RKIP could also be involved in triggering cellular signaling functions. In fact, it was reported previously that the phosphorylation of RKIP is involved in the regulation of MAPK signaling in the suprachiasmic nucleus (SCN) and photic entrainment of the SCN circadian clock (Antoun et al., 2012). In addition, our present study demonstrates that the phosphorylation of RKIP is important for triggering at least one form of long-term synaptic plasticity.
Footnotes
This work was supported by the World Class Institute (WCI) Program of the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology of Korea (Grant WCI 2009-003), a PRESTO grant from the Japan Science and Technology Agency, the Takeda Science Foundation, and the Research Foundation for Opto-Science and Technology. We thank Dr. George J. Augustine and Dr. C. Justin Lee for valuable comments on this paper, and Dr. Jinhyun Kim for helpful suggestions on immunohistochemical analysis.
The authors declare no competing financial interests.
References
- Antoun G, Cannon PB, Cheng HY. Regulation of MAPK/ERK signaling and photic entrainment of the suprachiasmatic nucleus circadian clock by Raf kinase inhibitor protein. J Neurosci. 2012;32:4867–4877. doi: 10.1523/JNEUROSCI.5650-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Dhami GK, Babwah AV, Sterne-Marr R, Ferguson SS. Phosphorylation-independent regulation of metabotropic glutamate receptor 1 signaling requires G protein-coupled receptor kinase 2 binding to the second intracellular loop. J Biol Chem. 2005;280:24420–24427. doi: 10.1074/jbc.M501650200. [DOI] [PubMed] [Google Scholar]
- Endo S, Launey T. ERKs regulate PKC-dependent synaptic depression and declustering of glutamate receptors in cerebellar Purkinje cells. Neuropharmacology. 2003;45:863–872. doi: 10.1016/s0028-3908(03)00210-7. [DOI] [PubMed] [Google Scholar]
- Finch EA, Tanaka K, Augustine GJ. Calcium as a trigger for cerebellar long-term synaptic depression. Cerebellum. 2012;11:706–717. doi: 10.1007/s12311-011-0314-x. [DOI] [PubMed] [Google Scholar]
- Frayne J, Ingram C, Love S, Hall L. Localisation of phosphatidylethanolamine-binding protein in the brain and other tissues of the rat. Cell Tissue Res. 1999;298:415–423. doi: 10.1007/s004419900113. [DOI] [PubMed] [Google Scholar]
- Granovsky AE, Rosner MR. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18:452–457. doi: 10.1038/cr.2008.43. [DOI] [PubMed] [Google Scholar]
- Hanawa H, Kelly PF, Nathwani AC, Persons DA, Vandergriff JA, Hargrove P, Vanin EF, Nienhuis AW. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transduce primitive hematopoietic cells from human blood. Mol Ther. 2002;5:242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- Hartell NA. cGMP acts within cerebellar Purkinje cells to produce long term depression via mechanisms involving PKC and PKG. Neuroreport. 1994;5:833–836. doi: 10.1097/00001756-199403000-00024. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Ito-Ishida A, Kakegawa W, Yuzaki M. ERK1/2 but not p38 MAP kinase is essential for the long-term depression in mouse cerebellar slices. Eur J Neurosci. 2006;24:1617–1622. doi: 10.1111/j.1460-9568.2006.05055.x. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Ruigrok TJ, Caffé R, Cozzari C, Levey AI, Mugnaini E, Voogd J. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res. 1997;114:67–96. doi: 10.1016/s0079-6123(08)63359-2. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Tabata T. Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos Trans R Soc Lond B Biol Sci. 2008;363:2173–2186. doi: 10.1098/rstb.2008.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Fujii H, Gotoh Y, Morooka T, Shimohama S, Nishida E, Hirano T. Requirement for mitogen-activated protein kinase in cerebellar long term depression. J Biol Chem. 1999;274:13498–13502. doi: 10.1074/jbc.274.19.13498. [DOI] [PubMed] [Google Scholar]
- Keller ET, Fu Z, Brennan M. The biology of a prostate cancer metastasis suppressor protein: Raf kinase inhibitor protein. J Cell Biochem. 2005;94:273–278. doi: 10.1002/jcb.20169. [DOI] [PubMed] [Google Scholar]
- Klysik J, Theroux SJ, Sedivy JM, Moffit JS, Boekelheide K. Signaling crossroads: the function of Raf kinase inhibitory protein in cancer, the central nervous system and reproduction. Cell Signal. 2008;20:1–9. doi: 10.1016/j.cellsig.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marmé D, Rapp UR. Protein kinase Cα activates RAF-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- Kondo T, Kakegawa W, Yuzaki M. Induction of long-term depression and phosphorylation of the δ2 glutamate receptor by protein kinase C in cerebellar slices. Eur J Neurosci. 2005;22:1817–1820. doi: 10.1111/j.1460-9568.2005.04319.x. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Schweighofer N, Kawato M. Exploration of signal transduction pathways in cerebellar long-term depression by kinetic simulation. J Neurosci. 2001;21:5693–5702. doi: 10.1523/JNEUROSCI.21-15-05693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TD, Shirai Y, Okamoto T, Tatsukawa T, Nagao S, Shimizu T, Ito M. Lipid signaling in cytosolic phospholipase A2α-cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proc Natl Acad Sci U S A. 2010;107:3198–3203. doi: 10.1073/pnas.0915020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCα confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY. Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron. 1997;18:1025–1038. doi: 10.1016/s0896-6273(00)80340-2. [DOI] [PubMed] [Google Scholar]
- Linden DJ. Long-term synaptic depression in the mammalian brain. Neuron. 1994;12:457–472. doi: 10.1016/0896-6273(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Linden DJ. Phospholipase A2 controls the induction of short-term versus long-term depression in the cerebellar Purkinje neuron in culture. Neuron. 1995;15:1393–1401. doi: 10.1016/0896-6273(95)90017-9. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109–112. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- Miyata M, Finch EA, Khiroug L, Hashimoto K, Hayasaka S, Oda SI, Inouye M, Takagishi Y, Augustine GJ, Kano M. Local calcium release in dendritic spines required for long-term synaptic depression. Neuron. 2000;28:233–244. doi: 10.1016/s0896-6273(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Morice C, Nothias F, König S, Vernier P, Baccarini M, Vincent JD, Barnier JV. Raf-1 and B-Raf proteins have similar regional distributions but differential subcellular localization in adult rat brain. Eur J Neurosci. 1999;11:1995–2006. doi: 10.1046/j.1460-9568.1999.00609.x. [DOI] [PubMed] [Google Scholar]
- Ohtsuki G, Piochon C, Hansel C. Climbing fiber signaling and cerebellar gain control. Front Cell Neurosci. 2009;3:4. doi: 10.3389/neuro.03.004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojika K, Mitake S, Tohdoh N, Appel SH, Otsuka Y, Katada E, Matsukawa N. Hippocampal cholinergic neurostimulating peptides (HCNP) Prog Neurobiol. 2000;60:37–83. doi: 10.1016/s0301-0082(99)00021-0. [DOI] [PubMed] [Google Scholar]
- Reynolds T, Hartell NA. Roles for nitric oxide and arachidonic acid in the induction of heterosynaptic cerebellar LTD. Neuroreport. 2001;12:133–136. doi: 10.1097/00001756-200101220-00034. [DOI] [PubMed] [Google Scholar]
- Schönwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Takács J, Gombos G, Görcs T, Becker T, de Barry J, Hámori J. Distribution of metabotropic glutamate receptor type 1a in Purkinje cell dendritic spines is independent of the presence of presynaptic parallel fibers. J Neurosci Res. 1997;50:433–442. doi: 10.1002/(SICI)1097-4547(19971101)50:3<433::AID-JNR9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Augustine GJ. A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron. 2008;59:608–620. doi: 10.1016/j.neuron.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Khiroug L, Santamaria F, Doi T, Ogasawara H, Ellis-Davies GC, Kawato M, Augustine GJ. Ca2+ requirements for cerebellar long-term synaptic depression: role for a postsynaptic leaky integrator. Neuron. 2007;54:787–800. doi: 10.1016/j.neuron.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Torashima T, Okoyama S, Nishizaki T, Hirai H. In vivo transduction of murine cerebellar Purkinje cells by HIV-derived lentiviral vectors. Brain Res. 2006;1082:11–22. doi: 10.1016/j.brainres.2006.01.104. [DOI] [PubMed] [Google Scholar]
- Verin AD, Liu F, Bogatcheva N, Borbiev T, Hershenson MB, Wang P, Garcia JG. Role of Ras-dependent ERK activation in phorbol ester-induced endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2000;279:L360–L370. doi: 10.1152/ajplung.2000.279.2.L360. [DOI] [PubMed] [Google Scholar]
- Wang SS, Augustine GJ. Confocal imaging and local photolysis of caged compounds: dual probes of synaptic function. Neuron. 1995;15:755–760. doi: 10.1016/0896-6273(95)90167-1. [DOI] [PubMed] [Google Scholar]
- Wang SS, Khiroug L, Augustine GJ. Quantification of spread of cerebellar long-term depression with chemical two-photon uncaging of glutamate. Proc Natl Acad Sci U S A. 2000;97:8635–8640. doi: 10.1073/pnas.130414597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang S, Tang X, Zhang A, Grabinski T, Guo Z, Hudson E, Berghuis B, Webb C, Zhao P, Cao B. Development and evaluation of monoclonal antibodies against phosphatidylethanolamine binding protein 1 in pancreatic cancer patients. J Immunol Methods. 2010;362:151–160. doi: 10.1016/j.jim.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Wen-Sheng W. Protein kinase C α trigger Ras and Raf-independent MEK/ERK activation for TPA-induced growth inhibition of human hepatoma cell HepG2. Cancer Lett. 2006;239:27–35. doi: 10.1016/j.canlet.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Wiegert JS, Bengtson CP, Bading H. Diffusion and not active transport underlies and limits ERK1/2 synapse-to-nucleus signaling in hippocampal neurons. J Biol Chem. 2007;282:29621–29633. doi: 10.1074/jbc.M701448200. [DOI] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–3085. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-κB-inducing kinase and TAK1 and inhibits NF-κB activation. Mol Cell Biol. 2001;21:7207–7217. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. Localization of diacylglycerol lipase- 128 around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Liu H, Coughlin J, Zheng J, Li L, Stone JC. Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood. 2005;105:3648–3654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]