

Abstract
The innate and adaptive immune systems are both crucial to oral disease mechanisms and their impact on systemic health status. Greater understanding of these interrelationships will yield opportunities to identify new therapeutic targets to modulate disease processes and/or increase host resistance to infectious or inflammatory insult. The topics addressed reflect the latest advances in our knowledge of the role of innate and adaptive immune systems and inflammatory mechanisms in infectious diseases affecting the oral cavity, including periodontitis and candidiasis. In addition, several potential links with systemic inflammatory conditions, such as cardiovascular disease, are explored. The findings elucidate some of the defense mechanisms utilized by host tissues, including the role of IL-17 in providing immunity to oral candidiasis, the antimicrobial defense of mucosal epithelial cells, and the pro-resolution effects of the natural inflammatory regulators, proresolvins and lipoxins. They also describe the role of immune cells in mediating pathologic bone resorption in periodontal disease. These insights highlight the potential for therapeutic benefit of immunomodulatory interventions that bolster or modulate host defense mechanisms in both oral and systemic disease. Among the promising new therapeutic approaches discussed here are epithelial cell gene therapy, passive immunization against immune cell targets, and the use of proresolvin agents.
Keywords: oropharyngeal candidiasis, bacterial invasion, fungal immunity, cyto-kines, periodontal bone resorption, lipoxins
Introduction
This article summarizes information presented during a session of the 50th Anniversary of the University at Buffalo Oral Biology Graduate Program, which took place June 12-14, 2013, in Buffalo, New York. The topics addressed reflect the latest advances in our understanding of the role of innate and adaptive immune systems and inflammatory mechanisms in infectious diseases affecting the oral cavity, including periodontitis and candidiasis. Several potential links with systemic inflammatory conditions, such as cardiovascular disease, and possible therapeutic implications of the data are also reviewed.
The recently recognized importance of Th17 cells and inter-leukin (IL)-17 in immunity to oral candidiasis is discussed. In addition, a new technique to bolster the antimicrobial defenses of mucosal epithelial cells by transient transfection of antimicrobial mRNAs is described. Research on the influence of the immune system on bone biology in periodontal disease is also reviewed. The findings reveal how interference with cell targets from immune cells involved in osteoclastogenesis may affect periodontal bone resorption. Finally, data are presented on the mechanistic links between periodontitis and systemic inflammation, particularly on cardiovascular disease. Potential mechanisms underlying this association include infection (bacteremia and infection at non-oral sites caused by oral bacteria or other direct bacterial actions), inflammation mediated by innate immune cells, activation of adaptive immunity, or a combination of these.
IL-17 in Oropharyngeal Candidiasis
Oropharyngeal candidiasis (OPC, also known as “oral thrush”) is a frequent opportunistic infection associated with severe immunocompromise. OPC is caused by the commensal fungus Candida albicans, which is found in the oral cavity and on the skin and other mucosal surfaces of most healthy individuals. OPC is especially common in HIV/AIDS and is considered an AIDS-defining illness (Fidel, 2011). Moreover, OPC severity tracks with declining CD4 T-cell numbers (Glocker and Grimbacher, 2010). Surprisingly, however, other forms of candidiasis are not overrepresented in AIDS, suggesting that CD4+ T-helper cells contribute disproportionately to oral immunity to Candida.
Since 1986 and for nearly 20 years, the dominant paradigm of Th cell immunity was that there were two subsets of Th cells: Th1 and Th2 (Steinman, 2007; Weaver and Murphy, 2007). These cells produced IFNγ and IL-4 and were driven to differentiate by IL-12 and IL-4, respectively. In 2005, a third subset of CD4+ T-cells was defined, characterized by production of the pro-inflammatory cytokine IL-17 (Harrington et al., 2005; Park et al., 2005). These cells were overlooked in large part because IL-23, a key maintenance cyto-kine for Th17 cells, shares its “p40” subunit with IL-12 (Oppmann et al., 2000); therefore, cells lacking IL-12p40 were deficient in not only Th1 but also Th17 cells.
The Role of IL-17 in Oropharyngeal Candidiasis
Historically, OPC was considered to be mainly a Th1-dependent response, because IL-12p40-deficient mice were susceptible to OPC (Farah et al., 2006). Accordingly, it was decided to test the hypothesis that Th17-based immunity might also be essential (Fig. 1), in a modified mouse model of OPC (Kamai et al., 2001; Solis and Filler, 2012). In this model, mice were infected sublingually with Candida (strain CAF2-1), and fungal load was assessed after 5 days. Susceptibility to OPC was compared in mice lacking just IL-12 (i.e., IL-12p35-/- mice) vs. mice lacking the IL-12p19 component. It was found that IL-23p19-/- and also IL-17R-/- mice were highly susceptible to disease, whereas wild-type (WT) and IL-12p25-/- mice were resistant (Conti et al., 2009). Therefore, contrary to the prevailing paradigm, IL-17-dependent responses are essential for immunity to oral candidiasis.
Figure 1.

Old vs. new model of Th-cell-based immunity to Candida albicans. Prior to the recognition of the Th17 cell subset, it was considered in the field that the dominant T-cell response to C. albicans was mediated by Th1 cells via IFNγ. This was premised on the observation that mice lacking the IL-12p40 subunit were susceptible to disease. However, IFNγ-deficient mice were not susceptible (Farah et al., 2006). The discovery of Th17 cells and the shared use of IL-12p40 with the cytokine IL-23 led to a revision of the paradigm, in which Th17 cells and IL-17 are essential for immunity to OPC (Conti et al., 2009).
The importance of IL-17 and Th17 cells to mucosal candidiasis has since been confirmed in humans (Huppler et al., 2012). Notably, various human diseases characterized by chronic mucocutaneous candidiasis (oral and dermal disease) have been found to be associated with defects in the IL-17 pathway. For example, patients with Job's Syndrome (Hyper-IgE Syndrome, HIES) have mutations in STAT3, leading to defects in IL-23 and IL-6 signaling and a paucity of Th17 cells (Holland et al., 2007; Milner et al., 2008). Similarly, humans with mutations in the IL-17R or with naturally occurring neutralizing antibodies against IL-17 also suffer chronic mucocutaneous candidiasis (Kisand et al., 2010; Puel et al., 2010, 2011). Thus, IL-17 is essential for immunity to C. albicans, in both rodent models and humans.
Considerable work has identified the pattern recognition receptor elements for fungi. These are dominantly C-type lectin receptors such as Dectin-1 and Dectin-2, which bind to cell wall carbohydrates of yeast (Fig. 2) (Brown, 2010; Hernández-Santos and Gaffen, 2012). Recognition of Candida by antigen-presenting cells (APCs) or epithelial cells leads preferentially to the production of IL-6, IL-12, IL-1β, and IL-23, which are inductive or maintenance cytokines for the Th17 lineage. IL-17 acts on target cells to induce expression of antimicrobial proteins (AMPS), such as β-defensins, and also chemokines that recruit neutrophils.
Figure 2.

IL-17-based immunity to Candida albicans. Antigen-presenting cells (mainly dendritic cells and macrophages) express pattern recognition receptors for fungi, including Toll-like receptors (TLRs), C-type lectin receptors (CLRs, such as Dectin-1 and Dectin-2), and NOD-like receptors, such as NLRP3. Signaling through these molecules, particularly the CLR adaptor CARD9, leads to the expression of IL-1β, IL-23, and IL-6, all of which are inductive for the Th17 lineage via the transcription factors STAT3 and RORγt. IL-17 and IL-22 production from Th17 cells, in turn, acts on target cells to induce expression of effector molecules that protect against candidiasis, including antimicrobial peptides such as defensins and neutrophil-recruiting chemokines.
Unlike humans, mice are naïve to Candida, and the standard 5-day model reflects a primarily naïve response, with almost no CD4+ Th17 cells in the draining lymph nodes. To better mimic the human condition, a re-challenge model was established in which mice were infected orally with fungus and then re-infected after 6 wk (Hernández-Santos et al., 2013). In this setting, a robust CD4+Th17 response was observed. There was no IL-17 in CD8 cells or CD4-CD8- (“double negative, DN”) cells. Surprisingly, CD4-/- mice were still immune to OPC. In CD4-/-mice, there was a compensatory IL-17 response in both CD8+ T-cells as well as the DN population, both of which could confer immunity to OPC upon adoptive transfer into a susceptible host (Hernández-Santos et al., 2013).
Protective Effects of IL-17
What is the mechanism by which IL-17 provides such strong immune responses to OPC? Microarray analyses were performed in WT and IL-17R-/- mice before and after infection. The most impaired gene in the IL-17R-/- mice was β-defensin 3 (BD3) (human orthologue is BD2) (Conti et al., 2009). Furthermore, saliva from Th17-deficient mice showed impaired killing of Candida in vitro. This was borne out in humans by studies of saliva samples provided by the National Institute of Allergy and Infectious Diseases from a cohort of patients with HIES. Strikingly, these patients’ saliva also exhibited reduced candidacidal activity in vitro, associated with reduced expression of BD2 and salivary histatins. Consistently, IL-17 can act directly on salivary gland cell cultures to induce histatin gene expression (Conti et al., 2011). Thus, IL-17 is a key cytokine involved in oral mucosal immunity against opportunistic Candida albicans infection.
In conclusion, evidence from animal and in vitro studies shows that IL-17-mediated immunity is essential for host defense against oral candidiasis (Conti and Gaffen, 2010). Clearly, more questions remain, such as the nature of the innate sources of IL-17 in the oral mucosa that help control oral thrush and the fundamental IL-17 signaling mechanisms that mediate immunity to this organism.
Augmenting Intracellular Epithelial Resistance to Invasive Pathogens
The mucosal epithelium is a major bodily barrier, protecting against environmental micro-organisms entering the body to cause systemic infections; mucosal breaches account for 80% to 90% of all infections (Asikainen et al., 2012; Deuring et al., 2013). When they invade the connective tissues and enter the systemic tissue compartments and blood, micro-organisms that typically colonize mucosal surfaces can be deadly. Preventing or treating infection at the mucosal epithelial interface is an important therapeutic strategy, but our understanding of how the epithelial cells of the mucosal epithelium protect the host against invasive micro-organisms is far from complete.
Epithelial Cell Defense Mechanisms
Mucosal epithelial cells protect against invasive pathogens by expressing cell autonomous innate immunity (Nisapakultorn et al, 2001; Champaiboon et al, 2009; Zaia et al, 2009; Sorenson et al, 2012). Innate immune effectors include antimicrobial peptides and proteins and production of reactive oxygen species (ROS) (Hsu et al, 2009). Yet normal oral epithelial cells scraped directly from buccal mucosa are laden with familiar oral commensal and pathogenic bacteria (Rudney et al, 2005a,b). Given the non-sterility of the epithelial cell interior, mechanisms must exist to limit the growth of invasive bacteria. Were it not for these mechanisms of cell autonomous immunity, we would suffer persistent mucosal abscesses with concomitant systemic dissemination and sepsis.
Unlike other antimicrobial proteins/peptides (AMPs), calprotectin (S100A8/A9) localizes in the cytoplasm of healthy, supra-basal mucosal epithelial cells. Upon infection and inflammation in oral and genital mucosae, S100A8/A9 up-regulates and can be found in all epithelial cell layers. Whereas S100A8/A9 is directly antimicrobial and effective against many bacterial and fungal species, cytoplasmic S100A8/A9 provides model epithelial cells with a unique, previously unrecognized function: resistance to invasion and intracellular growth by invasive pathogens (Nisapakultorn et al, 2001; Champaiboon et al, 2009; Zaia et al, 2009; Sorenson et al, 2012). As an intracellular innate immune molecule, S100A8/A9 is particularly interesting, because it acts directly and indirectly to contribute to antimicrobial defense.
Invading bacteria interact with constitutively expressed S100A8/A9 in the cytoplasm (Fig. 3, upper left, 10 o'clock). During encounters with epithelial cells, pathogen-associated molecular patterns (PAMPs) on invading bacteria facilitate key cellular responses to invasion (Fig. 3, upper left). The PAMPs activate NF-κB (a transcription factor, not shown), which up-regulates IL-1α among other pro-inflammatory cytokines. IL-1α is secreted by a non-canonical pathway. In an autocrine loop, secreted IL-1α binds to the epithelial cell membrane IL-1 receptor (IL-1R1) (Sorenson et al, 2012). The ligated IL-1R1 signals through p38 MAPK to up-regulate the transcription factor, C/EBPβ, which binds to promoters to up-regulate expression of S100A8 and S100A9 and other AMPs (e.g., cathelicidin antimicrobial protein, β-defensins) (Bando et al, 2013). By increasing the cytoplasmic content of S100A8/A9, the antibacterial capacity in the cytoplasm is thought to increase.
Figure 3.
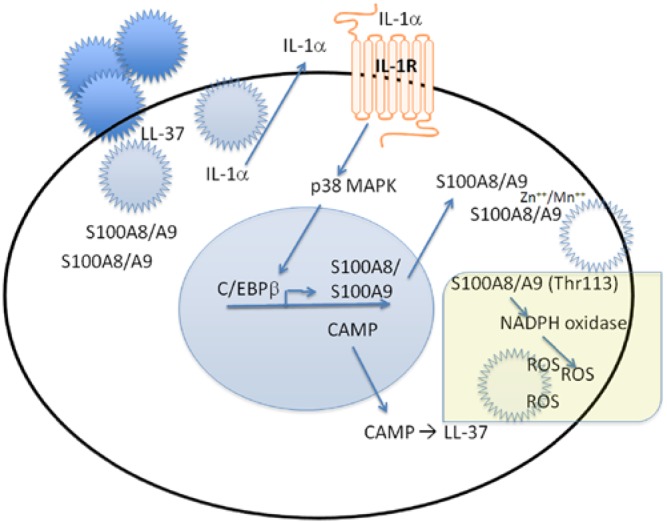
Autonomous intracellular innate immunity in mucosal epithelial cells. In this model, current evidence suggests that, upon entry into the cell, invading bacteria encounter S100A8/A9 in the cytoplasm and LL-37 in the endoplasmic reticulum. Bacterial invasion also stimulates the release of intracellular IL-1α to be bound by the IL-1 receptor (R) on the plasma membrane. Engagement of the IL-1R signals through p38 MAP kinase to promote binding of the transcription factor C/EBPβ and up-regulated transcription and translation of S100A8/A9 and cathelicidin antimicrobial peptide (CAMP). CAMP is cleaved to the more potent LL-37. S100A8/A9 in mucosal epithelial cells can be hypothesized to function as a direct antimicrobial peptide by chelating Zn++ and Mn++, both essential for bacterial growth. Likewise, S100A8/A9 is known to activate NADPH oxidase on the cytoplasmic face of the plasma membrane, resulting in increased production of reactive oxygen species (ROS) and other antimicrobial compounds. These several S100A8/A9- and CAMP-dependent mechanisms function to provide intra-epithelial innate immunity against invading pathogens.
Indeed, increasing the intra-epithelial content of S100A8/A9 and the other key intracellular anti-bacterial peptide, LL-37, may be a viable strategy to increase the resistance of mucosal epithelium to infection. With a few notable exceptions, such as HIV-1/AIDS, mucosal infections result in diseases of low to moderate morbidity and little if any mortality. Gene therapy would seem like an attractive approach. The antimicrobial pep-tide neo-genes could be permanently expressed in mucosal epithelial cells, avoiding ectopic expression and side-effects. However, gene therapy confers the risk of unwanted mutations that can result in lethal illnesses. Therefore, an approach was sought that would capture some of the advantages of gene therapy but avoid the key disadvantage.
Epithelial Cell Transfection with Antimicrobial mRnA
A more benign approach was developed by bypassing the incorporation of neo-genes into the chromosomes. As was recently reported by Zou et al. (2013), epithelial cells were transfected with mRNA constructs containing either cathelicidin antimicrobial protein (CAMP; pro-protein for LL-37), S100A8 and S100A9 open reading frames, A8-IRES-A9 (fusion), or A8-nIRES-A9 (fusion, native IRES). Depending on the construct, CAMP, S100A8, and S100A9 protein levels generally peaked between 16 and 44 hr after mRNA transfection. CAMP was effectively processed to LL-37 over time. Following mRNA transfection, CAMP and S100A8/A9 each independently increased resistance of epithelial cells to invasion by Listeria and Salmonella for up to 48 hr, and tandem constructs of S100A8/S100A9 were also effective. The mRNA packaging system used in the transfections reduced cell viability after 48 hr by 20%; only 2% of cell death was attributable to apoptosis.
Analysis of these data shows that transient transfection of antimicrobial mRNAs is effective in increasing epithelial cell resistance to microbial invasion, although experimental and technical challenges need to be addressed before it will be clinically useful. The increased intracellular production of AMPs appears to augment epithelial cell autonomous immunity. Furthermore, the great advantage of this technique is that it avoids any real or theoretical risk of unwanted mutations. To the extent that the increased resistance is effective against many mucosal pathogens, the strategy of mRNA transfections of AMPs may provide a new therapeutic tool in the management of infections. Indeed, the approach may be useful for any mRNA cargo that might have therapeutic benefit.
Is There a Role for Passive Immunity in the Treatment of Periodontal Disease?
Immune therapy (passive immunization) to disease-related targets is successfully used to ameliorate numerous diseases, including osteoporosis and rheumatoid arthritis (McClung et al., 2006; Hoff et al., 2013). It was therefore decided to investigate the effects of passive antibody treatment on periodontal bone resorption (PBR) in rodent models of periodontitis. Targets from immune cells involved in osteoclastogenesis and PBR were used for the production of antibody. These targets included immune-cell-mediated Receptor Activator of NF-KappaB Ligand (RANKL) and TACE (TNF-α converting enzyme). Polypeptides from RANKL and TACE cell targets were used for immunization of rabbits and syngeneic rats, respectively, and the prepared antibodies were applied locally to the gingival tissues.
Anti-RANKL Studies
Antibody to sRANKL was produced in rabbits to rat 19.6-kDa 174-amino-acid recombinant sRANKL polypeptide, which was >87% homologous with human sRANKL polypeptide. The F(ab')2 antibody was found to react with rat and human sRANKL and to block sRANKL-induced osteoclastogenesis.
Initially, the antibodies were tested in vitro on stimulated human peripheral blood mononuclear cells (PBMC) co-cultured with RAW osteoclast precursor cells that produce TRAP+ staining of multinucleated osteoclast-like cells (osteoclastogenesis). Treatment of PBMC with anti-TACE antibody resulted in a 90% reduction of osteoclast-like cells. Similarly, treatment of the cell supernatant after culture with anti-RANKL antibody resulted in an 80% reduction of osteoclast-like cells.
The effects of passive administration were investigated on immune T-cell-mediated PBR (Kawai et al., 2000) and on PBR induced by tail vein injection of these antigen-specific T clone cells. F(ab')2 anti-RANKL antibody was injected into the gingival papillae (Lin et al., 2011). PBR was significantly diminished and sRANKL concentration significantly reduced by injection of anti-RANKL F(ab')2 antibody. Osteoclasts on the alveolar bone surface were also significantly diminished after antibody injection. Furthermore, gingival sRANKL concentration and bone loss were significantly correlated in animals receiving anti-RANKL F(ab')2 antibody. These results suggest that antibody to RANKL can inhibit specific T-cell-induced PBR by RANKL function blockade and reduction of gingival tissue sRANKL, providing an immunological approach to ameliorate immune-cell-mediated PBR.
In a second study, Rowett rats infected orally with P. gingivalis were injected with anti-RANKL antibody, osteoprotegerin fusion protein (OPG-Fc), or control fusion protein (L6-Fc) (Han et al., 2013). Following the intervention, robust serum IgG/ salivary IgA antibody responses (p < .01) and T-cell proliferation (p < .05) to P. gingivalis were detected. PBR and RANKL concentration in gingival tissues were significantly elevated at Day 28 (p < .01 and p < .05, respectively) in P. gingivalis-infected vs. uninfected rats. Meanwhile, RANKL-expressing T-/B-cells in gingival tissues were also significantly increased in the P. gingivalis-infected group (p < .01). Importantly, injection of anti-RANKL antibody (p < .05) or OPG-Fc (p < .01), but not L6-Fc, after P. gingivalis infection resulted in significantly reduced PBR (16% and 19%, respectively). This study demonstrated that anti-RANKL antibody can significantly ameliorate infection-induced PBR and diminish the number of osteoclast-like cells on the periodontal alveolar bone surface.
Anti-TACE Studies
Antibody to human TACE was produced in inbred Rowett rats to a 170-amino-acid recombinant polypeptide of the extracellular domain corresponding to AA 235-404, which was 94% homologous with rat TACE. The isolated IgG reacted with both human and rat recombinant polypeptide of TACE, as well as with human mature active TACE and immature TACE, and blocked TACE enzymatic activity in humans and mice.
Several periodontitis models were used to determine the effect of passive antibody to TACE on PBR in disease control of Th1-specific (OMP29) cell clone transfer and bacterial challenge by A. actinomycetemcomitans (bearing OMP29). PBR was significantly reduced by 22% in the group receiving antibody with transfer and challenge, compared with the disease control group. sRANKL and TNF-α were also significantly reduced in gingival tissues (by 64% and 45%, respectively), as were osteoclast-like cells on the alveolar bone (by 85%).
In an infection-induced (P. gingivalis) model of PBR, the infected anti-TACE group showed 40% less PBR compared with the infected group without anti-TACE. Gingival tissue concentrations of sRANKL and TNF-α were also reduced by 69% and 60%, respectively. Furthermore, mRNA expression of RANKL, TNF-α, and TACE in gingival mononuclear cells decreased by 80%, 70%, and 50%, respectively.
A ligature-induced PBR model – a faithful representation of human clinical periodontitis – was also used to investigate the effects of anti-TACE administration. After 10 days, ligated rats injected with TACE demonstrated 33% and 78% reductions in PBR, when compared with ligated rats injected with buffer or with control IgG, respectively. Also, anti-TACE injection produced an 80% reduction in alveolar bone TRAP+ osteoclast-like cells, compared with control IgG injection.
These studies suggest that RANKL and TACE are potential therapeutic targets for the modification of periodontitis. Furthermore, local injection of antibody to these targets might serve as a proof of principle for biological treatment to ameliorate periodontal bone resorption.
To conclude, new treatments for PD should also address the major immune cell contributions to periodontal bone resorption (PBR). B- and T-lymphocytes in the gingival tissues are clearly involved in the pathogenesis of PD (Kawai et al., 2006) and express proteins that are involved in PBR (Kanzaki et al., 2010a). Cleavage of RANKL by TACE from lymphocytes is known to play a role in human periodontitis (Kanzaki et al., 2010b). TACE can cleave not only RANKL, but also TNF-α, possibly an alternative osteoclastogenic cytokine (Kobayashi et al., 2000). Increased TNF-α has been reported in periodontitis (Rossomando et al., 1990). Since synergy between sRANKL and TNF-α on osteoclastogenesis has been reported (Fuller et al., 2002), blockade or release of one or both of these cytokines could effectively inhibit cytokine-dependent osteoclastogenesis. Another recently described technique to block antigens that are involved in the pathogenesis of PBR would be to immunize with whole genomic DNA of the bacteria involved which encoded virulence components. We have recently tested this in rodent preclinical studies where such specific antibody is formed (Han et al., 2014).
Resolution of Inflammation and the Potential for Control of Inflammatory Diseases
Inflammation and Cardiovascular Disease
Severe periodontal disease affects 10-15% of the general population and has been linked to cardiovascular disease in cross-sectional and cohort studies (Janket et al., 2003). Studies have reported that elevated cell- and cytokine-mediated markers of inflammation, including C-reactive protein (CRP), fibrinogen, and various cytokines, are associated with periodontal disease (Black, 2004). The same pro-inflammatory markers in periodontal disease have also been linked with atherothrombogenesis (Danesh et al., 2000). By reducing the progression of periodontal disease, levels of inflammatory markers common to both diseases (i.e., IL-6, TNF-α, and CRP) are decreased, which might in turn decrease vascular disease (Mustapha et al., 2007). It is still unknown whether inhibiting/reducing inflammation in general or CRP in particular will decrease the rates of vascular effects.
In several atherosclerosis studies in animal models, periodontal disease was shown to be a contributing factor (Jain et al., 2003; Gibson et al., 2004). Activated immune cells in the atherogenic plaque produce inflammatory cytokines (interferon, interleukin-1, and TNF-α), which induce the production of substantial amounts of IL-6. These cytokines are also produced in various tissues in response to infection and in the adipose tissue of patients with the metabolic syndrome (Hansson, 2005). IL-6, in turn, stimulates the production of large amounts of acute-phase reactants, including CRP, serum amyloid A, and fibrinogen, especially in the liver. Although cytokines at all steps have important biologic effects, their amplification at each step of the cascade makes the measurement of downstream mediators such as CRP particularly useful for clinical diagnosis (Hansson, 2005). Increased hsCRP plasma levels in patients with prehyper-tension and patients with established hypertension may link these two conditions. Major depression, physical inactivity, family histories of CVD and periodontal disease, advancing age, and male gender are other risk factors for atherosclerotic CVD that are commonly found in patients with periodontitis and also may serve as confounders (Friedewald et al., 2009).
Lipid Mediators of Inflammation
Prostaglandins (PGs) are derived from hydrolysis of membrane phospholipids. Phospholipase A2 cleaves the sn-2 position of membrane phospholipids to free arachidonic acid, a precursor of a group of small lipids known as eicosanoids. Arachidonic acid is metabolized by two major enzyme pathways. Lipoxygenases (LO) catalyze the formation of hydroxyeicosatetraenoic acids (HETEs), leading to the formation of leukotrienes (LT). Cyclooxygenases (COX-1 and COX-2) catalyze the conversion of arachidonic acid into prostaglandins, prostacyclins, and thromboxanes. Prostaglandins have 10 sub-classes, of which D, E, F, G, H, and I are the most important in inflammation. Inflamed gingiva synthesizes significantly larger amounts of prostaglandins when incubated with arachidonic acid than does healthy gingiva. Prostaglandin E2 (PGE2) is a potent stimulator of alveolar bone resorption (Goodson et al., 1974). Periodontal ligament cells also produce PGE2 even when unstimulated. This secretion is enhanced by IL-1β, TNF-α, and parathyroid hormone. LO and COX products (LTB4, thromboxanes and PGE2, respectively) play important roles in systemic inflammation, endothelial cell activation and vascular endothelial growth factor (VEGF) expression, and platelet aggregation.
Natural Regulation of Innate Inflammation
Periodontal inflammation begins as a protective response to bacterial biofilm. In susceptible individuals, periodontal inflammation fails to resolve, and chronic inflammation becomes periodontal pathology with systemic impact. The rapid and complete elimination of leukocytes from a lesion is the ideal outcome following an inflammatory event. However, failure to return tissue to homeostasis results in neutrophil-mediated destruction and chronic inflammation, with destruction of bone and extracellular matrix, scarring, and fibrosis.
To date, efforts to control inflammation have been focused on the use of pharmacological agents that inhibit pro-inflammatory mediator pathways, e.g., non-steroidal anti-inflammatory drugs (NSAIDs). NSAIDs target COX-1- and COX -2-dependent pathways, inhibiting the generation of prostanoids. Newer classes of inhibitors target lipoxygenase pathways and leukotriene (LT) production or TNF-α. The side-effect profiles of these agents prohibit their extended use in periodontal therapy and have been shown to have negative impact on the progression of systemic inflammatory conditions including CVD, diabetes, and rheumatoid arthritis (RA).
More recent discoveries have uncovered eicosanoid pathways that signal the physiologic end of the acute inflammatory phase (for complete review, see Serhan et al., 2008). Lipoxins, the eicosanoid product, are receptor agonists that stimulate the resolution of inflammation and promote the restoration of tissue homeostasis by limiting PMN migration into sites of inflammation, modulating the phenotype of macrophages, and stimulating the uptake of apoptotic PMN without secretion of pro-inflammatory cytokines.
Lipoxins are natural pro-resolving molecules derived from endogenous fatty acids. Dietary fatty acids of the omega-3 class are also metabolized by similar pathways, and the products (resolvins) have biologic activity similar to that of lipoxins. Resolvins stimulate the resolution of inflammation through multiple mechanisms, including preventing neutrophil penetration, phagocytosis of apoptotic neutrophils, and enhancing clearance of inflammation within the lesion to promote tissue regeneration (Hasturk et al., 2007) (Fig. 4).
Figure 4.
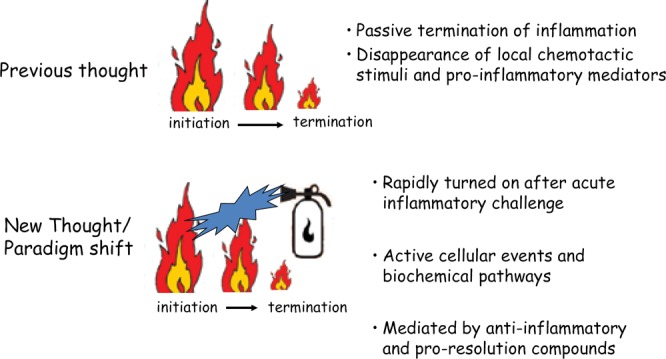
For the past two millennia, physicians have understood the cardinal signs of inflammation; for the past 1,000 years, they have understood the concept of pro-inflammatory mediators’ regulation of the inflammatory response. The new discovery of the late 20th century was the pathways of resolution of inflammation, an active process mediated by de novo synthesis of eicosanoids that bind to specific receptors.
Lipoxin levels in circulation have been directly linked to susceptibility to periodontitis and CVD in rabbits (Jain et al., 2003; Serhan et al., 2003), providing longitudinal evidence that periodontitis affects the progression of early atherogenesis. Importantly in the context of CVD, statins, which are known to have anti-inflammatory as well as cholesterol-lowering actions, exert their anti-inflammatory actions through the production of resolvins and lipoxins.
The Link between periodontal and Cardiovascular Inflammation
In a recent report investigating vascular inflammation and periodontal inflammation in the same subjects, atorvastatin was used to control inflammation (Subramanian et al., 2013). In this study, the uptake of fluorodeoxyglucose (FDG) was monitored by positron emission tomography (PET scan) to measure inflammation of the carotid and the periodontal tissues before and after a 12-wk course of two doses (high, 80 mg; and low, 10 mg) of atorvastatin. Significant reductions in FDG signals were seen after statin therapy (Fig. 5). The reduction is greater with 80-mg, compared with 20-mg, doses of atorvastatin, providing evidence for a dose response. Interestingly, reductions with 80-mg atorvastatin were seen even in patients who were previously treated with low-dose statins. The changes were noted as early as 4 wk. The results of the study suggest that agents that pharmacologically control inflammation through natural resolution mechanisms have the potential for the treatment of both CVD and periodontitis.
Figure 5.
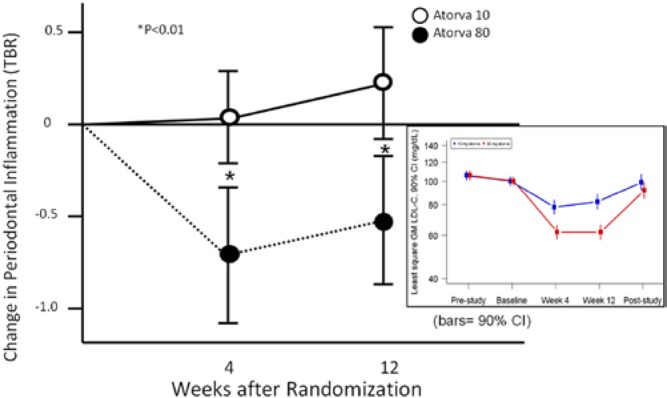
Reduction of periodontal inflammation by high-dose atorvastatin. Periodontal inflammation was measured via FDG uptake by PET scan before and after dosing with 80-mg vs. 10-mg atorvastatin for 12 wk. The change in periodontal inflammation with 80-mg atorvastatin was significantly greater than that with 10 mg and correlated with reductions in carotid inflammation (r = 0.58, p = .001) (figure derived from Subramanian et al., 2013). Statin anti-inflammatory activity is mediated, at least in part, by the production of resolvins (Spite and Serhan, 2010). Compliance with medication regimens in the trial were tracked through reductions in serum cholesterol (inset).
With our increased understanding of the natural pathways of resolution of inflammation, the therapeutic potential of agonists of resolution of inflammation begins to emerge. As pharmacological candidate molecules move into clinical trials, the potential of resolvins, lipoxins, statins, and perhaps other new compounds for the treatment of inflammatory disease will start to unfold.
Conclusion
Periodontal diseases remain a widespread health problem and cause of tooth loss and are now known to be caused in part by disordered host immune and inflammatory responses. Thus, in addition to the traditional approach of reducing and modifying microbial populations in the oral biofilm, therapeutic modulation of the host response may soon be regarded as a logical and potentially effective component of an integrated periodontal treatment strategy. Recent research confirms the importance of the innate and passive immune systems in disease development and has revealed promising results with experimental immuno-modulatory techniques such as transfection of microbial mRNA into epithelial cells and passive immunization to immune cell targets. Furthermore, the associations identified between periodontal and systemic disease processes suggest that manipulation of aspects of host immune and inflammatory responses has the potential to benefit both oral and overall health.
Acknowledgements
The authors were supported by National Institute of Dental and Craniofacial Research grants: DE19938 and 15566 (TVD); DE021831, DE018499 (MT); and grant DE 03420 (MT) and EUREKA grant R01DE021206 (MH) from the National Institutes of Health. MT acknowledges the contributions of Drs. Hirouki Kanzaki, Maiko Suzuki, Toshihisa Kawai, Xiaozhe Han, Xiaoping Lin, Yukiko Asami, and Ms. Kelly Miao. The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Asikainen P, Ruotsalainen TJ, Mikkonen JJ, Koistinen A, Ten Bruggenkate C, Kullaa AM. (2012). The defence architecture of the superficial cells of the oral mucosa. Med Hypotheses 78: 790–792. [DOI] [PubMed] [Google Scholar]
- Bando M, Zou X, Hiroshima Y, Kataoka M, Ross KF, Shinohara Y, et al. (2013). Mechanism of interleukin-1alpha transcriptional regulation of S100A9 in a human epidermal keratinocyte cell line. Biochim Biophys Acta 1829: 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA. (2004). TIMP3 checks inflammation. Nat Genet 36: 934–935. [DOI] [PubMed] [Google Scholar]
- Brown GD. (2010). How fungi have shaped our understanding of mammalian immunology. Cell Host Microbe 7: 9–11. [DOI] [PubMed] [Google Scholar]
- Champaiboon C, Sappington KJ, Guenther BD, Ross KF, Herzberg MC. (2009). Calprotectin S100A9 calcium-binding loops I and II are essential for keratinocyte resistance to bacterial invasion. J Biol Chem 284: 7078–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Gaffen SL. (2010). Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect 12: 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Baker O, Freeman A, Jang W, Li R, Holland S, et al. (2011). New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol 4: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesh J, Whincup P, Walker M, Lennon L, Thomson A, Appleby P, et al. (2000). Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses. BMJ 22: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring JJ, de Haar C, Kuipers EJ, Peppelenbosch MP, van der Woude CJ. (2013). The cell biology of the intestinal epithelium and its relation to inflammatory bowel disease. Int J Biochem Cell Biol 45: 798–806. [DOI] [PubMed] [Google Scholar]
- Farah C, Hu Y, Riminton S, Ashman R. (2006). Distinct roles for interleukin-12p40 and tumour necrosis factor in resistance to oral candidiasis defined by gene targeting. Oral Microbiol Immunol 21: 252–255. [DOI] [PubMed] [Google Scholar]
- Fidel PL, Jr., (2011). Candida-host interactions in HIV disease: implications for oropharyngeal candidiasis. Adv Dent Res 23: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, Libby P, et al. (2009). American Journal of Cardiology, Journal of Periodontology, The American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: periodontitis and atherosclerotic cardiovascular disease. J Periodontol 80: 1021–1032. [DOI] [PubMed] [Google Scholar]
- Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ. (2002). TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology 143: 1108–1118. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Gonzalez DA, Wong J, Genco CA. (2004). Porphyromonas gingivalis-specific immunoglobulin G prevents P. gingivalis-elicited oral bone loss in a murine model. Infect Immun 72: 2408–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker E, Grimbacher B. (2010). Chronic mucocutaneous candidiasis and congenital susceptibility to Candida. Curr Opin Allergy Clin Immunol 10: 542–550. [DOI] [PubMed] [Google Scholar]
- Goodson JM, Dewhirst FE, Brunetti A. (1974). Prostaglandin E2 levels and human periodontal disease. Prostaglandins 6: 81–85. [DOI] [PubMed] [Google Scholar]
- Han X, Lin X, Yu X, Lin J, Kawai T, LaRosa KB, et al. (2013). Porphyromonas gingivalis infection-associated periodontal bone resorption is dependent on receptor activator of NF-κB ligand. Infect Immun 81: 1502–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Larosa KB, Kawai T, Taubman MA. (2014). DNA-based adaptive immunity host from protects host from infection associated periodontal bone resorption via recognition of Pophyromonas gingivalis virulence component. Vaccine 32: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK. (2005). Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352: 1685–1695. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6: 1123–1132. [DOI] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. (2007). Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol 179:7021–7029. [DOI] [PubMed] [Google Scholar]
- Hernández-Santos N, Gaffen SL. (2012). Th17 cells in immunity to Candida albicans. Cell Host Microbe 11:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Santos N, Huppler AR, Peterson AC, Khader SA, McKenna KC, Gaffen SL. (2013). Th17 cells confer long term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol 6:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff M, Kavanaugh A, Haugeberg G. (2013). Hand bone loss in patients with psoriatic arthritis: posthoc analysis of IMPACT II data comparing Infliximab and placebo. J Rheumatol 40:1344–1348. [DOI] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. (2007). STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619. [DOI] [PubMed] [Google Scholar]
- Hsu K, Champaiboon C, Guenther BD, Sorenson BS, Khammanivong A, Ross KF, et al. (2009). Anti-infective protective properties of S100 calgranulins. Antiinflamm Antiallergy Agents Med Chem 8:290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppler AR, Bishu S, Gaffen SL. (2012). Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthritis Res Ther 14:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Batista EL, Jr, Serhan C, Stahl GL, Van Dyke TE. (2003). Role for periodontitis in the progression of lipid deposition in an animal model. Infect Immun 71:6012–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janket SJ, Baird AE, Chuang SK, Jones JA. (2003). Meta-analysis of peri-odontal disease and risk of coronary heart disease and stroke. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 95: 559–569. [DOI] [PubMed] [Google Scholar]
- Kamai Y, Kubota M, Kamai Y, Hosokawa T, Fukuoka T, Filler S. (2001). New model of oropharyngeal candidiasis in mice. Antimicrob Agents Chemother 45: 3195–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki H, Han X, Lin X, Kawai T, Taubman MA. (2010a). Is RANKL shedding involved in immune cell-mediated osteoclastogenesis? In: Interface Oral Health Science 2009. Sasano T, Suzuki O, editors. Tokyo: Springer, pp. 403–405. [Google Scholar]
- Kanzaki H, Han X, Kawait T, Taubman MA. (2010b). TACE, a therapeutic target in periodontal disease. J Dent Res 89(Spec Iss B):1144(abstract). [Google Scholar]
- Kawai T, Eisen-Lev R, Seki M, Eastcott JW, Wilson ME, Taubman MA. (2000). Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. J Immunol 164: 2102–2109. [DOI] [PubMed] [Google Scholar]
- Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux N Y, et al. (2006). B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol 169: 987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisand K, Bøe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. (2010). Chronic mucocutaneous candidiasis in APECED or thy-moma patients correlates with autoimmunity to Th17-associated cyto-kines. J Exp Med 207: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. (2000). Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med 191: 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Han X, Kawai T, Taubman MA. (2011). Antibody to receptor activator of NF-κB ligand ameliorates T cell-mediated periodontal bone resorption. Infect Immun 79: 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, AMG 162 Bone Loss Study Group, et al. (2006). Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354: 821–831. [DOI] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. (2008). Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452: 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapha IZ, Debrey S, Oladubu M, Ugarte R. (2007). Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol 78: 2289–2302. [DOI] [PubMed] [Google Scholar]
- Nisapakultorn K, Ross KF, Herzberg MC. (2001). Calprotectin expression in vitro by oral epithelial cells confers resistance to infection by Porphyromonas gingivalis. Infect Immun 69: 4242–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13: 715–725. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. (2010). Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. (2011). Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332: 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossomando EF, Kennedy JE, Hadjimichael J. (1990). Tumour necrosis factor alpha in gingival crevicular fluid as a possible indicator of periodon-tal disease in humans. Arch Oral Biol 35: 431–434. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Sedgewick GJ. (2005a). Actinobacillus actinomycetem-comitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. J Dent Res 84:59–63. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Zhang G. (2005b). Streptococci dominate the diverse flora within buccal cells. J Dent Res 84:1165–1171. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, et al. (2003). Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol 171: 6856–6865. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis NV, Filler SG. (2012). Mouse model of oropharyngeal candidiasis. Nat Protoc 7: 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson BS, Khammanivong A, Guenther BD, Ross KF, Herzberg MC. (2012). IL-1 receptor regulates S100A8/A9-dependent keratinocyte resistance to bacterial invasion. Mucosal Immunol 5: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Serhan CN. (2010). Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 107: 1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. (2007). A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med 13: 139–145. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Emami H, Vucic E, Singh P, Vijayakumar J, Fifer KM, et al. (2013). High dose atorvastatin reduces periodontal inflammation: a novel pleiotropic effect of statins. J Am Coll Cardiol 62: 2382–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Murphy KM. (2007). T-cell subsets: the more the merrier. Curr Biol 17: R61–R63. [DOI] [PubMed] [Google Scholar]
- Zaia AA, Sappington KJ, Nisapakultorn K, Chazin WJ, Dietrich EA, Ross KF, et al. (2009). Subversion of antimicrobial calprotectin (S100A8/ S100A9 complex) in the cytoplasm of TR146 epithelial cells after invasion by Listeria monocytogenes. Mucosal Immunol 2: 43–53. [DOI] [PubMed] [Google Scholar]
- Zou X, Sorenson BS, Ross KF, Herzberg MC. (2013). Augmenting epithelial resistance to invading bacteria using mRNA transfections. Infect Immun 81: 3975–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]