

Abstract
Somatically acquired uniparental disomies (aUPDs) are frequent events in solid tumors and have been associated with cancer‐related genes. Studies assessing their functional consequences across several cancer types are therefore necessary. Here, we aimed at integrating aUPD profiles with the mutational status of cancer‐related genes in a tumor‐type specific manner. Using TCGA datasets for 1,032 gastrointestinal cancers, including colon (COAD), rectum (READ), stomach (STAD), esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC), we show a non‐random distribution of aUPD, suggesting the existence of a cancer‐specific landscape of aUPD events. Our analysis indicates that aUPD acts as a “second hit” in Knudson's model in order to achieve biallelic inactivation of tumor suppressor genes. In particular, APC, ARID1A and NOTCH1 were recurrently inactivated by the presence of homozygous mutation as a consequence of aUPD in COAD and READ, STAD and ESCC, respectively. Furthermore, while TP53 showed inactivation caused by aUPD at chromosome arm 17p across all tumor types, copy number losses at this genomic position were also frequent. By experimental and computationally inferring genome ploidy, we demonstrate that an increased number of aUPD events, both affecting the whole chromosome or segments of it, were present in highly aneuploid genomes compared to near‐diploid tumors. Finally, the presence of mosaic UPD was detected at a higher frequency in DNA extracted from peripheral blood lymphocytes of patients with colorectal cancer compared to healthy individuals. In summary, our study defines specific profiles of aUPD in gastrointestinal cancers and provides unequivocal evidence of their relevance in cancer.
Keywords: uniparental disomy, copy‐number alterations, gastrointestinal cancers, single nucleotide variants, ploidy, mosaicism
Short abstract
What's new?
Somatically acquired uniparental disomies (aUPDs), in which two copies of a chromosome originate from the same parent, have been documented in various human cancers. Here, the authors examined the frequency of aUPDs in different gastrointestinal cancer types. Events involving aUPDs were found to occur at high incidence in gastrointestinal cancers and at increased frequency particularly in highly aneuploid genomes. The data also reveal a nonrandom distribution of aUPDs, with evidence of biallelic inactivation of tumor suppressor genes and activation of oncogenes in a tumor type‐specific manner. The findings suggest that aUPDs are functionally relevant in gastrointestinal malignancies.
Abbreviations
- ASCAT
Allele‐Specific Copy Number Analysis of Tumors
- aUPD
somatically acquired uniparental disomy
- BAF
B allele frequency
- CEP
Centromeric FISH probes
- CIN
chromosome instability
- CNAs
copy number alterations
- cnLOH
copy‐number neutral loss of heterozygosity
- COAD
colon adenocarcinoma
- CRC
colorectal cancer
- EAC
esophageal adenocarcinoma
- ESCA
esophageal carcinoma
- ESCC
esophageal squamous cell carcinoma
- FISH
Fluorescence in situ hybridization
- GI
gastrointestinal
- IBD
identical‐by‐descent
- INDELs
short insertions and deletions
- LOH
loss of heterozygosity
- LRR
Log R Ratio
- MAD
Mosaic Alteration Detection
- MAF
Mutation Annotation Format
- MLPA
Multiplex Ligation‐dependent Probe Amplification
- PSCBS
Parent‐Specific Circular Binary Segmentation
- READ
rectum adenocarcinoma
- RNAi
RNA interference.
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
- STAD
stomach adenocarcinoma
- TCGA
The Cancer Genome Atlas
- TMA
tissue microarray
- TSGs
tumor suppressor genes
- UPD
(constitutive) uniparental disomy
Introduction
Copy number alterations (CNAs) are the hallmark of human cancers, including gastrointestinal (GI) cancers; these CNAs result in a recurrent, tumor‐type specific landscape of DNA gains and losses.1 The application of SNP arrays has allowed the identification of loss of heterozygosity (LOH) and copy‐number neutral LOH (cnLOH), defined as LOH not affected by a reduction in copy number, which typically appears as a consequence of somatically acquired uniparental disomies (hereafter referred to as aUPDs).2, 3, 4 UPD was first described as a constitutional event, consisting in the inheritance of two copies of chromosomes from the same parental origin caused by a meiotic error which may lead to developmental disorders.5 Lately, mosaic UPD has been also associated with aging and cancer.6 Furthermore, aUPDs have been recurrently observed in several human malignancies, including both hematological neoplasms and solid tumors.3, 7
An important challenge in genome‐wide cancer studies is to distinguish driver CNAs, which lead to the development, progression and maintenance of tumors, from those considered passenger events, resulting as a consequence of high rates of chromosome instability (CIN).8 Thus, several efforts have been made in order to identify oncogenes and tumor suppressor genes (TSGs) in recurrently gained and deleted regions, respectively, which could provide insight into driver CNAs. In the “two‐hit” Knudson's hypothesis, one copy of a TSG is inactivated by a non‐synonymous mutation, while the other copy is inactivated by a similar mutation or by a loss of heterozygosity (LOH), acting as the “second hit.”9 Therefore, the integration of the mutational status of genes with regions frequently involved in genomic imbalances has become essential in order to identify functional inactivation of genes.10 In this context, aUPD can arise as an alternative mechanism to act as the “second hit” in the “two‐hit” Knudson's model. Similar to genomic losses, the mutational status of genes located at recurring regions of aUPD can provide evidence to determine the extent to which a certain aUPD event is a driver alteration if biallelic inactivation is achieved. In fact, UPD events have been already associated with driver genes in several cancer types.11, 12, 13, 14, 15, 16, 17, 18, 19 Moreover, some authors have also reported gain‐of‐function of homozygous mutations affecting oncogenes in regions of aUPD.20, 21 Yet, only few studies have focused on profiling such events in a genome‐wide tumor‐type specific manner.
Additionally, whole genome duplication, giving rise to highly unstable tetraploid genomes, has been accepted as a common event in several tumor types, and has been postulated as a driver event in the progression of cancer.22 The consequence of a genome tetraploidization includes the acquisition of numerical chromosome instability, which is defined by the increasing rate of mitotic segregation errors.23 Recently, the use of bioinformatic tools has allowed the assessment of allele‐specific copy number and, consequently, the identification of the tumor ploidy.24, 25 Therefore, it has been systematically established that highly aneuploid genomes resulting from whole genome duplications (i.e., tetraploidization) are common in cancer, in particular in epithelial tumors.8 In such a polyploid scenario, it is feasible to hypothesize that genomic gains and losses of the same parental chromosome could result in recurrent aUPDs.
In the present study, we aimed at integrating patterns of aUPD of GI tumors with the mutational status of genes located at these regions. Moreover, by inferring tumor ploidy, we also determined the extent to which highly aneuploid genomes displayed increased frequency of aUPD and, finally, interrogated the presence of constitutive UPD in colorectal cancer (CRC) patients.
Materials and Methods
Clinical samples
Level 1 data from the five major GI tumor‐types were obtained from The Cancer Genome Atlas (TCGA) project through the NCI Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov/). TCGA cohorts included samples for each tumor‐type and their matched normal paired samples, which consisted of 434 colon adenocarcinomas (COAD), 155 rectum adenocarcinomas (READ), 325 stomach adenocarcinomas (STAD) and 118 esophageal (ESCA) carcinomas, including 58 esophageal adenocarcinoma (EAC) and 60 esophageal squamous cell carcinoma (ESCC) samples.
Additionally, genotyping data from genome‐wide association studies of the EPICOLON cohort, which comprised 747 CRC patients and 503 controls ascertained through a prospective, multicenter, nationwide study in Spain, were used.26 For a subset of individuals of the EPICOLON cohort, DNA from paraffin‐embedded primary tumors and their associated normal mucosa were obtained using standard DNA extraction kits (Qiagen, Hilden, Germany) to perform validation experiments.
Finally, 20 colorectal adenocarcinomas provided by the Hospital Clínic of Barcelona/IDIBAPS Biobank were included in a tissue microarray (TMA). Clinical features of each patient were previously described.18 All patients signed the corresponding informed consent and the sample collection was approved by the institutional review board of the hospital.
SNP‐array data analysis
TCGA tumor and normal‐matched Affymetrix SNP 6.0 array data were used for this analysis. To identify CNAs and LOH, segmentation was performed using the Paired Parent‐Specific Circular Binary Segmentation (Paired PSCBS) method implemented in the PSCBS package.27 PSCBS uses a parametric bootstrapping technique to estimate the different allelic mean levels. Somatically acquired UPD calls are tested on the segments that are not in allelic balance, and is positive on those cases where the allelic CNA is under a certain threshold, derived from data considered background signal. Unfortunately, the background signal estimation may fail on tumors that do not present LOH, so we discarded those samples with a background signal ΔLOH >0.75. After the segmentation, segments smaller than 2.5 Mb were discarded. Two segments were considered the same if the gap between them was shorter than 2.5 Mb. Out of all paired samples examined in our study, only those that presented less than 300 segments were considered for further analyses. Details on data analysis have been previously published.18 Moreover, in order to identify the global ploidy of a previously reported dataset (GSE64114) and TCGA samples, we used the recommended pipeline of the Allele‐Specific Copy Number Analysis of Tumors (ASCAT) method.24 Affymetrix CEL files were preprocessed using PennCNV‐Affy to generate Log R Ratio (LRR) and B Allele Frequency (BAF) matrices, which were used as input files in ASCAT.
Previously published Affymetrix SNP 6.0 genotyping data from genome‐wide association studies of the EPICOLON cohort were used to infer mosaic CNAs and UPDs by applying the Mosaic Alteration Detection (MAD) method.28, 29 This algorithm detects the deviation of the B allele frequency (BAF) signal from the expected values typical for non‐altered homozygous (1 or 0) or non‐altered heterozygous (0.5) probes. BAF values for heterozygous SNPs were used to estimate the percentage of cells with the rearrangement.30 Unless specifically mentioned, segments smaller than 2 Mb and pericentromeric regions were not included in the analysis. In order to discriminate identical‐by‐descent regions (also known as IBD alleles), which may mimic UPD alleles, from constitutive mosaic UPD, segments with BAF value of 1 (complete LOH) were discarded.
Whole exome sequencing data analysis
When available, whole exome sequencing data were extracted from all cohorts from the GDC portal. For this analysis, single nucleotide variants (SNVs) and short insertions and deletions (INDELs) were considered. According to GATK Best Practices, MuTect2 was selected as somatic variant caller.31 Annotated variants were downloaded using the Mutation Annotation Format (MAF) as tab‐delimited text files. Regarding pathogenicity, truncating variants (nonsense, frameshift and those affecting splicing variants) were directly considered. Additionally, missense variants were assessed with six different prediction tools: PhyloP (deleteriousness threshold of phyloP46way_placental score ≥ 1.6), SIFT (prediction of damaging), PolyPhen2 (HumVar prediction of probably damaging or possibly damaging), MutationTaster (prediction of disease‐causing or disease‐causing‐automatic), LRT (prediction of deleterious) and CADD (Phred score ≥ 15). Only those missense variants predicted as pathogenic by at least three algorithms were considered for further analysis. In order to allow an accurate annotation of missense variants with Oncotator (http://portals.broadinstitute.org/oncotator/), genomic coordinates were switched from genome build GRCh38 (hg38) to GRCh37 (hg19) using the NCBI Genome Remapping Service (https://www.ncbi.nlm.nih.gov/genome/tools/remap). Only variants showing at least a 20% of alternative allele frequency and a coverage of 20× were considered for analysis. Comparison with PSCBS results was performed through an in‐house R pipeline.32
Fluorescence in situ hybridization
Centromeric Fluorescence in situ hybridization (FISH) probes (CEP) for chromosomes 7 and 15 labeled in green and for chromosomes 18 and 20 labeled in orange were used according the manufacturer's recommendations (Vysis Inc., Downers Grove, IL). FISH analyses were performed in 4–5 μm thickness sections of the TMA containing two replicates of both tumor and normal adjacent mucosa for each sample. Pretreatment included three xylene incubations, increasing concentration of ethanol series, permeabilization with EDTA and treatment with pepsin. Next, slides were incubated in 1× PBS with MgCl2 and fixed with 1% paraformaldehyde. Denaturation was performed in a Thermo Brite (Vysis) at 78 °C during 6 min for panel one (CEP7 and 18) and 85 °C during 3 min for panel two (CEP15 and 20). Hybridization was performed at 37 °C overnight. Post‐hybridization washes were performed in 0.4× SSC/0.3% NP40 at 74 °C for 2 min and 2× SSC/0.1% NP40 at room temperature during 1 min. A minimum of 100 cells were imaged with a Nikon Eclipse 50i fluorescence microscope using the Isis Fluorescence Imaging System (MetaSystems, Altlussheim, Germany). In order to infer ploidy, the weighted mean copy number of all chromosomes analyzed in each sample was calculated. A threshold for considering a highly aneuploid genome was set at 2.5, which corresponded to a hypotriploid genome (i.e., 57 chromosomes).
Multiplex ligation‐dependent probe amplification (MLPA)
MLPA analysis was performed using Salsa MLPA probemix P043‐D1 and Salsa MLPA probemix P037‐B1 CLL‐1 according to manufacturer's recommendations (MRC‐Holland, Amsterdam, Netherlands). Electrophoresis was performed using the ABI 3100 genetic analyzer (Applied Biosystems, Foster City, CA) with GeneScan 500 ROX dye Size Standard. Results were evaluated using the SeqPilot software version 4.0.1 (JSI Medical Systems GmbH, Kippenheim, Germany).
Microsatellite analysis
Multiplexed PCR amplification of polymorphic short tandem repeat (STR) loci was performed using commercial STR marker panels for chromosomes 11 and 17 (ABI PRISM® Linkage Mapping Set, Version 2.5, Applied Biosystems). For chromosome 11, a total of seven STRs were selected, three within the region of interest (D11S4046, D11S1338, D11S902), and four outside this region used as normal control region (D11S935, D11S904, D11S987, D11S937). For chromosome 17, eight STRs were selected, including four within the LOH region (D17S831, D17S938, D17S1852, D17S799) and four outside this region (D17S798, D17S1868, D17S944, D17S785). All forward primers were fluorescently labeled with different fluorochromes (FAM, VIC and NED). The PCR amplification was performed under standard conditions using fluorescently labeled primers. The PCR products were run on an ABI3100 Genetic Analyzer (ABI, Foster City, CA), and the results were analyzed with the GeneMapper v3.5 software. Single peaks were considered uninformative.
Statistical analysis
A permutation test was implemented to assess the statistical significance of “second hit” events. For each gene and patient we defined the probability of having a “second hit” event as the probability of having one or more mutations in a gene multiplied by the fraction of the genome with copy‐number aberrations in each patient. The mutation probability was calculated based on the size of each gene and the number of mutations in each patient. We performed 109 tests for all genes that showed more than one “second hit” event in our dataset. The resulting p values were adjusted for multiple testing using Benjamini‐Hochberg's method. Only genes with q values <0.1 were considered.
The Mann–Whitney sum‐rank test was used in order to compare the number of aUPD events between tumor‐types and also between highly aneuploid and near‐diploid genomes. Correlation analysis was applied when comparing ploidy values extracted from FISH and ASCAT methods. The software GraphPad Prism 6.0 (GraphPad) was used to assess statistical significance and to plot graphs.
Results
aUPD profiling in GI cancers
Genome‐wide aUPD analysis was performed in 265 colon (COAD), 105 rectum (READ), 121 stomach (STAD) and 57 esophageal (ESCA, including 18 EAC and 39 ESCC) tumors and their corresponding normal‐matched samples extracted from level 1 TCGA data portal by applying the algorithm PSCBS. Most of the samples showed at least one genomic region affected by aUPD (96.49% in ESCA, 91.74% in STAD, 88.89% in READ and 85.38% in COAD cohorts). Specifically, ESCA showed the highest number of regions with aUPD, with a median of five events per sample (p < 0.05). In contrast, COAD showed a median of three aUPD events per sample being the tumor type with statistically significant lowest number of aUPD (p < 0.05) (Supporting Information Fig. S1A). We then classified aUPD in two types of events: whole chromosome and segmental aUPDs, the last including both telomeric and interstitial fragments. Overall, segmental aUPDs were the most frequent alterations with a mean of 3.39 events per sample across all cohorts compared to 0.87 whole chromosome aUPD events (p < 0.0001). Moreover, we observed a higher amount of segmental aUPD in the upper compared to the lower GI tract (mean of 4.65 for ESCA and STAD vs. mean of 2.84 for COAD and READ; p < 0.0001) (Supporting Information Fig. S1B).
In order to establish an overview of the overlap between regions recurrently affected by aUPD and CNA in a tumor‐type specific manner, genome‐wide circos plots were generated for each individual cohort (Supporting Information Fig. S2A‐E). Significant positive correlations between the frequency of regions with aUPD and copy number losses were identified (0.253 in COAD, 0.334 in READ, 0.285 in EAC, 0.105 in ESCC and 0.304 in STAD; p < 0.0001); however, the correlations between the frequency of regions with aUPD and copy number gains were all negative (−0.206 in COAD, −0.223 in READ, −0.072 in EAC, −0.029 in ESCC and − 0.156 in STAD; p < 0.0001). The most frequently genomic regions affected by aUPD in at least 10% of the samples for each cohort are listed in Supporting Information Table 1. Our analysis indicated that the chromosome region 17p13.3‐p13.1 was affected across all GI tumor types (Fig. 1). In particular, the highest frequency of aUPD events affecting this region was observed in EAC (up to 55.56%), and it was the second most frequently affected region in ESCC, READ and COAD, with frequencies up to 53.85%, 26.67% and 17.36%, respectively. When including CNAs in the analysis, chromosome arm 17p also showed high frequencies of copy number losses in all cohorts, thus becoming the most altered genomic region throughout all tumor types. The second most prevalent genomic region affected by aUPD was the chromosome arm 9q. Specifically, aUPDs affecting 9q21.11‐q34.3 were present in 64.10%, 27.78% and 21.48% of ESCC, EAC and STAD samples, respectively. Notably, the region at 9q22.32‐q34.3 in ESCC showed the highest frequency of aUPD across all cohorts (Fig. 1). Finally, UPD at 5q12.1‐q35.3 was the most common event in READ and COAD, affecting up to 32.38% and 20.38% of the samples, respectively. While this alteration was also present in a maximum frequency of 22.22% in EAC, it was rarely detected in ESCC and STAD, showing a frequency of 10.26% and 9.09%, respectively (Fig. 1). In contrast, these two latest cohorts showed high frequencies of copy number losses at this region of chromosome 5q (61.54% in ESCC and 38.84% in STAD) (Supporting Information Fig. S2A and C). Additional regions of interest were those that showed high frequency of aUPD but rarely involved any chromosome loss, such as 6p25.3‐p21.1 (15.09%) and 17q21.32‐q25.3 (19.05%) in COAD and READ, respectively (Supporting Information Table 1).
Figure 1.
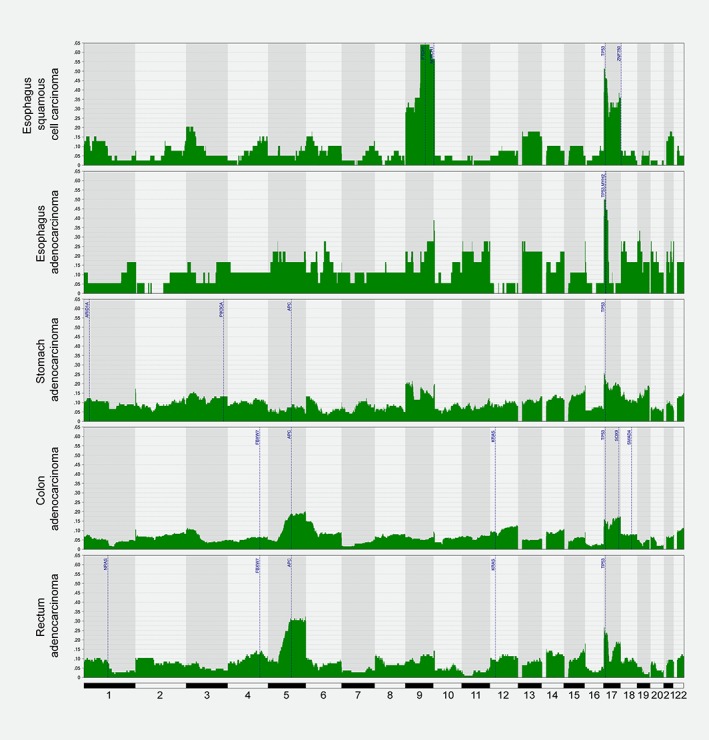
Genomic profiles of aUPDs in GI cancers. Frequency plots showing the distribution of aUPDs along the whole genome in esophageal squamous cell carcinoma, esophageal adenocarcinoma, stomach adenocarcinoma, colon adenocarcinoma, and rectum adenocarcinomas. In blue are indicated the most frequently mutated genes affected by aUPD in each tumor type. [Color figure can be viewed at wileyonlinelibrary.com]
Functional consequences of aUPDs on cancer‐related genes
In order to explore whether aUPDs contributed to the inactivation of TSGs and the activation of oncogenes, we assessed whole exome sequencing data from all five tumor types. Only high‐impact variants (i.e., protein truncating and damaging missense mutations) were considered in this analysis. Our results indicated that the STAD cohort (N = 120) displayed the highest amount of mutations, including SNVs and INDELs, per sample (average of 173.33). The average number of mutations per sample corresponding to the COAD (N = 235), READ (N = 78), EAC (N = 18) and ESCC (N = 39) cohorts was 123.64, 72.1, 45.83 and 37.03, respectively. We then evaluated which of these variants were located in regions of aUPD. Our analysis indicated that ESCA showed the highest average percentage of mutations per patient in regions affected by aUPDs (11.79% in EAC and 10.75% in ESCC), while STAD, READ and COAD cohorts presented 10.60%, 8.15% and 6.74% of mutated genes simultaneously affected by aUPD, respectively.
Subsequently, we integrated the mutational status of genes with CNA and aUPD profiles for each tumor type (Table 1, Fig. 1 and Supporting Information Figures S3‐S7). To do this analysis, only genes with mutational frequencies over 7% in each specific tumor type and a minimum threshold of aUPD was set at 10% were considered. In addition, an adjusted p value was calculated to statistically assess the association of each mutated gene with its presence at the site of aUPD or CNA. Our results indicated that TP53 was simultaneously mutated and affected by aUPD across all GI cancers. For example, 69.23% of the EAC samples which showed a mutation at TP53 also presented aUPD affecting 17p13.1 (q value < 10−9). Moreover, a mutation in this TSG accompanied by a copy number loss was also detected in all GI cancers. In contrast to the aforementioned example in EAC, 73.6% of COAD samples with a mutation at TP53 also displayed simultaneous copy number loss at 17p13.1 (q value <10−9). Therefore, our analysis suggested that tumor samples with mutated TP53 showed either aUPD or genomic losses as “second hit” events ranging from 75% to 93.6% across all GI cancers. Additionally, our analysis also revealed several examples of aUPD‐mediated inactivation of TSGs in a tumor‐type dependent manner. Of note, 34.92% of rectum and 22.29% of colon adenocarcinomas showing an inactivating mutation at APC also displayed aUPD events at 5q22.2 (q value <10−9). In contrast, 15.86% and 20.57% of samples with mutated APC displayed copy number losses in READ and COAD, respectively (q value <10−9). Besides TP53, NOTCH1 and PTCH1 were the most frequently mutated genes involved in regions of aUPD in ESCC. While 75% of the samples showing an inactivating mutation in NOTCH1 also presented aUPD at 9q34.3 (q value = 5.5 × 10−9), aUPD events at 9q22.32 were affecting all samples with mutated PTCH1 (q value <10−9). No samples with copy number losses affecting either of these two genes when mutated were detected. Finally, the second most frequently inactivated gene in STAD was ARID1A. The chromosome region containing ARID1A, 1p35.3, was rarely affected by copy number losses (3.70%, n.s.), but often affected by aUPD (14.81%, q value = 0.019). Although APC was also mutated in STAD (7.76%), the frequency of aUPD was lower than genomic copy number losses (22.22%, n.s. vs. 44.44%, q value = 0.0073) in this cancer type.
Table 1.
Most frequently mutated genes accompanied by aUPD or copy number losses in GI cancers
| Cohort | Gene | Cytoband | “1st hit” mutation1 % | “2nd hit” aUPD2 , 3 % (qval) | “2nd hit” CN Loss3 % (qval) |
|---|---|---|---|---|---|
| COAD | APC | 5q22.2 | 74.47 | 22.29 (< 10−9) | 20.57 (< 10−9) |
| TP53 | 17p13.1 | 53.19 | 20 (< 10−9) | 73.6 (< 10−9) | |
| KRAS | 12p12.1 | 39.57 | 15.05 (< 10−9) | 3.23 (0.0061) | |
| FBXW7 | 4q31.3 | 13.62 | 12.5 (0.0098) | 12.5 (0.0772) | |
| SMAD4 | 18q21.2 | 10.64 | 12 (0.00061) | 68 (< 10−9) | |
| SOX9 | 17q24.3 | 10.21 | 16.67 (< 10−9) | 8.33 (0.000087) | |
| READ | APC | 5q22.2 | 80.77 | 34.92 (< 10−9) | 15.87 (< 10−9) |
| TP53 | 17p13.1 | 73.08 | 31.58 (< 10−9) | 57.89 (< 10−9) | |
| KRAS | 12p12.1 | 39.74 | 19.35 (< 10−9) | 3.23 (0.004) | |
| FBXW7 | 4q31.3 | 14.10 | 18.18 (0.0036) | 9.09 (0.031) | |
| NRAS | 1p13.2 | 11.54 | 22.22 (8.4 × 10−6) | 0 | |
| STAD | TP53 | 17p13.1 | 45.00 | 31.48 (< 10−9) | 59.26 (< 10−9) |
| ARID1A | 1p36.11 | 22.50 | 14.81 (0.019) | 3.70 (n.s.) | |
| PIK3CA | 3q26.32 | 11.67 | 21.43 (0.0728) | 0 | |
| APC | 5q22.2 | 7.50 | 22.22 (n.s.) | 44.44 (0.0073) | |
| ESCC | TP53 | 17p13.1 | 71.79 | 53.57 (< 10−9) | 21.43 (< 10−6) |
| NOTCH1 | 9q34.3 | 10.26 | 75 (5.5 × 10−9) | 0 | |
| PTCH1 | 9q22.32 | 10.26 | 100 (< 10−9) | 0 | |
| ZNF750 | 17q25.3 | 7.69 | 66.67 (1.64 × 10−8) | 33.33 (0.000048) | |
| EAC | TP53 | 17p13.1 | 72.22 | 69.23 (< 10−9) | 23.08 (< 10−9) |
| MYH2 | 17p13.1 | 11.11 | 100 (1.41 × 10−8) | 0 |
n.s., not significant.
Only genes that reached a 7% threshold of mutation by cohort have been considered.
Only genes that reached a 10% threshold of “second hit” driven by an aUPD event have been considered.
Genomic events detected in only one sample were not considered for this analysis.
Despite the high prevalence of TSGs in regions of aUPD, our analysis also unveiled that several well‐known oncogenes were affected by aUPD. This was the example of KRAS in COAD and READ, NRAS in READ and PIK3CA in STAD (Table 1). Indeed, the frequency of aUPD as “second hit” was statistically significant for KRAS in COAD and READ (15.05% and 19.35%, respectively, q value <10−9) and for NRAS in READ (22.22%, q value = 8.4 × 10−6), and showed a tendency for PIK3CA in STAD (21.43%, q value = 0.0728).
aUPD is frequently detected in highly aneuploid genomes
Aneuploid genomes are common in cancer; we therefore assessed to which extent the total DNA content contributes to the generation of aUPD events. First, we sought to infer the ploidy by performing FISH analysis in a set of 20 colorectal adenocarcinomas from Hospital Clínic of Barcelona/IDIBAPS Biobank. We quantified copy numbers of chromosomes 7, 15, 18 and 20 using centromere specific probes. Our results showed that chromosomes 7 and 20 were recurrently gained with a median of 3.48 copies for chromosome 7 (range from 2.02 to 5.07) and 3.24 copies for chromosome 20 (range from 1.88 to 6.01). On the other hand, chromosome 18 was mostly lost showing a median of 1.49 copies (range from 1.09 to 3.45). Chromosome 15 showed a median of 1.91 copies (range from 1.18 to 3.16) (Supporting Information Fig. S8A). In order to infer the genome ploidy based on the FISH counts, we used the weighted mean copy number of all chromosomes analyzed in each sample. By applying this, we detected that 60% of samples showed highly aneuploid genomes, which were defined by ploidy values higher than 2.5 (Table 2). Likewise, applying ASCAT to this sample set, we identified 50% of cases with a highly aneuploid genome. The correlation between FISH data and DNA ploidy provided by ASCAT was statistically tested (r = 0.6; p < 0.01) (Supporting Information Fig. S8B). Out of 20 cases, only four showed discrepancy between the two approaches. We then aimed at comparing the frequency of aUPD events in highly aneuploid genomes versus near‐diploid samples. We could show that aUPD events were more frequently observed in highly aneuploidy genomes (p < 0.01) (Fig. 2 a).
Table 2.
FISH assessment and ASCAT ploidy
| Sample | Chr. 71 | Chr. 151 | Chr. 181 | Chr. 201 | FISH ploidy2 | ASCAT ploidy |
|---|---|---|---|---|---|---|
| 2T | 3.21 | n.a. | 1.61 | n.a. | 2.41 | 2.24 |
| 5T | 4.42 | 1.79 | 1.63 | 2.30 | 2.53 | 1.93 |
| 6T | 3.73 | 3.16 | 1.53 | 4.39 | 3.20 | 3.87 |
| 8T | 3.38 | 1.18 | 1.16 | 2.83 | 2.14 | 2.89 |
| 9T | 3.56 | 1.48 | 1.56 | 3.75 | 2.59 | 2.14 |
| 10T | 2.95 | 1.90 | 1.09 | 1.99 | 1.98 | 1.98 |
| 11T | 3.92 | 2.24 | 1.49 | 5.39 | 3.26 | 3.59 |
| 12T | 2.38 | n.a. | 3.45 | n.a. | 2.92 | 4.04 |
| 13T | 3.76 | 1.81 | 1.45 | 4.37 | 2.85 | 4.29 |
| 33T | 3.12 | 1.83 | 1.19 | 1.88 | 2.01 | 2.10 |
| 37T | 3.59 | n.a. | 1.62 | n.a. | 2.61 | 3.29 |
| 40T | 2.68 | 1.80 | 1.48 | 2.07 | 2.01 | 1.97 |
| 41T | 5.07 | 2.10 | 1.32 | 6.01 | 3.63 | 2.37 |
| 42T | 4.63 | 1.46 | 1.67 | 3.24 | 2.75 | 3.33 |
| 43T | 3.21 | 2.11 | 1.19 | 2.90 | 2.35 | 2.37 |
| 44T | 2.22 | 3.01 | 1.74 | 3.69 | 2.67 | 2.68 |
| 45T | 3.99 | 2.16 | 1.47 | 4.95 | 3.14 | 4.24 |
| 46T | 3.39 | 2.15 | 1.35 | 2.69 | 2.39 | 2.15 |
| 47T | 2.02 | n.a. | 1.96 | n.a. | 1.99 | 2.45 |
| 50T | 3.81 | n.a. | 1.41 | n.a. | 2.61 | 3.31 |
n.a., not available.
Values indicated are weighted means of copy number for the corresponding chromosome.
Average of chromosomes 7, 15, 18 and 20.
Figure 2.
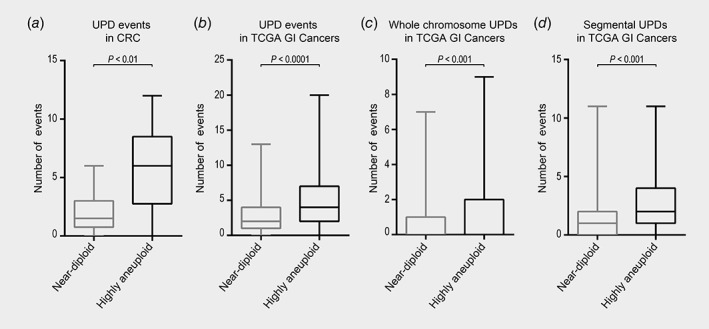
Quantification of aUPDs in highly aneuploid and near‐diploid tumors. Box‐plots showing significant differences between highly aneuploid and near‐diploid genomes for (a) all aUPD events in the FISH‐validated ploidy assessment CRC sample set, (b) all aUPD events in TCGA cohorts, (c) whole‐chromosome aUPDs in TCGA cohorts and (d) segmental aUPDs in TCGA cohorts. The Mann–Whitney sum‐rank test was used to compare number of aUPD events between the two groups of genome ploidies.
Next, we explored the ploidy computed by ASCAT in TCGA cohorts. We observed that COAD and STAD samples showed the lowest genome ploidy, with a median of 2.44 and 2.60, respectively. On the other hand, READ showed a median of 2.96, and ESCA showed a median of 3.07. When comparing the frequency of aUPD events in highly aneuploid versus near‐diploid genomes, our results confirmed that highly aneuploid tumors displayed a significantly higher amount of aUPD events than near‐diploid tumors (p < 0.001) (Fig. 2 b). In addition, when aUPD events were classified in whole chromosome and segmental aUPDs, we also detected that the number of these events was greater in highly aneuploid tumors (p < 0.001 for whole chromosome aUPDs and p < 0.0001 for segmental aUPDs) (Fig. 2 c and D).
Mosaic UPD events are present in CRC patients
Constitutive UPD was assessed using DNA extracted from peripheral blood lymphocytes of the EPICOLON cohort, which consisted of 1,250 individuals, including 503 healthy controls and 747 CRC patients. By estimating LRR and BAF values from genome‐wide association studies SNP 6.0‐array data, our analysis resulted in the identification of 13 clonal mosaic events, four corresponding to healthy controls (0.795%) and nine to CRC patients (1.204%) (Table 3). By performing the same analysis in the colon and rectum adenocarcinoma datasets from TCGA, frequencies of mosaic structural rearrangements were 0.88 and 1.89, respectively. No significant differences were observed after adjusting for gender and age groups, most likely due to the small sample size. With the exception of a focal, 1.07 Mb deletion at 13q14, only alterations larger than 2 Mb were considered. In the EPICOLON cohort we detected five UPDs (four patients and one control), six genomic losses (four patients and two controls), one duplication (one control) and one trisomy (one patient). The most clonal event was the loss of chromosome region 5q14.1‐q33.1 in a control individual, which appeared in 59% of cells (Fig. 3 a). Chromosome arms affected by UPD included 5q, 9p, 11p and 17p in CRC patients, and 20q in a healthy control. On the other hand, mosaic deletions were identified at chromosome arms 1q, 18p, 10q and 13q in patients with CRC (Fig. 3 b), and 5q and 2p in healthy controls. When material was available, we attempted to validate the rearrangements, either by MLPA or microsatellite markers (Table 3). For example, UPD events at 11p15.5‐p15.1 and at 17p13.3‐p11.2 were confirmed by microsatellite analysis in the peripheral blood DNA (Fig. 3 c and d). Similarly, a deletion at 10q22.3‐q23.2 was validated by MLPA in the peripheral blood DNA, and identified in both the normal colon mucosa and in the corresponding primary tumor of the same patient. Genes reported in the COSMIC Cancer Gene Census (https://cancer.sanger.ac.uk/census) catalog that are located at genomic regions affected by mosaic rearrangements are displayed in Supporting Information Table 2.
Table 3.
Summary of mosaic rearrangements
| Case/control | Rearrangement | Cytoband1 | Chr | Start | Stop | Size (Mb) | Clonality | Validation |
|---|---|---|---|---|---|---|---|---|
| Control | Deletion | 5q14.1‐q33.1 | 5 | 82,926,332 | 151,852,840 | 68.93 | 59% | MLPA |
| Control | UPD | 20q11.23‐q13.33 | 20 | 36,224,534 | 62,912,463 | 26.69 | 17% | n.d. |
| Control | Duplication | 20q11.21‐q13.13 | 20 | 30,691,943 | 49,777,691 | 19.09 | 29% | n.d. |
| Control | Deletion | 2p24.1‐p23.3 | 2 | 23,298,851 | 25,421,803 | 2.12 | 57% | n.d. |
| Case | Trisomy | 12p13.33‐q24.33 | 12 | 0 | 133,851,895 | 133.85 | 15% | n.d. |
| Case | UPD | 17p13.3‐p11.2 | 17 | 6,689 | 17,344,122 | 17.34 | 36% | MS |
| Case | UPD | 11p15.5‐p15.1 | 11 | 198,510 | 21,002,580 | 20.80 | 30% | MS |
| Case | Deletion | 13q14.2‐q14.3 | 13 | 50,394,625 | 51,461,086 | 1.07 | 40% | MLPA |
| Case | Deletion | 10q22.3‐q23.2 | 10 | 81,685,024 | 89,167,880 | 7.48 | 49% | MLPA |
| Case | UPD | 5q14.3‐q23.1 | 5 | 89,863,279 | 120,267,945 | 30.40 | 12% | n.d. |
| Case | UPD | 9p24.3‐p24.1 | 9 | 46,587 | 5,731,315 | 5.68 | 14% | n.d. |
| Case | Deletion | 1q21.1‐q21.2 | 1 | 144,988,936 | 147,823,776 | 2.83 | 28% | n.d. |
| Case | Deletion | 18p11.21 | 18 | 12,033,735 | 14,920,039 | 2.89 | 23% | n.d. |
Cytobands and genomic coordinates according to genome build GRCh37/hg19.
Abbreviations: MLPA, multiplex ligation‐dependent probe amplification analysis; MS, microsatellite analysis; n.d., not determined.
Figure 3.
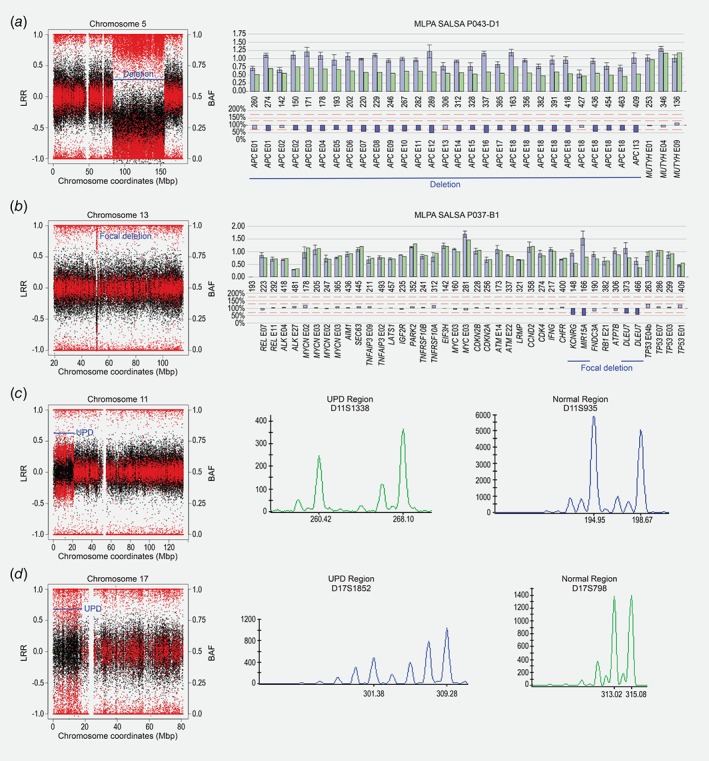
Representative mosaic rearrangements further validated by MLPA or microsatellite analysis. Plots resulting from the MAD method showing the signal intensity Log R ratio (LRR) as black dots (left Y‐axis) and the B allele frequency (BAF) as red dots (right Y‐axis) in the EPICOLON cohort. In the X‐axis are displayed the Mb position according to the Human GRCh37/hg19 genome assembly. (a) Mosaic deletion at 5q in a healthy control validated by MLPA SALSA P043‐D1 probemix in the peripheral blood DNA. (b) Focal deletion at 13q in a CRC patient validated by MLPA SALSA P037‐B1 probemix in the peripheral blood DNA. (c) Mosaic UPD at 11p in a CRC patient validated by microsatellite analysis in the peripheral blood DNA. The figure shows electrophoretograms of two STR markers, D11S1338‐VIC within the region of UPD at chromosome 11 and D11S935‐FAM in a normal region of this chromosome. (d) Mosaic UPD at 17p in a CRC patient validated by microsatellite analysis in the peripheral blood DNA. Electrophoretograms of two STR markers, D17S1852‐FAM within the region of UPD at chromosome 17 and D17S798‐VIC in a normal region of this chromosome, are indicated. In both panels, the X‐axis shows the length of the PCR products (bp) determined using the GeneScan 500 ROX dye Size Standard, and the Y‐axis shows fluorescence intensity in relative fluorescence units (RFU). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
In the present study, we have performed the largest systematic integrative analysis of somatically acquired UPD with the mutational status of genes located at regions affected by aUPD in the five most frequent GI cancers, including esophageal adenocarcinomas, esophageal squamous cell carcinomas, stomach, colon and rectum adenocarcinomas. Our results uncovered that GI cancers exhibit a high incidence of aUPD events. In particular, we could show that STAD, EAC and ESCC carried significantly different patterns of aUPD compared to COAD and READ, thus suggesting the existence of cancer‐specific landscapes of aUPD across GI cancers. As observed for CNAs, aUPD profiles in the COAD cohort matched with those identified in READ samples, which is in agreement with previous reports.11, 18, 33, 34, 35 Remarkably, the high rate of segmental aUPD in STAD and ESCA suggests higher levels of structural CIN in these cancer types compared to COAD and READ, which agrees with a previous study showing more focal amplification events in the upper than in the lower GI tract.36 Furthermore, most GI cancers contain aneuploid genomes.37 Our experimental and computational assessment of the genome ploidy confirmed that highly aneuploid tumors display an increased number of aUPD compared to near‐diploid tumors, affecting both whole chromosomes and segments of chromosomes. In fact, whole chromosome UPD may originate from pre‐ or postzygotic chromosome segregation defects.3 A recent report has speculated that inappropriate activation of RNA interference (RNAi) machinery and meiotic gene expression might induce UPD in fission yeast.38 Whether dysfunctional meiotic cohesins or genes involved in RNAi in cancer cells induce aUPD remains elusive. On the other hand, segmental aUPD appears to result from postzygotic mitotic recombination at the sites of high sequence homology.39 In fact, the positioning of chromosome breakpoints at 5q involving segmental aUPDs in the COAD and READ cohorts were located at 70–80 Mb (Supporting Information Fig. S9), which coincided with sites of meiotic recombination (data not shown). This association is in agreement with previous findings in familial adenomatous polyposis patients, were similar genomic breakpoints were detected and were also associated with mitotic recombination events.40 Therefore, it might be plausible to rationalize that in highly aneuploid genomes, nonrandom nuclear topology of homologous chromosomes enables higher chances of mitotic recombination events.
The integrative analysis of regions affected by aUPD and the mutational status of genes within these regions revealed evidence that aUPD acts as a “second hit” to inactivate TSGs. Specifically, our results confirmed that aUPD events represent an important mechanism to functionally inactivate APC in colon and rectum adenocarcinomas.11, 18, 34, 41, 42, 43 Therefore, these results further strengthen the hypothesis that colorectal tumor cells strive to maintain a disomy for chromosome 5 despite the benefit of inactivating APC. In contrast to CRC, the loss of 5q in STAD is twice as high as the frequency of aUPD, suggesting different mechanisms to achieve the biallelic inactivation of APC. Moreover, we also identified inactivation of NOTCH1 driven by aUPD at chromosome arm 9q as the “second hit” in ESCC. Although aUPD at chromosome arm 9q was also detected at high frequency in EAC, mutations in NOCTH1 were not observed in this tumor type, suggesting that mutations in other genes might contribute to the positive selection of this event in EAC. While SNVs affecting NOTCH1 have been previously reported in ESCC, they have not been widely described in EAC.44 Copy‐number neutral LOH affecting NOTCH1 has been described in head and neck squamous cell carcinoma; however, no association between aUPD and gene mutation has been previously established.45 In fact, the inactivation of NOTCH1 has been reported in about 10% of patients with tumors of squamous origin, including skin, oral cavity, esophageal and lung, suggesting that Notch signaling regulates these cancers.46 Other genes at 9q affected by aUPD in ESCC involved Patched 1 (PTCH1), a gene that has been previously found mutated in a region with aUPD in basal cell carcinomas.47 Furthermore, our results indicated that ARID1A is the second most frequently inactivated TSG in the STAD cohort, and is frequently affected by aUPD as the “second hit”. ARID1A is a subunit of the SWI/SNF chromatin remodeling family, regulating the transcription of MYC and other genes.48 Inactivating mutations of this TSG have been previously described, especially in gastric cancer.49 Despite the fact that we identified aUPD events in chromosome arms 11q and 12q in STAD, mutated genes were not present in these regions.50 Finally, we detected that TP53 was ubiquitously inactivated by aUPD across all GI cancers. Previous reports assessing LOH at the TP53 locus in ESCC have already shown copy‐number neutral LOH in TP53 mutant tumors.15 aUPD events at 17p have been identified in other cancer types such as glioblastoma,51 pediatric adrenocortical tumors,52 diffuse large B cell lymphoma,53 and in patients with newly diagnosed myelodysplastic syndromes.54 Interestingly, aUPD may also play a crucial role in activating cancer‐related genes. In our analysis, the well‐known oncogenes KRAS, NRAS and PIK3CA were frequently affected by aUPD. Copy number gains for KRAS and PIK3CA are common in COAD, READ and STAD; however, NRAS is barely gained in READ, suggesting that aUPD is the main genetic mechanism to achieve homozygous activation of this gene.55 Altogether, even though we cannot discard that a second mutation, epigenetic modifications or DNA conformational changes lead to loss‐of‐function of TSGs or activation of proto‐oncogenes, our results point to an unquestionable relevance of aUPD in cancer as “second hit”. Therefore, there is the need to perform integrative analysis of aUPD with methylation profiling and gene expression to unveil their functional consequences.
In addition to aUPD in tumor cells, we also observed constitutive UPD events in the peripheral blood lymphocytes of CRC patients and healthy individuals. This finding is in agreement with previously published data suggesting that large structural genetic mosaicism was associated with aging and various types of solid tumors.6, 56, 57 In fact, chromosomal mosaic events have been recently described in patients with cancer predisposing disorders, such as Fanconi anemia.58 Here, we show a possible relationship between clonal mosaicism and CRC. Using the EPICOLON cohort, we identified mosaic UPD in CRC patients affecting genomic regions 5q14.3‐q23.1, 11p15.5‐p15.1, 17p13.3‐p11.2 with known CRC‐related genes such as APC, IGF2 and TP53, respectively, and a copy number loss at 13q14.2‐q14.3 involving the putative TSGs DLEU7, DLEU1, DLEU2 and the microRNAs mir‐15a and miR‐16‐1, implicated in B‐cell chronic lymphocytic leukemia.59 Lastly, a previously identified ~7.4 Mb mosaic deletion at 10q22.3‐ q23.2 in a patient with early onset CRC involving the BMPR1A gene has been validated and confirmed in the normal colon mucosa and in the primary tumor.60 Whether these mosaic structural alterations identified in the peripheral blood lymphocytes play a causative role in the cancer etiology requires further exploration.
Supporting information
Supplementary Figure S1: Characterization of aUPDs event across GI datasets. (A) Box‐plot showing significant differences of aUPD events between tumor‐types. (B) segmental aUPD events between upper‐ and lower‐GI tract. The Mann–Whitney sum‐rank test was used to compare number of aUPDs events between the two groups of genome ploidies. *P < 0.05, ***P < 0.001, ****P < 0.0001.
Supplementary Figure S2: Genomic profiles of aUPDs and CNAs in GI cancers. Circos plots showing frequencies of aUPDs, losses and gains in (A) esophageal squamous cell carcinoma, (B) esophageal adenocarcinoma, (C) stomach adenocarcinomas, (D) colon adenocarcinoma, and (E) rectum adenocarcinoma. Chromosomes are represented in the exterior of the circos plots. Regions of aUPD are represented in green in the outer ring, and copy number losses and gains are depicted in blue and red, respectively, in the after rings.
Supplementary Figure S3: Integrative analysis of mutations with aUPD and copy number alterations in colon adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S4: Integrative analysis of mutations with aUPD and copy number alterations in rectum adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S5: Integrative analysis of mutations with aUPD and copy number alterations in stomach adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S6: Integrative analysis of mutations with aUPD and copy number alterations in esophageal squamous cell carcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S7: Integrative analysis of mutations with aUPD and copy number alterations in esophageal adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S8: Correlation between FISH and ASCAT in a cohort of colorectal adenocarcinomas. (A) Plot depicting weighted mean FISH signals of centromeric probes 7, 15, 18 and 20 in 20 normal and tumor samples. (B) Correlation plot between FISH signals and ASCAT ploidy.
Supplementary Figure S9: Location of telomeric aUPD and copy number loss breakpoints at the chromosome arm 5q. Histogram showing the distribution of breakpoints of telomeric aUPDs and copy number losses along chromosome 5q. Chromosome positions were binned in groups of 5 Mb.
Supplementary Table 1. Most frequently UPD events identified in GI cancers
Supplementary Table 2. Genes from the COSMIC Cancer Gene Census catalog located at regions of mosaic rearrangements 1
Acknowledgements
The authors would like to thank Dr. Javier del Rey for technical assistance, and Dr. Eva Hernández‐Illán for critical reading of the study. The authors also thank the Biobank Platform from Hospital Clínic‐IDIBAPS for preparing the samples and the EPICOLON consortium for providing access to GWAS Affymetrix SNP 6.0 data and paraffin‐embedded sample blocks when available.
Conflict of interest: The authors declare that they have no conflict of interest.
K.T. and P.E. contributed equally to this work.
I.P. and J.C. contributed equally to this work.
References
- 1. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature 2010;463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makishima H, Maciejewski JP. Pathogenesis and consequences of Uniparental Disomy in Cancer. Clin Cancer Res 2011;17:3913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med 2009;15:120–8. [DOI] [PubMed] [Google Scholar]
- 4. Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss‐of‐heterozygosity during human development and cancer. Biol Cell 2011;103:303–17. [DOI] [PubMed] [Google Scholar]
- 5. Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet 1980;6:137–43. [DOI] [PubMed] [Google Scholar]
- 6. Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 2012;44:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood 2010;115:2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zack TI, Schumacher SE, Carter SL, et al. Pan‐cancer patterns of somatic copy number alteration. Nat Genet 2013;45:1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knudsona G. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature 2010;463:893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Melcher R, Hartmann E, Zopf W, et al. LOH and copy neutral LOH ( cnLOH ) act as alternative mechanism in sporadic colorectal cancers with chromosomal and microsatellite instability. Carcinogenesis 2011;32:636–42. [DOI] [PubMed] [Google Scholar]
- 12. Tuna M, Ju Z, Smid M, et al. Prognostic relevance of acquired uniparental disomy in serous ovarian cancer. Mol Cancer 2015;14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dunbar AJ, Gondek LP, O'Keefe CL, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c‐Cbl, in myeloid malignancies. Cancer Res 2008;68:10349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carén H, Erichsen J, Olsson L, et al. High‐resolution array copy number analyses for detection of deletion, gain, amplification and copy‐neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene. BMC Genomics 2008;9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saeki H, Kitao H, Yoshinaga K, et al. Copy‐neutral loss of heterozygosity at the p53 locus in carcinogenesis of esophageal squamous cell carcinomas associated with p53 mutations. Clin Cancer Res 2011;17:1731–40. [DOI] [PubMed] [Google Scholar]
- 16. Hu N, Clifford RJ, Yang HH, et al. Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics 2010;11:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Puijenbroek M, Middeldorp A, Tops CMJ, et al. Genome‐wide copy neutral LOH is infrequent in familial and sporadic microsatellite unstable carcinomas. Fam Cancer 2008;7:319–30. [DOI] [PubMed] [Google Scholar]
- 18. Torabi K, Miró R, Fernández‐Jiménez N, et al. Patterns of somatic uniparental disomy identify novel tumor suppressor genes in colorectal cancer. Carcinogenesis 2015;36:1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segditsas S, Rowan AJ, Howarth K, et al. APC and the three‐hit hypothesis. Oncogene 2009;28:146–55. [DOI] [PubMed] [Google Scholar]
- 20. Tuna M, Smid M, Zhu D, et al. Association between acquired uniparental disomy and homozygous mutations and HER2/ER/PR status in breast cancer. PLoS One 2010;5:e15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanada M, Suzuki T, Shih L‐Y, et al. Gain‐of‐function of mutated C‐CBL tumour suppressor in myeloid neoplasms. Nature 2009;460:904–8. [DOI] [PubMed] [Google Scholar]
- 22. Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004;5:45–54. [DOI] [PubMed] [Google Scholar]
- 23. Camps J, Ponsa I, Ribas M, et al. Comprehensive measurement of chromosomal instability in cancer cells: combination of fluorescence in situ hybridization and cytokinesis‐block micronucleus assay. FASEB J 2005;19:828–30. [DOI] [PubMed] [Google Scholar]
- 24. Van Loo P, Nordgard SH, Lingjaerde OC, et al. Allele‐specific copy number analysis of tumors. Proc Natl Acad Sci 2010;107:16910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castellvi‐Bel S, Ruiz‐Ponte C, Fernandez‐Rozadilla C, et al. Seeking genetic susceptibility variants for colorectal cancer: the EPICOLON consortium experience. Mutagenesis 2012;27:153–9. [DOI] [PubMed] [Google Scholar]
- 27. Olshen AB, Bengtsson H, Neuvial P, et al. Parent‐specific copy number in paired tumor–normal studies using circular binary segmentation. Bioinformatics 2011;27:2038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandez‐Rozadilla C, Cazier J‐B, Tomlinson IP, et al. A colorectal cancer genome‐wide association study in a Spanish cohort identifies two variants associated with colorectal cancer risk at 1p33 and 8p12. BMC Genomics 2013;14:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. González JR, Rodríguez‐Santiago B, Cáceres A, et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics 2011;12:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodríguez‐Santiago B, Malats N, Rothman N, et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet 2010;87:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. R Core Team . R: A Language and Environment for Statistical Computing. 2017.
- 33. Lips EH, de Graaf EJ, Tollenaar RAEM, et al. Single nucleotide polymorphism array analysis of chromosomal instability patterns discriminates rectal adenomas from carcinomas. J Pathol 2007;212:269–77. [DOI] [PubMed] [Google Scholar]
- 34. Andersen CL, Wiuf C, Kruhøffer M, et al. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis 2007;28:38–48. [DOI] [PubMed] [Google Scholar]
- 35. Yam YY, Hoh BP, Othman NH, et al. Somatic copy‐neutral loss of heterozygosity and copy number abnormalities in Malaysian sporadic colorectal carcinoma patients. Genet Mol Res 2013;12:319–27. [DOI] [PubMed] [Google Scholar]
- 36. Dulak AM, Schumacher SE, van Lieshout J, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and Oncogenesis. Cancer Res 2012;72:4383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 2018;33:721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Folco HD, Chalamcharla VR, Sugiyama T, et al. Untimely expression of gametogenic genes in vegetative cells causes uniparental disomy. Nature 2017;543:126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stephens K, Weaver M, Leppig KA, et al. Interstitial uniparental isodisomy at clustered breakpoint intervals is a frequent mechanism of NF1 inactivation in myeloid malignancies. Blood 2006;108:1684–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Howarth K, Ranta S, Winter E, et al. A mitotic recombination map proximal to the APC locus on chromosome 5q and assessment of influences on colorectal cancer risk. BMC Med Genet 2009;10:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Middeldorp A, Van Eijk R, Oosting J, et al. Increased frequency of 20q gain and copy‐neutral loss of heterozygosity in mismatch repair proficient familial colorectal carcinomas. Int J Cancer 2012;130:837–46. [DOI] [PubMed] [Google Scholar]
- 42. Zarzour P, Boelen L, Luciani F, et al. Single nucleotide polymorphism array profiling identifies distinct chromosomal aberration patterns across colorectal adenomas and carcinomas. Genes Chromosomes Cancer 2015;54:303–14. [DOI] [PubMed] [Google Scholar]
- 43. Zauber P, Marotta S, Sabbath‐Solitare M. Copy number of the adenomatous polyposis coli gene is not always neutral in sporadic colorectal cancers with loss of heterozygosity for the gene. BMC Cancer 2016;16:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Agrawal N, Jiao Y, Bettegowda C, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov 2012;2:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marescalco MS, Capizzi C, Condorelli DF, et al. Genome‐wide analysis of recurrent copy‐number alterations and copy‐neutral loss of heterozygosity in head and neck squamous cell carcinoma. J Oral Pathol Med 2014;43:20–7. [DOI] [PubMed] [Google Scholar]
- 46. Sakamoto K. Notch signaling in oral squamous neoplasia. Pathol Int 2016;66:609–17. [DOI] [PubMed] [Google Scholar]
- 47. Teh M‐T, Blaydon D, Chaplin T, et al. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils Uniparental Disomy as a key somatic event. Cancer Res 2005;65:8597–603. [DOI] [PubMed] [Google Scholar]
- 48. Wu R‐C, Wang T‐L, Shih I‐M. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther 2014;15:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet 2011;43:1219–23. [DOI] [PubMed] [Google Scholar]
- 50. Arakawa N, Sugai T, Habano W, et al. Genome‐wide analysis of DNA copy number alterations in early and advanced gastric cancers. Mol Carcinog 2017;56:527–37. [DOI] [PubMed] [Google Scholar]
- 51. Lo KC, Bailey D, Burkhardt T, et al. Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization. Genes Chromosomes Cancer 2008;47:221–37. [DOI] [PubMed] [Google Scholar]
- 52. Pinto EM, Chen X, Easton J, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun 2015;6:6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sebastián E, Alcoceba M, Martín‐García D, et al. High‐resolution copy number analysis of paired normal‐tumor samples from diffuse large B cell lymphoma. Ann Hematol 2016;95:253–62. [DOI] [PubMed] [Google Scholar]
- 54. Svobodova K, Zemanova Z, Lhotska H, et al. Copy number neutral loss of heterozygosity at 17p and homozygous mutations of TP53 are associated with complex chromosomal aberrations in patients newly diagnosed with myelodysplastic syndromes. Leuk Res 2016;42:7–12. [DOI] [PubMed] [Google Scholar]
- 55. Cancer T, Atlas G. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Machiela MJ, Zhou W, Sampson JN, et al. Characterization of large structural genetic Mosaicism in human autosomes. Am J Hum Genet 2015;96:487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 2012. [cited 2018 Feb 9];44:642–50. 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reina‐Castillón J, Pujol R, López‐Sánchez M, et al. Detectable clonal mosaicism in blood as a biomarker of cancer risk in Fanconi anemia. Blood Adv 2017;1:319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Palamarchuk A, Efanov A, Nazaryan N, et al. 13q14 deletions in CLL involve cooperating tumor suppressors. Blood 2010;115:3916–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fernandez‐Rozadilla C, Brea‐Fernández A, Bessa X, et al. BMPR1A mutations in early‐onset colorectal cancer with mismatch repair proficiency. Clin Genet 2013;84:94–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Characterization of aUPDs event across GI datasets. (A) Box‐plot showing significant differences of aUPD events between tumor‐types. (B) segmental aUPD events between upper‐ and lower‐GI tract. The Mann–Whitney sum‐rank test was used to compare number of aUPDs events between the two groups of genome ploidies. *P < 0.05, ***P < 0.001, ****P < 0.0001.
Supplementary Figure S2: Genomic profiles of aUPDs and CNAs in GI cancers. Circos plots showing frequencies of aUPDs, losses and gains in (A) esophageal squamous cell carcinoma, (B) esophageal adenocarcinoma, (C) stomach adenocarcinomas, (D) colon adenocarcinoma, and (E) rectum adenocarcinoma. Chromosomes are represented in the exterior of the circos plots. Regions of aUPD are represented in green in the outer ring, and copy number losses and gains are depicted in blue and red, respectively, in the after rings.
Supplementary Figure S3: Integrative analysis of mutations with aUPD and copy number alterations in colon adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S4: Integrative analysis of mutations with aUPD and copy number alterations in rectum adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S5: Integrative analysis of mutations with aUPD and copy number alterations in stomach adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S6: Integrative analysis of mutations with aUPD and copy number alterations in esophageal squamous cell carcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S7: Integrative analysis of mutations with aUPD and copy number alterations in esophageal adenocarcinoma. Plot showing the 100 most frequently mutated genes in the Y‐axis and cases in the X‐axis (light gray squares). “First hit” mutations are indicated with black squares. Red, copy number gain; blue, copy number loss; green, aUPD; dark gray, overlapping alterations.
Supplementary Figure S8: Correlation between FISH and ASCAT in a cohort of colorectal adenocarcinomas. (A) Plot depicting weighted mean FISH signals of centromeric probes 7, 15, 18 and 20 in 20 normal and tumor samples. (B) Correlation plot between FISH signals and ASCAT ploidy.
Supplementary Figure S9: Location of telomeric aUPD and copy number loss breakpoints at the chromosome arm 5q. Histogram showing the distribution of breakpoints of telomeric aUPDs and copy number losses along chromosome 5q. Chromosome positions were binned in groups of 5 Mb.
Supplementary Table 1. Most frequently UPD events identified in GI cancers
Supplementary Table 2. Genes from the COSMIC Cancer Gene Census catalog located at regions of mosaic rearrangements 1