

Abstract
Glomerular arteriolar vasoconstriction and tubulointerstitial injury are observed before glomerular damage occurs in models of hypertension. High interstitial ATP concentrations, caused by the increase in arterial pressure, alter renal mechanisms involved in the long-term control of blood pressure, autoregulation of glomerular filtration rate and blood flow, tubuloglomerular feedback (TGF) responses, and sodium excretion. Elevated ATP concentrations and augmented expression of P2X receptors have been demonstrated under a genetic background or induction of hypertension with vasoconstrictor peptides. In addition to the alterations of the microcirculation in the hypertensive kidney, the vascular actions of elevated intrarenal angiotensin II levels may be mitigated by the administration of broad purinergic P2 antagonists or specific P2Y12, P2X1, and P2X7 receptor antagonists. Furthermore, the prevention of tubulointerstitial infiltration with immunosuppressor compounds reduces the development of salt-sensitive hypertension, indicating that tubulointerstitial inflammation is essential for the development and maintenance of hypertension. Inflammatory cells also express abundant purinergic receptors, and their activation by ATP induces cytokine and growth factor release that in turn contributes to augment tubulointerstitial inflammation. Collectively, the evidence suggests a pathophysiological activation of purinergic P2 receptors in angiotensin-dependent hypertension. Coexistent increases in intrarenal angiotensin II and activates Ang II AT1 receptors, which interacts with over-activated purinergic receptors in a complex manner, suggesting convergence of their post-receptor signaling processes.
Keywords: Hypertension, ATP, P2X antagonists, Purinergic P2X receptors, Angiotensin II, Renal hemodynamics, AT1 receptor antagonists
Introduction
Renal injury in the setting of hypertension is thought to be due, at least partially, to inappropriate renal hemodynamic changes, which initially damage afferent arterioles and the glomeruli, and eventually lead to tubulointerstitial inflammation [1]. However, the inflammatory process precedes the glomerular injury [2], and it is currently recognized as an early response leading to the development and persistence of hypertension [3]. The infiltration of leukocytes, lymphocytes, and macrophages, along with smooth muscle cell proliferation in afferent arterioles and myofibroblast-like cells in the tubulointerstitium induce renal dysfunction and sodium and water retention, leading to salt-sensitive hypertension [3–6]. Accumulating evidence indicates that activation of purinergic receptors plays an important role in the pathophysiology of salt-sensitive hypertension by maintaining the production of vasoactive mediators [7, 8], exacerbating tubulointerstitial inflammation [9], and by impairing pressure natriuresis [5].
Mechanisms of elevation of interstitial ATP
Sheer stress, particularly on endothelial cells, is an important mediator of ATP release which increases intracellular calcium concentrations [10, 11] by causing an influx of extracellular calcium through P2X receptors and, where present, voltage gated Ca++ channels [12, 13]. In elegant studies, Yamamoto el al. [10] developed a chemiluminescence image method that allowed the visualization of ATP release on the surface of human cells from the pulmonary artery. When sheer stress was induced, ATP was released from the entire surface of the cell, and the ATP signals were higher at the edges of the cells, which were rich in caveolin-1.
Under physiological conditions, there is a direct association between increases of renal perfusion pressure and the autoregulatory associated rise of renal vascular resistance with an augmentation of renal interstitial fluid ATP concentrations [14]. In this context, high ATP concentrations result from the stimulation of endothelial cells by sheer stress, via activation of P2X4 receptors, along with the tubuloglomerular feedback-mediated release of ATP from the macula densa cells [15–17]. This association suggests a relationship between high blood pressure and sustained augmentation of interstitial ATP concentrations [14]. However, the inflammatory cells in the tubulointestitium induce non-specific release of ATP in response to cell injury and lymphocytes and macrophages’ release of cytokines and chemoattractant factors that exacerbate the inflammatory process [18–20]. Under this milieu, the elevated interstitial ATP concentrations coincide with increased expression and atypical distribution of purinergic receptors [21]. The mechanisms involved in the change of expression and localization of the purinergic receptors remains unclear [22], but these changes occur when tissues are under the influence of hypoxia and inflammation.
Macula densa cells release ATP via a large conductance anion channel located in the basolateral membrane in response to increased NaCl concentration and other solutes in the tubular fluid [15]. In addition, connexins 37 and 40 allow ATP release via gap junctions in the juxtaglomerular apparatus [23], both could be associated with acute increases of ATP concentrations in the renal interstitial fluid in a setting of sustained high perfusion pressure [14, 16].
Purinergic receptors in hypertension
The activation of P2 receptors by increased concentrations of ATP [24] has been demonstrated in the genesis and maintenance of salt-sensitive hypertension [25] and angiotensin II (Ang II)-induced hypertension [21, 24], which in turn contribute to salt-sensitive hypertension [4, 26–29]. Higher expression of P2X7 receptors has been demonstrated in the glomeruli of hypertensive renin transgenic rats [30], as well as in Dahl salt-sensitive rats [25]. In addition, overexpression of P2X1, P2Y1, P2X4, and P2X7 receptors was described in the intrarenal arteries and afferent arterioles of angiotensin II-infused hypertensive rats [21]. Purinergic P2 receptors are essential for the regulation of several intrarenal mechanisms that impact long-term control of blood pressure [31–33], such as pressure natriuresis [29, 34–37], autoregulation of glomerular filtration rate and blood flow [28, 38–41], and regulation of sodium excretion [29, 36, 39]. Studies using Ang II-induced hypertensive rats [9, 21, 24] have provided more evidence supporting the importance of P2 receptors in the kidneys of hypertensive models and their possible interaction with Ang II AT1 receptors [21, 28]. The glomerular microcirculation in this model is characterized by high afferent and efferent arterial resistances, elevation of glomerular capillary pressure, and reductions of glomerular blood flow and filtration coefficient, resulting in a diminished single-nephron glomerular filtration rate [21, 31]. While short-term elevations of ATP levels in renal interstitium help to protect the intrarenal microvasculature from pressure-induced injury and hyperfiltration [14, 40, 41], the persistent intrarenal release of nucleotides activates inflammatory pathways and inflammasome NLRP3 and exerts proliferative responses in vascular smooth muscle cells [42, 43] and interstitial cells, resulting in hypertrophy and hyperplasia of renal arterioles [7, 24]. Furthermore, ATP-mediated activation of the renal interstitial inflammasome [44] would be a key step in the initiation of the proliferative reaction and fibrosis that develop during sustained hypertension. Such conditions are associated with tubulointerstitial infiltration of lymphocytes and macrophages [45–47], glomerular mesangial cell proliferation [24], myofibroblast expression, capillary rarefaction, and afferent arteriolar hypertrophy [47]. It is likely that these abnormalities are mediated by coexistent activation of Ang II AT1 receptors and purinergic P2X receptors in a pathophysiological condition such as hypertension [21, 24, 47, 48].
In the hemodynamic pattern observed in the setting of Ang II-dependent hypertension, the acute infusion of a broad P2 antagonist, such as PPADS (specific for P2X and P2Y receptors), restored afferent and efferent resistances, glomerular plasma flow, ultrafiltration coefficient, and single-nephron glomerular filtration rate (SNGFR) to near normal values [31]. In the same context, co-administration of PPADS or a P2Y12 antagonist (clopidogrel), during the Ang II infusion, prevented the characteristic tubulointerstitial lesions and afferent arteriolar hypertrophy [24]. PPADS and the P2Y12 antagonist inhibited clearly the effects of P2 and P2Y12 receptors on renal hemodynamics [24, 31] and prevented renal injury while renin activity and hypertension remained unchanged [24]. Thus, the blockade of P2X or P2Y receptors have beneficial effects in the glomerular microcirculation and reduce renal damage [49] induced by chronic infusion of Ang II without changes in systemic blood pressure [21].
Adverse effects of purinergic receptors in the microcirculation in hypertension
Activation of P2X receptors has deleterious effects on the renal circulation [35]. Among the P2X receptors, P2X1, P2X4, P2X7, or P2X7/P2X4 trimers [50] have pro-inflammatory activity [25, 51–53]; P2X7 is the most active receptor in the release of cytokines such as IL1β, IL18, TNFα, and MPC-1 [49, 52] which may have vasoactive properties that modify the glomerular microcirculation.
In Ang II-induced hypertension, Ang II concentrations are high in the renal cortex, since the kidney captures the circulating Ang II infused via a miniosmotic pump and increases endogenous Ang II production [54]. Under these conditions, acute blockade of P2X1 and P2X7 allowed evaluation of the alterations in glomerular microcirculation induced by Ang II [20]. The specific blockade of P2X1 (NF449) [55] and P2X7 (A438079) [56] returned the arteriolar resistances, plasma flow, Kf, and SNGFR to near normal levels [21], (Fig. 1). The P2X1 [57, 58] and P2X7 receptors are located in the vascular smooth muscle cells of renal vessels and are overexpressed in the AngII hypertensive rats [21]; although vasodilation was induced by both antagonists, they blocked different receptors on the same vessels. In addition, the inhibition of P2X7 receptors increased renal perfusion in the Ang II hypertensive rat [28] and P2X7 deficiency reduced the renal injury in experimental glomerulonephritis [59]. These findings support an important post-receptor convergence between Ang II and P2X receptor signaling pathways in the setting of hypertension. The finding that both AT1 and P2X receptors are activated simultaneously raises the issue of interactive post receptor signaling which deserves to be studied further (Fig. 2).
Fig. 1.
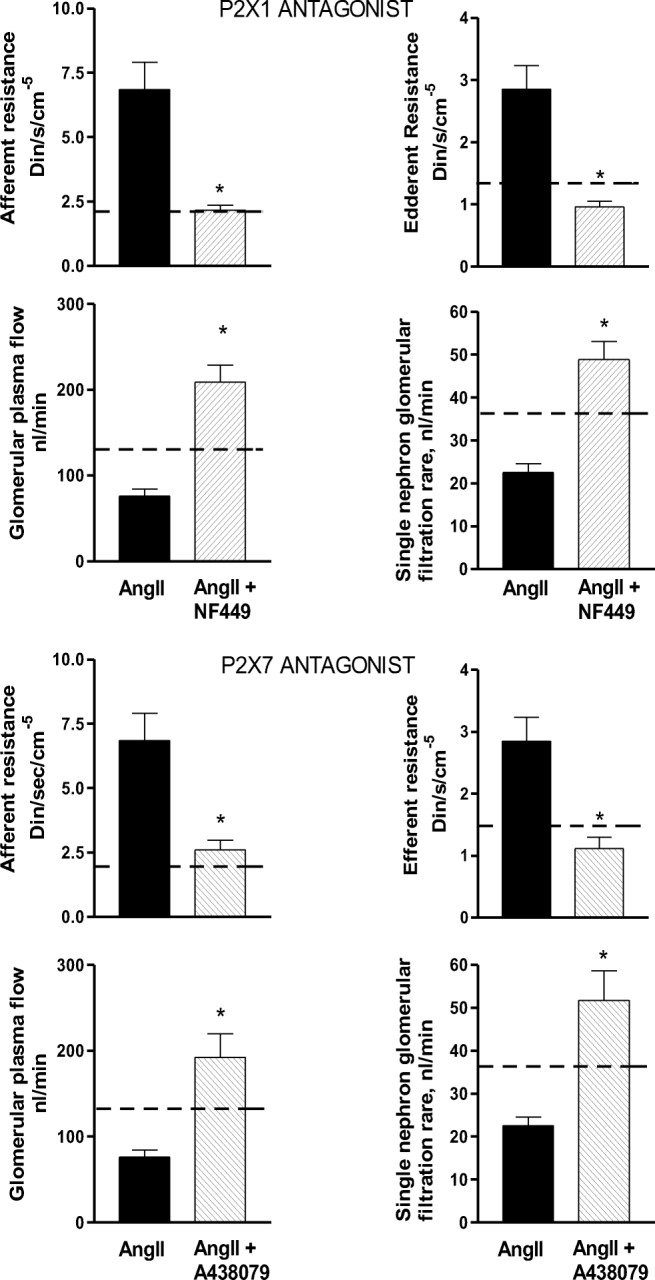
Renal hemodynamics in rats that received 14 days of Angiotensin II (Ang II) (435 ng/kg/min) during an acute infusion of P2X1 antagonist (NF449) and a P2X7 antagonist (A438079). Only the groups that received Ang II and the AngII + antagonists are shown. The dash line represents the normal values obtained in a Sham rat + vehicle. As observed, the groups that received the antagonists of P2X1 or P2X7 showed a significant decrease of afferent and efferent arteriolar resistances (*< from 0.05 to 0.01) the leads to a significant increase in renal plasma flow; as a consequence of these changes, the single-nephron glomerular filtration rate returned to near normal values, similar to that of the Sham rat. These data clearly demonstrate that in the Ang II-dependent hypertension, the renal vasoconstriction induced by Ang II is associated with an important P2X1 and P2X7 receptor-mediated contribution. In addition, these findings suggest a convergence of their post-receptor signaling pathways
Fig. 2.

Proposed mechanism for the effect of the Ang II infusion during 14 days and the P2X1 and P2X7 induced vasoconstriction. Ang II produces systemic hypertension and a rise of interstitial fluid concentrations of ATP as well as Ang II. Renal vasoconstriction is induced by both, a direct effect of Ang II and as a result of the regulatory response to hypertension. Glomerular hemodynamics shows an increase in afferent and efferent resistances (AR, ER) which leads to a decrease of renal blood flow (GBF) and a reduced filtration coefficient (KF); all these changes result in a fall of the single-nephron glomerular filtration rate (SNGFR). The dotted lines represent the values in Sham rats + vehicle for comparison. These alterations induce renal ischemia leading to an overexpression of P2X receptors in the smooth muscle cells of the intrarenal arterioles. Concomitantly, tubulointerstitial inflammatory cell infiltration results and intrarenal ATP is elevated leading to activation of P2X receptors in the intrarenal arteries and on the surface of the inflammatory cells. Collectively, these changes results in cytokines, growth and chemoattractant factors production, which exacerbate the inflammatory infiltration and intensify renal vasoconstriction. Oxidative stress, increases of adenosine (ADO), decreases in nitric oxide (NO), increases in local production of Ang II, and stimulation of the sympathetic tone (SNS) occur. These alterations modify sodium excretion and impair natriuresis, resulting in decreased Na+ excretion relative to the expected for the elevation of blood pressure
Pathways of AT1R and purinergic P2R post-receptor convergence
The AT1 and P2X receptors represent a point of convergence downstream of many signaling pathways for vasoconstriction including PIP2-PLC-IP3 pathway, RhoA/ROCK-dependent pathway, and voltage-dependent calcium channels (VDCC). P2XR activation stimulates IP3 receptors in smooth muscle cells and releases calcium from the endoplasmic reticulum [60, 61]. Studies in the triple IP3 receptor knockout mice demonstrated that aortic contraction induced by Ang II is decreased due to the lack of IP3 receptor-activation, [62]. In diabetic rats overexpressing P2X7 in several tissues [30, 63, 64], mesenteric contraction induced by ATP was completely abolished by losartan [64]; these findings suggest the close intracellular signaling crosstalk between both pathways. These concepts are supported by studies in sensorial neurons, in which xestoporin C (IP3 receptor antagonist), strongly inhibited the BzATP-triggered [Ca2+]i [65], suggesting the importance of IP3R receptors in the actions of BzATP (preferentially stimulating P2X7R). Further studies from the same neurons demonstrated that PPADS, (a broad purinergic antagonist) blocks the ATP-evoked intracellular Ca2+ release induced by IP3R receptors [66].
The evidence mentioned above suggest that the IP3 receptors have a common pathway shared during activation of AT1 and P2X receptors [61], which may explain why P2X antagonists are able to block the effects of Ang II in hypertensive rats. In this regard, P2X receptors also mediate IP3 receptor-dependent Ca2+ release by an intrinsic mechanism involving phospholipase C, since phospholipase inhibition also decreases calcium release to a similar degree than those induced by α,β,meATP production [67]. In addition, Gómez et al. [68] proposed that Ang II induces cell damage via RhoA/ROCK-dependent pathway, since the blockade of this pathway prevented the increase of membrane permeability of mesangial cells [68] and in vascular smooth muscle cells [67] (Fig. 3). Rho-kinase inhibition prevented the Ang II-mediated augmentation o angiotensinogen in cultured preglomerular vascular smooth muscle cells [68]. Inscho et al. [69] used the juxtamedulla nephron preparation to demonstrate that Rho-kinase-modulated autoregulatory adjustments in renal microvascular resistance since Rho-kinase inhibition blunted afferent arteriolar responses to Ang II and P2X1R agonists. Thus RhoA/ROCK-dependent pathway may be an important point of convergence signaling.
Fig. 3.

Potential synergic interactions between purinergic P2X receptors and AT1R receptors. ATP-activation of P2X receptors opens the ligand-gated Na+/Ca2+ channel, inducing both, an increase of Ca2+ concentration within the cytoplasm and local membrane depolarization. As a consequence, the voltage-gated Ca2+ channels (VGCC) adopts its open conformation that contributes to further increase the Ca2+ concentrations. Simultaneously, the stimulation of AT1R by Ang II activates the phospholipase C (PLC) and the RhoA/ROCK pathways. PLC pathway leads to inositol-1,4,5-triphosphate (IP3) formation. IP3 in turn induces the opening of the ligand-gated Ca2+ channels (IP3R) in the endoplasmic reticulum. The increase of the Ca2+ concentration in the cytosol results in a positive feedback of the ryanodine receptor (RyR). The overall effect of the interaction of ATP and Ang II with their corresponding receptors is a conveyance leading to an increase of Ca2+ concentration in the cytosol, enough to induce muscular contraction with the consequent reduction of the arteriolar diameter. On the other hand, the RhoA/ROCK pathway leads to phosphorylation of myosin phosphatase target subunit (P-MYPT) thus inhibiting the activity of myosin light chain phosphatase (MLCP), with the consequent vascular smooth muscle cell (VSMC) contractility
In renal vascular smooth muscle cell, ATP activates P2X1R-induced Ca++ entry via VDCC which then evokes further IP3 receptor-mediated Ca2+ release from sarcoplasmic reticulum [61]. Also, Ang II induced Ca2+ release from sarcoplasmic reticulum and enhanced Ca2+ channel currents via AT1R [70]. These data indicate that both, AT1R and P2XR, share the voltage-dependent calcium channels (VDCC) signaling pathway to elevate intracellular Ca2+ concentration. Moreover, since the action of ATP to increase intracellular calcium seems to be greater than the actions induced by Ang II [68], it is reasonable to assume that ATP antagonists may prevent the vasoconstriction induced by Ang II. However, this crosstalk needs to be clarified with further studies at the cellular level.
Inflammation and purinergic receptors
P2X1 receptors are located on intrarenal arteries as previously described [57, 58], but P2X7 are overexpressed in the smooth muscle of intrarenal arteries of Ang II-induced hypertensive rats [21]. The stimulation of these receptors in the intrarenal arteries and afferent arterioles by ATP explains the alterations in glomerular hemodynamics observed in this model of hypertension [21, 28].
When ATP is released from the cells through pannexin or connexin hemichannels [23, 29], its concentration increases in the interstitial space; if the elevated ATP levels are sustained by a continuous production and release, it becomes one of the main promoters of inflammation associated with tissue injury [71, 72], which occurs in ischemia, hypoxia, and necrotic or apoptotic processes [53, 73]. Furthermore, inflammatory cells express P2X and P2Y receptors (P2X7, P2X1, P2X4, P2Y2, P2Y6, and P2Y12) [74, 75] and ATP functions as a chemotactic signal for phagocytes by activation of purinergic receptors in the inflammatory cells. Acute increases of extracellular ATP induce ROS production and further release of ATP [51, 75, 76].
During acute inflammation, the extracellular concentrations of ATP are limited by ectoenzymes (apyrase, ATPase, alkaline phosphatase, ectonucleotidases, etc.) that metabolize ATP to ADP and adenosine [75, 77]. Such enzymatic activities contribute to the resolution of the inflammatory process [71, 78, 79]. However, in Ang II-induced hypertension, a decrease of ecto-adenosine deaminase is associated with elevated adenosine concentrations [7], which could induce an imbalance between A1 and A2 receptor activation that may influence the vasoconstriction induced by Ang II and ATP, since adenosine A1 receptors induce vasoconstriction and A2 induce vasodilation [7, 80]. In the kidney, A1 receptors are activated at low concentrations of adenosine, but A2 receptors are predominantly activated at higher concentrations and induce vasodilation [80].
Nevertheless, the effects of chronic elevation of extracellular renal ATP in the tubulointerstitium remain incompletely understood [3, 78]. The relevance of tubulointerstitial inflammation becomes evident when the infiltration of lymphocytes and macrophages is prevented in different models of hypertension [24, 81, 82]. Administration of immunosuppressors (i.e., mycophenolate mofetil) [5, 9, 81], antinflammatory compounds, or genetic manipulation [46, 72, 83] are associated with reduction of tubulointerstitial inflammation and decreased renal injury [46, 84, 85]. A common finding with these treatments is the prevention of blood pressure elevation [4, 86]. For instance, the interruption of co-administration of Ang II and mycophenolate mofetil after 14 days, followed by 5 weeks high-salt diet administration, modifies glomerular hemodynamics, particularly the increases in afferent and efferent resistances. However, the other determinants of single-nephron glomerular filtration rate returned to near normal levels, associated with a significant decrease of tubulointertitial infiltration and prevention of salt sensitive hypertension [9, 46, 86].
The available data provide the basis for proposing a compelling pathophysiological mechanism for the development of hypertension and sensitivity to salt. In a setting of hyperactivity of the sympathetic nervous system, stimulation of the renin-angiotensin system as well as a genetic susceptibility to hypertension [25, 85], an elevation of blood pressure above the upper limit of the renal autoregulatory mechanism leads to increases in interstitial fluid ATP concentrations and gradually to subtle injury. The transmission of the elevated blood pressure to the peritubular capillaries disrupts the capillary walls, allowing the leak of plasma and leucocytes into the tubulointerstitial space. Leucocytes subsequently mediate local inflammation [19, 87], which in turn, induces microvascular and tubulointerstitial injury, as well as capillary rarefaction [46]. These alterations produce focal ischemia, release of interleukins, and upregulation of adhesion molecules followed by more infiltration of monocytes, perpetuating the inflammatory response [84, 87]. In the presence of this milieu, the effects of elevated Ang II and ATP [24] and tubulointerstitial inflammation are critical factors for the progression to salt-sensitive hypertension [5, 47, 83, 88, 89], since they enhance the sensitivity of tubuloglomerular feedback and sodium retention [36, 90].
While blood pressure increases, cortical and medullary perfusion return to normal levels and tubular ischemia is alleviated [3]. Concomitant with these changes, the increase in blood flow stimulates nitric oxide production in the arteries, which then improves sodium excretion [28]; the blood pressure remains elevated as a result of the tubulointerstitial alterations mentioned above [3], whereas the pressure-natriuresis slope remains suppressed and [5], salt-sensitive hypertension develops since elevated blood pressure is necessary to maintain normal sodium excretion and preserve Na+ homeostasis [5, 9, 36]. Thus, tubulointerstitial injury without glomerular damage is a common feature during the early stages of salt-sensitive hypertension [9]. Vascular resistance initially increases in response to high blood pressure and afferent arteriolar hypertrophy ensues. In spite of these adaptive changes, hyperperfusion and glomerular hypertension is not completely normalized leading to damage to the glomerular capillary network with further decline in sodium excretion [91, 92].
Conclusions
Hypertension and renal vasoconstriction induce hypoxia, oxidative stress, autoimmunity, and inflammation, which are involved in the pathophysiological mechanisms that induce salt sensitivity. The particular combination of factors such as elevated sheer stress, high interstitial ATP concentration, activation of P2 receptors, and elevated renal interstitial Ang II collectively lead to the release of interleukins and growth factors all contributing to the development of hypertensive renal injury. In addition, the evidence presented in this review suggests that purinergic antagonists may help prevent the progression of renal damage to chronic kidney disease in hypertensive patients.
Funding information
This work was supported by grant number 219981 (to M Franco) from the National Council of Sciences and Technology (CONACYT) Mexico and L G Navar and Suppaporn Kulthinee were supported in part by a Center of Biomedical Research Excellence grant from the National Institute of General Medical Sciences (P30-GM-103337).
Compliance with ethical standards
Conflict of interest
Martha Franco declares that she has no conflict of interest.
Oscar Pérez-Mendez declares that he has no conflict of interest.
Supaporn Kulthinee declares that he has no conflict of interest.
L Gabriel Navar declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Martha Franco, Phone: (5255) 5573-6902, Email: marthafranco@lycos.com.
Oscar Pérez-Méndez, Email: opmendez@yahoo.com.
Supaporn Kulthinee, Email: skultini@gmail.com.
L. Gabriel Navar, Email: Navar@tulane.edu.
References
- 1.Tolins JP, Shultz P, Raij L. Mechanisms of hypertensive glomerular injury. Am J Cardiol. 1988;62:54G–58G. doi: 10.1016/0002-9149(88)90033-1. [DOI] [PubMed] [Google Scholar]
- 2.Johnson RJ, Schreiner GF. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension. Kidney Int. 1997;52:1169–1179. doi: 10.1038/ki.1997.442. [DOI] [PubMed] [Google Scholar]
- 3.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodríguez-Iturbe Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med. 2002;346:913–923. doi: 10.1056/NEJMra011078. [DOI] [PubMed] [Google Scholar]
- 4.Franco M, Martínez F, Rodríguez-Iturbe B, Johnson RJ, Santamaría J, Montoya A, Nepomuceno T, Bautista R, Tapia E, Herrera-Acosta J. Angiotensin II, interstitial inflammation and the pathogenesis of salt-sensitive hypertension. Am J Physiol Renal Physiol. 2006;291:F1281–F1287. doi: 10.1152/ajprenal.00221.2006. [DOI] [PubMed] [Google Scholar]
- 5.Franco M, Tapia E, Bautista R, Pacheco U, Santamaria J, Quiroz Y, Johnson RJ, Rodriguez-Iturbe B. Impaired pressure natriuresis resulting in salt-sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. Am J Physiol Renal Physiol. 2013;304:F982–F990. doi: 10.1152/ajprenal.00463.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg M, Fineberg NS. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension. 1991;18:67–71. doi: 10.1161/01.HYP.18.1.67. [DOI] [PubMed] [Google Scholar]
- 7.Franco M, Bautista R, Pérez-Méndez O, González L, Pacheco U, Sánchez-Lozada LG, Tapia E, Morreal R, Martínez F. Renal interstitial adenosine is increased in angiotensin II-induced hypertensive rats. Am J Physiol Renal Physiol. 2008;294:F84–F92. doi: 10.1152/ajprenal.00123.2007. [DOI] [PubMed] [Google Scholar]
- 8.Sealey JE, Blumenfield JD, Bell GM, Pecker MS, Sommers SC, Laragh JH. On the renal basis for essential hypertension: nephron heterogeneity with discordant rennin secretion and sodium excretion causing hypertensive vasoconstriction-volume relationship. J Hypertens. 1988;6:763–777. doi: 10.1097/00004872-198811000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Franco M, Tapia E, Santamaría J, Zafra I, García-Torres R, Gordon KL, Pons H, Rodríguez-Iturbe B, Johnson JR, Herrera-Acosta J. Renal cortical vasoconstriction contributes to the development of salt sensitive hypertension after angiotensin II exposure. J Am Soc Nephrol. 2001;10:2263–2271. doi: 10.1681/ASN.V12112263. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci. 2011;124:3477–34832. doi: 10.1242/jcs.087221. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto K, Sokabe T, Ohura N, Nakatsuka H, Kamiya A, Ando J. Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2003;285:H793–H803. doi: 10.1152/ajpheart.01155.2002. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca (2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res. 2000;87:385–391. doi: 10.1161/01.RES.87.5.385. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto K, Korenaga R, Kamiya A, Qi Z, Sokabe M, Ando J. P2X (4) receptors mediate ATP-induced calcium influx in human vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2000;279:H285–H292. doi: 10.1152/ajpheart.2000.279.1.H285. [DOI] [PubMed] [Google Scholar]
- 14.Nishiyama A, Majid DS, Taher KA, Miyatake A, Navar LG. Relation between interstitial ATP concentrations and autoregulation-mediated changes in vascular resistance. Circ Res. 2000;86:656–662. doi: 10.1161/01.RES.86.6.656. [DOI] [PubMed] [Google Scholar]
- 15.Bell PD, Lapoint JY, Sabirov R, Hayashi S, Peti-Peterdy J, Manabe KI, Kovacs G, Osaka Y. Macula densa signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci. 2003;100:4322–4327. doi: 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palygin O, Evans LC, Cowley AW, Jr, Staruschenko A. Acute in vivo analysis of ATP release in rat kidneys in response to changes of renal perfusion pressure. J Am Heart Assoc. 2017;6:e006658. doi: 10.1161/JAHA.117.006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peti-Peterdi J. Calcium wave of tubuloglomerular feedback. Am J Physiol Renal Physiol. 2006;291:F473–F480. doi: 10.1152/ajprenal.00425.2005. [DOI] [PubMed] [Google Scholar]
- 18.Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. 2018;19:1222. doi: 10.3390/ijms19041222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 20.Vitiello L, Gorini S, Rosano G, la Sala A. Immunoregulation through extracellular nucleotides. Blood. 2012;120:511–518. doi: 10.1182/blood-2012-01-406496. [DOI] [PubMed] [Google Scholar]
- 21.Franco M, Bautista-Pérez R, Cano-Martínez A, Pacheco U, Santamaría J, Del Valle-Mondragón L, Pérez-Méndez O, Navar LG. Physiopathological implications of P2X1 and P2X7 receptors in regulation of glomerular hemodynamics in angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2017;313(F):9–F19. doi: 10.1152/ajprenal.00663.2016. [DOI] [PubMed] [Google Scholar]
- 22.Robinson LE, Murrell-Lagnado The trafficking and targeting of P2X receptors. Front Cell Neurosci. 2013;7:233. doi: 10.3389/fncel.2013.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takenaka T, Inoue T, Kanno Y, Osaka H, Hill CE, Suzuki H. Conexin 37 and 40 transduce purinergic signals mediating renal autoregulation. Am J Physiol Integr Comp Physiol. 2008;294:R1–R11. doi: 10.1152/ajpregu.00269.2007. [DOI] [PubMed] [Google Scholar]
- 24.Graciano ML, Nishiyama A, Jackson K, Seth DM, Ortiz RM, Prieto-Carrasquero M, Kobori H, Navar LG. Purinergic receptors contribute to early mesangial transformation and renal vessel hypertrophy during angiotensin II induced hypertension. Am J Physiol Renal Physiol. 2008;294:F161–F169. doi: 10.1152/ajprenal.00281.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji X, Naito Y, Hirokawa G, Weng H, Hiura Y, Takahashi R, Iwai N. P2X(7) receptor antagonism attenuates the hypertension and renal injury in Dahl salt sensitive rats. Hypertens Res. 2012;35:173–179. doi: 10.1038/hr.2011.153. [DOI] [PubMed] [Google Scholar]
- 26.Guan Z, Inscho EW. Role of adenosine 5′triphosphate in regulating renal microvascular function and in hypertension. Hypertension. 2011;58:333–340. doi: 10.1161/HYPERTENSIONAHA.110.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menzies RI, Unwin RJ, Bailey MA. P2 receptors and hypertension. Acta Physiol (Oxf) 2015;213:S232–S241. doi: 10.1111/apha.12412. [DOI] [PubMed] [Google Scholar]
- 28.Menzies RI, Howarth AR, Unwin RJ, Frederick WK, Mullis JJ, Bailey MA. Inhibition of the purinergic P2X7 receptor improves renal perfusion in angiotensin-II-infused rats. Kidney Int. 2015;88:1079–1087. doi: 10.1038/ki.2015.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sipos A, Vargas SL, Toma I, Hanner F, Willecke K, Peti-Peterdi J. Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J Am Soc Nephrol. 2009;20:1724–1732. doi: 10.1681/ASN.2008101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vonend O, Turner CM, Chan CM, Loesch A, DellÄnna GC, Srai KS, Burnstock G, Unwin RJ. Glomerular expression of the ATP-sensitive P2X receptor in diabetic and hypertensive rat models. Kidney Int. 2004;66:157–166. doi: 10.1111/j.1523-1755.2004.00717.x. [DOI] [PubMed] [Google Scholar]
- 31.Franco M, Bautista R, Tapia E, Soto V, Santamaría J, Osorio H, Pacheco U, Sánchez-Lozada LG, Kobori H, Navar LG. Contribution of renal purinergic receptors to renal vasoconstriction in angiotensin II-induced hypertensive rats. Am J Physiol Renal Physiol. 2011;300:F1301–F1309. doi: 10.1152/ajprenal.00367.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guyton AC. Blood pressure control: special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 33.Guyton AC. The surprising kidney-fluid mechanism for pressure control, its infinite gain. Hypertension. 1990;16:725–730. doi: 10.1161/01.HYP.16.6.725. [DOI] [PubMed] [Google Scholar]
- 34.Ivy JR, Bailey MA. Pressure natriuresis and the renal control of arterial blood pressure. J Physiol. 2014;592:3955–3396. doi: 10.1113/jphysiol.2014.271676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menzies RI, Unwin RJ, Rash RK, Beard DA, Cowley AW, Jr, Callsin BE, Mullins JJ, Bailey MA. Effect of P2X4 and P2X7 receptor antagonism on the pressure diuresis relationship in rats. Front Physiol. 2013;4:235. doi: 10.3389/fphys.2013.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mironova E, Boiko N, Bugaj V, Stockand JD. Regulation of sodium excretion and arterial blood pressure by purinergic signaling intrinsic to the distal nephron: consequences and mechanisms. Acta Physiol (Oxf) 2015;213:213–221. doi: 10.1111/apha.12372. [DOI] [PubMed] [Google Scholar]
- 37.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 38.Majid DS, Inscho EW, Navar LG. P2 purinoceptor saturation by adenosine triphosphate impairs renal autoregulation in dogs. J Am Soc Nephrol. 1999;10:492–498. doi: 10.1681/ASN.V103492. [DOI] [PubMed] [Google Scholar]
- 39.Guan Z, Fellner RC, Van Beusecum J, Inscho EW. P2 receptors in renal autoregulation. Curr Vasc Pharmacol. 2014;12:818–828. doi: 10.2174/15701611113116660152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osmond DA, Zhang S, Polloch JS, Yamamoto T, De Miguel C, Inscho EW. Clopidogrel preserves kidney autoregulatory behavior in Ang II-induced hypertension. Am J Physiol Renal Physiol. 2014;306:F619–F628. doi: 10.1152/ajprenal.00444.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osmond DA, Inscho EW. P2X1 receptor blockade inhibits whole kidney autoregulation of renal blood flow in vivo. Am J Physiol Renal Physiol. 2010;298:F1360–F13641. doi: 10.1152/ajprenal.00016.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erlinge D, Yoo H, Edvinsson L, Reis DJ, Wahlestedt C. Mitogenic effects of ATP on vascular smooth muscle cells vs. other growth factors and sympathetic cotransmitters. Am J Physiol Heart Circ Physiol. 1993;265:H1089–H1097. doi: 10.1152/ajpheart.1993.265.4.H1089. [DOI] [PubMed] [Google Scholar]
- 43.Wang DJ, Huang NN, Heppel LA. Extracellular ATP and ADP stimulate proliferation of porcine aortic smooth muscle cells. J Cell Physiol. 1992;153:221–233. doi: 10.1002/jcp.1041530202. [DOI] [PubMed] [Google Scholar]
- 44.Gomvault A, Baron L, Couillin I. ATP release and purinergic signalling in NLRP3 inflammasome activation. Front Immunol. 2012;3:414. doi: 10.3389/fimmu.2012.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrison DG, Guzik TJ, Lob EH, Madur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez-Iturbe B, Johnson RJ. The role of renal microvascular disease and interstitial inflammation in salt-sensitive hypertension. Hypertens Res. 2010;33:975–980. doi: 10.1038/hr.2010.148. [DOI] [PubMed] [Google Scholar]
- 47.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Prinzl P, Floege J, Schwartz SM. Renal injury from angiotensin II-mediated hypertension. Hypertension. 1992;19:664–674. doi: 10.1161/01.hyp.19.5.464. [DOI] [PubMed] [Google Scholar]
- 48.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short term exposure to angiotensin II. Hypertension. 1999;33:1013–1019. doi: 10.1161/01.HYP.33.4.1013. [DOI] [PubMed] [Google Scholar]
- 49.Menzies RI, Tam Frederick WT, Unwin RJ, Bailey MA. Purinergic signaling in kidney disease. Kidney Int. 2017;91:315–316. doi: 10.1016/j.kint.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 50.Craigie E, Birch RE, Unwin RJ, Wildman SS. The relationship between P2X4 and P2X7: a physiologically important interaction? Front Physiol. 2013;4:216. doi: 10.3389/fphys.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–F742. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 53.Kawano A, Tsukimoto M, Mori D, Noguchi T, Harada H, Takenouchi T, Kitani H, Kojima S. Regulation of P2X7-dependent inflammatory functions by P2X4 receptor in mouse macrophages. Biochem Biophys Res Commun. 2012;420:102–107. doi: 10.1016/j.bbrc.2012.02.122. [DOI] [PubMed] [Google Scholar]
- 54.Shao W, Seth DM, Navar LG. Augmentation of endogenous intrarenal angiotensin II levels in Val5-AngII-infused rats. Am J Phys Renal Phys. 2009;296:F1067–F1071. doi: 10.1152/ajprenal.90596.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kassack MU, Braun K, Ganso M, Ullmann H, Nickel P, Böing B, Müller G, Lambrecht G. Structure-activity relationships of analogues of NF449 confirm NF449 as the most potent and selective known P2X1 receptor antagonist. Eur J Med Chem. 2004;39:345–357. doi: 10.1016/j.ejmech.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 56.Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br J Pharmacol. 2009;157:1203–1214. doi: 10.1111/j.1476-5381.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan CM, Unwin RJ, Bardini M, Oglesby IB, Ford AP, Townsend-Nicholson A, Burnstock G. Localization of P2X1 purinoceptors by autoradiography and immunohistochemistry in rat kidneys. Am J Physiol Renal Physiol. 1998;274:F799–F804. doi: 10.1152/ajprenal.1998.274.4.F799. [DOI] [PubMed] [Google Scholar]
- 58.Turner CM, Vonend O, Chan C, Burnstock G, Unwin RJ. The pattern of distribution of selected ATP- sensitive P2 receptor subtypes in normal rat kidney: an immunohistological study. Cell Tissues Organs. 2003;175:105–117. doi: 10.1159/000073754. [DOI] [PubMed] [Google Scholar]
- 59.Taylor SR, Turner CM, Elliott JI, McDaid J, Hewitt R, Smith J, Pickering MC, Whitehouse DL, Cook HT, Burnstock G, Pusey CD, Unwin RJ, Tam FW. P2X7 deficiency attenuates renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20:1275–1281. doi: 10.1681/ASN.2008060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernier LP, Ase AR, Séguéla P. Post-translational regulation of P2X receptor channels: modulation by phospholipids. Front Cell Neurosci. 2013;7:226. doi: 10.3389/fncel.2013.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Povstyan OV, Harhun MI, Gordienko DV. Ca2+ entry following P2X receptor activation induces IP3 receptor-mediated Ca2+ release in myocytes from small renal arteries. Br J Pharmacol. 2011;162:1618–1638. doi: 10.1111/j.1476-5381.2010.01169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, Jing R, Liu J, Lederer WJ, Chen J, Ouyang K (2016) IP3 receptors regulate vascular smooth muscle contractility and hypertension. J Clin Invest Insight (17):e89402 [DOI] [PMC free article] [PubMed]
- 63.Solini A, Jacobini C, Ricci C, Chiozzi P, Amadio L, Pricci F, Di Mario U, Di Virgilio F, Pugliese G. Purinergic modulation of mesangial extracellular matrix production: role in diabetic and other glomerular diseases. Kidney Int. 2005;67:875–885. doi: 10.1111/j.1523-1755.2005.00152.x. [DOI] [PubMed] [Google Scholar]
- 64.Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Mechanisms underlying altered extracellular nucleotide-induced contractions in mesenteric arteries from rats in later-stage type 2 diabetes: effect of ANG II type 1 receptor antagonism. Am J Physiol Heart Circ Physiol. 2011;301:H1850–H1861. doi: 10.1152/ajpheart.00502.2011. [DOI] [PubMed] [Google Scholar]
- 65.Chao CC, Huang CC, Lu DY, Wong KL, Chen YR, Chen TH, Leung YM. Ca2+ store depletion and endoplasmic reticulum stress are involved in the P2X7 receptors-mediated neurotoxicity in differentiated NG108.15 cells. J Cell Biochem. 2012;113:1377–1385. doi: 10.1002/jcb.24010. [DOI] [PubMed] [Google Scholar]
- 66.Hoesch RE, Yinger K, Weinreich D, Kao JP. Coexistence of functional IP(3) receptor and ryanodine receptors in vagal sensory neurons and their activation by ATP. J Neurophysiol. 2002;88:1212–1219. doi: 10.1152/jn.2002.88.3.1212. [DOI] [PubMed] [Google Scholar]
- 67.Sukhanova KY, Bouryi VA, Gordienko DV. Convergence of ionotropic and metabotropic signal pathways upon activation of P2X receptors in vascular smooth muscle cells. Neurophysiololy. 2014;46:398–404. doi: 10.1007/s11062-015-9464-7. [DOI] [Google Scholar]
- 68.Gómez GI, Fernández P, Velarde V, Sáenz JC. Angiotensin II-induced mesangial cell damage is preceded by cell membrane permeabilization due to upregulation of non-selective channels. Int J Mol Sci. 2018;19:e957. doi: 10.3390/ijms19040957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inscho EW, Cook AK, Webb RC, Jin LM. Rho-kinase inhibition reduces pressure-mediated autoregulatory adjustments in afferent arteriolar diameter. Am J Physiol Renal Physiol. 2009;296:F590–F597. doi: 10.1152/ajprenal.90703.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fuller AJ, Benjamin C, Hauschild BC, Gonzalez-Villalobos R, Awayda MS, Imig JD, Inscho EW, Navar LG. Calcium and chloride channel activation by angiotensin II-AT1 receptors in preglomerular vascular smooth muscle cells. Am J Physiol-Renal physiol. 2005;289:F760–F767. doi: 10.1152/ajprenal.00422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bours MJ, Dagnelie PC, Giuliani AL, Wesselius A, Di Virgilio F. P2 receptors and extracellular ATP: a novel homeostatic pathway in inflammation. Front Biosci (Schol Ed) 2011;3:1443–1456. doi: 10.2741/235. [DOI] [PubMed] [Google Scholar]
- 72.Luttikhuizen DT, Harmsen MC, de Leij LF, van Luy MJ. Expression of P2 receptors at sites of chronic inflammation. Cell Tissue Res. 2004;317:289–298. doi: 10.1007/s00441-004-0939-x. [DOI] [PubMed] [Google Scholar]
- 73.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signaling during inflammation. Nature. 2014;509:310–317. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L, Jacobsen SE, Bengtsson A, Erlinge D. P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+ stem and progenitor cells. BMC Immunol. 2004;5:16. doi: 10.1186/1471-2172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Di Virgilio F. P2X receptors and inflammation. Curr Med Chem. 2015;22:866–877. doi: 10.2174/0929867322666141210155311. [DOI] [PubMed] [Google Scholar]
- 78.Jacob F, Pérez Novo C, Bachert C, Van Crombruggen K. Purinergic signaling in inflammatory cells. P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal. 2013;9:285–306. doi: 10.1007/s11302-013-9357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burnstock G. P2X ion channel receptors and inflammation. Purinergic Signal. 2016;12:59–67. doi: 10.1007/s11302-015-9493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feng MG, Navar LG. Afferent arteriolar vasodilator effect of adenosine predominantly involves A2B receptor activation. Am J Physiol Renal Physiol. 2010;299:F310–F315. doi: 10.1152/ajprenal.00149.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension. 2006;48:149–156. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 82.Pechman KR, Basile DP, Lund H, Mattson DL. Immune suppression blocks sodium-sensitive hypertension following recovery from ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1234–R1239. doi: 10.1152/ajpregu.00821.2007. [DOI] [PubMed] [Google Scholar]
- 83.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–R1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quiroz Y, Pons H, Gordon KL, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ, Rodríguez-Iturbe B. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol. 2002;282:F191–F201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 85.Rodríguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Pyhsiol Renal Physiol. 2004;286:F606–F616. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- 86.Franco M, Martínez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodríguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293:R251–R256. doi: 10.1152/ajpregu.00645.2006. [DOI] [PubMed] [Google Scholar]
- 87.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582–593. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- 88.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lara LS, McCormack M, Semprum-Prieto LS, Shenouda S, Majid DS, Kobori H, Navar LG, Prieto MC. AT1 receptor-mediated augmentation of angiotensinogen, oxidative stress, and inflammation in ANG II-salt hypertension. Am J Physiol Renal Physiol. 2012;302:F85–F94. doi: 10.1152/ajprenal.00351.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song J, Lu Y, Lai EY, Wei J, Wang L, Chandrashekar K, Wang S, Shen C, Juncos LA, Liu R. Oxidative status in the macula densa modulates tubuloglomerular feedback responsiveness in angiotensin II-induced hypertension. Acta Physiol (Oxf) 2015;213:249–258. doi: 10.1111/apha.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matavelli LC, Zhou X, Varagic J, Susic D, Frohlich ED. Salt loading produces severe renal hemodynamic dysfunction independent of arterial pressure in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H814–H819. doi: 10.1152/ajpheart.00671.2006. [DOI] [PubMed] [Google Scholar]
- 92.Sannai T, Kimura G. Renal function reserve and sodium sensitivity in essential hypertension. J Lab Clin Med. 1996;128:89–97. doi: 10.1016/S0022-2143(96)90117-1. [DOI] [PubMed] [Google Scholar]