

Abstract
Lysine N-pyrrolation, converting lysine residues to Nϵ-pyrrole-l-lysine, is a recently discovered post-translational modification. This naturally occurring reaction confers electrochemical properties onto proteins that potentially produce an electrical mimic to DNA and result in specificity toward DNA-binding molecules such as anti-DNA autoantibodies. The discovery of this unique covalent protein modification provides a rationale for establishing the molecular mechanism and broad functional significance of the formation and regulation of Nϵ-pyrrole-l-lysine–containing proteins. In this study, we used microbeads coupled to pyrrolated or nonpyrrolated protein to screen for binding activities of human serum-resident nonimmunoglobin proteins to the pyrrolated proteins. This screen identified apolipoprotein E (apoE) as a protein that innately binds the DNA-mimicking proteins in serum. Using an array of biochemical assays, we observed that the pyrrolated proteins bind to the N-terminal domain of apoE and that oligomeric apoE binds these proteins better than does monomeric apoE. Employing surface plasmon resonance and confocal microscopy, we further observed that apoE deficiency leads to significant accumulation of pyrrolated serum albumin and is associated with an enhanced immune response. These results, along with the observation that apoE facilitates the binding of pyrrolated proteins to cells, suggest that apoE may contribute to the clearance of pyrrolated serum proteins. Our findings uncover apoE as a binding target of pyrrolated proteins, providing a key link connecting covalent protein modification, lipoprotein metabolism, and innate immunity.
Keywords: post-translational modification (PTM), apolipoprotein E (apoE), protein-protein interaction, serum, innate immunity, inflammation, damage-associated molecular pattern (DAMP), hyperlipidemia, Lysine N-pyrrolation, Nϵ-pyrrole-l-lysine
Introduction
Proteins undergo covalent modification by reactive metabolites, such as oxidized fatty acids and intermediates of glycolysis, under physiological and pathophysiological conditions (1). The modification of proteins exerts a major impact on their chemical properties, leading to important functional and regulatory consequences. The ϵ-amino group of lysine, one of the three basic residues critical for protein structure and function, is a major target of covalent modification. Modification of lysine residues results in the gain of physicochemical characteristics, such as an increased negative charge caused by neutralization of the lysine residues. These host-derived, modified proteins are also categorized as damage-associated molecular patterns (DAMPs),2 which represent endogenous danger molecules as distinct from pathogen-associated molecular patterns. Oxidized low-density lipoproteins (LDLs) are one of the DAMPs, in which lysine residues are predominantly modified, and there are numerous claims of their presence in vivo. DAMPs can bind to pattern recognition receptors, such as Toll-like receptors, and mediate inflammatory responses by triggering the release of a variety of mediators (2). DAMPs, possessing an exposed epitope, are also recognized by the soluble pattern recognition receptors, such as innate antibodies (3, 4). It has been proposed that the neutralization of positive charges through covalent modification of the lysine residues might be involved in the interaction with the antibodies (5).
The conversion of the lysine residues into various types of pyrrole derivatives has previously been shown to take place upon the reaction of proteins with various lipid peroxidation-derived aldehydes (6–11). Using an LC-ESI-MS/MS with a stable isotope dilution method, we have recently established the presence of proteins containing Nϵ-pyrrole-l-lysine (pyrK) in the human and mouse sera (12). This finding provided direct evidence, for the first time, that the pyrrolation of proteins converting lysine residues to pyrK (Fig. 1) occurs in vivo. The pyrrole ring is an important structural motif found in a wide range of biologically active natural products and is highly susceptible to electrophilic attack and oxidation (13). It also displays a variety of chemical properties, including π–π stacking and hydrogen-bonding interactions, that may contribute to the closer packing and higher density of pyrrole-containing molecules. Because of these chemical characteristics, the conversion of lysine residues to pyrK confers unique and interesting properties onto proteins. We have previously shown that the lysine N-pyrrolation is associated with an increase in the net negative charges of the proteins because of the modification of the lysine residues and formation of amino charge-neutralized structures (12). In addition, the pyrrolated proteins show a ligand specificity toward the DNA-binding molecules, such as the anti-DNA autoantibodies and DNA intercalators. The production of multispecific antibodies that recognize both pyrrolated proteins and DNA is indeed accelerated in systemic lupus erythematosus, a systemic autoimmune disease characterized by the elevated production of autoantibodies. Thus, lysine N-pyrrolation is a physiologically relevant post-translational modification and could be an endogenous source of DNA mimic proteins. These findings provide a rationale for establishing a molecular mechanism and broad functional significance of the formation and regulation of the pyrrolated proteins. In this context, we speculated the presence of an innate regulatory mechanism for the pyrrolated proteins having structural properties similar to DNA and tried to identify serum pyrrole-binding proteins.
Figure 1.

The scheme of lysine N-pyrrolation.
Results
Identification of serum pyrrole-binding proteins
To determine the presence of serum nonimmunoglobin protein(s) that can bind pyrrolated proteins, normal human serum was incubated with beads coupled to either the native BSA or pyrrolated BSA (pyrBSA), and the bound proteins were eluted and then separated by SDS-PAGE under reducing conditions (Fig. 2A). Several unique bands, including the protein with a molecular mass of ∼30 kDa (indicated by the filled arrowhead in Fig. 2A), were exclusively found in the pulldown with the pyrBSA. A similar protein band was also detected in the pyrrolated poly-l-lysine pulldowns. To identify the putative pyrrole-binding protein, the band was excised and analyzed by MALDI-TOF MS. Nine peptides were found in the pulldown with pyrBSA, of which >29% could be attributed to apolipoprotein E (apoE) (Table S1).3 Another protein band with the molecular mass of ∼75 kDa (indicated by the open arrowhead in Fig. 2A) was also detected in the SDS-PAGE, which was identified as prothrombin (Table S2).
Figure 2.

Identification of an innate binding protein for pyrrolated proteins in human serum. A, pulldown assay for the detection of binding proteins for the pyrrolated proteins in human serum. The human serum was incubated with the ligand-coupled beads for 1 h at room temperature. The ligands used were BSA, pyrBSA, poly-l-lysine (PLL), and pyrrolated PLL. The proteins bound to the beads were eluted by adding the sample buffer and heating (80 °C for 10 min) and separated by SDS-PAGE. The proteins indicated by the filled and open arrowheads represent apoE and prothrombin, respectively, identified by MALDI-TOF MS. B, binding of pyrrolated proteins to the apoE isoforms. The apoE isoforms (50 μg/ml) were immobilized on a plate and incubated with biotin-labeled BSA or pyrBSA (20 μg/ml) at 4 °C for 12 h. The data are from single experiments, performed in triplicate wells, representative of three individual experiments. The results shown are means ± S.D. (n = 3). C, pulldown assay for the binding of pyrrolated proteins to the apoE isoforms. Three apoE isoforms were separately incubated with the ligand-coupled beads for 1 h at room temperature. The ligands used were BSA and pyrBSA. The proteins bound to the beads were eluted by adding the sample buffer and heating (80 °C, 10 min) and separated by SDS-PAGE. The apoE isoforms were detected by immunoblotting (IB) with anti-apoE mAb E6D7 (Abcam). D, surface plasmon resonance measurements. The interactions of pyrrolated BSA and an apoE isoform immobilized by amine coupling in the lanes 2–4 of a BIAcore sensor chip CM5 were monitored by the single-cycle kinetics method (n = 3). Lane 1 was used as the blank.
Binding of apoE isoforms to the pyrrolated proteins
apoE, a single polypeptide chain composed of 299 amino acid residues, exists in three major isoforms: apoE2, apoE3, and apoE4, which differ at amino acid residues 112 and 158 (Fig. S1) (14). Because the MALDI-TOF MS analysis of the serum pyrrole-binding proteins did not allow isoform identification, we investigated whether variation in these amino acids might contribute to their differences in the binding preferences for the pyrrolated proteins. The solid-phase binding and pulldown assays showed that pyrBSA was almost equally bound to the three apoE isoforms (Figs. 2, B and C). The result was also confirmed by the surface plasmon resonance measurements (Fig. 2D and Table 1).
Table 1.
Kinetic parameters of the interaction between pyrrolated BSA and apoE isoforms measured by surface plasmon resonance
ka, association rate constant; kd, dissociation rate constant; KD, equilibrium dissociation constant; Rmax, analyte binding capacity of the surface; χ2, measure of the average squared residual (the difference between the experimental data and the fitted curve); U value, estimate of the uniqueness of the calculated values for rate constants and Rmax.
| ka | kd | KD | Rmax | χ2 | U value | |
|---|---|---|---|---|---|---|
| 104 1/Ms | 10−5 1/s | 10−10 m | RU | RU2 | ||
| apoE2 | 6.77 ± 0.63a | 8.17 ± 0.75 | 12.1 ± 0.57 | 22.7 ± 2.60 | 0.200 ± 0.144 | 14.0 ± 6.56 |
| apoE3 | 8.74 ± 0.68 | 15.6 ± 4.71 | 18.1 ± 6.19 | 15.8 ± 2.43 | 0.245 ± 0.178 | 16.0 ± 9.54 |
| apoE4 | 6.64 ± 0.24 | 15.2 ± 2.64 | 22.8 ± 3.80 | 25.4 ± 0.86 | 0.189 ± 0.056 | 4.67 ± 0.58 |
a The experiments were performed in triplicate, and the means ± S.D. are shown.
Pyrrole-binding sites in apoE3
apoE contains two independently folded functional domains, a 22-kDa N-terminal domain (amino acids 1–191) and a 10-kDa C-terminal domain (amino acids ∼225–299), separated by a hinge region (15). The N-terminal domain contains the LDL receptor–binding region located at residues 134–150 and Arg-172 and forms a four-helix bundle of amphipathic α-helices. The C-terminal domain contains amphipathic α-helices that are involved in the binding to lipoprotein particles. apoE also contains two heparin-binding sites: a high-affinity binding site in the N-terminal domain located at sequence 142–147 and a low-affinity binding site in the C-terminal domain located at sequence 211–218 (16, 17). Heparin indeed competed in a concentration-dependent manner for the binding of apoE3 to pyrBSA (Fig. 3A), suggesting that heparin and the pyrrolated proteins might share a binding region on apoE. To further characterize the pyrrole-binding site, apoE3 was cleaved with thrombin, and the fragments were tested for their ability to bind to the biotin-labeled pyrBSA. Thrombin cleavage resulted in the generation of one major fragment of 22 kDa (E22) corresponding to residue 1–191 and three fragments of 25 (E25), 12 (E12), 10 kDa (E10) corresponding to residues 1–216, 191–299, and 216–299, respectively (Fig. 3B). Ligand blot analysis showed that the pyrrolated BSA bound to the two larger thrombolytic fragments (E25 and E22), whereas no significant binding was observed with the two smaller fragments (E12 and E10) (Fig. 3C). The further cyanogen bromide hydrolysis of the E22 fragment resulted in the loss of ligand binding (data not shown), suggesting that a relatively large region, containing the sequence 142–147 (one of the heparin-binding sites), may be required to display the binding with the pyrrolated proteins.
Figure 3.

Binding of apoE isoforms to the pyrrolated proteins. A, effect of heparin on the binding of pyrrolated proteins to apoE3. The biotin-labeled BSA or biotin-labeled pyrrolated BSA was preincubated by serial dilutions of heparin before the addition to the apoE3-coated ELISA plates. Differences were analyzed by Tukey's HSD test among the BSA- or pyrBSA-coated group. Different letters above the bars indicate a significant difference (p < 0.01). NS, not significant. The results shown are means ± S.D. (n = 6). B, the schematic representation of the structural domains of apoE3 and four thrombin-digested peptides. C, ligand blot assay for the binding of the pyrrolated proteins to thrombin-digested apoE3. apoE3 was digested with thrombin for 3 h at 37 °C, and the resultant peptides were separated by 15% gel nonreducing SDS-PAGE. The gels were either stained with Coomassie Brilliant Blue R-250 (CBB) or transferred to PDVF membrane followed by ligand blotting with biotin-labeled (Bt) BSA or biotin-labeled pyrrolated BSA. The data are representative of three individual experiments. The four peptides (E10, E12, E22, and E25) were identified by MALDI-TOF peptide mass mapping.
We also questioned whether apoE could recognize the pyrrolated lysine itself as a ligand. When apoE3 was incubated with the authentic pyrK (Fig. 4A) and analyzed by native gel electrophoresis, a slight mobility shift of the protein was observed (Fig. 4B). In addition, although apoE3 showed a weak affinity for pyrK (Fig. 4C), pyrK inhibited the binding of apoE3 to the pyrrolated proteins in a dose-dependent manner (Fig. 4D). Using a biotin-labeled N-pentylpyrrole, we determined the binding of the pyrrole probe to the apoE and the 22-kDa fragment (Figs. 4, E and F).
Figure 4.

Binding of apoE to pyrK. A, chemical structure of pyrK. B, mobility shift of apoE upon incubation with pyrK. C, surface plasmon resonance measurements. pyrK was subjected to an exchange reaction to preload it and tested for an interaction with apoE3 immobilized on a Biacore chip. Sensorgrams shows an increase in response units (RU) reflective of pyrK binding (association) and a slow decrease in response consistent with a loss of mass from washout (dissociation) after each injection (arrows). D, effect of pyrK on the binding of pyrrolated proteins to apoE3. apoE3 was preincubated by serial dilutions of pyrK before addition to the pyrrolated protein-coated ELISA plates. The experiment was performed in duplicate wells. Shown are the means ± S.D. of three independent experiments. E, chemical structures of biotin-labeled N-pentylamine (Bt-PA) and N-pentylpyrrole (Bt-Ppyr). F, ligand blot assay for the binding of biotin-labeled probes to thrombin-digested apoE. apoE was digested with thrombin for 3 h at 37 °C, and the resultant peptides were separated by 15% gel nonreducing SDS-PAGE. The gels were either stained with Coomassie Brilliant Blue R-250 (CBB) or transferred to PDVF membrane followed by ligand blotting with biotin-labeled N-pentylamine or biotin-labeled N-pentylpyrrole. The data are representative of three individual experiments.
Binding characteristics of apoE to pyrrolated molecules
apoE binds to the phospholipid dimyristoylphosphatidylcholine (DMPC) to form discoidal particles containing an average of four apoE molecules per particle (18). The binding capacity of the pyrrolated proteins and pyrK to the lipid (DMPC)–bound apoE3 was lower than that to the lipid-free form (Fig. 5A). The data suggest that the pyrrolated molecules might interact mainly with the lipid-free apoE.
Figure 5.
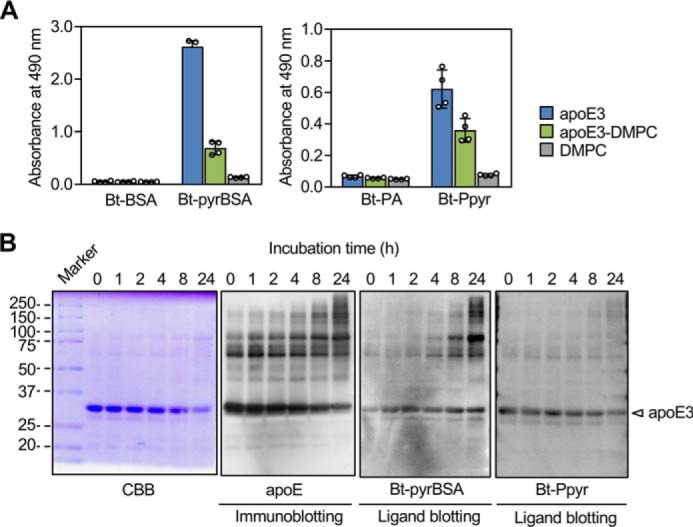
Binding characteristics of apoE to pyrrolated molecules. A, binding capacity of the pyrrolated proteins to DMPC-bound apoE. The biotin-labeled (Bt) BSA or biotin-labeled pyrrolated BSA was added to the ELISA plates that had been coated with DMPC, apoE, or apoE preincubated with DMPC. The experiment was performed in duplicate wells. Shown are the means ± S.D. of two independent experiments. B, ligand blot assay for the binding of the biotin-labeled pyrBSA or N-pentylpyrrole to polymerized apoE3. apoE3 preincubated for 0–24 h at 37 °C was separated by 10% gel nonreducing SDS-PAGE. The gels were either stained with Coomassie Brilliant Blue R-250 (CBB) or transferred to PDVF membrane followed by immunoblotting with anti-apoE mAb E6D7 (Abcam) or by ligand blotting with biotin-labeled pyrBSA or N-pentylpyrrole.
On the other hand, the lipid-free apoE is known to form oligomers through its C-terminal domain (19). It has also been suggested that the oligomerization of the apoE molecules alters their physiological functions (20, 21). The better binding of the oligomeric apoE3 compared with that of the monomeric form was indeed shown by ligand blotting (Fig. 5B). In contrast to the binding of the pyrrolated proteins to the oligomeric forms of apoE3, the biotin-labeled N-pentylpyrrole was rather specific to the monomeric form of apoE3. Thus, the apoE3 monomer can weakly recognize pyrK and its related analogs, whereas the self-association of the apoE3 monomer to oligomers may be essential for the strong binding to the pyrrolated proteins. Despite significant binding to pyrBSA and pyrK, apoE3 had no binding potential to other pyrrole-containing small molecules, such as hemin, bilirubin, pyrrole-2-carboxylic acid, and tryptophan (data not shown).
Lysine N-pyrrolation in apoE-deficient mice
The apoE deficiency is associated with a series of pathological conditions, including atherosclerosis. To gain an insight into the connection between the pyrrolation of serum proteins and apoE, we analyzed pyrK in the sera from spontaneously hyperlipidemic apoE-deficient (C.KOR/StmSlc-Apoesh) mice with a genetic background of BALB/c using LC-ESI-MS/MS. The total triglyceride and cholesterol levels were significantly elevated in the hyperlipidemia mice compared with the controls, whereas no significant differences were observed in the body weight and glucose level (data not shown). The measurement of pyrK showed that there are significant differences in the serum pyrrolated protein levels between the control and apoE-deficient mice (Fig. 6, A and B). Of interest, the pyrrolated protein levels in the hyperlipidemia males were significantly higher than in the females. We also examined the antibody titer against the pyrrolated proteins in the sera from the male control and hyperlipidemia mice and observed that there is an increased tendency for the serum IgG titers to the pyrrolated proteins in the hyperlipidemia mice as compared with the control mice (Fig. 6C). In addition, the levels of the anti-DNA titers in the hyperlipidemia mice were notably higher than those in the control mice. Thus, the high abundance of the pyrrolated proteins was found in the sera from the apoE-deficient hyperlipidemia mice compared with the controls, which was associated with the enhanced immune response to the pyrrolated proteins and DNA. These data suggest the relevance of the lysine N-pyrrolation, generating DNA mimic proteins, for the immune response in hyperlipidemia.
Figure 6.

Acceleration of lysine N-pyrrolation and immune response in apoE-deficient mice. A, LC-ESI-MS/MS analysis of pyrK in the hydrolysate of sera from male control (apoE+/+) (lower panels) and male spontaneously hyperlipidemic (apoE−/−) (upper panels) mice. The ion current tracings with selected reaction monitoring (SRM) are shown. The two left panels represent the SRM of pyrK in the hydrolysate of sera from apoE+/+ (upper panel) and apoE−/− (lower panel) mice. The two right panels represent the SRM of isotope-labeled [13C6,15N2]pyrK added in the serum samples of apoE+/+ (upper panel) and apoE−/− (lower panel) mice as the internal standard (I.S.). B, the levels of serum pyrrolated proteins in the sera from apoE+/+ and apoE−/− mice. **, p < 0.01. C, the levels of the antibody titers against antigens (BSA, pyrBSA, and DNA) in the sera from the male control and hyperlipidemia mice. Elevation of immune response to BSA (left panels), pyrrolated BSA (middle panels), and dsDNA (right panels) in the sera from control and hyperlipidemic mice is shown. The levels of the IgG Abs in the plasma samples were measured by ELISA using native and pyrrolated BSAs and calf-thymus dsDNA as the coating antigens. *, p < 0.05; **, p < 0.01. The measurements of pyrK (B) and antibody titer (C) were performed separately using different group of animals.
Given the significant increase in the serum levels of the pyrrolated protein in the apoE-deficient mice, we attempted to identify the direct molecular target of the lysine N-pyrrolation in the sera of hyperlipidemia mice. To this end, we separated and fractionated the sera by anion exchange chromatography on a HitrapQ FF 5 ml column, and the level of the pyrrolated proteins in each fraction was evaluated by the pyrK measurement. In both the control and hyperlipidemia mice, pyrK was mainly detected in the fractions eluted from 10 to 25 min (Fig. 7). In addition, we selected one fraction eluted from 14 to 16 min in the apoE-deficient mice and analyzed by MALDI-TOF MS (Fig. S2). From three independent proteomic experiments, the protein contained in the fraction was identified as serum albumin. The same protein was identified as the pyrrolation target in the control mice.
Figure 7.
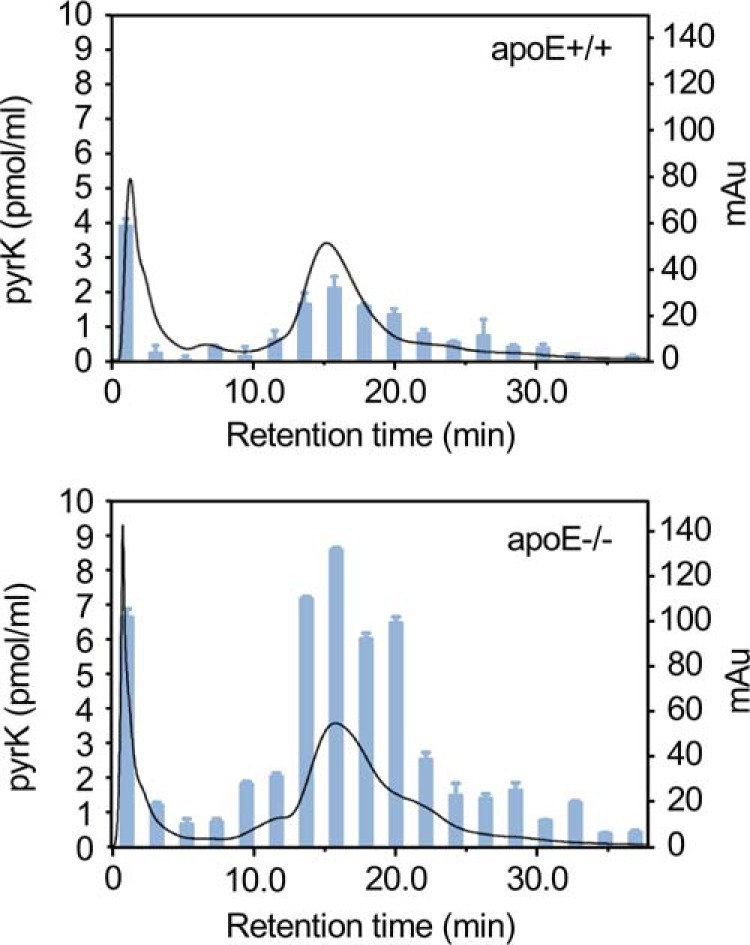
Identification of serum pyrrolation targets in apoE-deficient mice. The sera from apoE+/+ (upper panel) and apoE−/− (lower panel) mice were separated and fractionated by anion exchange chromatography on a HitrapQ FF 5-ml column. The level of the pyrrolated proteins in each fraction was evaluated by the measurement of pyrK using LC-ESI-MS/MS. Solid line, profile of UV absorbance at 280 nm. Bars, amount of pyrK. The results shown are means ± S.D. of two independent experiments.
Based on the findings obtained from the animal study regarding hyperlipidemic mice, we further determined the relationship between the lysine N-pyrrolation and apoE in human hyperlipidemia (Table S3). Nineteen hyperlipidemia patients meeting these specific criteria and twenty healthy individuals were analyzed to measure the pyrK. The hyperlipidemia patients showed slightly, but significantly elevated levels of serum pyrrolated proteins compared with the normal control individuals (normal (n = 20): 6.82 ± 2.14 pmol/mg protein; hyperlipidemia (n = 19): 8.35 ± 1.66 pmol/mg protein (mean ± S.D.); p < 0.05 by Student's unpaired t test) (Fig. 8A). The apoE levels of the hyperlipidemia patients were ∼3-fold higher than those of the control subjects (Fig. 8B). These data suggest a link between apoE and protein N-pyrrolation in hyperlipidemia.
Figure 8.
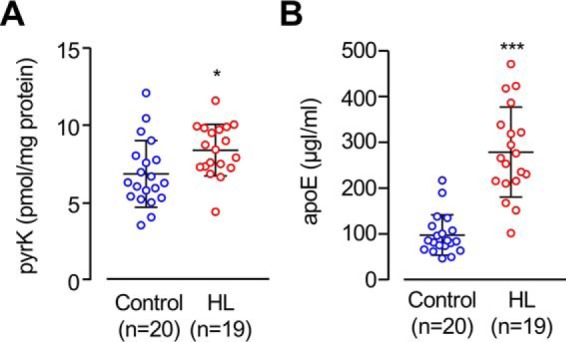
Serum levels of pyrK and apoE in hyperlipidemia. A, the levels of serum pyrrolated proteins were measured in the sera from normal subjects (n = 20) and patients with hyperlipidemia (HL, n = 19). *, p < 0.05. B, the levels of serum apoE were measured in the sera from normal subjects (n = 20) and patients with hyperlipidemia (n = 19). ***, p < 0.005.
apoE mediates cellular binding of pyrrolated proteins
apoE plays an important role in the lipoprotein metabolism through its binding to lipoprotein particles and with members of the LDL receptor family (22, 23). To address whether apoE affects the binding of pyrrolated proteins to the cell surface, we carried out a binding assay in the presence or absence of apoE. The pyrrolated proteins, even in the absence of apoE3, exhibited significant binding to RAW264.7 macrophages (Fig. 9, A and B). Notably, the binding of pyrrolated proteins to the cells was dramatically enhanced by apoE3 (Fig. S3). Both apoE2 and apoE4 also showed a similar effect (Fig. 9C and Fig. S4). Of interest, despite the significant inhibition of the binding of pyrrolated proteins by acetylated LDL, the apoE3-mediated binding of the pyrrolated proteins was barely affected by the native and modified LDLs (Fig. 9D). These data and the observation that apoE3 alone could also show a significant binding to the cells (Fig. S5) suggest that apoE may contribute to the clearance of the pyrrolated proteins.
Figure 9.
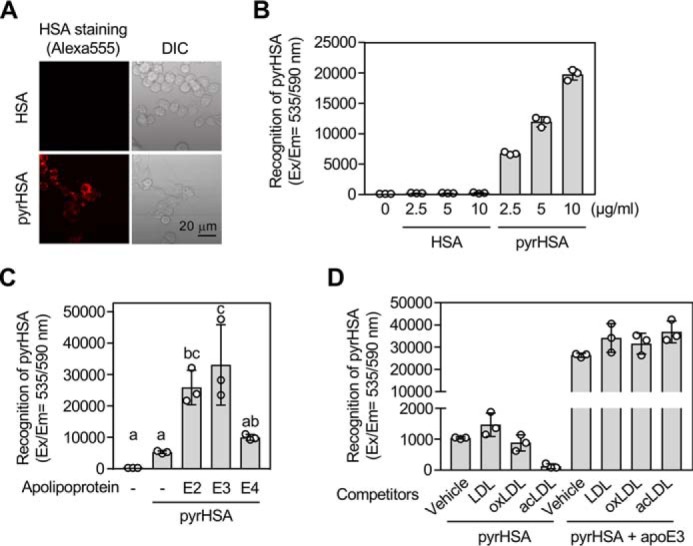
apoE3 enhances cellular recognition of pyrrolated proteins. A and B, binding/uptake of pyrHSA in RAW264.7 cells. A, representative fluorescence microscopy images of the cells treated with 1 μg/ml of Alexa Fluor® 555–labeled HSA (upper panels) or 1 μg/ml of Alexa Fluor® 555–labeled pyrHSA (lower panels) for 1 h. Left, Alexa Fluor® 555; right panels, brightfield. DIC, differential interference contrast. B, the cells were treated with 0–10 μg/ml of Alexa Fluor® 555–labeled HSA (n = 3) or Alexa Fluor® 555–labeled pyrHSA (n = 3) for 1 h. The results shown are means ± S.D. (n = 3). C, effect of apolipoproteins on binding/uptake of pyrHSA. RAW264.7 cells were treated with 10 μg/ml of Alexa Fluor® 555–labeled pyrHSA together with or without 50 μg/ml of apoE (apoE2, apoE3, or apoE4) for 1 h. The results shown are means ± S.D. (n = 3). The differences were analyzed by Tukey's HSD test. Different letters above bars indicate a significant difference (p < 0.05). D, effect of lipoproteins on binding/uptake of pyrHSA in RAW264.7 cells. After treatment of lipoproteins (100 μg/ml) for 1 h, RAW264.7 cells were treated with 10 μg/ml of Alexa Fluor® 555–labeled pyrHSA together with or without 50 μg/ml of apoE3 for 1 h. The results shown are means ± S.D. (n = 3).
Discussion
In the current study, we examined the presence of serum nonimmunoglobin proteins that are capable of binding the pyrrolated proteins and identified apoE as a hitherto unrecognized innate binding protein for the DNA mimic proteins in human serum. To the best of our knowledge, this is the first report describing a previously unknown function of apoE as a binding protein for covalently modified proteins. apoE is known to play multiple roles in the regulation of lipid metabolism and shows a diverse array of biological functions by interacting with multiple molecules, including lipoproteins, lipid-soluble vitamins, and cholesterol (15, 24). apoE binds with a high affinity to heparin and cell-surface heparan sulfate proteoglycan (25). Both apoE3 and E4 isoforms, containing the helix–loop–helix secondary structure as well as a high arginine content found in many DNA-binding protein, have been shown to bind dsDNA with high affinity and functions as a transcription factor (26). The presence of the specific binding protein for the pyrrolated proteins is thought to be an important indication that the processes leading to biologically relevant pyrrole molecules are taking place. Thus, the discovery of apoE as a pyrrole-binding protein offers new insights into the mechanisms and biological roles of lysine N-pyrrolation.
The solid-phase binding assay and surface plasmon resonance measurement demonstrated that all three apoE isoforms tightly bind the pyrrolated proteins (Fig. 2). We also showed that heparin competed with the pyrrolated proteins upon binding to apoE3 and that the pyrrolated proteins bound to the 22-kDa fragment correspond to the N-terminal domain (Fig. 3). These results and the fact that lysine N-pyrrolation is associated with an increase in the net negative charges of the proteins (12) suggest that the binding of apoE to the pyrrolated proteins may involve an electrostatic interaction, probably the same mechasnim proposed for apoE–heparin interaction (25). However, because biological significance of pyrrolated proteins remains to be completely established, it is still too early to conclude that this interaction mediates a common mechanism in apoE biology.
An interesting finding in this study is that apoE3 binds some pyrrole-containing small molecules as ligands. The mobility shift and ligand-binding assays revealed that apoE3 directly interacted with pyrK and its analog, N-pentylpyrrole (Fig. 4), suggesting the presence of a recognition site for an N-alkylpyrrole moiety. The ligand blot analysis of the thrombin-digested human apoE3 also demonstrated the binding of the biotinylated pentylpyrrole to the N-terminal domain, corresponding to the binding site of the pyrrolated proteins. In addition, heparin hardly blocked the binding of N-pentylpyrrole to the immobilized apoE3. These lines of evidence allow us to postulate the presence of a unique N-alkylpyrrole-binding site in the N-terminal domain, which may also contribute to the interaction of apoE3 with the pyrrolated proteins. Another interesting observation is that the pyrrolated proteins and N-pentylpyrrole preferentially interact with the oligomeric and monomeric forms of apoE3, respectively (Fig. 5), indicating that the apoE monomer needs to associate with oligomers before binding to pyrrolated proteins. It is tempting to speculate that the oligomerization of apoE may enhance the electrostatic interaction with pyrrolated proteins to stabilize them in a manner similar to the increased binding of the apoE dimer to heparan sulfate compared with the monomeric form (25).
To obtain in vivo evidence for a link between apoE and lysine N-pyrrolation, we determined pyrK in the sera from spontaneously hyperlipidemic apoE-deficient mice using LC-ESI-MS/MS. The data revealed that the deficiency of apoE led to a significant elevation of the pyrrolated proteins (Fig. 6). Interestingly, the serum pyrrolated protein levels were much higher in male than in female apoE-deficient mice. However, the precise mechanism for the gender and species difference in the formation of the pyrrolated proteins remains unknown. It is reasonable that the loss of function of apoE favors the accumulation of pyrrolated proteins in the apoE-deficient mice. Based on the enhanced pyrrolation of serum proteins in the apoE-deficient mice, we separated the mouse serum by gel filtration and identified serum albumin as one of the major targets of the lysine N-pyrrolation (Fig. 7). The identification of serum albumin as the major target of pyrrolation may be reasonable because of its relative abundance in human blood serum. It may also be associated with the fact that serum albumin is a binding protein for numerous chemical compounds, including fatty acids, amino acids, and hormones. These physiologically relevant protein-bound ligands may generate metabolites that could be involved in the lysine N-pyrrolation of serum albumin. It can be speculated that the pyrrolation of lysine residues in serum albumins may lead to the structural and functional alteration of the proteins. Because serum albumin is the most abundant plasma protein and the major blood stream carrier, it will be interesting and exciting to study how pyrrolation of the protein can affect the transport and availability of numerous chemical compounds and molecules in the blood vascular system. On the other hand, only a small difference in the serum levels of the pyrrolated proteins was observed between the control subjects and hyperlipidemia patients (Fig. 8). This result might be attributed to the high levels of serum apoE in the hyperlipidemia patients compared with the controls. Thus, a sufficient amount of apoE may be crucial for maintaining normal serum levels of the pyrrolated proteins in vivo. A previous study has shown that a significant proportion of plasma apoE resides within an intermediate-sized remnant-like lipoprotein fraction in both normolipidemic and hyperlipidemic, correlating with various proatherogenic lipid parameters (27). The data led us to speculate that apoE may bind pyrrolated proteins generated within the lipoprotein fraction.
apoE deficiency is associated with the production of autoantibodies against antigens including neuronal nuclei, cardiolipin, and oxidized LDL (28, 29). The most likely scenario is that the altered lipid and lipoprotein metabolism caused by the deficiency of apoE may accelerate the immune response, leading to the overproduction of these autoantibodies. On the other hand, our previous study suggested that a physiologically relevant oxidized metabolite of fatty acids might be involved in the formation of pyrrolated proteins (12). Thus, the alternative hypothesis is that an accelerated peroxidation of fatty acids in LDL and/or cardiolipin followed by the formation of pyrrolated proteins may lead to the overproduction of the autoantibodies not only to pyrrolated proteins but also to other autoantigens in apoE-deficient mice. However, causal relationship between protein pyrrolation caused by apoE deficiency and autoimmunity remains unclear.
A previous study has also detected splenomegaly and significantly increased serum titers of the anti-DNA autoantibody in the apoE-deficient mice as compared with the control mice (30). We observed in this study that the high abundance of pyrrolated proteins in the apoE-deficient mice was accompanied by the enhanced immune responses to the DNA and pyrrolated proteins (Fig. 6). This result may be associated with our previous findings that the pyrrolation transforms self-molecules into autoantigens, accelerating the production of autoantibodies against pyrrolated proteins and DNA (12). In addition, prominent increases in the antibody titers to both pyrrolated proteins and DNA in patients with systemic lupus erythematosus have been observed. Thus, the accumulation of pyrrolated proteins caused by apoE deficiency might be directly or indirectly involved in the production of autoantibodies against pyrrolated proteins and DNA. However, it is still unclear whether the pyrrolated proteins could be a trigger of production of other autoantibodies.
Innate immunity is stimulated by a wide range of modified biological molecules, such as oxidized low-density lipoproteins (3). These health- and disease-associated molecules have been shown to accelerate a pro-inflammatory response through receptors, including scavenger receptors (2). The previous finding (12) that the pyrrolated proteins are a ligand of natural IgM antibodies suggests that they represent DAMPs that trigger immune responses. Our preliminary study has also shown that the binding of the pyrrolated proteins was enhanced in the cells overexpressed with a scavenger receptor, LOX-1, and the anti-LOX-1 neutralizing antibody partially attenuated the NF-κB response triggered by the pyrrolated proteins. These data suggest that the pyrrolated proteins may also be a ligand of LOX-1. On the other hand, we showed that the cellular recognition of the pyrrolated proteins was dramatically facilitated by apoE3 (Fig. 9 and Fig. S3). These findings and the observation that the deficiency of apoE favored the accumulation of the pyrrolated proteins (Fig. 6) suggest the functional significance of apoE as a binding protein in the defense against the pyrrolated proteins. On the other hand, based on the observations that the apoE3-mediated binding of pyrrolated proteins was barely affected by the modified LDLs and that apoE3 alone could also bind to the cells, we speculated that apoE might serve as a bridging molecule for the cellular binding of the pyrrolated proteins and play a role in providing homeostatic responses against pyrrolated proteins ubiquitously generated in biological systems (Fig. 10). However, the details of the molecular mechanism for the apoE-mediated cellular recognition of the pyrrolated proteins remain unknown. Future work is therefore needed to understand the interplay between a cell surface molecule and apoE in mediating the biological effects of pyrrolated proteins and to determine whether the pyrrolated protein–apoE3 complex could contribute to vascular inflammation in vivo.
Figure 10.
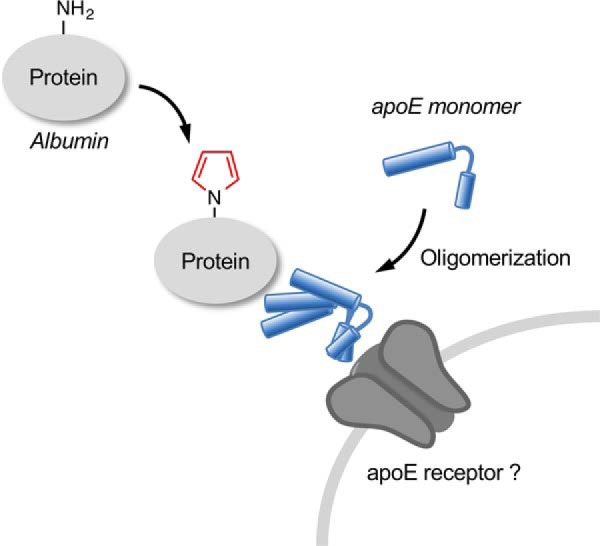
Schematic illustration of the formation and regulation of lysine N-pyrrolation.
Experimental procedures
Materials
BSA was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Recombinant human apoE isoforms (E2, E3, and E4) were obtained from PEPRO TECH. Dynabeads M-270 carboxylic acid were obtained from Invitrogen. EZ-Link® Sulfo-NHS-biotin, EZ-Link® biotin-LC-hydrazide, and EZ-Link® pentylamine biotin were obtained from Thermo Fisher Scientific. Purified thrombin from human plasma and dsDNA and PLL were obtained from Sigma. Serum apoE was measured using the human apoE ELISA kit (Assaypro). 4′,6-Diamidino-2-phenylindole was obtained from Dojindo (Kumamoto, Japan). All of the other reagents used in the study were of analytical grade and obtained from commercial sources.
Human serum samples
Serum samples were obtained from 20 healthy individuals and 19 patients with hyperlipidemia who underwent diagnostic evaluation at the Nagoya University Hospital (Nagoya, Japan). Serum samples from hyperlipidemic patients who were also diagnosed as autoimmune diseases, including rheumatoid arthritis, have been removed. This study was approved by the Ethical Committee of the Nagoya University School of Medicine. Patients were considered to have this lipid disorder based on clinical and biochemical criteria: high total cholesterol (>220 mg/dl), triglyceride (>150 mg/dl), and LDL (>140 mg/dl) levels at the baseline.
Animals
BALB/c and apoE-deficient mice (C.KOR/StmSlc-Apoeshl) were purchased from Japan SLC (Hamamatsu, Japan). The mice were housed in a temperature-controlled pathogen-free room with light from 7:00 a.m. to 7:00 p.m. (daytime) and had free access to standard food and water. All procedures were approved by the Animal Experiment Committee in the Graduate School of Bioagricultural Sciences of Nagoya University. For quantification of pyrK in sera, BALB/c mice (male, n = 6; female, n = 5) and apoE-deficient mice (male, n = 6; female, n = 5) were used as sera donors at 10 weeks old. For measurement of antibody titer, male BALB/c mice (n = 5) and male apoE-deficient mice (n = 5) were used as sera donors at 10 weeks old. Blood was collected from the tail vein and allowed to stand for 2 h at room temperature, after which the sera were collected by centrifugation at 3,500 rpm for 10 min and stored at −80 °C until used. The all mice were euthanized by cervical dislocation at 10 weeks old. Plasma/serum glucose, triglyceride, low-density lipoprotein, and total cholesterol were determined by the Nagoya University Clinical Laboratory.
Immunoblot analysis and ligand blotting
The samples were run on 15% SDS-polyacrylamide gels for the thrombin-digested samples and 10% SDS-polyacrylamide gels for the analysis of the apoE isoforms and dimer. After electrophoresis, the gel was transblotted onto a polyvinylidene difluoride membrane (GE Healthcare), incubated with skim milk for blocking, washed, and then incubated overnight with an anti-apoE antibody (E6D7, abcam), biotin-labeled BSA (50 μg), or biotin-labeled pyrrolated BSA (50 μg) in TBS with Tween 20 at 4 °C. After washing, the membrane was incubated with 1000× HRP-linked secondary Ab or HRP-NeutrAvidin for 1 h at room temperature. This procedure was followed by the addition of the ECL reagents. The bands were visualized using a Cool Saver AE-6955 (ATTO, Tokyo, Japan).
Preparation of pyrrolated compounds
The pyrrolated proteins were prepared by incubating BSA or HSA (1.0 mg/ml) with 1 mm butanedial in PBS for 24 h at 37 °C. pyrK was prepared as previously reported (12). The biotin-labeled N-pentylpyrrole was prepared upon incubation of 1 mm EZ-Link® pentylamine biotin with 1 mm butanedial for 24 h at 37 °C.
Pulldown assay
BSA or PLL coupled to the Dynabeads was incubated with 1 mm butanedial in 0.2 m PBS for 24 h to obtain the pyrrolated protein-coupled beads. The beads (2 × 107) were then added to microcentrifuge tubes and incubated with 400 μl of 2× dilution human serum in PBS/Tween for 3 h at room temperature. After washing three times with PBS/Tween, the binding protein was eluted by adding the sample buffer and heating (80 °C for 10 min). Protein separated by SDS-PAGE performed under reducing conditions was processed for tryptic digestion and MALDI-TOF/TOF MS (AB SCIEX 5800). Proteins were identified by the MASCOT (Matrix Science, London, UK) searching algorithms using the NCBInr database.
Thrombin digestion of apoE3
The apoE3 (25 μg) was digested with thrombin (1% w/w) in 72 μl of PBS for 3 h at 37 °C and subjected to SDS-PAGE. For identification of the apoE peptides, gel pieces were treated with reducing and alkylating agents followed by proteolysis with the Protease MAX surfactant (Promega) and Trypsin Gold (Promega) in 50 mm NH4HCO3 buffer. The supernatant was collected and desalted by ZipTip C18 reverse-phase microcolumns (Millipore). Peptide mass fingerprints were generated with MALDI-TOF/TOF MS (AB SCIEX 5800). Peptides were identified with the MASCOT searching algorithms using the NCBInr database.
Surface plasmon resonance
The surface plasmon resonance assays were performed using a Biacore T100 instrument (GE Healthcare). The apoE isoforms were immobilized on a sensor chip CM5 (GE Healthcare) at a density of 1000 response units. The interaction between the immobilized apoE isoforms and pyrrolated BSA (6.25–100 nm) was examined at 25 °C with a flow rate of 30 μl/min by a single-cycle kinetics analysis program. PBS was used as the running buffer. The response curves obtained from injecting buffer only and from the control flow cell (without immobilized apoE isoforms) were subtracted from the apoE-immobilized cell to correct for any nonspecific binding. BIAevaluation software (version 4.1) was used to perform the kinetic analysis.
LC-ESI-MS/MS analysis of pyrK
pyrK was measured using a stable isotope dilution-based LC-ESI-MS/MS technique as previously reported (12).
ELISA
Binding of pyrrolated proteins to the apoE isoforms was examined by ELISA in which the apoE isoforms (50 μg/ml) were immobilized on a plate and incubated with biotin-labeled BSA or pyrBSA (20 μg/ml) at 4 °C for 12 h. Competition assays were performed by ELISA in which binding of either biotin-labeled BSA or pyrrolated BSA to the coated apoE3 was competed by small molecules, such as heparin, DMPC, and pyrK. Biotin-labeled BSA or pyrrolated BSA was preincubated with indicated concentrations of the competitors before addition to the apoE3-coated ELISA plate. Bound biotin-labeled protein was detected with NeutrAvidin coupled to HRP. Substrate was measured as described above.
Measurement of antibody titer
The antibody titer against the pyrrolated proteins and DNA in the sera from male BALB/c mice and male apoE-deficient mice (n = 5 each) was analyzed by ELISA as previously described (12). BSA, pyrrolated BSA, and calf-thymus dsDNA (50 μg/ml, 100 μl/well) were used as the coating antigens.
Confocal microscopy analysis
The RAW264.7 cells were incubated with Alexa Fluor® 555–conjugated HSA or pyrHSA (1 or 10 μg/ml) together with or without apoE proteins (50 μg/ml) for 1 h. After washing with PBS, the cells were fixed in PBS containing 4% paraformaldehyde for 15 min and permeabilized with 0.5% Triton X-100 in PBS for 15 min. The cells were then sequentially incubated in PBS solutions containing 1% BSA (for 1 h) and anti-apoE mAb E6D7 (Abcam) containing 0.1% BSA (overnight, 4 °C). The cells were treated with Alexa Fluor® 488–conjugated anti-mouse IgG (Invitrogen) for 1 h, rinsed with PBS, and covered with anti-fade solution (Invitrogen). Images of the cellular immunofluorescence were acquired using a confocal laser scanning microscope (LSM5 PASCAL; Zeiss).
Fluorescent measurement
The RAW264.7 cells in a 96-well plate were treated with the indicated concentrations of Alexa Fluor® 555–labeled HSA or Alexa Fluor 555®–labeled pyrHSA together with or without 50 μg/ml of apoE (apoE2, apoE3, or apoE4) in FBS-free DMEM. After 1h, the cells were washed with PBS and the fluorescence (Ex/Em = 535/590 nm) was measured using a microplate reader (Spark 10M, Tecan).
Competitive assay
Human LDL (1.019<r<1.063 g/ml) was isolated from the serum of a healthy donor by sequential ultracentrifugation and extensively dialyzed agaist PBS containing 0.1 mm EDTA. LDL was sterilized by filtration through a 0.22-μm filter and stored under argon at 4 °C. The RAW264.7 cells were preincubated with 100 μg/ml of each competitor (LDL, acetylated LDL, or oxidized LDL) for 1 h at 37 °C in a 5% CO2 humidified atmosphere, followed by incubation with 10 μg/ml of Alexa Fluor® 555–labeled pyrHSA together with or without 50 μg/ml of apoE3 for 1 h. The cells were then washed with PBS, and the fluorescence was measured using a microplate reader (Spark 10M, Tecan).
Statistical analysis
The data represent the means ± S.D. where indicated. Statistical significance was evaluated using unpaired Student's t test or, when appropriate, Tukey's HSD test.
Author contributions
S. H., Y. H., H. M., N. H., J. Y., T. S., M. C., M. I., M. Z., and K. N. investigation; T. S., R. K., T. M., M. C., M. I., M. Z., K. N., and K. U. validation; T. S. and K. U. writing-review and editing; R. K. and T. M. resources; K. U. conceptualization; K. U. data curation; K. U. formal analysis; K. U. supervision; K. U. funding acquisition; K. U. writing-original draft; K. U. project administration.
Supplementary Material
Acknowledgment
We thank Yuki Hondoh for excellent editorial support.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 20117007, 21248016, and 24658122. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1–S5.
In this paper, the notation designating apoE refers to the sequence of apoE3, the most common isoform of human apoE.
- DAMP
- damage-associated molecular pattern
- apoE
- apolipoprotein E
- DMPC
- phospholipid dimyristoylphosphatidylcholine
- LDL
- low-density lipoprotein
- PRR
- pattern recognition receptor
- pyrK
- Nϵ-pyrrole-l-lysine
- HSD
- honestly significant difference
- ESI
- electrospray ionization
- pyrBSA
- pyrrolated BSA
- HSA
- human serum albumin
- HRP
- horseradish peroxidase.
References
- 1. Moellering R. E., and Cravatt B. F. (2013) Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 10.1126/science.1238327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janeway C. A. Jr., and Medzhitov R. (2002) Innate immune recognition. Annu Rev Immunol. 20, 197–216 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- 3. Miller Y. I., Choi S. H., Wiesner P., Fang L., Harkewicz R., Hartvigsen K., Boullier A., Gonen A., Diehl C. J., Que X., Montano E., Shaw P. X., Tsimikas S., Binder C. J., and Witztum J. L. (2011) Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 108, 235–248 10.1161/CIRCRESAHA.110.223875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weismann D., and Binder C. J. (2012) The innate immune response to products of phospholipid peroxidation, Biochim. Biophys. Acta 1818, 2465–2475 10.1016/j.bbamem.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chikazawa M., Otaki N., Shibata T., Miyashita H., Kawai Y., Maruyama S., Toyokuni S., Kitaura Y., Matsuda T., and Uchida K. (2013) Multispecificity of immunoglobulin M antibodies raised against advanced glycation end products: involvement of electronegative potential of antigens. J. Biol. Chem., 288, 13204–13214 10.1074/jbc.M113.452177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sayre L. M., Arora P. K., Iyer R. S., and Salomon R. G. (1993) Pyrrole formation from 4-hydroxynonenal and primary amines. Chem. Res. Toxicol. 6, 19–22 10.1021/tx00031a002 [DOI] [PubMed] [Google Scholar]
- 7. Itakura K., Osawa T., and Uchida K. (1998) Structure of a fluorescent compound formed from 4-hydroxy-2-nonenal and Nα-hippuryllysine: A model for fluorophores derived from protein modifications by lipid peroxidation. J. Org. Chem. 63, 185–187 10.1021/jo971239+ [DOI] [PubMed] [Google Scholar]
- 8. Hidalgo F. J., and Zamora R. (1993) Fluorescent pyrrole products from carbonyl-amine reactions. J. Biol. Chem. 268, 16190–16197 [PubMed] [Google Scholar]
- 9. Hidalgo F. J., and Zamora R. (1995) Epoxyoxoene fatty esters: key intermediates for the synthesis of long chain pyrrole and furan fatty esters. Chem. Phys. Lipids 77, 1–11 10.1016/0009-3084(95)02449-S [DOI] [Google Scholar]
- 10. Zhang W. H., Liu J., Xu G., Yuan Q., and Sayre L. M. (2003) Model studies on protein side chain modification by 4-oxo-2-nonenal. Chem. Res. Toxicol. 16, 512–523 10.1021/tx020105a [DOI] [PubMed] [Google Scholar]
- 11. Salomon R. G., Subbanagounder G., O'Neil J., Kaur K., Smith M. A., Hoff H. F., Perry G., and Monnier V. M. (1997) Levuglandin E2-protein adducts in human plasma and vasculature. Chem. Res. Toxicol. 10, 536–545 10.1021/tx960157y [DOI] [PubMed] [Google Scholar]
- 12. Miyashita H., Chikazawa M., Otaki N., Hioki Y., Shimozu Y., Nakashima F., Shibata T., Hagihara Y., Maruyama S., Matsumi N., and Uchida K. (2014) Lysine pyrrolation is a naturally-occurring covalent modification involved in the production of DNA mimic proteins. Sci. Rep. 4, 5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walsh C. T., Garneau-Tsodikova S., and Howard-Jones A. R. (2006) Biological formation of pyrroles: nature's logic and enzymatic machinery. Nat. Prod. Rep. 23, 517–531 10.1039/b605245m [DOI] [PubMed] [Google Scholar]
- 14. Mahley R. W., Weisgraber K. H., and Huang Y. (2009) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J. Lipid Res. 50, S183–S188 10.1194/jlr.R800069-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hatters D. M., Peters-Libeu C. A., and Weisgraber K. H. (2006) Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 31, 445–454 10.1016/j.tibs.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 16. Weisgraber K. H., Rall S. C. Jr., Mahley R. W., Milne R. W., Marcel Y. L., and Sparrow J. T. (1986) Human apolipoprotein E. Determination of the heparin binding sites of apolipoprotein E3. J. Biol. Chem. 261, 2068–2076 [PubMed] [Google Scholar]
- 17. Cardin A. D., Hirose N., Blankenship D. T., Jackson R. L., Harmony J. A., Sparrow D. A., and Sparrow J. T. (1986) Binding of a high reactive heparin to human apolipoprotein E: identification of two heparin-binding domains. Biochem. Biophys. Res. Commun. 134, 783–789 10.1016/S0006-291X(86)80489-2 [DOI] [PubMed] [Google Scholar]
- 18. Pitas R. E., Innerarity T. L., and Mahley R. W. (1980) Cell surface receptor binding of phospholipid–protein complexes containing different ratios of receptor-active and -inactive E apoprotein. J. Biol. Chem. 255, 5454–5460 [PubMed] [Google Scholar]
- 19. Perugini M. A., Schuck P., and Howlett G. J. (2000) Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J. Biol. Chem. 275, 36758–36765 10.1074/jbc.M005565200 [DOI] [PubMed] [Google Scholar]
- 20. Garai K., Baban B., and Frieden C. (2011) Dissociation of apoE oligomers to monomers is required for high affinity binding to phospholipid vesicles. Biochemistry 50, 2550–2558 10.1021/bi1020106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garai K., Baban B., and Frieden C. (2011) Self-association and stability of the apoE isoforms at low pH: Implications for apoE-lipid interactions. Biochemistry 50, 6356–6364 10.1021/bi2006702 [DOI] [PubMed] [Google Scholar]
- 22. Mahley R. W. (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 10.1126/science.3283935 [DOI] [PubMed] [Google Scholar]
- 23. Weisgraber K. H. (1994) Apolipoprotein E: structure-function relationships. Adv. Protein Chem. 45, 249–302 10.1016/S0065-3233(08)60642-7 [DOI] [PubMed] [Google Scholar]
- 24. Frieden C., Wang H., and Ho C. M. W. (2017) A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc. Natl. Acad. Sci. U.S.A. 114, 6292–6297 10.1073/pnas.1705080114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamauchi Y., Deguchi N., Takagi C., Tanaka M., Dhanasekaran P., Nakano M., Handa T., Phillips M. C., Lund-Katz S., and Saito H. (2008) Role of the N- and C-terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: a surface plasmon resonance study. Biochemistry 47, 6702–6710 10.1021/bi8003999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Theendakara V., Peters-Libeu C. A., Spilman P., Poksay K. S., Bredesen D. E., and Rao R. V. (2016) Direct transcriptional effects of apolipoprotein E. J. Neurosci. 36, 685–700 10.1523/JNEUROSCI.3562-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cohn J. S., Tremblay M., Amiot M., Bouthillier D., Roy M., Genest J. Jr., and Davignon J. (1996) Plasma concentration of apolipoprotein E in intermediate-sized remnant-like lipoproteins in normolipidemic and hyperlipidemic subjects. Arterioscler. Thromb. Vasc. Biol. 16, 149–159 10.1161/01.ATV.16.1.149 [DOI] [PubMed] [Google Scholar]
- 28. Zhou Y., Cheshire A., Howell L. A., Ryan D. H., and Harris R. B. (1999) Neuroautoantibody immunoreactivity in relation to aging and stress in apolipoprotein E-deficient mice. Brain Res. Bull. 49, 173–179 10.1016/S0361-9230(99)00052-0 [DOI] [PubMed] [Google Scholar]
- 29. Ma Z., Choudhury A., Kang S. A., Monestier M., Cohen P. L., and Eisenberg R. A. (2008) Accelerated atherosclerosis in apoE deficient lupus mouse models. Clin Immunol. 127, 168–175 10.1016/j.clim.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y., Huang Z., Lu H., Lin H., Wang Z., Chen X., Ouyang Q., Tang M., Hao P., Ni J., Xu D., Zhang M., Zhang Q., Lin L., and Zhang Y. (2012) Apolipoprotein E-knockout mice show increased titers of serum anti-nuclear and anti-dsDNA antibodies. Biochem. Biophys. Res. Commun. 423, 805–812 10.1016/j.bbrc.2012.06.044 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.