

Abstract
Glycogen synthase kinase-3 (GSK-3), a Ser/Thr protein kinase abundantly expressed in neurons, plays diverse functions in physiological and neurodegenerative conditions. Our recent study shows that upregulation of GSK-3 suppresses long-term potentiation and presynaptic release of glutamate; however, the underlying mechanism is elusive. Here, we show that activation of GSK-3β retards the synaptic vesicle exocytosis in response to membrane depolarization. Using calcium imaging, whole-cell patch-clamp, as well as specific Ca2+ channel inhibitors, we demonstrate that GSK-3β phosphorylates the intracellular loop-connecting domains II and III (LII-III) of P/Q-type Ca2+ channels, which leads to a decrease of intracellular Ca2+ rise through the P/Q-type voltage-dependent calcium channel. To further illustrate the mechanisms of GSK-3β's action, we show that activation of GSK-3β interferes with the formation of the soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) complex through: (1) weakening the association of synaptobrevin with SNAP25 and syntaxin; (2) reducing the interactions among the phosphorylated LII-III and synaptotagmin, SNAP25, and syntaxin; and (3) inhibiting dissociation of synaptobrevin from synaptophysin I. These results indicate that GSK-3β negatively regulates synaptic vesicle fusion events via interfering with Ca2+-dependent SNARE complex formation.
Introduction
Glycogen synthase kinase-3 (GSK-3) is a protein kinase with diverse physiological functions in mediating intracellular signaling, regulating neuronal plasticity, gene expression, and cell survival (Grimes and Jope, 2001). Of the two isoforms of GSK-3, GSK-3α and GSK-3β, GSK-3β has been found to play a crucial role in several neurological disorders, such as Alzheimer's disease (Takashima, 2006), bipolar disorder (Gould et al., 2004), and schizophrenia (Kozlovsky et al., 2002), all of which show synaptic abnormalities, and it is a prime drug target for a variety of neurological disorders (Eldar-Finkelman, 2002). GSK-3β also plays a central role in neuronal polarity during development (Jiang et al., 2005). Upregulation of GSK-3β in mice not only impairs the spatial learning and memory, but also inhibits long-term potentiation (Hernández et al., 2002; Hooper et al., 2007). Similar results have also been detected by simultaneous inhibition of phosphatidyl inositol 3 kinase and/or protein kinase C (PKC) in rats (Liu et al., 2003; Zhu et al., 2007). Activation of GSK-3 reduces presynaptic glutamate release (Zhu et al., 2007), and inhibition of GSK-3 by lithium increases release of serotonin and cholinergic neurotransmission (Treiser et al., 1981; Fujii et al., 2000). These observations suggest that GSK-3β may play a role in learning and memory through regulating presynaptic release of neurotransmitters. But it is not known how GSK-3β regulates the presynaptic neurotransmission.
In presynaptic nerve terminals, neurotransmitter release is mediated by the exocytosis of transmitter-filled vesicles (Sudhof, 2004). This process is triggered by Ca2+ that enters a nerve terminal via voltage-dependent Ca2+ channels (VDCCs) (Stevens, 2003). At synapse, the precise control of synaptic exocytosis is achieved by a tight coupling of the Ca2+ channel and synaptic vesicle release sites (Pumplin et al., 1981). The sub-millisecond delay between Ca2+ influx and exocytosis of synaptic vesicles implies that the Ca2+ channels involved are in close proximity to the release sites (Llinás et al., 1981). Among various VDCCs, N-type and P/Q-type Ca2+ channels play a major role in regulating the presynaptic neurotransmitter release (Reid et al., 1997).
For an efficient neurotransmitter release, the formation of soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) complex is required (Kim and Catterall, 1997). This process involves (1) binding of the synprint sites on N-type or P/Q-type Ca2+ channels with presynaptic membrane proteins synaptotagmin and t-SNARE (i.e., SNAP25 and syntaxin), (2) dissociation of synaptobrevin (Syb) (also called VAMP2, vesicle associated membrane protein 2), a vesicular SNARE protein responsible for synaptic vesicle fusion in synapse) from synaptophysin I (SypI), and (3) association of Syb with t-SNARE (Pennuto et al., 2003), all of which are tightly regulated by intracellular Ca2+ concentration ([Ca2+]i) and the phosphorylation of the synprint sites on N-type and P/Q-type Ca2+ channels located within the intracellular loop-connecting domains II and III (LII-III) (Yokoyama et al., 1997). Phosphorylation of the synprint sites by cyclin-dependent protein kinase 5 (cdk5) inhibits the association of LII-III with SNAP25 and synaptotagmin (Tomizawa et al., 2002). With elevation of [Ca2+]i, the complex of SypI and Syb dissociates. These data together suggest that the interactions of several synaptic proteins play a crucial role in exocytosis, and that these interactions are tightly regulated by [Ca2+]i and the phosphorylation of the Ca2+ channel proteins. Until now, it is has not been known whether GSK-3 can phosphorylate the Ca2+ channels, and whether and how GSK-3 activation may affect the interaction of the vesicle exocytosis-related proteins.
In the present study, we demonstrate that GSK-3β regulates presynaptic vesicle exocytosis. Upregulation of GSK-3β retards presynaptic exocytosis by phosphorylating and inhibiting the P/Q-type calcium channel, thus suppressing Ca2+ influx and Ca2+-dependent SNARE complex formation.
Materials and Methods
Chemicals and antibodies.
FM4-64 was from Invitrogen. Fluo3Am was from Calbiochem. ω-CgTx GVIA and ω-Aga IVA were from Alomone Labs. Recombinant GSK-3β and glutathione-Agarose were from Sigma. Monoclonal antibody (mAb) against total GSK-3β (1:1000 for Western blotting), polyclonal antibody (pAb) against phosphorylated GSK-3β at Ser9 (1:1000 for Western blotting), mAb against HA-tag (1:200 for immunofluorescence), and pAb against phosphothreonine (1:1000 for Western blotting) were from Cell Signaling Technology. pAb against Syb (1:1000 for Western), mAb against SNAP25 (1:1000 for Western blotting), and mAb against synaptotagmin I (1:1000 for Western blotting) were from Abcam. pAb against Syntaxin 1a (1:1000 for Western blotting) was from Millipore. mAb against SypI (1:1000) and mAb against phosphoserine (1:500 for Western blotting) were from Sigma. Neurobasal and B27 were from Invitrogen. Wild-type and dominant-negative GSK-3β plasmids were gifts from Dr. J. R. Woodgett at Toronto University (Toronto, ON, Canada), and the cDNAs encoding GSK-3β were subcloned to enhanced green fluorescent protein (EGFP)-C1 vector. HA-pcDNA3.0 plasmid was a gift from Dr. K. Marcelo at the University of Pennsylvania School of Medicine (Philadelphia, PA). pHluroin-Syb plasmid was a gift from Dr. Gero Miesenboeck at Yale University School of Medicine (New Haven, CT). pECFP-Syb and pEYFP-SypI plasmids were gifts from Dr. Flavia Valtorta at San Raffaele Scientific Institute and Vita-Salute University (Milan, Italy).
Cell culture and transfection.
Primary embryonic hippocampal neurons of the rats were cultured according to the procedure described previously (Meberg and Miller, 2003; Kaech and Banker, 2006). Briefly, the hippocampi of the embryonic rats (18 d, ∼10 embryos/litter) were collected and incubated with 4 ml of 0.125% trypsin in calcium- and magnesium-free Hanks' balanced salt solution for 15 min, and then centrifuged at 80 × g for 5 min after addition of 4 ml of the neuronal plating medium containing DMEM/F12 with 10% FBS, the cells in the pellet were triturated and plated onto a 60 mm plastic culture dish (400∼600 neurons/mm2) and incubated in a humidified incubator with 5% CO2 at 37°C for 2∼4 h. Then, the neurobasal medium supplemented with 2% B27 (maintenance medium) was replaced and the cells were cultured for 8 days in vitro (DIV). The medium was changed every 3 d with replacement of half the volume with fresh maintenance medium each time. Neurons were transfected at 6 DIV using Lipofecatamine 2000 according to the manufacturer's instructions. N2a cells were cultured as described previously (Zhu et al., 2007).
Production and purification of LII-III antibody.
The peptide ALRQTARPRESARDPDARR that corresponds to rat sequence (844-862) is located in a highly variable region within the intracellular loop LII-III of the α1A subunit of P/Q-type calcium channel proteins (National Center for Biotechnology Information reference sequence, NP_037050.2). For immunization, the peptide was conjugated to keyhole limpet hemocyanin (KLH) according to the established procedure. New Zealand rabbits were immunized subcutaneously with 500 μg of KLH-conjugated peptide emulsified in complete Freund's adjuvant. Two booster doses at 21 d intervals, with 500 μg of antigen emulsified in incomplete Freund's adjuvant, were injected subcutaneously. The rabbits were bled 8 d after the second boost, and the isolated serum was titrated for peptide binding by ELISA. The polyclonal rabbit IgG was affinity purified on a peptide-coupled column using a SulfoLink kit.
Purification and phosphorylation of LII-III peptide.
Complementary DNA encoding LII-III (amino acids 724–983) cloned from rat brain of the 1A subunit of P/Q-type Ca2+ channel [glutathione S-transferase (GST)-LII-III] was a gift from Dr. Kazuhito Tomizawa at the Department of Physiology, Okayama University Graduate School of Medicine and Dentistry (Okayama, Japan). GST-tagged protein was expressed in Escherichia coli (BL21) and purified as described previously (Lee et al., 1996). The purified GST proteins were dialyzed with phosphate buffer. Phosphorylation of GST-LII-III peptide by recombinant GSK-3β was performed in buffer containing 20 mm 4-morpholinepropane sulfonic acid, pH 7.4, 30 mm MgCl2, 100 μm [γ-32P]ATP (300 dpm/pmol), and 1 mm dithiothreitol. GST-LII-III was incubated with or without GSK-3β (20∼40 mU/μl final) at 32°C for 1 h. The reaction was terminated by boiling in 2× Laemmli sample buffer. The Western blotting and autoradiography analysis were performed as described previously (Tomizawa et al., 2002).
Preparation and phosphorylation of synaptosome, immunoprecipitation, and binding assay.
The synaptosome (P2 fraction) was prepared according to a previously established method (McGahon and Lynch, 1996). The hippocampal CA3 region was excavated and homogenized in 320 mm ice-cold sucrose and centrifuged at 800 × g for 5 min at 4°C. The resulting supernatant was further centrifuged at 20,000 × g for 15 min at 4°C, and P2 fraction containing synaptosome was collected. Synaptosomes (0.5 mg of protein) were incubated with streptolysin O to permeabilize the plasma membrane following the administration of DMSO, wortmannin (Wort), Wort plus SB216763 (SB), and SB alone for 30 min at 37°C (Reisinger et al., 2004). After adding the anti-SypI or anti-Syb antibody to the synaptosome, the reaction mixture was gently rocked at 4°C overnight. Then, 20 μl of protein G agarose was added to the immunocomplex and gently rocked at 4°C for another 2 h. The agarose beads were collected by pulsing and resuspended in 2×Laemmli sample buffer for Western blotting analysis.
For binding assay, GST-LII-III proteins were first incubated with recombinant GSK-3β as above by adding 100 μm cold ATP for 1 h at 32°C, and then incubated with glutathione–agarose for 1 h at 4°C. The mixture was added to a 10 ml column and washed with buffer containing Tris-HCl, pH 7.4, 150 mm NaCl, and 0.4% Triton X-100, then washed with PBS once. The solubilized synaptosome protein (300 μg) was loaded onto the column and washed three times with buffer containing Tris-HCl, pH 7.4, 150 mm NaCl, 20 μm CaCl2, and 0.1% Triton X-100. The protein complex collected from the column was denatured with boiled SDS-PAGE buffer and centrifuged. Then, the supernatants were used for Western blotting using antibodies against SNAP25, synaptotagmin I, and syntaxin 1, respectively.
FM4-64 recycles.
FM4-64 experiments were performed on a Zeiss LSM510 confocal microscope with a 40× objective lens as reported previously (Gaffield and Betz, 2006). In brief, neurons at 8 DIV were loaded in 10 μm FM4-64 in a solution containing 45 mm K+ for 1 min and then washed with a solution containing 3 mm K+ for 15 min. Neurons were subjected to destaining in 90 mm K+ solution for 5 min, and the time-lapse recording was performed.
Fluo3Am Ca2+ imaging.
Neurons at 8 DIV were loaded with 10 μm Fluo3Am (Invitrogen), pH 7.4, containing 135 mm NaCl, 1 mm MgCl2, 1.5 mm CaCl2, 5 mm HEPES, and 10 mm glucose for 30 min at 37°C followed by a 30 min wash. Images were acquired before and after perfusion with saline buffer containing 90 mm KCl at a rate of one image per 6 s.
Patch clamp.
Patch pipettes were fabricated from borosilicate glass capillaries (outer diameter, 1.5 mm; inner diameter, 1 mm; Science Products) on a Narishige P83 puller. Pipettes usually had a resistance of 2–3 MΩ when filled with a pipette solution containing 80 mm cesium methanesulfonate, 20 mm TEA, 1 mm CaCl2, 5 mm MgCl2, 11 mm EGTA, 10 mm HEPES, 10 mm ATP, and 0.5 mm GTP, and the osmolarity was adjusted to 275–280 mOsm with sucrose. Tight-seal whole-cell recordings were obtained at room temperature (21−24°C). Membrane currents were recorded with a patch-clamp amplifier (EPC9, HEKA Elektronik) and collected on-line with the TIDA for Windows acquisition and analysis program (HEKA Elektronik). Command voltages were adjusted for 5 to 6 mV liquid junction potentials (Neher, 1992). The membrane capacitance was measured using the EPC9 capacitance cancellation. The mean membrane capacitance was 11.0 ± 2.7 pF. The input resistance of the examined neurons was determined by short, hyperpolarizing voltage steps from a 50 mV holding potential and was >1 GΩ in most neurons with the recording solution (see above). The series resistance estimated by exponential fitting of the capacitance artifacts during brief hyperpolarizing voltage commands from 50 to 60 mV conformed well to the values obtained with the EPC9 capacitance cancellation paradigm (Sigworth et al., 1995). Series resistance compensation was used to improve the voltage-clamp control (60–80%). The maximal residual voltage error estimated from the product of series resistance and the maximal current Imax did not exceed 5.5 mV.
Image fitting and analyses.
For the fluorescence image fitting, a rectangular region of interest (ROI) was labeled on the spot where the event occurred. Changes in the fluorescence intensities of FM4-64 or pHluorin were monitored over time, and the intensity was corrected to the area of ROI before fitting. For FM4-64 curve, single exponential decay functions were fitted to the diagrams with fluorescence intensity changes using ImageJ software (Wayne Rasband, National Institutes of Health, Bethesda, MD), and time constant τ was evaluated (Vardjan et al., 2007). For pHluorin, a sigmoidal curve, {F = F 0 + F max/[1 + (t/t 1/2)c]}, was fitted and half-time (t 1/2) was analyzed. F is the fluorescence ratio, F 0 is the initial fluorescence ratio, t is time, F max is the maximal change in fluorescence ratio, and c is a constant fitted to the data by regression analysis using SigmaPlot 9 (Systat Software) (Lehane and Kirk, 2008). For Fluo3Am, the maximum slope of the curve in the elevating phase and the increment of fluorescent intensity were calculated (Loughrey et al., 2004).
Total internal reflection fluorescence microscopy.
N2a cells were cotransfected with pHluorin-Syb and wtGSK-3β, dnGSK-3β, or pcDNA3.0 for 24 h. Then, the cells were plated on a round coverslip (refractive index n488 = 1.80; thickness = 0.17 mm; Olympus) mounted on an observation chamber and incubated in a 1:1 mixture of DMEM and Optimem for time-lapse imaging by total internal reflection fluorescence microscopy (TIRFM). The components include IX-70 invert microscope with a 1.65 numerical aperture objective lens (100+Apo O HR; Olympus), Polychrome system for epifluorescence illumination (TILL Photonics), argon ion laser system (Melles Griot), Kine FLEX kinematic single-mode fiber and fiber coupler (Point Source), Sensi Cam super VGACCD camera and frame grabber (PCO), piezo Z-drive and E-622 LVPZT amplifier/servo control unit (Physik Instrumente), GFP filter set (Chroma), and an IBM personal computer. Fluorescence light was filtered by a GFP filter set and collected through the lateral port of the inverted microscope. Immersion oil (refractive index n488 = 1.81; Cargille Laboratories) was used to bridge the optical contact between the object and the coverslip. Images were recorded by a cooled CCD camera that was controlled by Polychrome, frame grabber, and image software TILL, version 4.0 (TILL Photonics). Exposure time (100–300 ms) and interval time (4–7 s) were adjusted according to the fluorescence intensity of the images. Recording was started at 1 min before 90 mm KCl stimulation and lasted for 5 min. Single and time-lapsed images were recorded and analyzed by TILL version 4.0 and Photoshop 7.0 (Adobe). The dynamics of image data were analyzed using Igor Pro 4.03 (Wave Metrics).
Fluorescence resonance energy transfer.
Expression vectors encoding fluorescent proteins were cotransfected into the hippocampal neurons (6 DIV) or N2a cells at a ratio of 1:3 (donor/acceptor). The transfected neurons (8 DIV) or N2a cells (24 h after transfection) were washed once with KRH/EGTA and incubated at 37°C (5% CO2) in the KRH/EGTA solution containing 0.1 nm α-latrotoxin for 30 min, then washed twice with KRH/EGTA. Fluorescence resonance energy transfer (FRET) between cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) was measured as follows: channel 1 = donor emission by excitation of the donor (excitation, 458 nm; emission, 465–485 nm); channel 2 = FRET emission by excitation of the donor (excitation, 458 nm; emission, 525–585 nm); and channel 3 = acceptor emission by excitation of the acceptor (excitation, 514 nm; emission, 525–585 nm) (Latz et al., 2007). The efficiency of FRET was analyzed by a FRET macro of Furoview500 (Olympus).
Statistics.
Data were analyzed by using SPSS 11.0 statistical software, and the one-way ANOVA procedure followed by Student's t tests was used to determine the differences between the groups.
Results
Upregulation of GSK-3β retards presynaptic vesicle exocytosis
To explore the role of GSK-3β in presynaptic exocytosis, we expressed in hippocampal neurons (6 DIV) EGFP-labeled wtGSK-3β, the dominant-negative (dnGSK-3β), or the vector (C1). Two days after the transfection, we measured vesicle exocytosis using the destaining of FM4-64 triggered by high K+ solution (90 mm). The FM4-64 was almost completely depleted in the neurons expressing C1 (Fig. 1 A, red) or dnGSK-3β (Fig. 1 C, red), whereas strong FM4-64 staining was still detected in the neurons expressing wtGSK-3β at 5 min after the K+ stimulation (Fig. 1 E, red). Using time-lapse recording, we analyzed the kinetics of the FM4-64 destaining after the K+ stimulation. A rapid decline of the fluorescence was detected in the neurons expressing EGFP-C1 (Fig. 1 B, red) and the decline curve was significantly sharper in the neurons expressing dnGSK-3β (Fig. 1 D, red), however, only minor decrease was detected in the neurons expressing wtGSK-3β (Fig. 1 F, red). Under the same conditions, the EGFP intensity did not change much (Fig. 1 A–F, green), which not only confirmed the expression of the plasmids but also provided an internal reference to rule out the artificial quench of the fluorescence. By fitting the destaining curve of FM4-64 to a single exponential decay (Fig. 1 G), we found that the time constant τ value was 120.8 ± 14.9 s in control neurons. Interestingly, the τ value increased to 179.7 ± 44.7 s in the neurons expressing wtGSK-3β and dropped to 33.7 ± 5.7 s in the neurons expressing dnGSK-3β (Fig. 1 H). These data suggest that GSK-3β regulates presynaptic vesicle exocytosis.
Figure 1.

A–H, GSK-3β inhibits presynaptic exocytosis. K+-evoked release of FM4-64 was recorded by confocal microscope. Dissociated hippocampal neurons transfected with EGFP-vector or EGFP-dnGSK-3β or EGFP-wtGSK-3β were measured by decline of FM4-64 for the vesicle release at the axon terminals (boxes). A, C, E, The images were captured before (Pre) and 5 min after (Post) K+ treatment. Scale bar, 50 μm. B, D, F, The kinetic curves were recorded from ∼100 s before to ∼5 min after K+ stimulation. G, H, Single-exponential decay functions were fitted to the diagrams with fluorescence intensity changes of FM4-64 analyzed by ImageJ software (G) and the time constant τ (H). I–L, Mean ± SD, ANOVA, Student's t test; **p < 0.01 versus EGFP; ##p < 0.01 versus wtGSK (n = 10∼12). I, J, K+-evoked release of pHluorin-Syb recorded by TIRFM: N2a cells were cotransfected with pHluorin-Syb and pcDNA3.0 (I) or wtGSK-3β (J) for 24 h. I, J, Consecutive images with enhanced fluorescence intensity after K+ stimulation were collected and amplified (from the boxes). Scale bar: 10 μm; 1 μm for amplification. K, L, The intensity of pHluorin was plotted and fitted to an exponential growth curve. The results show that overexpression of wtGSK-3β retards the K+-stimulated exocytosis.
To further verify the inhibitory role of GSK-3β in exocytosis, we cotransfected into the N2a cells, with HA-wtGSK-3β and pHluorin-Syb, a pH-sensitive form of GFP fused to the Syb that provides a sensitive optical probe to track the exocytosis (Miesenbock et al., 1998). The coexpression of wtGSK-3β and Syb was confirmed by HA and pHluorin (GFP) staining at 24 h after the transfection (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Then, we evoked the cells with K+ and recorded the changes of fluorescence intensity using the TIRFM system. As shown in Figure 1, I and J, the fluorescence increased rapidly in response to K+ stimulation, and it almost achieved the highest intensity at 0.5 s in pcDNA-transfected cells (Fig. 1 I); however, it took at least 9 s to reach a relatively saturated fluorescence level in the cells expressing wtGSK-3β (Fig. 1 J). After fitting to the sigmoidal curve (Fig. 1 K,L), we found that the t 1/2 for the elevating phase of pHluorin fluorescence was ∼0.10 s in control neurons, whereas it increased to ∼8.7 s in the neurons expressing wtGSK-3β. These results further confirm an inhibitory role of GSK-3β in exocytosis.
To ensure that the overexpression of GSK-3 led to an increase in the kinase activity, we measured GSK-3 activity in N2a cells with a kinase assay. Compared with the vector-transfected control, the activity of GSK-3 increased to ∼244% in the cells expressing wtGSK-3β, whereas it decreased to ∼73% when dnGSK-3β was expressed (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Upregulation of GSK-3β suppresses P/Q-type VDCC
The exocytosis is tightly correlated with the intracellular Ca2+ microdomain induced by Ca2+ influx (Becherer et al., 2003). To investigate whether GSK-3 affects the [Ca2+]i, we cotransfected DsRed and wtGSK-3β (1:3) in hippocampal neurons and measured the Ca2+ influx at 8 DIV with time-lapse imaging. We loaded the transfected (red) and the untransfected neurons with Fluo3Am (Fig. 2 A, Pre), a fluorescence Ca2+ indicator. After being evoked by K+, the fluorescence intensity was enhanced rapidly in both control neurons and neurons expressing wtGSK-3β (Fig. 2 A, Post). However, the response was much weaker in the neurons expressing wtGSK-3β (Fig. 2 A, arrows; B, dashed green line) than in the neurons without wtGSK-3β (Fig. 2 A, arrowheads; B, solid green line). Meanwhile, no significant change was detected in Dsred that could be used as control (Fig. 2 A, red; B, red line). Quantitative analysis showed that the maximum velocity (V max), and the increment of Fluo3AM in wtGSK-3β-transfected cells decreased to 59.5 ± 2.1% and 59.1 ± 4.4% of the control levels, respectively. These data suggest that GSK-3 activation retards the high K+-evoked Ca2+ influx.
Figure 2.
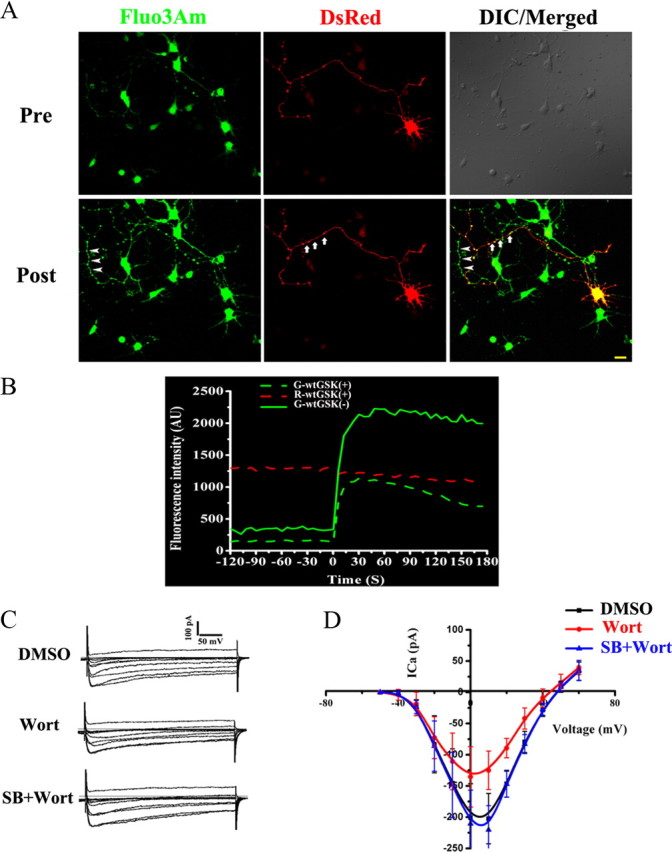
A, B, GSK-3β inhibits high voltage-dependent Ca2+ influx. Calcium imaging: the hippocampal neurons with (arrows) or without (arrowheads) expression of wtGSK-3β were loaded with 10 μm Fluo3Am, the images (A) and fluorescence intensity at the axons (B) were recorded from ∼2 min before (Pre) to 3 min after (Post) the K+ stimulation. C, D, The Ca2+ current recorded by patch clamp: neurons were cultured for 8 DIV and then treated with Wort, SB, or vehicle (DMSO) 30 min before recording. The representative Ca2+ current (C) and the I–V curves were analyzed by EPC 10 (D). The results show that upregulation of GSK-3β suppresses the calcium influx.
The Ca2+ influx is controlled by VDCCs; therefore, we studied the effect of GSK-3 on VDCCs by using a whole-cell patch clamp. When a Ca2+ current was evoked by applying a 100-ms-duration pulse depolarizing from a −90 mV holding potential to −10 mV, an inward current with gradual decay was obtained in 8 DIV neurons (Fig. 2 C). The peak Ca2+ current decreased to 70% of the control level after GSK-3 activation by wortmannin, and simultaneous inhibition of GSK-3 by SB216763 restored the Ca2+ current (Fig. 2 C,D). Further analysis revealed that a conspicuous upward shift of the current–voltage (I–V) curve was only shown in the wortmannin-treated neurons (Fig. 2 D). These results suggest that GSK-3 activation inhibits the VDCCs.
Among various VDCCs, P/Q-type (CaV2.1) and N-type Ca2+ channels (CaV2.2) are the ones most often implicated in presynaptic neurotransmitter release (Sudhof, 2004). To identify the subtype of the VDCCs involved in the GSK-3β-regulated Ca2+ influx, we expressed dnGSK-3β into the hippocampal neurons and measured the FM4-64 destaining in the presence or absence of the P/Q- or N-type Ca2+ channel inhibitors at 8 DIV. Compared with the untransfected neurons in the same dish, the neurons expressing dnGSK-3β showed a more rapid decline of the FM4-64 fluorescence intensity (supplemental Fig. 3, available at www.jneurosci.org as supplemental material, solid red line vs dashed red line), indicating a faster release by GSK-3β inhibition. As expected, inhibition of either N-type (ω-CgTx GVIA) or P/Q-type (ω-Aga IVA) VDCCs could retard FM4-64 depletion, whether with or without dnGSK-3β (supplemental Fig. 3, available at www.jneurosci.org as supplemental material, green and blue lines vs red). However, only the inhibition of P/Q-type VDCC by Aga abolished the dnGSK-3β-enhanced destaining of FM4-64 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material, dashed and solid blue lines), implying a predominant role of P/Q-type VDCC in the GSK-3β-regulated Ca2+ influx.
Then, we further verified the role of P/Q-type VDCC in the GSK-3β-regulated Ca2+ influx by Ca2+ imaging. As mentioned above, we expressed dnGSK-3β to increase the Ca2+ entry in hippocampal neurons and measured the intracellular Ca2+ level by Fluo3Am in the presence or absence of the VDCC inhibitors at 8 DIV. In the absence of the inhibitors (Fig. 3 A, DMSO), all the neurons showed enhanced fluorescence in response to K+ stimulation; however, the increment was much higher in the dnGSK-3β-expressing neurons (Fig. 3 A, red, arrows) than in the untransfected ones, especially in the neuronal processes (Fig. 3 A, arrowheads). Quantitative analysis showed that the increment was ∼112% in dnGSK-3β-expressing neurons over the untransfected ones (Fig. 3 B, DMSO), and the V max increased from ∼3.3 AU/s (arbitrary unit per second) to ∼9.5 AU/s (Fig. 3 C, DMSO). When the N-type Ca2+ channel was inhibited, the neurons, whether with or without dnGSK-3β, showed lower increment (Fig. 3 A, Cgtx; B, 64.2% with dnGSK-3β vs 45.3% without) and slower V max (Fig. 3 C, 5.9 AU/s with dnGSK-3β vs 2.0 AU/s without) than the controls (DMSO), suggesting that N-type Ca2+ channel participates in the presynaptic Ca2+ influx. However, the inhibition of N-type Ca2+ channel did not reduce the dnGSK-3β-induced increment (Fig. 3 A–C, Cgtx, ∼1.1-fold with dnGSK-3β vs without, p > 0.05), suggesting that the N-type Ca2+ channel may not contribute to the GSK-3β-regulated Ca2+ entry. Under the same condition, the inhibition of P/Q-type Ca2+ channel abolished the dnGSK-3β-induced increment of the fluorescence intensity (Fig. 3 A, Aga; B, 53.6% with dnGSK-3β vs 58.5% without) and the increased V max (Fig. 3 C, 2.9 AU/s with dnGSK-3β vs 2.4 AU/s without). These results further validate the role of P/Q-type VDCC in GSK-3β-regulated Ca2+ influx.
Figure 3.

GSK-3β inhibits Ca2+ influx through P/Q-type calcium channels. A, Calcium images were recorded as described in Figure 2, except that the P/Q-type (Aga) or N-type (CgTx) Ca2+ channel inhibitors were added 30 min before recording. The Fluo3Am increment (ΔF) were calculated by subtracting the preimage from the postimage of Fluo3Am and transferred into the fluorescence gradient images using Image Pro Plus 4.5 and LSM510. Scale bar, 20 μm. B, C, Quantitative analysis of the relative increment. ΔF/Δt (F, fluorescence intensity; t, time). The results show that only inhibition of P/Q-type calcium channel retards the dnGSK-3β-induced calcium influx. Mean ± SD, ANOVA, Student's t test; **p < 0.01 untransfected versus dnGSK-3β-transfected; ## p < 0.01 DMSO versus inhibitors in untransfected neurons; $$ p < 0.01 DMSO versus inhibitors in dnGSK-3β-transfected neurons (n = 6).
GSK-3β phosphorylates calcium channel at the synprint site
Phosphorylation of Ca2+ channel at the synprint site inhibits the channel activity through interfering with the interaction of synaptic proteins (Yokoyama et al., 1997; Carnes et al., 2007). To explore whether GSK-3β can phosphorylate P/Q-type VDCC, we prepared GST-labeled LII-III of the P/Q-type Ca2+ channel protein that contains the synprint sites and produced the antibody against LII-III, which reacts with the purified LII-III peptide dose dependently (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material). After in vitro incubation of the GST-LII-III with GSK-3β in the presence of 100 μm [γ-32P]ATP, we detected a specific radioactive band at ∼60 kDa, which was also positively labeled by anti-LII-III and anti-phosphothreonine (pThr) antibodies (Fig. 4 A). No immunoreaction was detected by using anti-pSer, although a positive band was detected in rat brain cortex extracts that validated the efficacy of the antibody (Fig. 4 A, lane N). These in vitro data demonstrate that GSK-3β can phosphorylate the Ca2+ channel protein at threonine residue.
Figure 4.

GSK-3β phosphorylates P/Q-type calcium channels. A, In vitro phosphorylation of LII-III by GSK-3β: the purified LII-III (20 μg) was incubated with GSK-3β in the presence of 100 μm [γ-32P]ATP for autoradiography and Western blotting with the antibodies against GSK-3β, LII-III, pThr, and phosphoserine (pSer), respectively (N= rat brain cortex extract). B, Phosphorylation of LII-III by GSK-3 in synaptosome: the purified synaptosome (P2) from rat brain hippocampi was incubated with DMSO, Wort, or SB, or Wort plus SB for 30 min, then the suspension was immunoprecipitated with anti-LII-III antibody and the supernatant (S), and the pellets were probed by anti-pThr or anti-LII-III (M = marker). X = 20. Note that GSK-3β can phosphorylate P/Q-type calcium channel at Thr residues.
To further verify the role of GSK-3 in phosphorylating Ca2+ channel, we prepared the synaptosome (P2) from rat hippocampi and incubated the P2 fraction with Wort, an indirect activator of GSK-3 (Liu et al., 2003), or simultaneously with SB216763 (SB), a specific inhibitor of GSK-3. Then, we performed immunoprecipitation using anti-LII-III antibody and measured the phosphorylation of LII-III. We found that GSK-3 activation significantly enhanced the phosphorylation of LII-III at pThr, whereas simultaneous inhibition of GSK-3 abolished the Wort-induced phosphorylation (Fig. 4 B). These results further validate the role of GSK-3 in phosphorylating P/Q-type Ca2+ channel at Thr residues. The slow migrating bands shown in Fig. 4 B might be produced by alternative splicing of α1A subunit, where the synprint sites are located, according to previous studies (Martin-Moutot et al., 1996; Bourinet et al., 1999).
Upregulation of GSK-3β interrupts the SNARE complex formation through disrupting the interaction of presynaptic proteins
Formation of SNARE complex by association of Syb with t-SNARE proteins (SNAP25 and syntaxin) is a critical step for exocytosis (Schoch et al., 2001); therefore, we first measured whether GSK-3 affects this association in synaptosome (P2) fraction. By immunoprecipitation, we observed that the levels of syntaxin and SNAP25 coprecipitated with anti-Syb decreased when GSK-3 was activated by Wort, while simultaneous inhibition of GSK-3 by SB restored the t-SNARE levels (Fig. 5 A,B), suggesting that activation of GSK-3 interrupts formation of SNARE complex.
Figure 5.

A, B, GSK-3 disrupts the interactions of synaptic proteins. GSK-3 activation inhibits association of Syb with SNAP25 and syntaxin: the purified hippocampal synaptosome (P2) was incubated with DMSO, or Wort, or SB, or Wort plus SB for 30 min, then immunoprecipitated with anti-Syb and probed by anti-SNAP25, syntaxin Ia, and Syb (A) and quantitative analysis (B). Mean ± SD, ANOVA, Student's t test; **p < 0.01 versus DMSO; ##p < 0.01 versus Wort (n = 3). C, D, Phosphorylated LII-III by GSK-3β is incompetent in association with synaptotagmin, SNAP25, and syntaxin: purified LII-III peptide was phosphorylated by GSK-3β and then coimmunoprecipitated with P2 fraction for Western blotting (C) and quantitative analysis (D). E–G, GSK-3 activation hinders dissociation of Syb from SypI by FRET: the hippocampal neurons (6 DIV) were cotransfected with EGFP-labeled GSK-3β and ECFP-Syb or EYFP-SypI as indicated, then the dissociation of Syb from SypI was detected at 8 DIV. The images were recorded before (Pre) and after (Post) K+ stimulation, and the axon terminals as marked were amplified. H–K, GSK-3 activation inhibits dissociation of Syb from SypI by immunoprecipitation: the purified P2 was incubated with DMSO, Wort, or Wort plus SB for 30 min, then immunoprecipitated with anti-Syp I (H) and probed by anti-Syb, or vice versa (J), and quantitative analysis was performed (I, K). Note that GSK-3 activation disturbed the interaction of synaptic proteins during exocytosis.
The association of the Ca2+ channel protein with synaptotagmin and t-SNARE proteins is important for the formation of SNARE complex (Pfeffer, 1996). Therefore, we tested whether phosphorylation of the synprint peptide of P/Q-type channel (LII-III) by recombinant GSK-3β affects its association with these synaptic proteins prepared from synaptosome (P2). We found that the levels of synaptotagmin, SNAP25, and syntaxin in P2 fraction bound with the synprint peptide decreased prominently after being incubated with recombinant GSK-3β (Fig. 5 C,D), suggesting that phosphorylation of LII-III by GSK-3β blocks its association with synaptotagmin and the t-SNARE proteins.
Syb needs to dissociate from SypI before it can bind to syntaxin and SNAP25 to form the SNARE complex (Pennuto et al., 2003). To explore whether GSK-3 affects the affinity of SypI/Syb complex, we used the FRET technique. Hippocampal neurons were cotransfected with expression vectors encoding ECFP-Syb or EYFP-SypI and wtGSK-3β, dnGSK-3β, or pcDNA, and then the changes in FRET efficiency before and after K+ stimulation were measured using a confocal microscope at 8 DIV. A significantly decreased YFP intensity with mild (in pcDNA-transfected neurons) (Fig. 5 E) or more dramatic (in dnGSK-3β-transfected neurons) (Fig. 5 G) enhancement of CFP was observed, especially in the distal axon, suggesting an enhanced dissociation of Syb from SypI by GSK-3β inhibition. However, no obvious change of the fluorescence intensity for both CFP and YFP was detected in the neurons expressing wtGSK-3β in response to K+ stimulation (Fig. 5 F), suggesting an inhibited dissociation of Syb from SypI by GSK-3β activation.
We also quantitatively analyzed the alteration of FRET efficiency in N2a cells. Similar results as in the primary neurons were observed, and furthermore, we also found that the cells with expression of wtGSK-3β did not show any obvious change of the fluorescence intensity up to 10 min after K+ stimulation (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material). With Youvan's FRET calculation, we found that the proportion of the cells showing high FRET efficiency (0.75–1) decreased from ∼75% to ∼20% in both pcDNA- and dnGSK-3β-transfected groups at 5 min after K+ stimulation, and the proportion of the cells showing low FRET efficiency (0–0.25) was much higher in dnGSK-3β-expressing cells than the pcDNA control cells. However, no obvious alteration of the proportions was detected in the cells expressing wtGSK-3β after K+ stimulation for up to 10 min (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material).
To further validate the effect of GSK-3 on the SypI/Syb complex, we performed immunoprecipitation by incubating P2 fraction with anti-SypI or anti-Syb antibodies. We found that the level of SypI bound to anti-Syb or vice versa was significantly increased when GSK-3 was activated, whereas simultaneous inhibition of GSK-3 abolished the increment (Fig. 5 H–K).
These data together reveal that GSK-3 can regulate the association or dissociation of Syb with SNARE components and that activation of GSK-3 interrupts the interactions.
Discussion
GSK-3 activation decreases presynaptic release of glutamate (Zhu et al., 2007), but the mechanism is not clear. The aim of the current article was to study whether and how GSK-3 affects exocytosis, the basic machinery for the presynaptic release of transmitter-filled vesicles. The major findings are as follows: (1) upregulation of GSK-3β retards the presynaptic vesicle exocytosis; (2) GSK-3β phosphorylates the LII-III (synprint site) of P/Q-type Ca2+ channel and upregulation of GSK-3 decreases Ca2+ influx from P/Q-type VDCC; and (3) GSK-3β decreases the association of LII-III with synaptotagmin, SNAP25, and syntaxin, the association of Syb with SNAP25 and syntaxin, and the dissociation of Syb from SypI, all of which are required for SNARE complex formation and thus for an efficient exocytosis. We speculate that GSK-3β retards presynaptic vesicle exocytosis by phosphorylating P/Q-type Ca2+ channel, arresting Ca2+ influx and thus inhibiting the Ca2+-dependent SNARE complex formation, which shows the mechanisms underlying the impaired presynaptic neurotransmitter release induced by GSK-3 activation.
VDCCs are essential elements of fast stimulus-secretion coupling in presynaptic termini of the neurons. In response to membrane depolarization, a rapid and localized Ca2+ signal is produced that interacts with Ca2+ sensors on the exocytotic apparatus to initiate vesicle fusion and neurotransmitter release (Zucker, 1996). Previous studies have shown that N-type and P/Q-type but not L-type calcium channels are involved in presynaptic exocytosis (Hirning et al., 1988; Wheeler et al., 1994). Furthermore, N-type VDCCs regulate neurotransmitter release at both peripheral and central synapses (Westenbroek et al., 1992), whereas the P/Q-type VDCCs play a primary role only at the central synapses (Westenbroek et al., 1995; Catterall, 1998). To identify which subtype of the VDCCs is involved in the GSK-3-regulated Ca2+ influx, we used specific inhibitors of N-type and P/Q-type Ca2+ channels. By expression of dnGSK-3β to enhance Ca2+ influx, we could measure the suppression of the Ca2+ entry by the inhibitors. As both N-type and P/Q-type inhibitors could suppress the enhanced Ca2+ influx, although the suppression of P/Q-type inhibitor was more prominent, we were unable to clarify the subtype of the VDCCs that contributed to the GSK-3-regulated Ca2+ influx. To solve this problem, we designed the study to compare the differences in the increment of the Ca2+ influx between the neurons with and without expression of dnGSK-3β in response to the exposure of the inhibitors. We found that inhibition of P/Q-type Ca2+ channel almost completely abolished the dnGSK-3β-enhanced Ca2+ entry, whereas inhibition of N-type Ca2+ channel resulted in an increment similar to that for the control neurons. These data suggest that both N-type and P/Q-type Ca2+ channels contribute to the high K+-evoked Ca2+ influx; however, the Ca2+ entry from P/Q-type VDCCs plays a more important role than N-type VDCCs in GSK-3-regulated neuronal vesicle exocytosis.
Then we asked, how might GSK-3 affect the activity of the Ca2+ channel? Previous studies show that phosphorylation of L-type Ca2+ channel increases the Ca2+ current in skeletal muscle (Curtis and Catterall, 1985), whereas phosphorylation of N-type Ca2+ channel inactivates the channel activity in bullfrog sympathetic neurons (Werz et al., 1993). Additionally, the synprint site of the N-type Ca2+ channels can be phosphorylated by PKC, Ca2+/calmodulin protein kinase II, and cdk5, and the phosphorylation inhibits its association with SNARE proteins (Yokoyama et al., 1997), which causes inhibition of neurotransmitter release (Schoch et al., 2001). In the present study, we found that GSK-3, a widely distributed Ser/Thr kinase in the brain, could phosphorylate the LII-III of P/Q-type Ca2+ channel and the phosphorylation suppressed its binding with the SNARE proteins. By using general phosphorylation-predicting tools, such as “Disphos” and “Netphos,” we analyzed the possible phosphorylation motifs of the LII-III peptide. It was shown that Ser854, Ser867, Ser882, and Thr915 in the LII-III peptide had scores of >0.8 (Disphos) and 0.995 (Netphos). We then used kinase-specific phosphorylation-predicting tools, including PPSP, GPS, and CRPphos, for the analysis, and the results from all these three methods also pointed to Thr915 as the possible GSK-3β phosphorylation site. We also noticed that the GSK-3 phosphorylation motif containing Thr915 (PRRHTHRP) in LII-III has two prolines and three arginines, which may facilitate chemically the phosphorylation reaction. Additionally, the Ser867 (PSSP) in LII-III was also a potential phosphorylation site of GSK-3 according to the prediction and kinase motif analyses; however, we did not detect any positive signal by using anti-phosphoserine antibody. Furthermore, though there are 60 Ser/Thr residues in LII-III, but only Ser854, Ser867, Ser882, and Thr915 are the potential phosphorylation sites according to the theoretical prediction analyses, and we only detected ∼1.5 mol phosphate/mol LII-III peptide by activation of GSK-3β. The exact phosphorylation site(s) of LII-III by GSK-3β might be studied in the future by mass spectrometry.
Syb plays a crucial role in SNARE complex formation. In differentiated neurons, Syb forms two mutually exclusive protein complexes, i.e., the SNARE complex composed of Syb and the plasma membrane proteins syntaxin and SNAP25, and the complex of Syb and SypI (Pfeffer, 1996). We found in the present study that the association of Syb with SNAP25 and syntaxin decreased with activation of GSK-3. As the association of Syb with the SNARE components is tightly regulated by the [Ca2+]i, we speculate that the decreased Ca2+ influx induced by GSK-3 activation may at least partially contribute to the decreased association. Additionally, the Syb–SypI complex purified from synaptosome does not contain syntaxin or SNAP25 (Edelmann et al., 1995), and an inverse correlation between the basal rate of synaptic vesicle exocytosis and the extent of Syb–SypI association has also been observed (Bacci et al., 2001). Furthermore, inhibition of the binding of SypI with Syb by a peptide increases the mEPSC frequency in neurons (Bacci et al., 2001). These data suggest that Syb must dissociate from SypI before entering the SNARE complex for the exocytosis. Elevation of intracellular Ca2+ causes dissociation of the Syb–SypI complex (Reisinger et al., 2004). Our data in the present study show that activation of GSK-3 retards the dissociation of Syb–SypI complex, which is consistent with a previous study showing that suppressing GSK-3 activity in zebrafish retinal ganglion cell increases the size of Syb-positive puncta in axons terminal (Tokuoka et al., 2002). Based on these observations, we speculate that the retarded dissociation of Syb from SypI also interrupts the SNARE complex formation and thus impairs the exocytosis.
In summary, we found that GSK-3β inhibits presynaptic vesicle exocytosis. GSK-3 phosphorylates the synprint site of P/Q-type calcium channel that interrupts association of the Ca2+ channel with synaptic proteins. The decreased [Ca2+]i-induced GSK-3 activation also disrupts the association of Syb with t-SNARE and the dissociation of Syb from SypI. These effects together will lead to an impaired exocytosis (Fig. 6). Our findings reveal the cellular and molecular basis for dysfunctional regulation of excitatory synaptic transmission by GSK-3 signaling.
Figure 6.

A schematic diagram showing the mechanisms underlying the GSK-3-inhibited presynaptic exocytosis of vesicles. According to the results presented in the current study, we propose that activation of GSK-3β may inhibit the exocytosis by phosphorylating/inactivating P/Q-type VDCC and arresting Ca2+ influx, which disrupts the interaction of synaptic proteins, including decreasing the association of Syb with t-SNARE, decreasing the dissociation of Syb from SypI, and decreasing the association of LII-III with synaptotagmin and the t-SNARE (t-SNARE, syntaxin, and SNAP25).
Footnotes
This work was supported in part by grants from the National Natural Science Foundation of China (30800342 and 30971478) and the National Science and Technology Committee of China (2006CB500703). We thank Dr. J. R. Woodgett for GSK-3β plasmids, Dr. K. Marcelo for HA-pcDNA3.0 plasmid, Dr. Gero Miesenboeck for pHluroin-Syb plasmid, Dr. F, Valtorta for pECFP-Syb and pEYFP-SypI plasmids, Dr. H. Xu for N2a cell line, and Dr. K. Shen for proofreading of this paper.
References
- Bacci A, Coco S, Pravettoni E, Schenk U, Armano S, Frassoni C, Verderio C, De Camilli P, Matteoli M. Chronic blockade of glutamate receptors enhances presynaptic release and downregulates the interaction between synaptophysin-synaptobrevin-vesicle-associated membrane protein 2. J Neurosci. 2001;21:6588–6596. doi: 10.1523/JNEUROSCI.21-17-06588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherer U, Moser T, Stühmer W, Oheim M. Calcium regulates exocytosis at the level of single vesicles. Nat Neurosci. 2003;6:846–853. doi: 10.1038/nn1087. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Carnes CA, Janssen PM, Ruehr ML, Nakayama H, Nakayama T, Haase H, Bauer JA, Chung MK, Fearon IM, Gillinov AM, Hamlin RL, Van Wagoner DR. Atrial glutathione content, calcium current, and contractility. J Biol Chem. 2007;282:28063–28073. doi: 10.1074/jbc.M704893200. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- Curtis BM, Catterall WA. Phosphorylation of the calcium antagonist receptor of the voltage-sensitive calcium channel by cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1985;82:2528–2532. doi: 10.1073/pnas.82.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Fujii T, Nakai K, Nakajima Y, Kawashima K. Enhancement of hippocampal cholinergic neurotransmission through 5-HT1A receptor-mediated pathways by repeated lithium treatment in rats. Can J Physiol Pharmacol. 2000;78:392–399. [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- Gould TD, Zarate CA, Manji HK. Glycogen synthase kinase-3: a target for novel bipolar disorder treatments. J Clin Psychiatry. 2004;65:10–21. [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hernández F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem. 2002;83:1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- Hirning LD, Fox AP, McCleskey EW, Olivera BM, Thayer SA, Miller RJ, Tsien RW. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science. 1988;239:57–61. doi: 10.1126/science.2447647. [DOI] [PubMed] [Google Scholar]
- Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25:81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kim DK, Catterall WA. Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc Natl Acad Sci U S A. 1997;94:14782–14786. doi: 10.1073/pnas.94.26.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky N, Belmaker RH, Agam G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur Neuropsychopharmacol. 2002;12:13–25. doi: 10.1016/s0924-977x(01)00131-6. [DOI] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- Lee KY, Rosales JL, Tang D, Wang JH. Interaction of cyclin-dependent kinase 5 (Cdk5) and neuronal Cdk5 activator in bovine brain. J Biol Chem. 1996;271:1538–1543. doi: 10.1074/jbc.271.3.1538. [DOI] [PubMed] [Google Scholar]
- Lehane AM, Kirk K. Chloroquine resistance-conferring mutations in pfcrt give rise to a chloroquine-associated H+ leak from the malaria parasite's digestive vacuole. Antimicrob Agents Chemother. 2008;52:4374–4380. doi: 10.1128/AAC.00666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, Xu H, Wang JZ. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem. 2003;87:1333–1344. doi: 10.1046/j.1471-4159.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- Llinás R, Steinberg IZ, Walton K. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J. 1981;33:323–351. doi: 10.1016/S0006-3495(81)84899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughrey CM, Smith GL, MacEachern KE. Comparison of Ca2+ release and uptake characteristics of the sarcoplasmic reticulum in isolated horse and rabbit cardiomyocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1149–1159. doi: 10.1152/ajpheart.00060.2004. [DOI] [PubMed] [Google Scholar]
- Martin-Moutot N, Charvin N, Leveque C, Sato K, Nishiki T, Kozaki S, Takahashi M, Seagar M. Interaction of SNARE complexes with P/Q-type calcium channels in rat cerebellar synaptosomes. J Biol Chem. 1996;271:6567–6570. doi: 10.1074/jbc.271.12.6567. [DOI] [PubMed] [Google Scholar]
- McGahon B, Lynch MA. The synergism between metabotropic glutamate receptor activation and arachidonic acid on glutamate release is occluded by induction of long-term potentiation in the dentate gyrus. Neuroscience. 1996;72:847–855. doi: 10.1016/0306-4522(95)00579-x. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW. Culturing hippocampal and cortical neurons. Methods Cell Biol. 2003;71:111–127. doi: 10.1016/s0091-679x(03)01007-0. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Pennuto M, Bonanomi D, Benfenati F, Valtorta F. Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol Biol Cell. 2003;14:4909–4919. doi: 10.1091/mbc.E03-06-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR. Transport vesicle docking: SNAREs and associates. Annu Rev Cell Dev Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Pumplin DW, Reese TS, Llinás R. Are the presynaptic membrane particles the calcium channels? Proc Natl Acad Sci U S A. 1981;78:7210–7213. doi: 10.1073/pnas.78.11.7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Clements JD, Bekkers JM. Nonuniform distribution of Ca2+ channel subtypes on presynaptic terminals of excitatory synapses in hippocampal cultures. J Neurosci. 1997;17:2738–2745. doi: 10.1523/JNEUROSCI.17-08-02738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger C, Yelamanchili SV, Hinz B, Mitter D, Becher A, Bigalke H, Ahnert-Hilger G. The synaptophysin/synaptobrevin complex dissociates independently of neuroexocytosis. J Neurochem. 2004;90:1–8. doi: 10.1111/j.1471-4159.2004.02472.x. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Affolter H, Neher E. Design of the EPC-9, a computer-controlled patch-clamp amplifier. 2. Software. J Neurosci Methods. 1995;56:203–215. doi: 10.1016/0165-0270(94)00129-5. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Neurotransmitter release at central synapses. Neuron. 2003;40:381–388. doi: 10.1016/s0896-6273(03)00643-3. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2006;9:309–317. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- Tokuoka H, Yoshida T, Matsuda N, Mishina M. Regulation by glycogen synthase kinase-3β of the arborization field and maturation of retinotectal projection in zebrafish. J Neurosci. 2002;22:10324–10332. doi: 10.1523/JNEUROSCI.22-23-10324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa K, Ohta J, Matsushita M, Moriwaki A, Li ST, Takei K, Matsui H. Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J Neurosci. 2002;22:2590–2597. doi: 10.1523/JNEUROSCI.22-07-02590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiser SL, Cascio CS, O'Donohue TL, Thoa NB, Jacobowitz DM, Kellar KJ. Lithium increases serotonin release and decreases serotonin receptors in the hippocampus. Science. 1981;213:1529–1531. doi: 10.1126/science.6269180. [DOI] [PubMed] [Google Scholar]
- Vardjan N, Stenovec M, Jorgacevski J, Kreft M, Zorec R. Subnanometer fusion pores in spontaneous exocytosis of peptidergic vesicles. J Neurosci. 2007;27:4737–4746. doi: 10.1523/JNEUROSCI.0351-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werz MA, Elmslie KS, Jones SW. Phosphorylation enhances inactivation of N-type calcium channel current in bullfrog sympathetic neurons. Pflugers Arch. 1993;424:538–545. doi: 10.1007/BF00374919. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the α 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, Wang Q, Chen JG, Wang JZ. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci. 2007;27:12211–12220. doi: 10.1523/JNEUROSCI.3321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]