

Abstract
To interact with the egg, the spermatozoon must undergo several biochemical and motility modifications in the female reproductive tract, collectively called capacitation. Only capacitated sperm can undergo acrosomal exocytosis, near or on the egg, a process that allows the sperm to penetrate and fertilize the egg. In the present study, we investigated the involvement of cyclic adenosine monophosphate (cAMP)-dependent processes on acrosomal exocytosis. Inhibition of protein kinase A (PKA) at the end of capacitation induced acrosomal exocytosis. This process is cAMP-dependent; however, the addition of relatively high concentration of the membrane-permeable 8-bromo-cAMP (8Br-cAMP, 0.1 mmol l−1) analog induced significant inhibition of the acrosomal exocytosis. The induction of acrosomal exocytosis by PKA inhibition was significantly inhibited by an exchange protein directly activated by cAMP (EPAC) ESI09 inhibitor. The EPAC selective substrate activated AE at relatively low concentrations (0.02–0.1 μmol l−1), whereas higher concentrations (>5 μmol l−1) were inhibitory to the AE induced by PKA inhibition. Inhibition of PKA revealed about 50% increase in intracellular cAMP levels, conditions under which EPAC can be activated to induce the AE. Induction of AE by activating the actin severing-protein, gelsolin, which causes F-actin dispersion, was inhibited by the EPAC inhibitor. The AE induced by PKA inhibition was mediated by phospholipase C activity but not by the Ca2+-channel, CatSper. Thus, inhibition of PKA at the end of the capacitation process induced EPAC/phospholipase C-dependent acrosomal exocytosis. EPAC mediates F-actin depolymerization and/or activation of effectors downstream to F-actin breakdown that lead to acrosomal exocytosis.
Keywords: acrosomal exocytosis, exchange protein directly activated by cyclic adenosine monophosphate, protein kinase A, sperm
INTRODUCTION
Mammalian spermatozoa must reside in the female reproductive tract for several hours before gaining the ability to fertilize the egg. During this time, the sperm undergoes several biochemical modifications and motility changes, collectively termed capacitation. The capacitated spermatozoon can undergo the acrosome reaction or acrosomal exocytosis (AE), a process that enables it to penetrate and fertilize the egg. The AE is an exocytotic process in which the outer acrosomal membrane fuses with the overlying plasma membrane. AE triggers evoke Ca2+ and H+ efflux via the sperm plasma membrane, as well as Ca2+ mobilization from the acrosome. It is well established that the cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) mediates sperm capacitation, including indirect elevation of protein tyrosine phosphorylation,1 actin polymerization,2 and hyper-activated motility.3 Mice lacking the sperm-specific PKA catalytic subunit α2 are sterile, and their sperm cannot develop capacitation-dependent hyper-activated motility.4,5
For many years, it was thought that cAMP/PKA mediates the AE process. In the recent years, it was found that cAMP can mediate cellular processes via a PKA-independent mechanism, which includes the small GTPase Rap1 activated by the guanine nucleotide exchange factor, exchange protein directly activated by cAMP (EPAC).6,7,8 The best-characterized cAMP targets are PKA, EPAC, and cyclic-nucleotide-gated channels. The testis-specific Na+/H+ exchanger also contains a cAMP binding site.9 In other cell types, it was shown that PKA and EPAC are both involved in exocytic processes by facilitating Ca2+ release from intracellular Ca2+ stores. In mouse sperm, the PKA catalytic subunit α4 is not expressed in the sperm head, suggesting that PKA, in contrast to cAMP1,10,11,12,13 is probably not involved in the mechanism that mediates the AE.10
It was already shown that EPAC mediates human and mouse AE.13,14 In the present study, we showed that inhibition of PKA induces EPAC-dependent AE. Moreover, induction of AE by progesterone, angiotensin II, thapsigargin (TG) or Ca2+-ionophore is also mediated by EPAC suggesting a physiological role of EPAC in the AE mechanism.
MATERIALS AND METHODS
Reagents
A23187, TG, 1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione (U73122) and 2'5'-dideoxyadenosine (ddAdo) were purchased from Calbiochem (San Diego, CA, USA). All other chemicals were purchased from Sigma-Aldrich Israel Ltd. (Rehovot, Israel) unless otherwise stated.
Sperm preparation
Human semen from fertile men was liquefied by incubation for 30 min at room temperature; then, the semen was loaded on a 40%–80% gradient (PureCeption Lower and Upper Phase Gradient) and centrifuged (5810R, Eppendorf, Hamburg, Germany) for 10 min at 3000 g at room temperature. The lower layer containing the sperm was collected and resuspended twice in Ham's F-10 medium containing 21 mmol l−1 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 25 mmol l−1 sodium bicarbonate (Cat No. 144-55-8), 0.6% human serum albumin, 7.6 mmol l−1 sodium lactate (Cat No. 312-85-6) washed in Ham's F-10, then centrifuged again, and the sperm allowed to “swim up” after the last wash at 37°C. The motile cells (over 80% motile cells) were collected without the pellet and resuspended in capacitation medium. This procedure allowed motile sperm to be obtained without leukocyte contamination. All experimental protocols were approved and performed according to the relevant guidelines and regulations of the Helsinki Committee of Sheba Hospital, Ramat-Gan, Israel, and informed consent was obtained from all participants.
Sperm capacitation
Human sperm (1 × 107 cells per ml) were incubated in capacitation media, HAMF-10 at 37°C in 5% CO2 for 3 h as described previously.15
Assessment of sperm acrosomal exocytosis
Human sperm (1 × 107 cells per ml) were incubated under capacitation conditions for 160 min, and then various compounds as described for each experiment in the figure legends were added for an additional 20 min. The AE inducers described for each experiment in the figure legends were added for 1 h. The percentage of acrosome-reacted sperm was determined microscopically (Axio imager Z1, Zeiss, Jena, Germany) using fluorescein isothiocyanate (FITC)-conjugated Pisum sativum agglutinin (PSA). An aliquot of spermatozoa (106 cells per 10 μl) was smeared on a glass slide and allowed to air-dry. The sperm were then fixed with methanol for 15 min at room temperature and washed three times at 5-min intervals. The first and third washes were performed with distilled water (dH2O), and the second wash with Tris-buffered saline (TBS) (137 mmol l−1 NaCl [Cat No. 7647-14-5], 2.7 mmol l−1 KCl [Cat No. 7447-40-7] and 20 mmol l−1 Tris–HCl, pH 7.6). The slides were air-dried and then incubated in a moist environment with PSA-FITC (50 mg ml−1 in TBS) for 35 min, then washed twice with dH2O at 5-min intervals and sealed with ProLong Gold antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA). For each treatment, at least 100 cells per slide were evaluated on triplicate slides, using × 400 magnification under an Axio imagerZ1 fluorescence microscope. Cells with green staining over the acrosomal cap were considered acrosome intact; those with equatorial green staining or no staining were considered acrosome-reacted. The percent acrosome-reacted cells (5%–10%) at time zero was subtracted from each measurement.
Intracellular cAMP determination
The total cAMP concentration was determined by an enzyme-linked immunosorbent assay (ELISA) using a commercial kit from Arbor Assay (Cyclic AMP Direct EIA Kit) according to the manufacturer's instructions. Briefly, sperm (107 cells per ml) were incubated under capacitation conditions. At the end of the incubation, the sperm were lysed, and ELISA was performed according to the manufacturer's protocol. cAMP was determined relative to a standard curve.
Determination of actin polymerization
The method was described by us previously.16 Sperm cells were spread on microscope slides, air-dried, and fixed in 1.75% formaldehyde in TBS for 10 min, then washed once with dH2O for 5 min, placed in 0.2% Triton X-100 in TBS for 30 min, washed three times at 5-min intervals in TBS and air-dried. Afterward, sperms were incubated in a moist chamber with 4 μmol l−1 phalloidin-FITC in TBS for 60 min, washed four times with dH2O at 5-min intervals and mounted with ProLong Gold antifade reagent. Images were captured on Axio imagerZ1 fluorescence microscope at ×400 magnification. Sperm pictures were photographed within 24 h to minimize the loss of fluorescence, at the same exposure. The fluorescence intensity, which indicates the F-actin level, was quantified for whole cells using the “MetaMorp Image J” software (National Institutes of Health Universal Imaging Corp., West Chester, PA, USA) and the background intensity was subtracted. All experiments were carried out in duplicate slides, and at least 50 cells per slide were quantified for fluorescence intensity.
Statistical analyses
Data were analyzed for statistical significance by analysis of variance (ANOVA) followed by a Tukey's honestly significant difference (HSD) comparison for at least three replicates. Differences of P < 0.05 were considered statistically significant. Sample size appears in the figure legends.
RESULTS
The effect of PKA inhibition at the end of sperm capacitation on acrosomal exocytosis
In the present study, we investigated the involvement of cAMP/PKA in human sperm AE. Inhibition of PKA (using H89) at the end of the capacitation process revealed a 3-fold increase in the AE rate (Figure 1). Treatment of capacitated human sperm with TG or progesterone also induced the occurrence of AE and the augmentation of AE by H89, TG, or progesterone was significantly reduced when both soluble and the transmembrane adenylyl-cyclase (AC) were inhibited using 2-Hydroxy-2-methylpropiophenone (2-OH) ddAdo (Figure 1). Interestingly, the induction of the AE by H89 was almost completely blocked by adding a relatively high concentration of the membrane-permeable 8-bromo-cAMP (8Br-cAMP, 0.1 mmol l−1) or by increasing intracellular cAMP using 1-methyl-3-isobutylxanthine (IBMX); this agent inhibits cAMP-phosphodiesterase, the enzyme that degrades cAMP (Figure 1). Similar inhibition was observed when the AE was induced by TG or progesterone (Figure 1). TG induces AE by inhibiting the Ca2+-ATPase (Ca2+ pump) localized in the outer acrosomal membrane, resulting in a decrease of intra-acrosomal Ca2+ leading to the opening of the store-operated-Ca2+-channel (SOCC) of the plasma membrane.17,18,19 Progesterone activates CatSper, which is a sperm-specific Ca2+- channel in the plasma membrane.20
Figure 1.
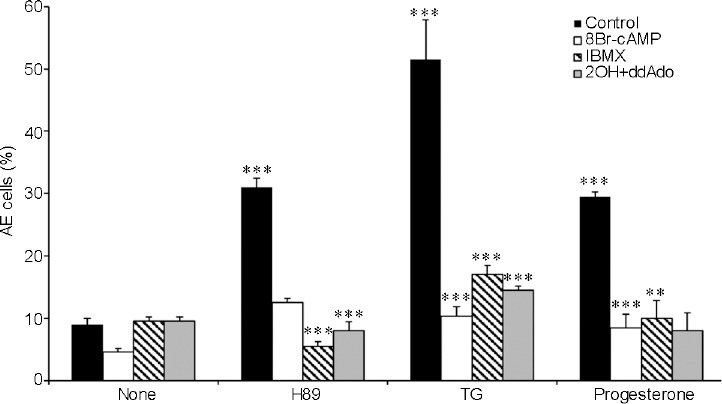
Effect of protein kinase A inhibition and 8-bromo-cAMP (8Br-cAMP) on AE. After 160 min of incubation (Control), 0.1 mmol l−1 (8Br-cAMP), 0.5 mmol l−1 1-methyl-3-isobutylxanthine (IBMX), or 20 μmol l−1 hydroxy-2-methylpropiophenone (2-OH-estradiol) + 100 μmol l−1 2'5'-dideoxyadenosine (2OH+ddAdo) (sAC/tmAC inhibitors) were added. The AE inducers are: N-[2-(p-Bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89; 50 μmol l−1), TG (3 μmol l−1), progesterone (5 μmol l−1) or antibiotic A23187 calcium ionophore (A23187; 10 μmol l−1). The values shown represent the mean ± standard deviation of duplicates from three experiments from three different donors. **P < 0.01 and ***P < 0.001, significant difference compared to the corresponding control. cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; sAC: soluble adenylyl cyclase; tmAC: transmembrane adenylyl cyclase; TG: thapsigargin.
A dose dependence analysis of the effect of 8Br-cAMP on the AE revealed that 5 μmol l−1 and 25 μmol l−1 8Br-cAMP stimulates AE, whereas higher concentrations showed no effect or even inhibition; nevertheless, all concentrations used inhibited the H89-induced AE (Supplementary Figure 1 (151.9KB, tif) ). The data in Figure 1 and Supplementary Figure 1 (151.9KB, tif) indicate that intracellular cAMP mediates the AE induced by PKA inhibition, while relatively high cAMP inhibits this effect. Thus, the different sensitivity to cAMP levels, and the fact that inhibition of PKA caused cAMP-dependent AE suggest that endogenous cAMP levels are probably enhanced when PKA is inhibited. Indeed, we found a 50% increase (from 2.6 to 3.9 pmol per 107 cells) in cAMP levels in cells treated with H89. Treatment with IBMX, a cAMP-phosphodiesterase (PDE) inhibitor, caused a much higher increase in [cAMP]i (up to 5.6 pmol per 107 cells), which is inhibitory to the AE process as mentioned above.
The possible involvement of EPAC in AE induced by PKA inhibition
Since inhibition of PKA (Figure 1) or low concentrations of 8Br-cAMP (Supplementary Figure 1 (151.9KB, tif) ) induced AE, we suggested that cAMP-dependent-EPAC might mediate this effect. We, therefore, tested the effect of various concentrations of 8-(4-Chlorophenylthio)-2'-O-methyladenosine-3′,5′-cyclic AMP (8pCPT), a selective activator of EPAC21 on AE. Activation of EPAC by addition of 8pCPT induced AE at relatively low concentrations of 8pCPT (0.02–10 μmol l−1), whereas at higher concentrations of 8pCPT (25–50 μmol l−1), the induction was much lower (Figure 2). These results indicate that AE is mediated by EPAC, and relatively high concentrations of EPAC substrate inhibit AE. Increasing intracellular cAMP using IBMX also show significant inhibition (75%) in AE induced by 0.1 μmol l−1 8pCPT (Figure 2).
Figure 2.
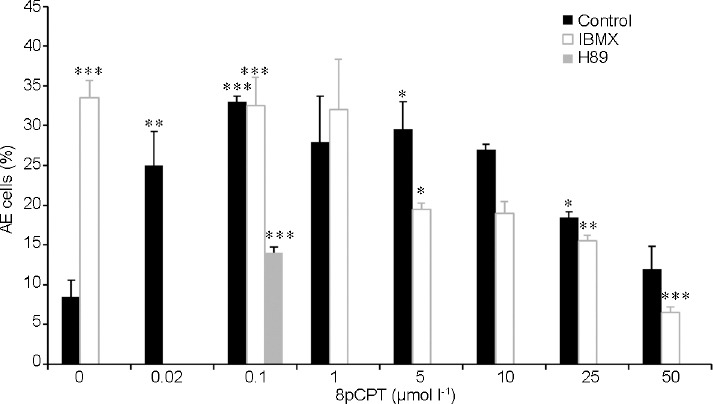
The effect of 8-(4-Chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic (8pCPT) on AE induced by protein kinase A inhibition. After 160 min of incubation (Control), various concentrations of 8pCPT were added and then 50 μmol l−1 N-[2-(p-bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89) or 0.5 mmol l−1 1-methyl-3-isobutylxanthine (IBMX) were added. The values represent the mean ± standard deviation of duplicates from three experiments from three different donors. *P < 0.05, **P < 0.01, and ***P < 0.001, significant difference compared to the corresponding control. AE: acrosomal exocytosis.
To further characterize the role of EPAC in H89-induced AE, we tested the effect of the EPAC-specific inhibitor, ESI0922,23 on AE induced by H89 and other inducers. As shown in Figure 3, ESI09 inhibited AE induced by 8pCPT, H89, TG, Angiotensin II, progesterone or the Ca2+-ionophore A23187, indicating that the AE induced by these six inducers is largely EPAC-dependent.
Figure 3.
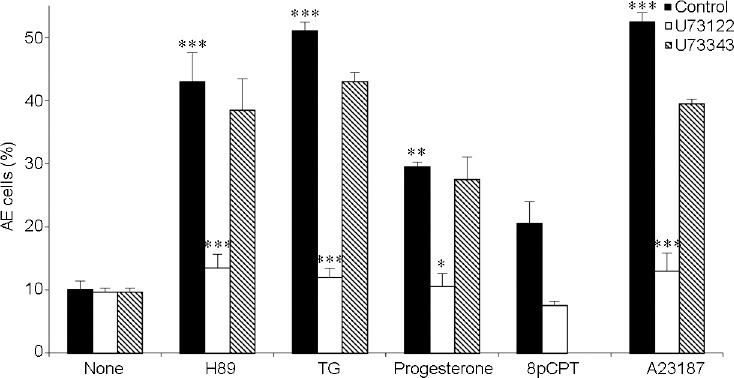
Inhibitory effect of the phospholipase C inhibitor 1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione (U73122) on AE. After 160 min of incubation (Control), 1 μmol l−1 U73122 (PLC inhibitor) or 1 μmol l−1 1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-2,5-pyrrolidinedione (U73343, inactive PLC inhibitor analogue) were added and then the AE inducers N-[2-(p-bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89; 50 μmol l−1), TG (3 μmol l−1), progesterone (5 μmol l−1), 8-(4-Chlorophenylthio)-2'-O-methyladenosine -3′,5′-cyclic (8pCPT; 0.1 μmol l−1) or antibiotic A23187 Calcium Ionophore (A23187; 10 μmol l−1) were added. The values represent the mean ± standard deviation of duplicates from three experiments from three different donors. *P < 0.05, **P < 0.01, and ***P < 0.001, significant difference compared to the corresponding control. PLC: phospholipase C; AE: acrosomal exocytosis; TG: thapsigargin.
Two isoforms of EPAC are present in sperm cells, EPAC1 and EPAC2;24,25 both are inhibited by ESI09.22,23 EPAC1 contains a binding site for phosphatidic acid (PA) which is important for its activation.26 Thus, the reduction of free PA in the cells by treatment with N-butanol will inhibit EPAC1 activity. PA is produced in cells from phosphatidyl-choline hydrolysis catalyzed by phospholipase D (PLD) activity. In the presence of the primary alcohol N-butanol, the free PA is converted back by PLD to phosphatidyl-butanol resulting in reduced levels of free PA in the cells.27 This system is active in sperm cells, as we showed that actin polymerization during sperm capacitation is inhibited by N-butanol treatment, which can be reversed by the addition of free PA to the cells.16 Here, we show that treatment with N-butanol inhibited AE induced by 8pCPT, H89, TG, or A23187 (Figure 4). No effect of 0.5% secondary-butanol which is not a PLD substrate, could be on AE or on sperm motility (not shown) indicating the specificity of the primary-butanol as an inhibitor of the PLD pathway. These results suggest that EPAC1 is the dominant isoform involved in AE.
Figure 4.
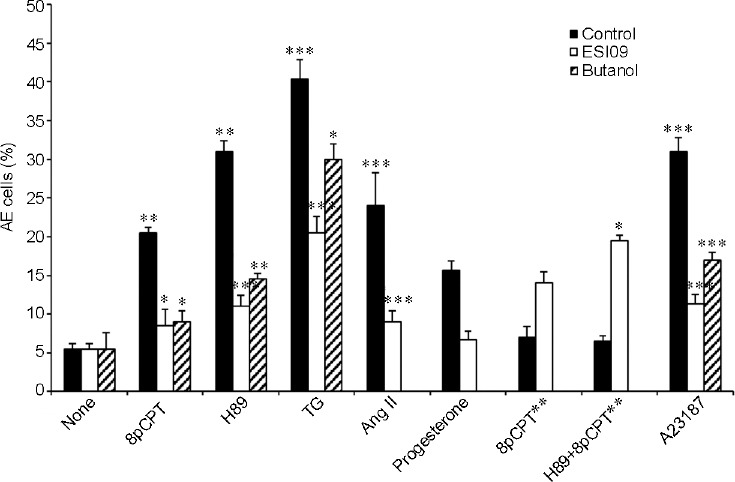
Effect of exchange protein directly activated by cAMP inhibitor and N-butanol on AE. After 160 min of incubation (Control), 0.5% butanol (N-butanol) or 10 μmol l−1 EPAC specific inhibitor 09 (ESI09) were added. The AE inducers are: 8-(4-Chlorophenylthio)-2'-O-methyladenosine-3′,5′-cyclic (8pCPT; 0.1 μmol l−1), N-[2-(p-bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89; 50 μmol l−1), TG (3 μmol l−1), angiotensin II (Ang II; 10 nmol l−1), polyphosphoinositide-binding-peptide (PBP10; 1 μmol l−1), 8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic (8pCPT**; 50 μmol l−1) and antibiotic A23187 calcium ionophore (A23187; 10 μmol l−1). The values represent the mean ± standard deviation of duplicates from three experiments from three different donors. *P < 0.05, **P < 0.01, and ***P < 0.001, significant difference compared to the corresponding control. EPAC: exchange protein directly activated by cAMP; cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; TG: thapsigargin.
The involvement of phospholipase C in AE induced by PKA inhibition
It was shown that EPAC-activated Rap GTPase is involved in phospholipase C (PLC) and Ca2+ signaling in the β2-adrenoceptor.28 PLC is an important factor involved in the AE mechanism.29,30 It catalyzes the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) to diacylglycerol, which activates protein kinase C (PKC), and inositol-1,4,5-triphosphate (IP3), which opens the Ca2+-channel in the outer-acrosomal membrane to release intra-acrosomal Ca2+,31 leading to induction of AE in human sperm.14,18,19 Reduction in intra-acrosomal Ca2+ by IP3 leads to SOCC activation17 and indeed, TG-induced AE, which occurs through SOCC activation, was over 90% inhibited by PLC inhibition using U73122 (Figure 3). AE induced by 8pCPT, H89, progesterone, or A23187 were also almost completely blocked by PLC inhibition (Figure 3). The specificity of PLC was determined by using the inactive analog 1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-2,5-pyrrolidinedione (U73343; 142878-12-4) which had very little effect on the degree of AE (Figure 3). These data demonstrated that AE, whether induced by activation of EPAC using 8pCPT, or by inhibition of PKA, activation of progesterone-R, or Ca2+-ionophore is mediated by PLC.
It was shown that EPAC is involved in actin modulation.32 During sperm capacitation, G-actin monomers are polymerized to F-actin, and before the AE, this F-actin should be dispersed to enable AE.2,33
F-actin formation during capacitation can occur only if the actin-severing protein gelsolin is inhibited by its binding to PIP2 and its tyrosine phosphorylation by Src.15 Before the AE, PIP2 is hydrolyzed by PLC, causing the release of p-gelsolin which undergoes dephosphorylation/activation resulting in F-actin dispersion.15
To examine the relationship between EPAC and actin in sperm, we looked for EPAC involvement in AE induced by activation of gelsolin using polyphosphoinositide-binding-peptide (PBP10), a peptide containing the PIP2-binding domain of gelsolin. Treatment with PBP10 will release gelsolin from PIP2, leading to its activation and F-actin dispersal, resulting in AE.15,33 It is seen in Supplementary Figure 2 (152.6KB, tif) that AE induced by PBP10 is almost completely blocked by 0.1 mmol l−1 8Br-cAMP or 50 μmol l−1 8pCPT or the EPAC inhibitor ES109, suggesting that EPAC mediates F-actin breakdown and/or the AE process downstream of F-actin dispersion. To solve this question, we tested the involvement of EPAC in the process of F-actin breakdown. It is shown in Figure 5 that TG, PBP10, H89, or 8pCPT induce F-actin breakdown and this breakdown is significantly inhibited by the EPAC inhibitor ESI09. These data clearly indicate that EPAC mediates F-actin depolymerization.
Figure 5.
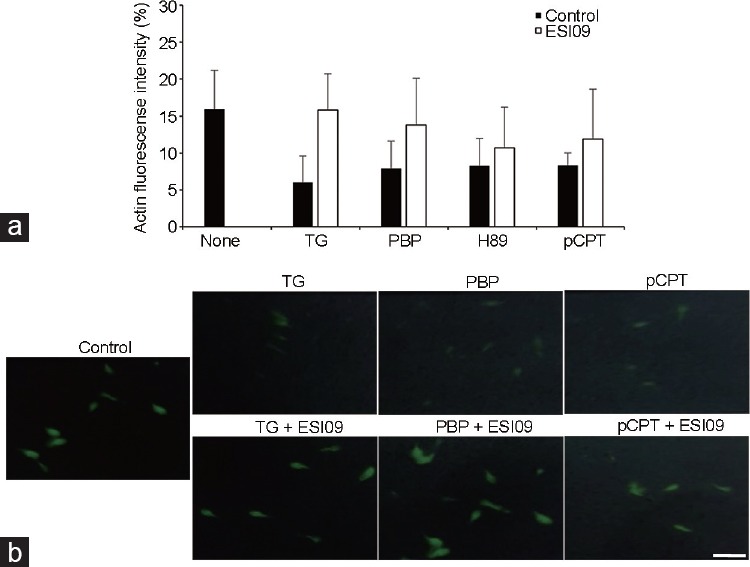
The actin florescence intensity. Human sperm (1 × 107 cells per ml) were incubated in Ham's F-10 capacitation medium for 160 min (Control). Next, 10 μmol l−1 EPAC specific inhibitor 09 (ESI09) was added for an additional 20 min. Then TG (3 μmol l− 1), polyphosphoinositide-binding-peptide (PBP10; 1 μmol l−1), N-[2-(p-bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89; 50 μmol l−1), 8-(4-Chlorophenylthio)-2′-O-methuladenosine-3′,5′-cyclic (8pCPT; 0.1 μmol l−1] were added for an additional 1 h. The values represent the mean ± standard deviation of duplicates from two experiments from two different donors. (a) F-actin fluorescence units. (b) Fluorescence microscope figures. Scale bars = 5 μm. EPAC: exchange protein directly activated by cAMP; cAMP: cyclic adenosine monophosphate; TG: thapsigargin.
Interestingly, the addition of TG to cells treated first with H89 or PBP10, there is 68% or 80% stimulation of AE, respectively (Supplementary Table 1).
Supplementary Table 1.
The effect of thapsigargin on acrosomal exocytosis in the presence of N-[2-(p-Bromocinnamylamino) ethyl]-5isoquinolinesulfonamide (H89) or polyphosphoinositide -binding-peptide
| Treatment | TG (AE cells [%]) | Cont (AE cells [%]) |
|---|---|---|
| Cont | 40.0±5.00** | 9.00±1.00 |
| H89 | 47.0±2.65* | 28.0±2.00* |
| PBP10 | 41.6±2.90* | 23.3±2.30 |
The values AE cells (%) represent the mean±s.d. of duplicates from three experiments from three different donors. *P<0.05, significant difference compared to the corresponding control; **P<0.01, significant difference compared to the corresponding control. TG: thapsigargin; AE: acrosomal exocytosis; PBP10: polyphosphoinositide - binding-peptide; s.d.: standard deviation; Cont: Control
The involvement of CatSper in AE induced by PKA inhibition
The sperm-specific Ca2+-channel CatSper is essential for the fertilizing ability of sperm.34 Since AE depends on Ca2+ influx into the sperm, we tested whether CatSper mediates AE induced by EPAC activation and by other inducers using the CatSper inhibitor, (1S,2S)-2-(2-(N-[(3-Benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-naphthyl cyclopropanecarboxylate dihydrochloride hydrate (NNC 55-0396). To find the optimal concentration of NNC, we tested its effect on AE-induced by progesterone, known to activate CatSper.20 We found that 1 μmol l−1 NNC caused complete inhibition of progesterone-induced AE. AE induced by TG was almost completely inhibited by NNC, while the AE induced by H89, 8pCPT or A23187 was only slightly reduced (Figure 6). The absence of an effect of NNC on Ca2+-ionophore A23187-induced AE, conditions under which the inhibition of Ca2+-channels is bypassed, indicates the specificity of NNC.
Figure 6.
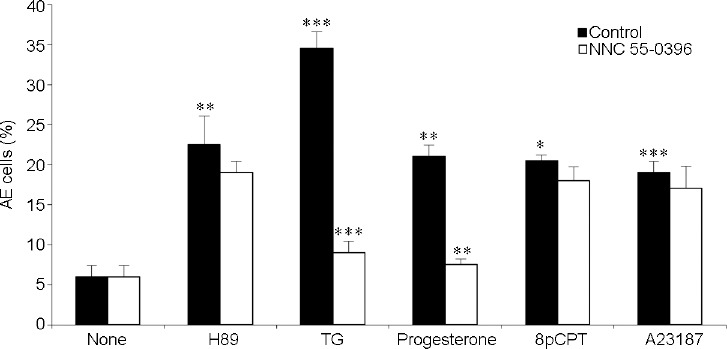
The effect of voltage-dependent calcium channel of sperm - CatSper inhibition on AE. After 160 min of incubation (Control), 1 μmol l−1 CatSper inhibitor (1S,2S)-2-(2-(N-[(3-Benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride hydrate (NNC 55-0396) were added and then the AE inducers N-[2-(p-Bromocinnamylamino)ethyl]-5isoquinolinesulfonamide (H89; 50 μmol l−1), TG (3 μmol l−1), progesterone (5 μmol l−1), 8-(4-Chlorophenylthio)-2'-O-methyladenosine-3′,5′-cyclic (8pCPT; 0.1 μmol l−1), or antibiotic A23187 calcium ionophore (A23187; 10 μmol l−1) were added. The values represent the mean ± standard deviation of duplicates from three experiments from three different donors. *P < 0.05, **P < 0.01, and ***P < 0.001, significant difference compared to the corresponding control. AE: acrosomal exocytosis; TG: thapsigargin.
DISCUSSION
In the present study, we investigated the involvement of cAMP/PKA in human sperm AE. Treatment of capacitated human sperm with the PKA inhibitor H89, TG, or progesterone induced the occurrence of AE which was significantly reduced when both soluble and the trans-membrane AC were inhibited indicating that cAMP-mediated this AE. However, the induction of the AE by H89, TG, or progesterone was significantly inhibited by adding a relatively high concentration of the membrane-permeable 8Br-cAMP (0.1 mmol l−1) or by increasing intracellular cAMP using IBMX. In human sperm, progesterone activates Rap1 in the acrosomal region in a cAMP-dependent manner, and Rap1 activates PLC, leading to Ca2+ release from the acrosome.35 TG causes also reduction of intra-acrosomal Ca2+ leading to the opening of SOCC. Since intracellular concentrations of cAMP are well regulated by its synthesis and degradation systems, it is reasonable to assume that elevated [cAMP]i would inhibit this Ca2+ mobilization process. Since the activation of AE by the PKA inhibitor H89 is also inhibited by 8Br-cAMP, it is possible that the AE induced by H89 also requires Ca2+ mobilization. This point is further discussed below.
The reduction in the AE induced by PKA- inhibition by the AC inhibitors indicates that a cAMP-dependent-PKA-independent mechanism mediates this effect. Moreover, the significant reduction of AE induced by PKA inhibition by relatively low concentration of 8Br-cAMP (25 μmol l−1), further supports such a cAMP-dependent-PKA-independent mechanism. A dose-dependence analysis of the effect of 8Br-cAMP on the AE revealed that 25 μmol l−1 8Br-cAMP stimulates AE, whereas higher concentrations showed no effect or even inhibition; nevertheless, all concentrations used inhibited the H89-induced AE. Thus, the AE induced by PKA inhibition is highly sensitive to increased cAMP concentrations. The data indicate that intracellular cAMP mediates the AE induced by PKA inhibition, while relatively high cAMP inhibits this effect. Thus, the different sensitivity to cAMP levels, and the fact that inhibition of PKA caused cAMP-dependent AE suggest that endogenous cAMP levels are probably enhanced when PKA is inhibited. Indeed, we found a 50% increase in [cAMP]i levels in cells treated with H89. Treatment with IBMX caused a much higher increase in [cAMP]i (up to 5.6 pmol per 107 cells), which is inhibitory to the AE process as mentioned above. Thus, 3.9 pmol cAMP per 107 cells is the suitable concentration for AE induction; however, 5.6 pmol cAMP per 107 cells is inhibitory to the AE process. The strong effects of small changes in cAMP levels emphasize the importance of tight regulation of intracellular cAMP levels by the sperm cell. The enhanced effect of PKA-inhibition on [cAMP]i is probably due to the fact the inhibition of PKA might decrease binding of cAMP to PKA, resulting in an increase in free cAMP in the cells.
Since inhibition of PKA or low concentrations of 8Br-cAMP-induced AE, we suggested that cAMP-dependent-EPAC might mediate this effect. It was shown previously that EPAC mediates AE in human sperm.14 Activation of EPAC by addition of 8pCPT induced AE at relatively low concentrations of 8pCPT (0.1–10 μmol l−1), whereas at higher concentrations (25–50 μmol l−1 8pCPT), the induction was much lower. These results indicate that AE is mediated by EPAC, and relatively high concentrations of EPAC substrate inhibit AE. The effect of 0.1–1.0 μmol l−1 8pCPT on AE is similar to the effect of H89, whereas higher concentrations of 8pCPT (5–50 μmol l−1) inhibit the induced effect of H89 on AE. Thus, at relatively high concentrations, 8pCPT inhibits AE induced by low 8pCPT or by H89. Moreover, AE induced by a low concentration of 8pCPT is inhibited by treatment with IBMX, suggesting that EPAC or its downstream effectors are inhibited by relatively high concentrations of intracellular cAMP.
To further prove the role of EPAC in H89-induced AE, we tested the effect of the EPAC-specific inhibitor, ESI0922,23 on AE. ESI09 inhibited AE induced by 8pCPT, H89, TG, Angiotensin II, progesterone or the Ca2+-ionophore A23187, indicating that the AE induced by these six inducers is largely EPAC-dependent. Thus, AE induced by PKA inhibition is mediated by EPAC; however, relatively high concentrations of EPAC substrate inhibit this activity. This finding is further supported by the inhibitory effect of high cAMP concentration on AE induced by PKA inhibition or by low concentrations of 8pCPT using IBMX.
The cAMP-dependent changes in EPAC dynamics are induced by allosteric rather than simple binding effects.36 These authors also show that EPAC contains multiple clusters of residues for which dynamics is either quenched or enhanced by cAMP. Thus, it is possible that at relatively high concentrations of cAMP, EPAC activity is reduced by allosteric effects, and when EPAC binds ESI09, this inhibitory effect is partially reversed, and its activity is rescued. Das et al.36 also showed that Gly-238 in EPAC is highly sensitive to cAMP although not to the antagonist Rp-cAMP, suggesting an allosteric role at Gly-238. These findings suggest differences in the affinities of cAMP and ESI09 to different regions of EPAC.
It has been suggested that the regulatory unit of EPAC2 contains high (B-site) and low (A-site) affinity sites for cAMP.37 Binding of cAMP to the B-site activates EPAC, whereas it is possible that binding to the A-site inhibits EPAC activity. This idea might explain why a relatively high level of cellular cAMP is inhibitory to EPAC-dependent AE. ESI09 is a competitive inhibitor of EPAC, which competes with cAMP binding.23,38 When low concentrations of 8pCPT (0.1 μmol l−1) were used to activate EPAC-dependent AE, ESI09 caused almost complete inhibition of AE. However, when 50 μmol l−1 8pCPT was used, which itself caused very slight stimulation of AE, a two-fold increase in AE was observed in the presence of ESI09, and an even greater increase in AE induced by ESI09 was seen when both 50 μmol l−1 8pCPT and H89 were present in the reaction mixture. We suggest that in the presence of 50 μmol l−1 8pCPT, the A-site of EPAC (the inhibitory site) is occupied by the cAMP analogue, and therefore no stimulation of AE is observed. The addition of ESI09 competes with cAMP for binding to this site, inducing its release and resulting in EPAC reactivation. These dual effects of ESI09, support our suggestion of high affinity activating, and low-affinity inhibitory sites for cAMP binding in EPAC. In conclusion, to achieve optimal EPAC-dependent AE, intracellular cAMP levels must be well regulated by its production and degradation systems in the cell.
Two isoforms of EPAC are present in sperm cells, EPAC1 and EPAC2;24,25 both are inhibited by ESI09.23,27 EPAC1 contains a binding site for PA which is important for its activation.26 Here, we show that treatment with N-butanol inhibited AE induced by 8pCPT, H89, TG, or A23187 indicating that EPAC1 is the dominant isoform involved in AE. The fact that AE induced by TG is only slightly inhibited by N-butanol (31%) suggests that AE-induced by TG is probably mediated by EPAC2 as well. Although N-butanol inhibits actin polymerization and increases spontaneous-AE when present during sperm capacitation,39 it does not cause any increase in the AE when added at the end of the capacitation, probably because of its inhibitory effect on EPAC1.
PLC is an important factor involved in the AE mechanism.29,30 TG-induced AE, which occurs through SOCC activation, was over 90% inhibited by PLC inhibition using U73122. AE induced by 8pCPT, H89, progesterone, or A23187 were also almost completely blocked by PLC inhibition. These data demonstrated that AE, whether induced by activation of EPAC using 8pCPT, or by inhibition of PKA, activation of progesterone-R, or Ca2+-ionophore is mediated by PLC. Thus, under these various conditions by which EPAC mediates AE, we see that PLC is also involved. The inhibition of 8pCPT-induced AE by the PLC inhibitor U73122 suggests that EPAC might activate PLC. Indeed, it was shown that the human sperm PLCε isoform is activated by EPAC-activated Rap1 and is required for Ca2+ mobilization from the acrosome.14 The fact that AE induced by the Ca2+-ionophore A23187 depends on EPAC and PLC indicates that high influx of Ca2+ is not sufficient to induce AE and that Ca2+ mobilization from the acrosome is necessary for AE to occur. This conclusion was supported by other studies working with permeabilized human sperm.13,14
To examine the relationship between EPAC and actin in sperm, we looked for EPAC involvement in AE induced by activation of gelsolin using PBP10, a peptide containing the PIP2-binding domain of gelsolin. Treatment with PBP10 release gelsolin from PIP2, leading to its activation and F-actin dispersal, resulting in AE.15,33 It was seen that AE induced by PBP10 is almost completely blocked by 0.1 mmol l−1 8Br-cAMP or 50 μmol l−1 8pCPT or the EPAC inhibitor ES109, suggesting that EPAC mediates F-actin breakdown and/or the AE process downstream of F-actin dispersion. It was shown that TG, PBP10, H89, or 8pCPT induce F-actin breakdown and this breakdown is significantly inhibited by the EPAC inhibitor ESI09. These data clearly indicate that EPAC mediates F-actin depolymerization.
We showed that AE induced by TG, H89, or 8pCPT is mediated by PLC activity. In our previous study in human sperm, we showed that hydrolysis of PIP2 by PLC leads to gelsolin activation and F-actin breakdown however F-actin breakdown induced by PBP10 is independent of PLC activity.15 It was suggested elsewhere that EPAC mediates PLC activation,14 which might be the mechanism by which EPAC leads to gelsolin activation. The fact that EPAC mediates PLC-independent F-actin breakdown induced by PBP10 indicate that EPAC has a direct effect on F-actin depolymerization in addition to its indirect effect due to PLC activation.
It is possible that other mechanisms not involving PLC are likely to mediate the effect of EPAC on the AE. It has been suggested that EPAC can activate the fusion component soluble-NSF-attachment protein receptor (SNARE)40 known to mediate the AE.40,41 SNARE is physically associated with F-actin leading to granule exocytosis in platelets.42 In MIN6 cells, F-actin negatively regulates insulin exocytosis via binding to the SNARE protein, syntaxin 4.43 Furthermore, in this insulin secretion system, gelsolin forms a complex with syntaxin 4 under basal conditions, which is relieved on stimulus-induced Ca2+ influx to activate gelsolin and induce its dissociation from syntaxin 4 to facilitate insulin exocytosis.44,45 Thus, it seems that syntaxin 4 activity is regulated by its binding to F-actin and gelsolin. To allow the outer acrosomal and the plasma membranes to fuse, the F-actin network produced between the two membranes during capacitation should be dispersed.46 Activation of gelsolin in sperm causes F-actin dispersion which would activate SNARE and other fusion proteins leading to AE.47,48 A working model suggested for the mechanism of AE indicates that EPAC might affect the AE through two mechanisms: one is EPAC-RAP-PLC-IP3 leading to Ca2+ release from the acrosome, and the second is EPAC-Rab3A-SNARE-AE.14,42 Since F-actin dispersion and AE induced by PBP10 is independent of PLC activity, we suggest that the inhibition of PBP10-induced AE by the EPAC inhibitor ESI09 is due to inhibition of the EPAC-Rab3-SNARE -AE cascade.
TG treatment further stimulates the AE in the presence of PBP10 or H89. These suggest that intracellular Ca2+ mobilization induced by TG is an important step for achieving AE, even under conditions whereby F-actin is already dispersed. The fact that AE induced by TG is inhibited by ESI09 and by U73122 suggest that EPAC involve in PLC-dependent Ca2+ mobilization which is supported by previous study.31
The sperm-specific Ca2+-channel CatSper is essential for the fertilizing ability of sperm.34 Since AE depends upon Ca2+ influx into the sperm, we tested whether CatSper mediates AE induced by EPAC activation and by other inducers using the CatSper inhibitor, NNC 55-0396. AE induced by TG was almost completely inhibited by NNC, while the AE induced by H89, 8pCPT, or A23187 was only slightly reduced. We suggested before that AE induced by H89 is mediated by EPAC. Here we showed that AE induced by H89 or by direct activation of EPAC using 8pCPT are not affected by CatSper inhibition. These data further support our model that inhibition of PKA activates EPAC leading to AE. Moreover, these data suggest that AE induced by EPAC is probably not mediated by Ca2+ influx into the cell via CatSper but rather by Ca2+ mobilization from the acrosome as a result of PLC activation by EPAC-activated GTPase, as described above. This conclusion is supported by other studies in human sperm, which show that EPAC activation induces PLC-dependent Ca2+ mobilization.14
The fact that AE-induced by progesterone is mediated by EPAC/PLC indicates that Ca2+ mobilization from the acrosome is a necessary step for AE even under conditions by which CatSper is activated. Moreover, the fact that AE induced by H89, 8pCPT, and A23187 is independent of CatSper activity, suggest that intracellular Ca2+ mobilization might be sufficient to induce AE, although it is possible that other plasma membrane Ca2+ channels might be active in Ca2+ influx.
In mouse sperm, CatSper34 and PKA10 are localized to the flagellum. Thus, it is not clear how these two factors are involved in AE. Regarding CatSper, it has been suggested that Ca2+ influx via this channel starts in the tail excluding initiation via Cav2.3 channel which localize in the head49 and lead to AE.50 It was recently shown that CatSper, as well as PKA, are conclusively linked to AE.51 Moreover, it has been shown that disruption of PKA from AKAP triggers AE in capacitated mouse sperm,51 which supports our findings.
CONCLUSIONS
In summary, the data presented here indicate that the inhibition of PKA results in an increase in intracellular cAMP and activation of EPAC-dependent AE. A further increase in cAMP levels is inhibitory for this process. We showed that low concentrations of the selective EPAC substrate added to the cells stimulate AE, whereas high concentrations show no effect on AE, and inhibit AE induced by PKA inhibition. It is well known that cellular levels of cAMP are precisely regulated by its production and decomposing systems. In this study, we provide one of the reasons why such regulation is important. Moreover, this study provides data regarding the involvement of cAMP, PKA, and EPAC in the AE mechanism.
AUTHOR CONTRIBUTIONS
DI performed the experiments, helped to write the manuscript, and performed the statistical analysis. YN made critical notes and helped to analyze the data. HB wrote the manuscript, interpreted the data, and supervised DI in her master studies. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
Dose response of the effect of 8-bromo-cAMP on AE induced by PKA inhibition. After 160 min incubation of (Control), 8-BromocAMP (8Br-cAMP( was added for an additional 20 min at the concentrations indicated and then 50 µmol l-1 N-[2-(p-Bromocinnamylamino)ethyl]- 5isoquinolinesulfonamide (H89) was added for 1 h. The values represent the mean ± s.d. of duplicates from three experiments from three different donors. **P < 0.01, significant difference compared to the corresponding control; ***P < 0.001, significant difference compared to the corresponding control. cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; PKA: protein kinase A; s.d.: standard deviation.
Effect of elevated 8-bromo-cAMP on AE induced by Gelsolin activation. After 160 min of incubation (Control), 50 µmol l-1 8-(4-chlorophenylthio)-2'-O-adenosine- 3' ,5'-cyclic (8pCPT), 0.1 mmol l-1 8-bromo-cAMP (γ 8BrcAMP), or 10 µM EPAC specific inhibitor 09 (ESI09) were added for an additional 20 min. Then, 1 µmol l-1 PBP10 was added for 1 h. The values represent the mean ± s.d. of duplicates from three experiments from three different donors. **P < 0.01, significant difference compared to the corresponding control; ***P < 0.001, significant difference compared to the corresponding control. cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; PBP10: polyphosphoinositide-binding-peptide; s.d.: standard deviation.
ACKNOWLEDGMENTS
We would like to thank Dr. Gili Band and Dr. Nir Etkovitz from Sheba Hospital, Ramat-Gan, Israel, for their help in obtaining sperm cells. We thank Dr. Maya Finkelstein from Wolfson Hospital, Holon, Israel, for helping in F-actin fluorescence study.
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Visconti PE, Bailey JL, Moore GD, Pan D, Olds-Clarke P, et al. Capacitation of mouse spermatozoa. I. Correlation between the capacitation state and protein tyrosine phosphorylation. Development. 1995;121:1129–37. doi: 10.1242/dev.121.4.1129. [DOI] [PubMed] [Google Scholar]
- 2.Brener E, Rubinstein S, Cohen G, Shternall K, Rivlin J, et al. Remodeling of the actin cytoskeleton during mammalian sperm capacitation and acrosome reaction. Biol Reprod. 2003;68:837–45. doi: 10.1095/biolreprod.102.009233. [DOI] [PubMed] [Google Scholar]
- 3.Shahar S, Wiser A, Ickowicz D, Lubart R, Shulman A, et al. Light-mediated activation reveals a key role for protein kinase A and sarcoma protein kinase in the development of sperm hyper-activated motility. Hum Reprod. 2011;26:2274–82. doi: 10.1093/humrep/der232. [DOI] [PubMed] [Google Scholar]
- 4.Morgan DJ, Weisenhaus M, Shum S, Su T, Zheng R, et al. Tissue-specific PKA inhibition using a chemical genetic approach and its application to studies on sperm capacitation. Proc Natl Acad Sci U S A. 2008;105:20740–5. doi: 10.1073/pnas.0810971105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nolan MA, Babcock DF, Wennemuth G, Brown W, Burton KA, et al. Sperm-specific protein kinase A catalytic subunit Calpha2 orchestrates cAMP signaling for male fertility. Proc Natl Acad Sci U S A. 2004;101:13483–8. doi: 10.1073/pnas.0405580101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–6. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–62. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–75. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Hu J, Bobulescu IA, Quill TA, McLeroy P, et al. A sperm-specific Na+/H+ exchanger (sNHE) is critical for expression and in vivo bicarbonate regulation of the soluble adenylyl cyclase (sAC) Proc Natl Acad Sci U S A. 2007;104:9325–30. doi: 10.1073/pnas.0611296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wertheimer E, Krapf D, de la Vega-Beltran JL, Sanchez-Cardenas C, Navarrete F, et al. Compartmentalization of distinct cAMP signaling pathways in mammalian sperm. J Biol Chem. 2013;288:35307–20. doi: 10.1074/jbc.M113.489476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leclerc P, Kopf GS. Mouse sperm adenylyl cyclase: general properties and regulation by the zona pellucida. Biol Reprod. 1995;52:1227–33. doi: 10.1095/biolreprod52.6.1227. [DOI] [PubMed] [Google Scholar]
- 12.Breitbart H, Spungin B. The biochemistry of the acrosome reaction. Mol Human Rep. 1997;3:195–202. doi: 10.1093/molehr/3.3.195. [DOI] [PubMed] [Google Scholar]
- 13.Branham MT, Mayorga LS, Tomes CN. Calcium-induced acrosomal exocytosis requires cAMP acting through a protein kinase A-independent, Epac-mediated pathway. J Biol Chem. 2006;281:8656–66. doi: 10.1074/jbc.M508854200. [DOI] [PubMed] [Google Scholar]
- 14.Branham MT, Bustos MA, De Blas GA, Rehmann H, Zarelli VE, et al. Epac activates the small G proteins Rap1 and Rab3A to achieve exocytosis. J Biol Chem. 2009;284:24825–39. doi: 10.1074/jbc.M109.015362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkelstein M, Etkovitz N, Breitbart H. Role and regulation of sperm gelsolin prior to fertilization. J Biol Chem. 2010;285:39702–9. doi: 10.1074/jbc.M110.170951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen G, Rubinstein S, Gur Y, Breitbart H. Crosstalk between protein kinase A and C regulates phospholipase D and F-actin formation during sperm capacitation. Dev Biol. 2004;267:230–41. doi: 10.1016/j.ydbio.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 17.Dragileva E, Rubinstein S, Breitbart H. Intracellular Ca2+-Mg2+-ATPase regulates calcium influx and acrosomal exocytosis in bull and ram spermatozoa. Biol Reprod. 1999;61:1226–34. doi: 10.1095/biolreprod61.5.1226. [DOI] [PubMed] [Google Scholar]
- 18.De Blas G, Michaut M, Trevino CL, Tomes CN, Yunes R, et al. The intraacrosomal calcium pool plays a direct role in acrosomal exocytosis. J Biol Chem. 2002;277:49326–31. doi: 10.1074/jbc.M208587200. [DOI] [PubMed] [Google Scholar]
- 19.Lopez CI, Pelletan LE, Suhaiman L, De Blas GA, Vitale N, et al. Diacylglycerol stimulates acrosomal exocytosis by feeding into a PKC-and PLD1-dependent positive loop that continuously supplies phosphatidylinositol 4,5-bisphosphate. Biochim Biophys Acta. 2012;1821:1186–99. doi: 10.1016/j.bbalip.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Strunker T, Goodwin N, Brenker C, Kashikar ND, Weyand I, et al. The CatSper channel mediates progesterone-induced Ca2+ influx in human sperm. Nature. 2011;471:382–6. doi: 10.1038/nature09769. [DOI] [PubMed] [Google Scholar]
- 21.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–6. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Ding C, Wild C, Liu H, Wang T, et al. Efficient synthesis of ESI-09, a novel non-cyclic nucleotide EPAC antagonist. Tetrahedron Lett. 2013;54:1546–9. doi: 10.1016/j.tetlet.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Chen H, Boulton S, Mei F, Ye N, et al. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sehrawat S, Cullere X, Patel S, Italiano J, Jr, Mayadas TN. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell. 2008;19:1261–70. doi: 10.1091/mbc.E06-10-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeong HW, Li Z, Brown MD, Sacks DB. IQGAP1 binds Rap1 and modulates its activity. J Biol Chem. 2007;282:20752–62. doi: 10.1074/jbc.M700487200. [DOI] [PubMed] [Google Scholar]
- 26.Consonni SV, Gloerich M, Spanjaard E, Bos JL. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc Natl Acad Sci U S A. 2012;109:3814–9. doi: 10.1073/pnas.1117599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han S, Huh J, Kim W, Jeong S, Min do S, et al. Phospholipase D activates HIF-1-VEGF pathway via phosphatidic acid. Exp Mol Med. 2014;46:e126. doi: 10.1038/emm.2014.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, et al. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol. 2001;3:1020–4. doi: 10.1038/ncb1101-1020. [DOI] [PubMed] [Google Scholar]
- 29.Etkovitz N, Tirosh Y, Chazan R, Jaldety Y, Daniel L, et al. Bovine sperm acrosome reaction induced by G-protein-coupled receptor agonists is mediated by epidermal growth factor receptor transactivation. Dev Biol. 2009;334:447–57. doi: 10.1016/j.ydbio.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Breitbart H, Finkelstein M. Regulation of sperm capacitation and the acrosome reaction by PIP2 and actin modulation. Asian J Androl. 2015;17:597–600. doi: 10.4103/1008-682X.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruete MC, Lucchesi O, Bustos MA, Tomes CN. Epac, Rap and Rab3 act in concert to mobilize calcium from sperm's acrosome during exocytosis. Cell Commun Signal. 2014;12:43. doi: 10.1186/s12964-014-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–9. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 33.Finkelstein M, Megnagi B, Ickowicz D, Breitbart H. Regulation of sperm motility by PIP2(4,5) and actin polymerization. Dev Biol. 2013;381:62–72. doi: 10.1016/j.ydbio.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Ren D, Navarro B, Perez G, Jackson AC, Hsu S, et al. A sperm ion channel required for sperm motility and male fertility. Nature. 2001;413:603–9. doi: 10.1038/35098027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucchesi O, Ruete MC, Bustos MA, Quevedo MF, Tomes CN. The signaling module cAMP/Epac/Rap1/PLCepsilon/IP3 mobilizes acrosomal calcium during sperm exocytosis. Biochim Biophys Acta. 2016;1863:544–61. doi: 10.1016/j.bbamcr.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Das R, Mazhab-Jafari MT, Chowdhury S, SilDas S, Selvaratnam R, et al. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J Biol Chem. 2008;283:19691–703. doi: 10.1074/jbc.M802164200. [DOI] [PubMed] [Google Scholar]
- 37.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, et al. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–36. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 38.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, et al. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–8. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shabtay O, Breitbart H. CaMKII prevents spontaneous acrosomal exocytosis in sperm through induction of actin polymerization. Dev Biol. 2016;415:64–74. doi: 10.1016/j.ydbio.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 40.De Blas GA, Roggero CM, Tomes CN, Mayorga LS. Dynamics of SNARE assembly and disassembly during sperm acrosomal exocytosis. PLoS Biol. 2005;3:e323. doi: 10.1371/journal.pbio.0030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez F, Zanetti MN, Mayorga LS, Tomes CN. Munc18-1 controls SNARE protein complex assembly during human sperm acrosomal exocytosis. J Biol Chem. 2012;287:43825–39. doi: 10.1074/jbc.M112.409649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woronowicz K, Dilks JR, Rozenvayn N, Dowal L, Blair PS, et al. The platelet actin cytoskeleton associates with SNAREs and participates in alpha-granule secretion. Biochemistry. 2010;49:4533–42. doi: 10.1021/bi100541t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jewell JL, Luo W, Oh E, Wang Z, Thurmond DC. Filamentous actin regulates insulin exocytosis through direct interaction with syntaxin 4. J Biol Chem. 2008;283:10716–26. doi: 10.1074/jbc.M709876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalwat MA, Wiseman DA, Luo W, Wang Z, Thurmond DC. Gelsolin associates with the N terminus of syntaxin 4 to regulate insulin granule exocytosis. Mol Endocrinol. 2012;26:128–41. doi: 10.1210/me.2011-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramos LS, Zippin JH, Kamenetsky M, Buck J, Levin LR. Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J Gen Physiol. 2008;132:329–38. doi: 10.1085/jgp.200810044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spungin B, Margalit I, Breitbart H. Sperm exocytosis reconstructed in a cell-free system. Evidence for the involvement of phospholipase C and actin filaments in membrane fusion. J Cell Sci. 1995;108:2525–35. doi: 10.1242/jcs.108.6.2525. [DOI] [PubMed] [Google Scholar]
- 47.Breitbart H, Cohen G, Rubinstein S. Role of actin cytoskeleton in mammalian sperm capacitation and the acrosome reaction. Reproduction. 2005;129:263–8. doi: 10.1530/rep.1.00269. [DOI] [PubMed] [Google Scholar]
- 48.Tomes CN, Michaut M, De Blas G, Visconti P, Matti U, et al. SNARE complex assembly is required for human sperm acrosome reaction. Dev Biol. 2002;243:326–38. doi: 10.1006/dbio.2002.0567. [DOI] [PubMed] [Google Scholar]
- 49.Escoffier J, Boisseau S, Serres C, Chen CC, Kim D, et al. Expression, localization and functions in acrosome reaction and sperm motility of Cav3.1 and Cav3.2 channels in sperm cells: an evaluation from Cav3.1 and Cav3.2 deficient mice. J Cell Physiol. 2007;212:753–63. doi: 10.1002/jcp.21075. [DOI] [PubMed] [Google Scholar]
- 50.Cohen R, Buttke DE, Asano A, Mukai C, Nelson JL, et al. Lipid modulation of calcium flux through CaV2.3 regulates acrosome exocytosis and fertilization. Dev Cell. 2014;28:310–21. doi: 10.1016/j.devcel.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stival C, Ritagliati C, Xu X, Gervasi MG, Luque GM, et al. Disruption of protein kinase A localization induces acrosomal exocytosis in capacitated mouse sperm. J Biol Chem. 2018;293:9435–47. doi: 10.1074/jbc.RA118.002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dose response of the effect of 8-bromo-cAMP on AE induced by PKA inhibition. After 160 min incubation of (Control), 8-BromocAMP (8Br-cAMP( was added for an additional 20 min at the concentrations indicated and then 50 µmol l-1 N-[2-(p-Bromocinnamylamino)ethyl]- 5isoquinolinesulfonamide (H89) was added for 1 h. The values represent the mean ± s.d. of duplicates from three experiments from three different donors. **P < 0.01, significant difference compared to the corresponding control; ***P < 0.001, significant difference compared to the corresponding control. cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; PKA: protein kinase A; s.d.: standard deviation.
Effect of elevated 8-bromo-cAMP on AE induced by Gelsolin activation. After 160 min of incubation (Control), 50 µmol l-1 8-(4-chlorophenylthio)-2'-O-adenosine- 3' ,5'-cyclic (8pCPT), 0.1 mmol l-1 8-bromo-cAMP (γ 8BrcAMP), or 10 µM EPAC specific inhibitor 09 (ESI09) were added for an additional 20 min. Then, 1 µmol l-1 PBP10 was added for 1 h. The values represent the mean ± s.d. of duplicates from three experiments from three different donors. **P < 0.01, significant difference compared to the corresponding control; ***P < 0.001, significant difference compared to the corresponding control. cAMP: cyclic adenosine monophosphate; AE: acrosomal exocytosis; PBP10: polyphosphoinositide-binding-peptide; s.d.: standard deviation.