

Summary
Interferon (IFN)-free direct-acting antiviral agents (DAAs) have revolutionized chronic hepatitis C virus (HCV) treatment; early studies suggest excellent efficacy in acute HCV. However, changes in innate immune responses during DAA therapy for acute HCV are unknown. We studied interferon-stimulated gene (ISG) expression and related cytokines/chemokines in HIV-infected patients with acute HCV receiving sofosbuvir plus ribavirin (SOF+RBV) as part of the A5327 clinical trial. ISG expression was determined from PBMCs, and circulating cytokines/chemokines were quantified from serum from study participants. The overall sustained virologic response (SVR) was 57%; all treatment failures were due to virologic relapse. Apart from NOS2a, baseline ISG/chemokine/cytokine levels were similar irrespective of treatment outcome. Downregulation of ISGs was observed at treatment week four and end of treatment (EOT), implicating HCV in establishing elevated ISGs early during HCV infection. Levels of many of these ISGs increased at post-treatment week 12 (PTW12) in relapsers only, coinciding with recurrent HCV RNA. Eleven ISGs were differentially expressed in responders vs relapsers. On-treatment viral suppression was also associated with a reduction in IP-10, CXCL11 and MIP-1β levels. In contrast, circulating IFN-α levels were significantly higher at EOT and PTW12 in responders vs relapsers. Upregulation of peripheral ISG expression is established early in the course of HCV infection during acute HCV infection, but did not predict subsequent treatment outcome with SOF+RBV. ISGs were downregulated during therapy and increased post-therapy in relapsers. IFN-α levels were higher in responders at EOT/PTW12, suggesting that impaired type I IFN production/secretion may contribute to relapse.
Keywords: acute hepatitis C, cytokines/chemokines, direct-acting antiviral (DAA) therapy, HCV/HIV coinfection, IFN-free, interferon-stimulated gene (ISG) expression, treatment response
1 |. INTRODUCTION
Hepatitis C virus (HCV) infects over 71 million people worldwide and is a significant etiological agent in the development of chronic liver disease.1 HCV establishes chronic infection in the majority of patients, while only a minority of patients (15%−25%) with acute HCV spontaneously clear the virus. Long-term chronic HCV infection results in persistent liver injury that can progress to cirrhosis after 15–20 years and risks of complications of end-stage liver disease such as decompensated liver failure, hepatocellular carcinoma and death.2 Human immunodeficiency virus (HIV) coinfection with HCV occurs in approximately 30% of patients owing to similar transmission routes and has been associated with accelerated liver fibrosis progression and more frequent liver-related complications, such as decompensation.3 In the postantiretroviral era, liver disease is now the second leading cause of non-AIDS-related mortality in HIV patients and therefore represents a significant health issue in these individuals.3
The development of direct-acting antiviral agents (DAAs) that target specific steps in the HCV replication cycle have revolutionalized antiviral therapy for chronic HCV, allowing combination interferon (IFN)-free DAA regimens that are not only highly efficacious (>95% sustained virologic response [SVR] rate) but also have a significantly improved adverse event profile. However, studies of their efficacy in acute HCV are limited. In addition, there is little known about the host-virus interactions in the setting of DAAs and how the immune system will respond to the inhibition of replication without concomitant exogenous interferon administration. It is well established that chronic HCV is associated with changes in innate immune responses characterized by the upregulation of interferon-stimulated genes (ISGs) 4–6 and elevations in several relevant IFN-sensitive chemokines and cytokines.7–10 The degree of host immune dysregulation has been associated with treatment outcomes with IFN-based therapy, and HCV eradication with IFN-based regimens has been shown to restore ISG and type I IFN responses.4–6 There are extremely limited data regarding reversal of impaired immune responses in the context of DAA therapy. Several small studies have demonstrated improvement in liver ISG expression, restoration of type I IFN signalling, and natural killer and T cell function following IFN-free DAA therapy in the setting of chronic infection.11–14
The aims of this study were to characterize the effect of viral antigen removal on peripheral ISG and interferon-effector gene (IEG) expression and circulating chemokines/cytokines in participants enrolled in the A5327 study who were receiving sofosbuvir (SOF) and ribavirin (RBV) for the treatment of acute HCV infection. Furthermore, we planned to assess levels of ISGs/IEGs or chemokines/cytokines for any association with viral eradication and treatment response. In the parent study, only 58% of individuals achieved an SVR, with the remaining patients relapsing following the completion of IFN-free therapy, representing a unique and well-characterized cohort to study differences in immune profiles between patients who ultimately clear virus and those who relapse following DAA therapy in the context of acute HCV infection.
2 |. MATERIALS AND METHODS
2.1 |. Patients and study design
A5327 was an open-label multicenter clinical trial to determine the safety and efficacy of IFN-free DAA therapy in HIV-1 subjects with confirmed acute HCV infection (NCT02128217). This study focuses on cohort one of this trial, where patients were treated with sofosbuvir plus weight-based ribavirin for 12 weeks. Key inclusion/exclusion criteria for entry into this study are summarized in Table S1 (see NCT02128217 for additional detail). All patients were HIV-1 positive on antiretroviral therapy (ART) and demonstrated HIV viral suppression throughout the study.
All subjects consented to collection of serum and peripheral blood mononuclear cells (PBMCs) at baseline, on-treatment week 4 (TW4), end of treatment (EOT) and at post-treatment week 12 (PTW12). Immune profiling was completed at each time point. Per the parent protocol, SVR was defined as HCV RNA less than the lower limit of quantification (<LLoQ) target not detected (TND) at PTW12, and relapse was defined as HCV RNA undetectable (<LLoQ and TND) at EOT but quantifiable HCV RNA (≥LLoQ) at any time during post-treatment follow-up. Participants achieving SVR will be referred to as responders in this study. The study was approved by each local institutional review board, and all participants provided written consent. The study was conducted according to the Declaration of Helsinki.
2.2 |. IFNL3 rs12979860 genotyping
Genomic DNA was extracted from PBMCs using the QuickExtract DNA Extraction Protocol (EPICENTRE® Biotechnologies, Madison, WI, USA), as per the manufacturer’s instructions. rs12979860 genotyping was performed by real-time PCR and direct PCR Sanger sequencing (primers: sense 5′-AGAAGCAGAGATGCGGC −3′ and antisense 5′-TCTGGGATTCCTGGACG −3′). Sequence alignment was performed using CodonCode v5.1.5 (CodonCode Corporation, Centerville, MA, USA).
2.3 |. HCV RNA quantification and HIV RNA quantification
HCV RNA was measured using the AmpliPrep/COBAS® assay (Roche, California, USA), where the lower limit of detection (LLoD) is 15 IU/mL. HIV RNA was measured using the LCx HIV RNA quantitative assay (Abbott Molecular, Rungis, France), with a dynamic range of 50–1 000 000 copies/mL.
2.4 |. RNA extraction and gene expression profiling
RNA was extracted from rapidly thawed cryopreserved PBMCs using the RNeasy Mini Kit (Qiagen, Hilden, Germany), as per the manufacturer’s instructions. Using 300 ng of total RNA, mRNA expression of a custom nCounter® codeset of 47 ISGs and nine IEGs was measured (NanoString® Technologies, Seattle, WA, USA). These ISGs/IEGs were chosen based on previously published literature indicating their relevance in the context of chronic HCV infection.4–6,15 Data were normalized to four housekeeping genes using the nSolver software v2.6 (NanoString® Technologies). Baseline log2 normalized mRNA expression and differential gene expression at each treatment time point were compared to baseline levels and according to subsequent treatment response. Gene expression profiles were also compared at each time point to baseline, both as normalized gene expression levels and expressed as fold change from baseline and time point of interest.
2.5 |. Cytokine/chemokine quantification
Plasma collected at each study time point and immediately frozen at −80°C was used to quantify a panel of chemokines and relevant cytokines. Levels and fold change in levels at each treatment time point were compared to baseline levels and also according to subsequent treatment response. Interleukin (IL)-6, IL-8, IL-10, IL-12p70, CXCL11, macrophage inflammatory protein (MIP)-1α, IFN-γ and TNF-α were measured using the human high-sensitivity T cell panel kit (Millipore, Billerica, MA, USA). Interferon-gamma-induced protein 10 (IP-10), IFN-α, monocyte chemoattractant protein-1 (MCP-1) and MIP-1β were measured using the human cytokine/chemokine magnetic bead kit (Millipore). ENA78 and IL-18 were measured using the human ProcartaPlex custom panel (Thermo Fisher Scientific, Waltham, MA), and chemokine (C-C motif) ligand five (RANTES) was measured using the human RANTES ELISA kit (eBioscience, San Diego, CA, USA).
2.6 |. Statistical analyses
Change in immune profiles from baseline to each study time point was calculated. Categorical data are presented as frequencies, and continuous data are presented as medians with interquartile ranges. The chi-squared and Fisher’s exact t tests were used to compare categorical data, and continuous data were compared with the MannWhitney U test and the Wilcoxon signed-rank test for unpaired and paired samples, respectively. GraphPad Prism v6 (GraphPad Software, Inc., La Jolla, CA) and STATA v11 were used to compare baseline demographics and levels of chemokines/cytokines. Peripheral gene expression data were generated using the nSolver software (NanoString Technologies), and R Studio v0.99.902 (RStudio, Inc., Boston, MA) and STATA v11 (StataCorp LP, College Station, TX) were used to determine differentially expressed genes in responders vs relapse. A P-value of <.05 was considered significant, and a P-value with Bonferroni correction was set at .001.
3 |. RESULTS
3.1 |. Patients
Fourteen of the 17 patients enrolled in the original parent study were included in this substudy. Three patients were excluded for the following reasons: two patients were found to have detectable HCV RNA at screening but at baseline the HCV RNA was subsequently found to be undetectable or extremely low at baseline (<100 IU/mL) and therefore these patients were excluded as they were felt likely to have different immune profiles to the remaining patients with high HCV RNA levels; the third patient did not have stored paired PBMC and plasma samples available for all study time points.
The baseline demographics and patient characteristics are presented in Table 1. In brief, all patients were male, with a median age of 46 years, and predominantly infected with HCV genotype one (93%). The majority of patients had the non-C/C IFNL3 genotype (8% C/C, 71% C/T and 21% T/T). Median baseline HCV RNA level was 6.39 log10 IU/mL. All patients were receiving ART with suppression of HIV RNA <50 copies/mL, and the median CD4 count was 452 cells/mm3. There were no significant differences in baseline demographics and patient characteristics between responders and relapsers (Table 1).
TABLE 1.
Baseline demographics and patient characteristics
| Responders (n = 8) |
Relapsers (n = 6) |
P-value | |
|---|---|---|---|
| Age, years (median, range) | 46.5 (21–65) | 45.5 (29–48) | .6040 |
| Male sex (n, %) | 8 (100%) | 6 (100%) | - |
| ALT, mg/dL (median, range) | 206.5 (92–564) | 183.0 (201–284) | .5181 |
| AST, mg/dL (median, range) | 134.0 (56–395) | 103.5 (83–162) | .6982 |
| Total bilirubin, mg/dL (median, range) | 0.7 (0.4–2.1) | 0.7 (0.6–1.1) | .6455 |
| IFNL3 genotype (CC) (n, %) | 1 (12.5%) | 0 (0) | 1.0000 |
| IFNL3 genotype (CT/TT) (n, %) | 7/1 (88%/12%) | 6/0 (100%/0%) | .5590 |
| HIV viral load, copies/mL (median, range) | <50 (<50–80) | <50 (<50–<50) | .3865 |
| CD4 count, cells/mm3 (median, range) | 458.0 (331–652) | 473.5 (213–618) | .5186 |
| HCV genotype 1 (n, %) | 7 (88%) | 6 (100%) | 1.0000 |
| HCV viral load, log10 IU/mL (median, range) | 6.22 (4.48–6.87) | 6.50 (5.82–7.28) | .3662 |
In the parent study, the overall SVR rate for cohort one was only 58% (10 of 17), and all virologic failures were due to virologic relapse.16 In this substudy, two of the 14 patients had high HCV RNA levels at baseline, defined as HCV RNA levels >6 000 000 IU/mL. At TW4, 86% had HCV RNA levels below the LLoD, and all patients had undetectable HCV RNA levels at EOT (Figure 1). The SVR rate was similar to the overall study (57%, eight of 14) (Figure 1). As observed in the parent study, all treatment failures were virologic relapse and no patient experienced viral breakthrough or viral nonresponse.
FIGURE 1.
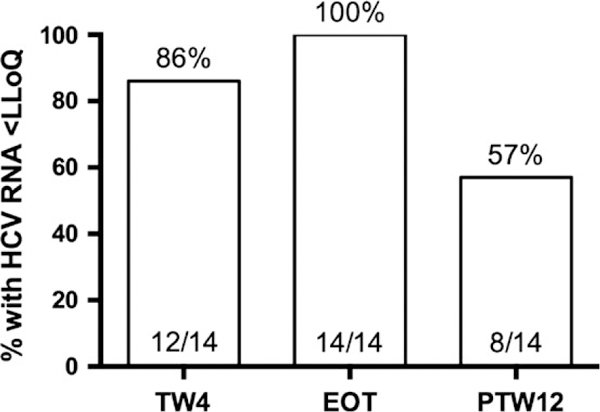
On-treatment and post-treatment virologie response. All virologie failures were relapses. HCV, hepatitis C virus; LLoQ, lower limit of quantification; TW4, treatment week 4; EOT, end of therapy; PTW12, post-treatment week 12
3.2 |. Gene expression
We determined the gene expression profiles of a panel of biologically relevant ISGs (n = 45) and lEGs (n = 9) previously reported to be important in HCV4–6,17,18 in PBMCs at baseline, TW4, EOT and PTW12. Of the 14 patients from this substudy, 13 were included in this gene expression analysis; one patient was excluded as their baseline RNA sample failed stringent NanoString quality control parameters.
3.2.1 |. Baseline ISG/IEG expression profiles
Figure 2 displays the heat map depicting the normalized log2-transformed peripheral ISG and IEG expression levels for all 13 patients at baseline. Of the 45 ISGs and nine lEGs examined, only NOS2a gene expression was differentially expressed >1.5-fold in responders compared to relapsers (P = .016); however, this failed to meet statistical significance when correcting for multiple testing. There were no other significant differences at baseline according to subsequent treatment response. Baseline ISG/IEG expression analysis according to IFNL3 genotype was not able to be performed as only one patient carried the C/C IFNL3 genotype.
FIGURE 2.
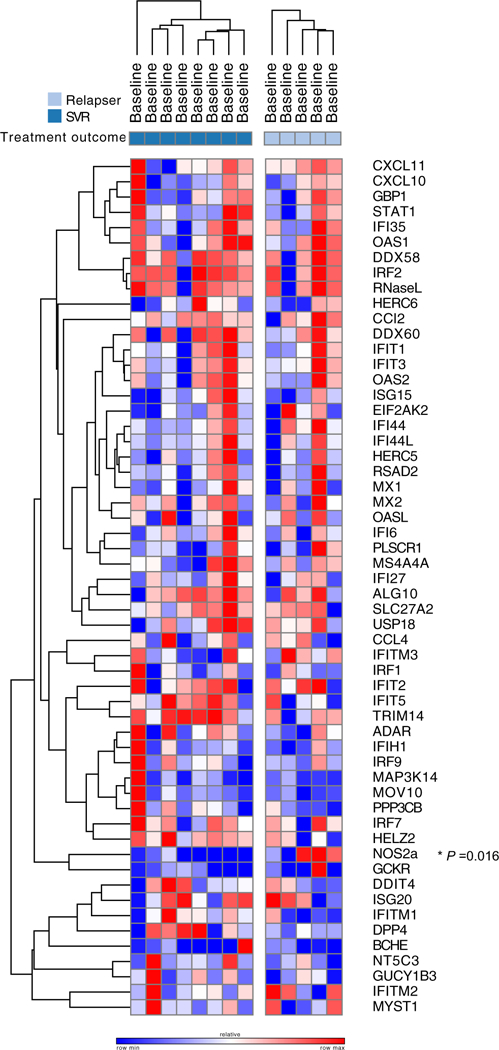
Heat map demonstrating baseline interferon-stimulated gene (ISG)/interferon-effector gene (IEG) log2-normalized gene expression among responders (SVR, dark blue) and relapsers (light blue). Red on the heat map represents upregulation and blue represents downregulation
3.2.2 |. On-treatment and post-treatment ISG/IEG expression profiles
Treatment week 4 (TW4)
At TW4, the majority of ISGs were downregulated (37 of 47) in the setting of on-treatment viral clearance in all patients, whereas only three of the nine IEGs were downregulated at TW4 (4 upregulated, two unchanged). However, only five genes (all ISGs) demonstrated >1.5-fold differential expression compared to baseline levels with a P-value <.05, including CXCL10, CXCL11, IFI44L, IFIT1 and IFIT3 (Figure 3, Table S2). In addition, 10 genes were upregulated at TW4, but none were differentially expressed >1.5-fold above baseline levels (Figure 3, Table S2). When stratified according to treatment outcome, only two genes were found to be differentially expressed >1.5-fold among responders and relapsers at TW4, including NOS2a (downregulated in relapsers) and GUCY1B3 (upregulated in relapsers) (all P-values <.05) (Table S3).
FIGURE 3.

Heat map demonstrating changes in interferon-stimulated gene (ISG)/interferon-effector gene (IEG) log2-normalized gene expression during sofosbuvir plus ribavirin therapy. Red on the heat map represents upregulation and blue represents downregulation
Note: SVR = sustained virologic response
TW4 = treatment week 4
EOT = end of treatment
PTW12 = post-treatment week 12
End of Treatment (EOT)
At EOT, the majority of ISGs were downregulated (40 of 47), 6 of which were >1.5-fold differentially expressed with a P-value <.05 (Figure 3, Table S2). Only four of the nine IEGs were downregulated, but none were differentially expressed >1.5-fold above baseline. Of the remaining IEGs, two IEGs were unchanged and three were upregulated but were <1.5-fold above baseline.
In addition, the gene expression levels of the ISGs DDIT4, IFI27, IFI44, IFIT2 and RSAD2, as well as the lEGs GCKR and GUCY1B3, were differentially expressed >1.5-fold between relapsers compared to responders (P < .05) (Table S3).
Post-Treatment Week 12 (PTW12)
In the context of viral relapse, gene expression profiles differed in responders compared to relapsers, with upregulation of 32 of the 47 ISGs in relapsers (Figure 3, Table S2), 11 of which were differentially expressed more than 1.5-fold with a P-value <.05. No lEGs were found to have differential expression according to treatment outcome at PTW12 (Figure 3, Table S3).
3.3 |. Changes in plasma levels of chemokines/cytokines with DAA treatment
3.3.1 |. Baseline
We determined the levels of a panel of circulating chemokines and cytokines from plasma samples at each time point reported to be associated with treatment response in chronic HCV infection. Figure 4 shows the median levels for six of the 14 quantified chemokines/cytokines according to treatment outcome (SVR vs relapsers) that demonstrated significant differences between responders and relapsers or significant changes during therapy. There was no difference in the median baseline level of any of the 14 chemokines or cytokines in responders compared to relapsers, including in IP-10 levels which have been associated with subsequent treatment outcome with IFN-based therapy in chronic HCV (Figure 4).
FIGURE 4.
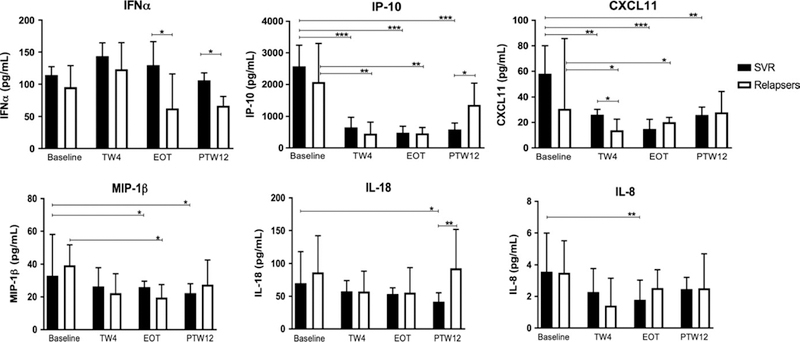
Levels of circulating cytokines and chemokines at baseline and during/after treatment according to treatment outcome. TW4, treatment week 4; EOT, end of treatment; PTW12, post-treatment week 12; *P < .05, **P < .005, ***P < .0005
3.3.2 |. On-treatment and post-treatment
IFNα levels
Interestingly, although levels of IFNα did not significantly change during treatment in responders, there was a trend towards increased IFNα at TW4 in responders, and levels were significantly higher in responders at EOT and PTW12 compared to relapsers (Figure 4), suggesting impaired IFNα production or secretion in patients that subsequently relapse with SOF+RBV.
IP-10, CXCL11, MIP-1β, IL-18 and IL-8 levels
IP-10 levels were significantly lower at TW4 and EOT compared to baseline in both responders and relapsers (Figure 4). IP-10 levels remained low at PTW12 in responders, but returned to pretreatment levels in the context of relapse (Figure 4). Similarly, CXCL11 levels were significantly lower at TW4 and EOT compared to baseline in responders and nonresponders, and significantly lower at PTW12 than at baseline in responders (Figure 4). Compared to baseline, circulating levels of MIP1β were significantly lower at EOT and PTW12 in responders and at EOT only in relapsers (Figure 4). There was trend towards lower IL-18 levels at TW4 and EOT, with significantly lower levels at PTW12 compared to baseline in responders (Figure 4). In addition, levels were also significantly lower in responders compared to relapsers at PTW12. At TW4, levels of IL-8 were significantly lower in responders compared to baseline (Figure 4). There were no other differences in IL-8 at other time points.
4 |. DISCUSSION
There are limited data regarding peripheral ISG/IEG expression and circulating cytokine/chemokine profiling in the context of acute HCV infection in patients coinfected with HIV, especially in the context of the newer DAA-based therapies. It has been well established that intrahepatic ISG expression is associated with subsequent treatment outcome during IFN-based therapy for chronic HCV.4,6 In contrast, baseline peripheral ISG/IEG expression is not always associated with subsequent treatment outcome; however, changes in ISG expression in PBMCs during IFN-based therapy of chronic HCV mirror on-treatment changes in the intrahepatic compartment.6 In addition, levels of several circulating chemokines/cytokines, in particular IP-10, have been associated with subsequent treatment response to IFN-based therapy.7–9,19 In the present study, 14 HIV-1-positive ART-treated patients with confirmed acute HCV deemed unlikely to spontaneously clear (>12 weeks acute HCV infection) received SOF+RBV IFN-free DAA therapy for 12 weeks. All patients were male and demonstrated adequate HIV RNA suppression with ART. The SVR rate with this IFN-free DAA regimen was unexpectedly low at only 57% (8/14), due to a high relapse rate, representing a unique cohort to interrogate the effect of DAA-induced viral suppression and re-appearance on innate immune responses in HIV-1 patients with acute HCV.
Contrary to the available data in chronic HCV patients, baseline IP-10 levels and levels of other circulating chemokines and cytokines did not differ significantly according to subsequent treatment outcome in this study. Peripheral NOS2a expression, an ISG previously shown to be a restriction factor for HCV replication,20 was the only ISG/IEG that was differentially expressed in patients who subsequently relapsed compared to responders. These data suggest that the equilibrium patterns of differential cytokine/chemokine induction and ISG expression demonstrated in CHC may not yet be fully established in acute HCV. However, we did observe that the majority of ISGs did become down-regulated at TW4 and EOT compared to baseline in the setting of on-treatment viral suppression, suggesting that upregulation of ISGs that is observed in CHC may already be established very early on in the natural history of infection during acute HCV that is able to be modulated and reversed following successful viral eradication with IFN-free DAA therapy. This is further supported by an increase in ISGs at PTW12 in patients who experienced relapse, suggesting a virally mediated increase in ISG expression.
The timing of ISG downregulation that we observed during DAA therapy in this study was different to patterns reported with prior standard of care (IFN-based) therapy for CHC where upregulation of ISGs was observed at TW4 in individuals who subsequently achieved SVR. In contrast, we observed downregulation of the majority of ISGs at TW4 (37 of 47 ISGs) and EOT (40 of 47 ISGs). Although the DAA regimen of SOF+RBV resulted in an SVR rate of only 58%, this regimen was still more effective in rapidly suppressing HCV than IFN-based therapy. We observed that 86% of our acute HCV patients treated with SOF+RBV had an HCV RNA level less than the LLoD compared to historical rates of only 20%−25% during IFN-based therapy. This increased early potency of therapy may therefore have altered changes in patterns of ISG expression with more rapid normalization of ISGs with more rapid viral suppression. Therefore, earlier time points during DAA therapy may be useful to follow changes in patterns of ISGs and determine if sets of ISGs may predict relapse with IFN-free DAA therapy. Interestingly, patterns of IEG expression were different to ISGs where there was less alteration in IEG expression during viral suppression, although it should be noted that only nine IEGs were examined.
As previously reported in chronic HCV patients, we also observed a reduction in levels of several circulating chemokines/cytokines in the context of on-treatment viral suppression, several of which rebounded to pretreatment levels in relapsers but remained low in responders, suggestive of a direct viral-mediated response. Circulating IFNα levels demonstrated a different pattern, where we observed that IFNα levels were significantly lower in relapsers at EOT and PTW12 compared to responders. The downregulation of ISGs at TW4 and their rebound at PTW12, accompanied by reduced IFNα levels, suggests that the ISG phenotype is only partially reversed, and that IFNα production or secretion may be impaired in relapsers, which may in turn impair type I IFN responses that are critical for optimal cell-mediated viral clearance and prevention of viral relapse. Conversely, the ISG downregulation of ISGs at TW4 that is sustained, together with greater circulating IFNα levels in responders, suggests that there is reversal of the ISG phenotype and that there is resetting of IFN-responsiveness that enables long-term viral eradication. These results are supported by prior findings in the intrahepatic compartment in chronic HCV patients treated with the same IFN-free regimen, where successful HCV clearance was associated with strong and early intrahepatic ISG induction, but increased type I IFN responses at EOT, suggesting HCV eradication and prevention of relapse were facilitated by restoring type I IFN responses in the liver. 11
This study is limited by the small number of patients and serves primarily as proof-of-concept that reversal of the “exhausted” ISG phenotype does occur during IFN-free DAA therapy for acute HCV, and that improved IFNα secretion or production at EOT and PTW12 may be important to prevent relapse. In addition, patients in this study were all coinfected with HIV, and although all maintained controlled HIV RNA levels on ART, immune reconstitution is not universal in all compartments, and normal immune health may not be achieved with HIV suppression alone. This study requires validation in larger cohorts of acute HCV patients with HIV coinfection, and similar studies should be conducted in acute HCV patients without HIV infection to determine the contribution of HIV to these findings. It would also be important to compare peripheral and intrahepatic ISG/IEG expression profiles, although with the advent of noninvasive biomarkers of fibrosis and the morbidity/mortality associated with liver biopsy, it is becoming increasingly difficult to obtain liver tissue for intrahepatic studies, particularly in acute HCV where there is no clinical indication for liver biopsy. Owing to the small number of patients and the low frequency of the “good-responder” IFNL3 genotype in this cohort, we were unable to perform a dedicated subanalysis according to IFNL3 genotype, which is relevant given the historical association between IFNL3 genotype, baseline circulation chemokine/cytokine levels and ISG expression. Given the exploratory nature of this study, and the fact that only 56 ISGs/IEGs were tested, many of which are known to be antiviral within limited pathways, a more comprehensive panel of ISGs/IEGs should be examined in larger cohorts where more robust statistical analyses can be performed.
This study is the first to demonstrate that peripheral ISGs are already elevated in the context of acute HCV and can be reversed with DAAs. Taken together, our data suggest that IFN-free DAA therapy induces innate immune responses in the periphery of HIV-1 patients with acute HCV infection, and that ISGs and IEGs may play important and potentially differing roles in DAA-induced viral clearance, suggesting their antiviral biological functions may be distinctly different. We also demonstrate that despite the high relapse rate with this weak DAA combination, patterns of changes in ISG expression differ from those previously observed in chronic HCV patients treated with the suboptimal prior standard of care therapy (IFN + RBV), whereby there was downregulation of the vast majority of ISGs by TW4. Apart from NOS2a, we did not identify any differences in ISG/IEG expression or circulating cytokine/chemokine levels according to subsequent treatment response in our acute HCV cohort. The sustained prolonged downregulation of ISGs in responders suggests that there is reversal of the exhausted ISG phenotype and improved type I IFN secretion/production at EOT/PTW12 with DAAs even in the context of acute HCV infection. Similar studies in larger acute HCV cohorts and with more potent regimens should be performed to confirm these findings.
Supplementary Material
Acknowledgments
Funding information
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number UM1 AI068634, UM1 AI068636 and UM1 AI106701. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. RTC was supported by NIH DK098079, AI082630 and DK078772. JAH is supported by an AASLD/LIFER Clinical and Translational Research Fellowship and a National Health and Medical Research Council Early Career Fellowship.
Abbreviations:
- ALT
alanine aminotransferase
- ART
antiretroviral therapy
- AST
aspartate aminotransferase
- DAAs
direct-acting antivirals
- EOT
end of treatment
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- IEG
interferon-effector gene
- IFN
interferon
- IL
interleukin
- IP-10
interferon-gamma-induced protein 10
- ISG
interferon-stimulated gene
- LLoD
lower limit of detection
- LLoQ
lower limit of quantification
- MCP-1
monocyte chemoattractant protein-1
- MIP
macrophage inflammatory protein
- PBMCs
peripheral blood mononuclear cells
- PTW12
post-treatment week 12
- RBV
ribavirin
- SOF
Sofosbuvir
- SVR
sustained virologic response
- TND
target not detected
- TW4
treatment week 4
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- 1.WHO. Global Hepatitis Report 2017. In: Hepatitis; 2017. [Google Scholar]
- 2.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:S35–S46. [DOI] [PubMed] [Google Scholar]
- 3.Smith CJ, Ryom L, Weber R, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;384:241–248. [DOI] [PubMed] [Google Scholar]
- 4.Honda M, Sakai A, Yamashita T, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. [DOI] [PubMed] [Google Scholar]
- 5.Urban TJ, Thompson AJ, Bradrick SS, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105:7034–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagging M, Askarieh G, Negro F, et al. Response prediction in chronic hepatitis C by assessment of IP-10 and IL28B-related single nucleotide polymorphisms. PLoS ONE. 2011;6:e17232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fattovich G, Covolo L, Bibert S, et al. IL28B polymorphisms, IP-10 and viral load predict virological response to therapy in chronic hepatitis C. Aliment Pharmacol Ther. 2011;33:1162–1172. [DOI] [PubMed] [Google Scholar]
- 9.Jablonska J, Pawlowski T, Laskus T, et al. The correlation between pretreatment cytokine expression patterns in peripheral blood mononuclear cells with chronic hepatitis C outcome. BMC Infect Dis. 2015;15:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han ZQ, Huang T, Deng YZ, Zhu GZ. Expression profile and kinetics of cytokines and chemokines in patients with chronic hepatitis C. Int J Clin Exp Med. 2015;8:17995–18003. [PMC free article] [PubMed] [Google Scholar]
- 11.Meissner EG, Wu D, Osinusi A, et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J Clin Invest. 2014;124:3352–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serti E, Chepa-Lotrea X, Kim YJ, et al. Successful interferon-free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology. 2015;149:e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin B, Hennecke N, Lohmann V, et al. Restoration of HCV-specific CD8 + T cell function by interferon-free therapy. J Hepatol. 2014;61:538–543. [DOI] [PubMed] [Google Scholar]
- 14.Larrubia JR, Moreno-Cubero E, Miquel J, Sanz-de-Villalobos E. Hepatitis C virus-specific cytotoxic T cell response restoration after treatment-induced hepatitis C virus control. World J Gastroenterol. 2015;21:3480–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Borozan I, Sun J, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138:e1121–e1123. [DOI] [PubMed] [Google Scholar]
- 16.Naggie S, Marks KM, Hughes M, et al. Sofosbuvir plus ribavirin without interferon for treatment of acute hepatitis C virus infection in HIV-1-infected individuals: SWIFT-C. Clin Infect Dis. 2017;64:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Borozan I, Feld J, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. [DOI] [PubMed] [Google Scholar]
- 18.Fusco DN, Brisac C, John SP, et al. A genetic screen identifies interferon-alpha effector genes required to suppress hepatitis C virus replication. Gastroenterology. 2013;144:1438–1449, 1449 e1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoneda S, Umemura T, Katsuyama Y, et al. Association of serum cytokine levels with treatment response to pegylated interferon and ribavirin therapy in genotype 1 chronic hepatitis C patients. J Infect Dis. 2011;203:1087–1095. [DOI] [PubMed] [Google Scholar]
- 20.Metz P, Dazert E, Ruggieri A, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012;56:2082–2093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.