

Abstract
Background and Purpose
Impaired endothelium‐dependent relaxation (EDR) is a hallmark of endothelial dysfunction. A deficiency of tetrahydrobiopterin (BH4) causes endothelial NOS to produce ROS rather than NO. PPARδ is an emerging target for pharmacological intervention of endothelial dysfunction. Thus, the present study examined the role of PPARδ in the regulation of dihydrofolate reductase (DHFR), a key enzyme in the BH4 salvage pathway.
Experimental Approach
Gene expression was measured by using qRT‐PCR and western blotting. Biopterins and ROS were determined by using HPLC. NO was measured with fluorescent dye and electron paramagnetic resonance spectroscopy. Vasorelaxation was measured by Multi Myograph System.
Key Results
The PPARδ agonist GW501516 increased DHFR and BH4 levels in endothelial cells (ECs). The effect was blocked by PPARδ antagonist GSK0660. Chromatin immunoprecipitation identified PPAR‐responsive elements within the 5′‐flanking region of the human DHFR gene. The promoter activity was examined with luciferase assays using deletion reporters. Importantly, DHFR expression was suppressed by palmitic acid (PA, a saturated fatty acid) but increased by docosahexaenoic acid (DHA, a polyunsaturated fatty acid). GSK0660 prevented DHA‐induced increased DHFR expression. Conversely, the suppressive effect of PA was mitigated by GW501516. In mouse aortae, GW501516 ameliorated the PA‐impaired EDR. However, this vasoprotective effect was attenuated by DHFR siRNA or methotrexate. In EC‐specific Ppard knockout mice, GW501516 failed to improve vasorelaxation.
Conclusion and Implications
PPARδ prevented endothelial dysfunction by increasing DHFR and activating the BH4 salvage pathway. These results provide a novel mechanism for the protective roles of PPARδ against vascular diseases.
Abbreviations
- Ad‐PPARδ
adenovirus expressing PPARδ
- ANGPTL4
angiopoietin‐like protein 4
- BAEC
bovine aortic endothelial cell
- BH2
dihydrobiopterin
- BH4
tetrahydrobiopterin
- ChIP
chromatin immunoprecipitation
- DAF‐FM DA
4‐amino‐5‐methylamino‐2′,7′‐diuorouorescein diacetate
- DHA
docosahexaenoic acid
- DHE
dihydroethidium
- DHFR
dihydrofolate reductase
- EDR
endothelium‐dependent relaxation
- eNOS
endothelial NOS
- EPR
electron paramagnetic resonance
- Fe(DETC)2
Fe2+ diethyldithocarbamate
- GTPCH1
GTP cyclohydrolase I
- L‐012
8‐amino‐5‐chloro‐7‐phenylpyridol[3,4‐d]pyridazine‐1,4(2H,3H)dione
- MTX
methotrexate
- PA
palmitic acid
- PDK4
pyruvate dehydrogenase kinase‐4
- PPRE
PPAR‐responsive element
- SNP
sodium nitroprusside
What is already known
PPARδ has a pivotal role in endothelial homeostasis.
What this study adds
PPARδ activation is involved in the up‐regulation of dihydrofolate reductase and mobilization of the tetrahydrobiopterin salvage pathway.
What is the clinical significance
This study will help us understand how dietary factors modify the risk of vascular disease.
1. INTRODUCTION
NO, produced in the endothelium by endothelial NOS (eNOS), is a critical regulator of vascular homeostasis (Li & Forstermann, 2013). Decreased NO bioavailability with elevated production of superoxide is a hallmark of major metabolic vascular diseases including diabetes, hypertension, and atherosclerosis (Zhao, Vanhoutte, & Leung, 2015). The balance between NO and superoxide production is determined by the availability of tetrahydrobiopterin (BH4), an essential cofactor of eNOS (Crabtree, Brixey, Batchelor, Hale, & Channon, 2013). With sufficient BH4, intact eNOS dimers couple their haem and O2 reduction to synthesize NO. When BH4 is deficient or the BH4 to dihydrobiopterin (BH2) ratio is low, eNOS becomes uncoupled and generates superoxide instead of NO, which is called “eNOS uncoupling” (Bendall, Douglas, McNeill, Channon, & Crabtree, 2014). Thus, eNOS uncoupling is a mechanism leading to endothelial dysfunction (Siragusa & Fleming, 2016). Intracellular BH4 results from the de novo biosynthesis and salvage pathways (Bendall et al., 2014). GTP cyclohydrolase I (GTPCH1) is the rate‐limiting enzyme in BH4 de novo biosynthesis (S. Cai, Khoo, & Channon, 2005). Dihydrofolate reductase (DHFR) is a key enzyme for the salvage pathway and catalyses the regeneration of BH4 from its oxidized form BH2 (Crabtree, Tatham, Hale, Alp, & Channon, 2009). The expression levels and activities of GTPCH1 and DHFR can be regulated at both transcriptional and post‐translational levels (Abali, Skacel, Celikkaya, & Hsieh, 2008; Huang, Zhang, Chen, Hatakeyama, & Keaney, 2005; Widder et al., 2007).
PPARδ is a member of ligand‐activated transcription factor belonging to the nuclear receptor superfamily. It is ubiquitously expressed in most metabolically active tissues, such as skeletal muscles and adipose tissues (Braissant, Foufelle, Scotto, Dauca, & Wahli, 1996). PPARδ controls the expression of many genes whose products are involved in fatty acid metabolism and glucose homeostasis. We and others have found that PPARδ was also present in endothelial cells (ECs) with beneficial effects on endothelial functions (Ehrenborg & Skogsberg, 2013; Fan et al., 2008; Tian et al., 2012). In ECs, PPARδ had potent anti‐inflammatory effects via a binary mechanism involving the induction of antioxidative genes and the suppression of pro‐inflammatory genes (Fan et al., 2008). Moreover, a PPARδ agonist restored the impaired endothelium‐dependent relaxation (EDR) in spontaneously hypertensive rats and deoxycorticosterone acetate‐salt rats (Zarzuelo et al., 2013; Zarzuelo et al., 2011). PPARδ activation increased Akt and eNOS phosphorylation and improved the NO‐dependent and insulin‐induced relaxation in aortae and mesenteric arteries from diabetic rats (Quintela et al., 2014). In endothelial progenitor cells, a PPARδ agonist increased the expression and enzymatic activity of GTPCH1 and BH4 levels (He, Smith, Lu, Joyner, & Katusic, 2011). Thus, we investigated whether a PPARδ agonist regulates DHFR expression and the BH4 salvage pathway.
2. METHODS
2.1. Animals
Male C57BL/6J mice, endothelium‐specific Ppard knockout mice (Ppard flox/flox, Cre+; Ppard EC−/−), and their wild‐type (WT) littermates (Ppard flox/flox, Cre−) aged 8–10 weeks and weighing 20–25 g were used for this study. C57BL/6J mice (RRID:IMSR_JAX:000664) were from the Experimental Animal Centre of Xi'an Jiaotong University. Endothelium‐specific Ppard knockout mice were generated by crossing the Ppard flox/flox mice, which possess loxP sites flanking exon 4 of Ppard gene (JAX stock #005897), with the Tie2‐Cre mice [B6.Cg‐Tg(Tek‐cre)12Flv/J, RRID:IMSR_JAX:004128] (Jackson Laboratory, Bar Harbor, ME, USA; Barak et al., 2002). Genomic DNA was extracted for PCR genotyping. In the present study, mice on C57BL/6 background were used. It is the species generally used in vascular biology. All animals were housed under standard laboratory conditions (12‐hr light/dark cycle, temperature 25°C, 55% humidity under specific pathogen‐free conditions). The mice were housed five per cage in plastic cages with corncob bedding and supplied with standard food and water ad libitum. All experiments involving animals conformed to the institutional and national guidelines for the care and use of animals with an institutional approval (XJTULAC2017‐729). Animal studies are reported in compliance with the ARRIVE guidelines (Karp et al., 2015; Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Mice were killed using the CO2 method following 2013 AVMA guidelines (Leary & Golab, 2013). All effort was taken to minimize the number of animals used and their suffering. The design of this study complies with the recommendations on experimental design in pharmacology (Curtis et al., 2018).
2.2. Preparation of BSA‐conjugated palmitic acid
BSA‐conjugated palmitic acid (PA) was prepared as described previously (Dai et al., 2017). Briefly, PA (P9767; Sigma Sigma‐Aldrich [St. Louis., MO, USA]) was dissolved in 50% ethanol at 65°C for 15 min to obtain a stock solution, concentration of 150 mmol·L−1. Aliquots of stock solution were complexed with fatty acid‐free BSA (A8806; Sigma) by incubation for 1 hr at 37°C. The final molar ratio of PA/BSA was 5:1. The control condition included a solution of vehicle (ethanol/water, 1:1, vol/vol) mixed with BSA at the same concentration as the PA solution.
2.3. Vascular reactivity
Mice were killed by CO2 inhalation, and the vessels were rapidly harvested and cleaned from adherent connective and fat tissue. Mouse thoracic aortic rings (2‐mm in length) were dissected quickly in cold Krebs solution and incubated in DMEM supplemented with 10% FBS. The aortic rings were treated with PA (300 μmol·L−1) in the presence or absence of GW501516 (1 μmol·L−1) and methotrexate (MTX, 2 μmol·L−1), incubated at 37°C continuously oxygenated with a gas mixture of 95% O2 plus 5% CO2. After incubation, the rings were transferred to a chamber filled with fresh Krebs solution and mounted in a myograph for measurement of changes in isometric force (Wang et al., 2015). The ACh‐induced EDRs were detected after the rings had been precontracted, induced by phenylephrine (10 μmol·L−1). Sodium nitroprusside (SNP)‐induced endothelium‐independent relaxations were also measured. The values of ACh and SNP doses are converted to log values to plot in the dose‐response curve, so that the best interpretation can be drawn.
2.4. Cell culture
HUVECs (RRID:CVCL_2959) were cultured in M199 containing heparin (0.1 mg·ml−1), FGF (10 ng·ml−1), l‐glutamine (2 mmol·L−1), penicillin (100 U·ml−1), streptomycin (100 U·ml−1), and 20% FBS. Bovine aortic endothelial cells (BAECs) were maintained in DMEM with 10% FBS and antibiotics.
2.5. Quantitative RT‐PCR
Total RNA was isolated using TRIzol reagent and reverse‐transcribed with the SuperScript reverse transcriptase and oligo (dT) primer. Quantitative RT‐PCRs were performed using SYBR Green Supermixes (Bio‐Rad) and a 7500 real‐time PCR detection system (Applied Biosystems, Foster City, CA, USA). Gene expression was normalized to GAPDH using the 2−ΔΔCt method. Primer sequences are listed in Table S1.
2.6. Western blotting
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. Protein was extracted with lysis buffer (50 mmol·L−1 Tris–HCl, pH 7.5, 15 mmol·L−1 EGTA, 100 mmol·L−1 NaCl, 0.1% Triton X‐100, and the protease inhibitors), separated on SDS‐PAGE gels, and then transferred to PVDF membranes. The blots were incubated with primary antibodies against DHFR (Santa Cruz Biotechnology Cat# sc‐136246, RRID:AB_2277247, 1:1,000 dilution), PPARδ (Santa Cruz Biotechnology Cat# sc‐7197, RRID:AB_2268420, 1:1,000 dilution), and β‐actin (Santa Cruz Biotechnology Cat# sc‐47778, RRID:AB_626632, 1:3,000 dilution), diluted in 0.1% TBST at 4°C overnight, and then incubated with HRP‐conjugated goat anti‐mouse IgG (Santa Cruz Biotechnology Cat# sc‐2005, RRID:AB_631736, 1:3,000 dilution) or goat‐anti rabbit IgG (Santa Cruz Biotechnology Cat# sc‐2004, RRID:AB_631746, 1:3,000 dilution) secondary antibodies for 1 hr at room temperature. After being washed, the blots were visualized by using the enhanced chemiluminescence (ECL) system. The eNOS dimerizations were assayed by using low‐temperature SDS‐PAGE as described previously (Xie et al., 2018). Briefly, protein lysates were mixed with loading buffer (without β‐mercaptoethanol) and without boiling before loading. Electrophoresis and blotting were kept at 4°C during the whole procedure. Primary antibody diluents were used three times for immunoblotting. The band intensities of the immunoblotting were quantified by using ImageJ (RRID:SCR_003070) and normalized to the levels of β‐actin.
2.7. Immunofluorescence
Cells were fixed with 4% paraformaldehyde, blocked with 10% goat serum for 1 hr, and incubated with DHFR antibody (1:200 dilution) at 4°C overnight, followed by the incubation with a TRITC‐conjugated secondary antibody for 1 hr at room temperature. Omission of primary antibody was used as negative controls. After the nuclei had been counterstained with DAPI, cells were examined under a confocal laser microscope (Nikon, Japan E‐C2) and analysed blinded to the treatments. The mean fluorescence intensity value was measured by Olympus Fluoview software. Pictures were taken in five different fields for each treatment.
2.8. Adenoviral vectors and infection
The adenovirus expressing PPARδ (Ad‐PPARδ) and the tetracycline‐responsive transactivator (Ad‐tTA) were prepared as previously described (Qin et al., 2008). Adenovirus‐mediated gene transfer was performed by exposing confluent HUVECs to the adenoviral vectors at a multiplicity of infection of 100 in the presence or absence of tetracycline (1 μg·ml−1) for 48 hr. Ad‐tTA was coinfected to induce the transgene expression in all adenoviral infections.
2.9. Measurement of intracellular biopterin content
HPLC was used for the measurement of total biopterin. Cell lysates were lysed in regular lysis buffer, centrifuged at 12,000 g at 4°C for 30 min, and then subjected to oxidation in acid and base. To a 100‐μl aliquot of supernatant, 50 μl of 1‐N HCL containing 1% iodine (I2) and 2% potassium iodide (KI) for acidic oxidation was added. To another aliquot, 50 μl of 1 mol·L−1 NaOH containing 1% I2 and 2% KI for basic oxidation was added. Then all samples were incubated in the dark at room temperature for 1 hr. Then for acidic oxidation, 10‐μl H2O was added, and for basic oxidation, 10‐μl of 5‐N HCl was added. Excess iodine was reduced by the addition of 5‐μl fresh 10% ascorbic acid. All samples were vortexed and centrifuged at 12,000 g for 30 min at 4°C. The supernatant (50 μl) was injected into the column by use of an HPLC system with an autosampler and a fluorescence detector (Agilent 1100). The C18 column (150 × 4.6 mm) was used for separation of biopterin with a mobile phase of ration of potassium phosphate buffer (50 mmol·L−1, pH = 3.0) running at a flow rate of 1.0 ml·min−1. The retention time of biopterin was approximately 10 min, and the excitation and emission wave lengths were 350 and 440 nm, respectively. Compounds were quantitated by their peak height in comparison with external standards. The amount of BH4 was determined from the difference between total (BH4 plus BH2 plus biopterin) and alkaline‐stable oxidized (BH2 plus biopterin) biopterin.
2.10. Luciferase reporter assay
The DNA segments of the 5′‐flanking region of the human DHFR gene were PCR amplified using TaKaRa LA Taq DNA polymerase from human genomic DNA isolated from Hela cells. These segments, 453 base pair (bp; −422 to +31, in relation to the transcription start site), 1,253 bp (−1,222, +31), and 1,911 bp (−1,880, +31), were subcloned into pGL3‐luc to create pGL3/hDHFR‐1911‐Luc, pGL3/hDHFR‐1253‐Luc, and pGL3/hDHFR‐453‐Luc plasmids. All constructs were confirmed by DNA sequencing. Plasmids expressing PPARδ were cotransfected with one of the three Luc‐reporter constructs into BAECs by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were also cotransfected with pRSV‐gal, a plasmid expressing β‐galactosidase, for the normalization of the transfection efficiency. The reporter activities were measured with the luciferase reporter assay system (Promega). The sequences of the PCR primers used to amplifying the promoter segments are shown in Table S2.
2.11. RNA interference
The siRNA sequence targeting mouse DHFR was as follows: 5′‐CCUCUUCAGUAGAAGGUAATT‐3′ (sense) and 5′‐UUACCUUCUACUGAAGAGGTT‐3′ (antisense). The siRNA with scrambled sequence was used as negative control (Scr siRNA). The double‐stranded RNAs (100 nmol·L−1) were transfected into thoracic aortic rings with Lipofectamine RNAi MAX (Invitrogen).
2.12. Chromatin immunoprecipitation assay
HUVECs were infected with Ad‐PPARδ for 48 hr. Cells were cross‐linked with 1% formaldehyde, harvested, and sheared by sonication. Immunoprecipitation was performed with anti‐PPARδ or normal rabbit IgG (Santa Cruz Biotechnology Cat# sc‐2027, RRID:AB_737197) as a control and protein A/G sepharose beads as described previously (Qin et al., 2008). Immunoprecipitated DNA was eluted and amplified with the primers flanking the putative PPARδ‐binding sites in the human DHFR genes by using quantitative RT‐PCR. Relative DNA binding was expressed as fold enrichment above those amplified from the IgG‐precipitated DNA. Primer sequences used are shown in Table S3.
2.13. Confocal fluorescence microscopy
Intracellular NO was examined in BAECs loaded with NO‐sensitive fluorescent dye 4‐amino‐5‐methylamino‐2′,7′‐diuorouorescein diacetate (DAF‐FM DA), with or without calcium ionophore A23187 treatment. BAECs were incubated with DAF‐FM DA (5 μmol·L−1) at 37°C in the dark for 30 min. The fluorescence intensity excited at 495 nm and emitted at 515 nm was determined using a fluorescence microscopy confocal system before or after the stimulation with A23187 (1 μmol·L−1). Changes in intracellular NO level were displayed as F1/F0, where F0 = average fluorescence signals before addition of A23187 and F1 = fluorescence signal at a defined time interval after addition of A23187.
Intracellular ROS was examined in BAECs loaded with the superoxide‐sensitive dye, dihydroethidium (DHE). BAECs were incubated with DHE (5 μmol·L−1) at 37°C for 30 min. The fluorescence imaging was observed with a fluorescent microscope at an excitation wavelength (Ex) of 510 nm and an emission wavelength (Em) of 580 nm.
2.14. HPLC analysis of DHE oxidation products
HPLC was used to separate the non‐specific ethidium and specific 2‐hydroxyethidium products of DHE oxidation in cells. Cells were lysed with 50% methanol and centrifuged at 12,000 g for 15 min. Supernatants were stored at −80°C in the dark until analysis. Samples (50 μl) were separated by NovaPak C18 column and monitored with a fluorescence detector (Ex/Em = 510/580 nm). 2‐Hydroxyethidium products were analysed with 98% mobile phase A (0.1% trifluoroacetic acid in H2O) and 2% mobile phase B (0.1% trifluoroacetic acid in acetonitrile). Flow rate was maintained at 1.0 ml·min−1. 2‐Hydroxyethidium peak areas were quantified and normalized with protein concentration. All values were normalized to the mean value of the control group in order to set the Y axis of the control group value to 100%.
2.15. Measurement of ROS by L‐012 chemiluminescence
Intracellular ROS was determined using the luminol derivative 8‐amino‐5‐chloro‐7‐phenylpyridol[3,4‐d]pyridazine‐1,4(2H,3H)dione (L‐012; Li et al., 2006). BAECs were plated in 96‐well plates (1 × 104 cells per well) and incubated with 100 μmol·L−1 L‐012 at 37°C for 30 min. The L‐012‐derived chemiluminescence was measured using a VICTOR™X2 luminescence microplate reader (PerkinElmer, Seattle, WA, USA). Results are expressed in relative light units.s‐1.
2.16. NO detection by electron paramagnetic resonance spectroscopy
Electron paramagnetic resonance (EPR) measurement of NO was performed using the colloid spin‐trap Fe2+ diethyldithocarbamate [Fe(DETC)2] (200 μmol·L−1) as previously described (Kleschyov et al., 2000). BAECs were incubated with Fe(DETC)2 at 37°C for 30 min in the presence of A23187 (1 μmol·L−1). Then the cells were collected and detected with an EMX X‐band spectrometer (Bruker Corporation, Billerica, MA, USA) using the following parameters: 10 mW; 9.86 GHz; modulation amplitude, 10 G; modulation frequency, 100 kHz; conversion time, 80 ms, and time constant, 81.92 ms; and scan time, 83.9 s. The EPR signal was indicated as the amplitude of the triplet signal.
2.17. Randomization and blinding
Blinding and random assignment of animals to different groups in this study were in accordance of the guidelines of BJP. The C57BL/6 mice were randomly divided to the control group and each experimental group. For Ppard EC−/− and their littermates, control mice, they were randomly divided to different subgroups and received different treatments. Mouse thoracic arteries were isolated and cut into rings. Then the rings were randomly assigned for different experimental treatments. Data collection and evaluation of all experiments were performed blindly of the group identity.
2.18. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). For qRT‐PCR, western blotting, chromatin immunoprecipitation (ChIP), luciferase reporter assay, immunofluorescence, and DAF‐FM fluorescence quantification, the results are expressed as “fold difference” compared to the corresponding control values, and the control values were set as 1.0. For the measurements of vascular reactivity, DHE fluorescence quantification, and DHE‐HPLC analysis, all values were normalized to the control group and the Y axis of the control group value was set to 100%. This normalization process was used to minimize the background variations derived from different experimental settings. Blinded data analysis was performed using GraphPad Prism 6.0 (RRID:SCR_002798). All data are expressed as mean ± SEM. Sample sizes in each group subjected to statistical analysis were determined based on previous studies, preliminary results, and the power analysis (Curtis et al., 2015). Student's unpaired t test (two groups) or one‐way ANOVA (three or more groups) followed by Dunnett or Newman–Keul comparisons were used when there was no significant variance inhomogeneity. The vascular reactivity experiments were analysed by using two‐way ANOVA followed by Bonferroni post‐tests. Post hoc tests were further conducted only if the F was significant. P < .05 was considered statistically significant.
2.19. Materials
PA, docosahexaenoic acid (DHA), BSA, MTX, phenylephrine, ACh, SNP, and DHE were from Sigma‐Aldrich. GSK0660 was from Tocris Bioscience (Bristol, UK). The antibodies against β‐actin, DHFR and PPARδ, normal rabbit IgG, HRP‐conjugated goat anti‐mouse IgG, goat‐anti rabbit IgG, and protein A/G sepharose beads were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA, RRID:SCR_008987). Antibody against eNOS (Cell Signaling Technology Cat# 9586, RRID:AB_2267266) was from Cell Signaling Technology (Danvers, MA, USA). DAF‐FM DA was from Life Science (Oregon, CA, USA). L‐012 was from Wako Pure Chemical Industries (Osaka, Japan).
2.20. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. Activation of PPARδ increased DHFR level in ECs
To examine the effect of PPARδ activation on DHFR expression, we treated HUVECs with the PPARδ agonist GW501516 (0, 0.1, and 1 μmol·L−1) for 24 hr or GW501516 (1 μmol·L−1) for different periods (0, 8, and 24 hr) and assessed the protein level of DHFR with the use of western blotting. As shown in Figure 1a,b, GW501516 increased DHFR protein level in dose‐ and time‐dependent manners. To ascertain the effect of GW501516 via PPARδ, we pretreated HUVECs with GSK0660, a selective PPARδ antagonist, before the exposure to GW501516. As shown in Figure 1c, the GW50516‐increased DHFR level was significantly attenuated by GSK0660. Further, overexpression of PPARδ with Ad‐PPARδ also increased the protein level of DHFR (Figure 1d). Consistently, immunofluorescence staining also showed that GW501516 increased DHFR level in a PPARδ‐dependent manner (Figure 1e).
Figure 1.
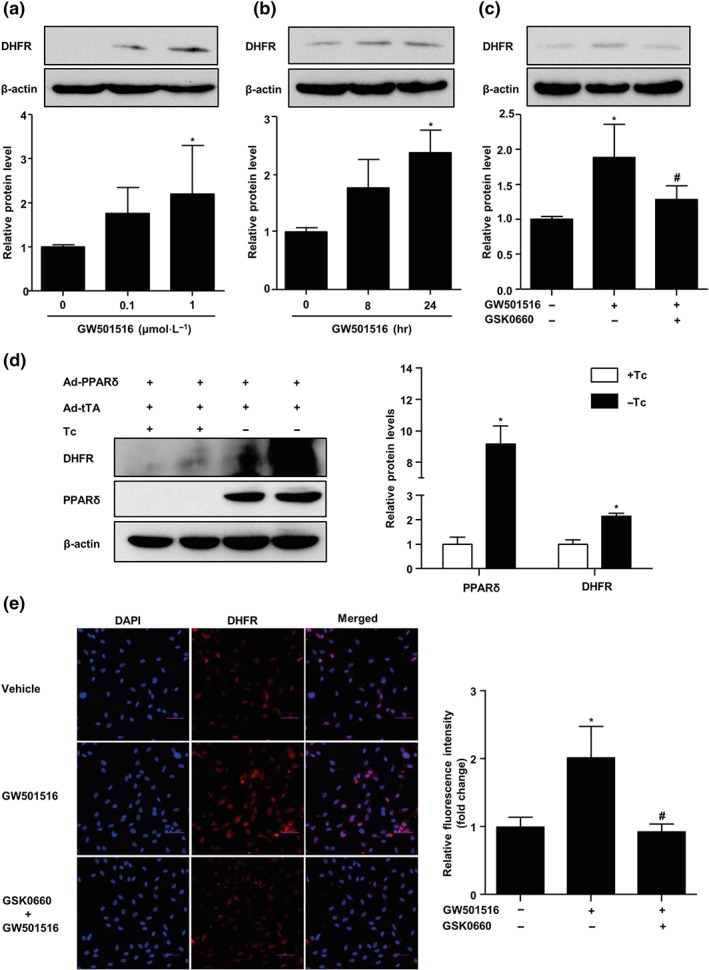
Activation of PPARδ increased DHFR level in ECs. (a) HUVECs were stimulated with GW501516 (0–1 μmol·L−1) for 24 hr; DHFR protein levels were assessed by western blotting (n = 5). (b) DHFR protein levels in HUVECs treated with GW501516 (1 μmol·L−1) for indicated times (0–24 hr, n = 5). (c) HUVECs were pretreated with GSK0660 (2 μmol·L−1, 1 hr) before the exposure to GW501516 (1 μmol·L−1, 24 hr). DHFR protein level was measured (n = 5). (d) HUVECs were coinfected with Ad‐PPARδ and Ad‐tTA in the presence or absence of tetracycline (Tc; 1 μg·ml−1) for 48 hr; DHFR and PPARδ protein levels were measured (n = 5). (e) Immunofluorescence staining for DHFR. Nuclei were counterstained with DAPI. The mean fluorescent intensity was evaluated. Scale bar: 50 μm (n = 5). Data are shown as mean ± SEM. *P < .05 versus control, and # P < .05 versus GW501516
We further determined the effects of PPARδ activation on DHFR mRNA level in HUVECs. As shown in Figure 2a,b, GW501516 also significantly increased the mRNA level of DHFR in dose‐ and time‐dependent manners. Meanwhile, angiopoietin‐like protein 4 (ANGPTL4) and pyruvate dehydrogenase kinase‐4 (PDK4), two PPARδ target genes, were also induced by GW501516 in dose‐ and time‐dependent manners. Conversely, selective PPARδ antagonist GSK0660 abolished GW501516‐induced mRNA levels of DHFR and PPARδ target genes (ANGPTL4 and PDK4; Figure 2c). In addition, overexpression of PPARδ also increased the mRNA levels of DHFR, ANGPTL4, and PDK4 (Figure 2d). Furthermore, incubation of ECs with GW501516 increased BH4 but decreased BH2 (oxidized forms of BH4) and thus augmented the ratio of BH4 to oxidized biopterin. However, these effects were blocked by GSK0660 (Figure 2e–j). Taken together, these results indicated that PPARδ activation increased the DHFR expression and BH4 salvage pathway in ECs.
Figure 2.
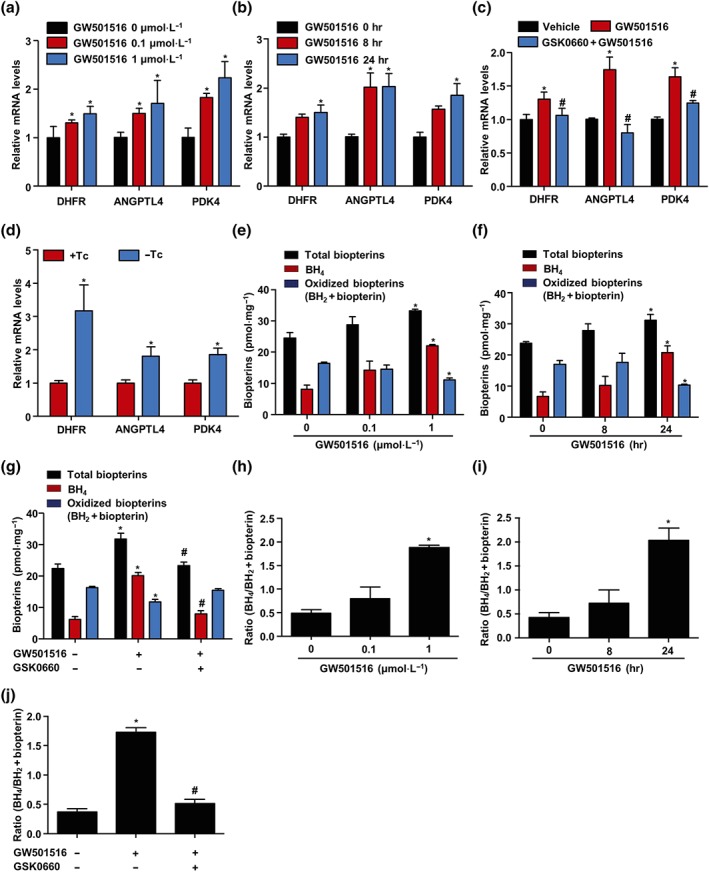
Activation of PPARδ increased DHFR mRNA level in ECs. (a) HUVECs were stimulated with GW501516 (0–1 μmol·L−1) for 24 hr; DHFR, ANGPTL4, and PDK4 mRNA levels were analysed by quantitative RT‐PCR (n = 5). (b) HUVECs were treated with GW501516 (1 μmol·L−1) for indicated times (0–24 hr); DHFR, ANGPTL4, and PDK4 mRNA levels were tested (n = 5). (c) HUVECs were pretreated with GSK0660 (2 μmol·L−1) for 1 hr and then exposed to GW501516 (1 μmol·L−1) for 24 hr; mRNA levels of DHFR, ANGPTL4, and PDK4 were assessed (n = 5). (d) HUVECs were coinfected with Ad‐PPARδ and Ad‐tTA in the presence or absence of tetracycline (Tc; 1 μg·ml−1) for 48 hr; ANGPTL4, PDK4, and DHFR mRNA levels were assessed (n = 5). ECs were (e) stimulated with GW501516 (0–1 μmol·L−1) for 24 hr or (f) treated with GW501516 (1 μmol·L−1) for indicated times (0–24 hr); total biopterins, BH4, and oxidized biopterins (BH2 + biopterin) were measured by HPLC (n = 5). (g) ECs were pretreated with GSK0660 (2 μmol·L−1) for 1 hr and then exposed to GW501516 (1 μmol·L−1) for 24 hr (n = 5). (h–j) BH4/BH2 levels were measured as described in panels (e) to (g); n = 5. Data are shown as mean ± SEM. *P < .05 versus vehicle, and # P < .05 versus GW501516
3.2. PPARδ transactivated the DHFR gene expression
As a transcription factor, the primary mechanism for PPARδ to regulate gene expression is through its binding to the specific recognition site, the PPAR‐responsive element (PPRE), in the regulatory regions of the target genes. Sequences analysis of the 5′‐flanking region of human DHFR gene by using our previously established prediction tool the PPARgene database (http://www.ppargene.org/; Fang et al., 2016) and the online database of transcription factor binding profiles (http://jaspar.genereg.net) revealed multiple putative PPREs within the 2,000‐bp region upstream of the transcription start site (Figure 3a). ChIP assay showed that PPARδ could directly bind to the PPREs located at −475/−460 (PPRE4), −663/−648 (PPRE5), −730/−715 (PPRE6), and −1,779/−1,764 (PPRE9) in the flanking region of the human DHFR gene (Figure 3b).
Figure 3.
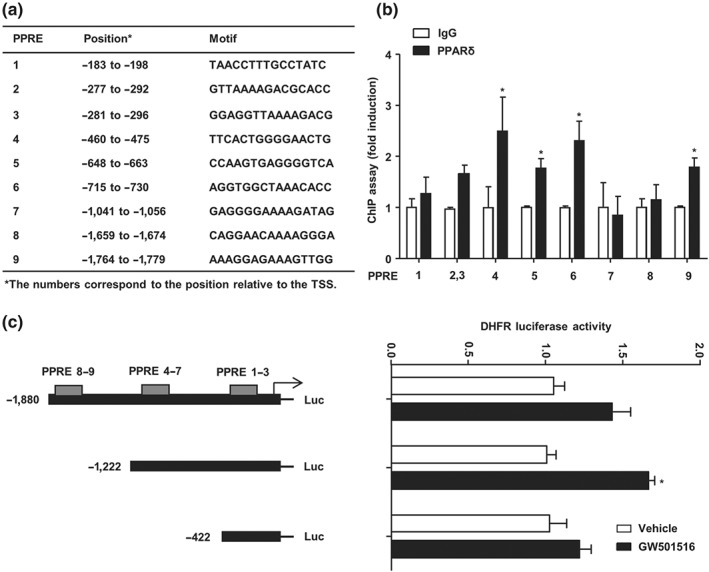
PPARδ transactivated the DHFR gene expression. (a) Putative PPREs in the human DHFR gene promoter are listed with their positions and core sequences. (b) HUVECs were infected with Ad‐PPARδ and Ad‐tTA in the presence or absence of tetracycline for 48 hr. The PPRE‐bound sequences were quantified by using quantitative RT‐PCR with the primers flanking the putative PPREs in the DHFR gene promoter (n = 5). (c) Luciferase reporter assay was performed by transfection of pGL3/hDHFR promoter‐luciferase reporters and pRSV‐β‐galactosidase into BAECs. After the treatment with GW501516 for 48 hr, luciferase activity was measured and normalized to β‐galactosidase activity (n = 5). Data are shown as mean ± SEM. *P < .05 versus vehicle
To further examine whether PPARδ transactivates the DHFR promoter, BAECs were transfected with pGL3/hDHFR‐1911‐Luc, pGL3/hDHFR‐1253‐Luc, or pGL3/hDHFR‐453‐Luc plasmids. As shown in Figure 3c, GW501516 significantly increased the luciferase reporter activity for pGL3/hDHFR‐1253‐Luc, which contains the PPRE 1–7 but lacks 8 and 9 motifs.
3.3. Opposite effects of PA and DHA on DHFR
As a saturated fatty acid, PA decreases NO production and impairs endothelial function (Ghosh, Gao, Thakur, Siu, & Lai, 2017). We examined the effect of PA on DHFR expression with quantitative RT‐PCR and western blotting. As shown in Figure 4a,b, PA significantly inhibited DHFR mRNA and protein levels in a dose‐dependent manner. In contrast, DHA, an unsaturated fatty acid and major component of fish oil, increased the expression of DHFR (Figure 4c,d).
Figure 4.
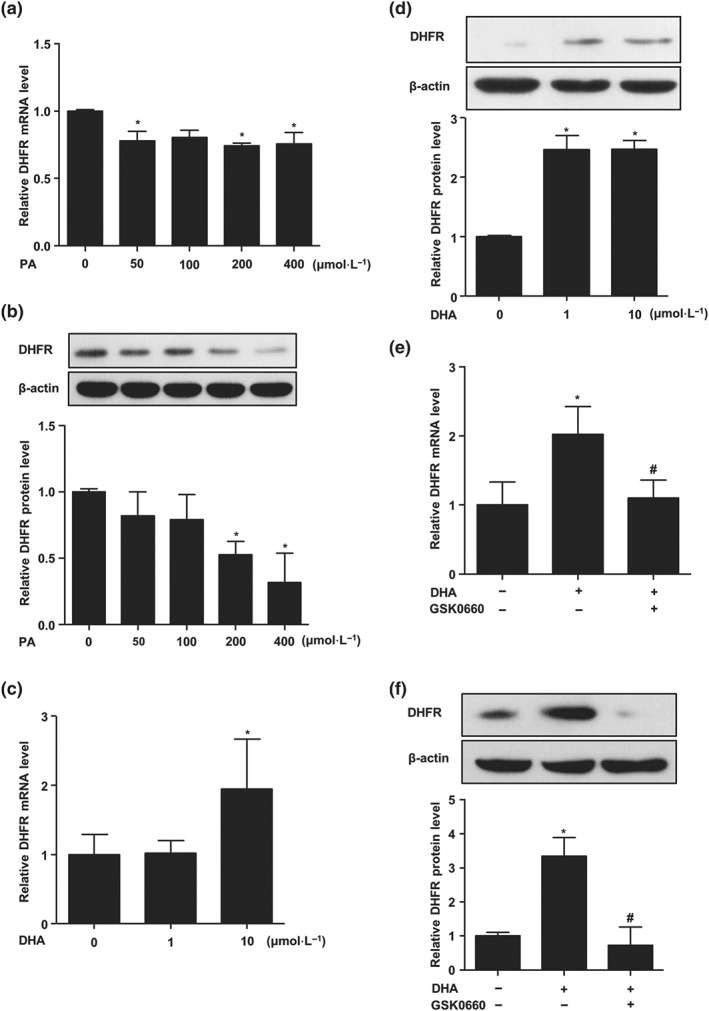
Opposite effects of PA and DHA on DHFR. ECs were treated with indicated concentrations of PA (0, 50, 100, 200, and 400 μmol·L−1) for 24 hr; DHFR (a) mRNA level and (b) protein level were analysed by quantitative RT‐PCR and western blotting (n = 5). HUVECs were treated with DHA (0–10 μmol·L−1) for 24 hr; DHFR (c) mRNA level and (d) protein level were assessed (n = 5). HUVECs were pretreated with GSK0660 (2 μmol·L−1, 1 hr) before the exposure to DHA (10 μmol·L−1, 24 hr); DHFR (e) mRNA level and (f) protein level were tested (n = 5). Data are shown as mean ± SEM. *P < .05 versus vehicle, and # P < .05 versus DHA
Since DHA is a natural agonist for PPARδ, we examined whether the DHA induction of DHFR was mediated by PPARδ activation. Both quantitative RT‐PCR and western blotting demonstrated that pretreatment with GSK0660 blocked the DHA‐induced expression of DHFR (Figure 4e,f).
3.4. PPARδ activation attenuated PA‐induced DHFR loss and eNOS uncoupling
Next, we investigated whether PPARδ activation attenuated the reduction of DHFR by PA. To this end, ECs were pretreated with different concentrations of GW501516 (0, 0.1, and 1 μmol·L−1) for 12 hr before the exposure to PA for 24 hr. As shown in Figure 5a, GW501516 dose dependently attenuated the decrease of DHFR by PA. However, GW501516 failed to restore DHFR level in the presence of the PPARδ antagonist GSK0660 (Figure 5b).
Figure 5.
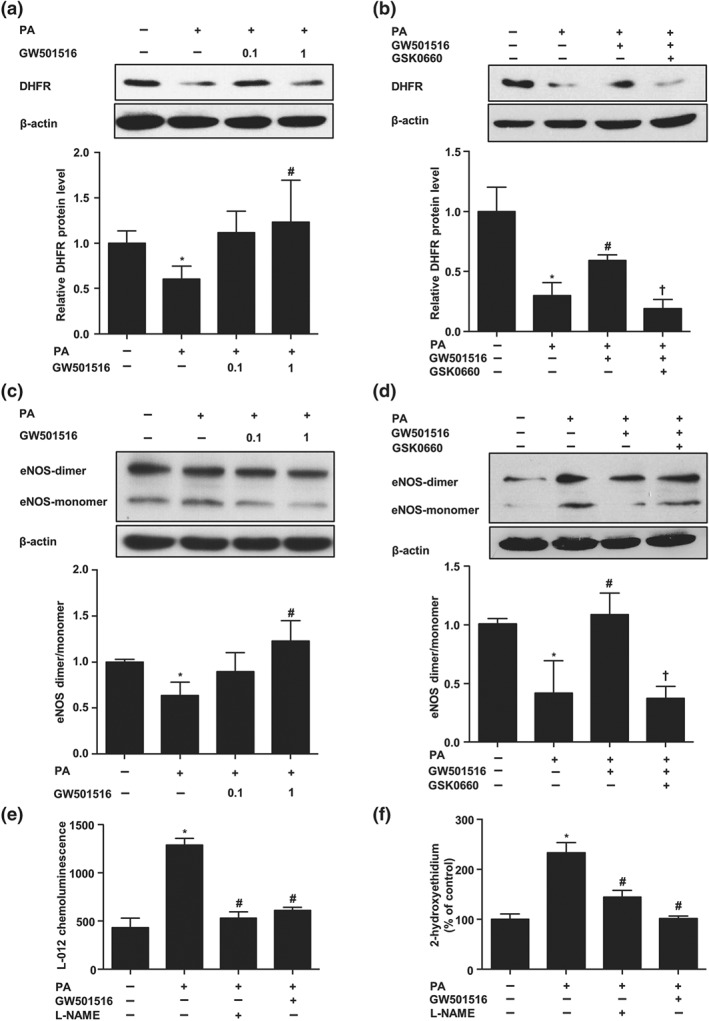
PPARδ activation attenuated PA‐induced DHFR loss and eNOS uncoupling. (a) ECs were pretreated with GW501516 (0–1 μmol·L−1, 12 hr) before the exposure to PA (200 μmol·L−1, 24 hr); DHFR protein level was measured by western blotting (n = 5). (b) ECs were pretreated with GSK0660 (2 μmol·L−1, 1 hr) and then incubated with GW501516 (1 μmol·L−1, 12 hr) before the exposure to PA (200 μmol·L−1, 24 hr); DHFR protein level was measured (n = 5). (c, d) Protein levels of eNOS dimers and monomers were detected by using low temperature SDS‐PAGE (n = 5). (e) ECs were pretreated with GW501516 (1 μmol·L−1, 12 hr) before the exposure to PA (200 μmol·L−1, 24 hr). Production of ROS was measured with L‐012 chemiluminescence in the absence or presence of L‐NAME (100 μmol·L−1, n = 5). (f) Superoxide production was measured with DHE‐HPLC (n = 5). Data are shown as mean ± SEM. *P < .05 versus vehicle; # P < .05 versus PA; † P < .05 versus PA + GW501516
The increased eNOS monomerization caused by BH4 deficiency is related to eNOS uncoupling and endothelial dysfunction (S. Cai et al., 2005). In order to examine the effect of PPARδ on PA‐induced eNOS uncoupling, ECs were pretreated with GW501516 (0, 0.1, and 1 μmol·L−1) for 12 hr before incubation with PA (200 μmol·L−1, 24 hr). As shown in Figure 5c, PA significantly decreased the level of eNOS dimers and the dimer:monomer ratio, which is an indicator of the uncoupling state. However, pretreatment with GW501516 prevented the uncoupling triggered by PA. Importantly, this beneficial effect of GW501516 on eNOS coupling was attenuated by GSK0660, suggesting a PPARδ‐specific mechanism (Figure 5d).
Since “uncoupled” eNOS produces ROS instead of NO (Kietadisorn, Juni, & Moens, 2012), we examined ROS level by using specific L‐012 chemiluminescence and DHE‐HPLC analysis in BAECs. We used BAECs as they were commonly used for the measurements of NO and ROS contents in the protocols we followed (Bernatchez et al., 2005; Fulton et al., 2008; Matsunaga et al., 2004). As shown in Figure 5e,f, PA increased cellular superoxide production. Importantly, the PA‐stimulated ROS production was largely blocked by L‐NAME, suggesting that eNOS is a major source of ROS. Meanwhile, PPARδ agonist GW501516 also had similar suppressive effect as L‐NAME.
3.5. Inhibition of DHFR attenuated the effects of PPARδ on EDR
Thoracic aortae isolated from C57BL/6J mice were used to measure the isometric tension. As shown in Figure 6a, PA (300 μmol·L−1) impaired vascular relaxation response to ACh. Notably, pretreatment of the arteries with GW501516 profoundly improved the relaxation. On the other hand, as an NO donor, SNP induced vasorelaxation, and there was no significant difference between PA‐treated and GW501516‐treated groups (Figure 6b), indicating that both PA‐impaired and GW501516‐improved effects were dependent on the NO bioavailability. To further examine the role of endothelial PPARδ, we compared the responses of the thoracic aortae isolated from Ppard EC−/− mice and their WT littermates. As shown in Figure 6c, in aortae from Ppard WT littermates, PA‐impaired vasorelaxation was prevented by GW501516. However, such beneficial effect was lost in the aortae from Ppard EC−/− mice. To further validate the role of DHFR in the beneficial effects of PPARδ on endothelial function, we used the siRNA to silence the expression of endogenous DHFR in mouse thoracic aortae. Compared with scrambled siRNA, DHFR siRNA effectively diminished the expression of DHFR (Figure 6d) and, as a result, compromised the protective effect of GW501516 against the PA‐impaired relaxation (Figure 6e). Similarly, preincubation the mouse aortae with MTX, a DHFR inhibitor, also diminished the effect of GW501516 on vasodilation (Figure 6f). Taken together, these results indicated that GW501516 attenuated the PA‐induced vasodilation in an endothelial PPARδ‐dependent mechanism and PPARδ improved vascular relaxation via a DHFR‐dependent mechanism.
Figure 6.
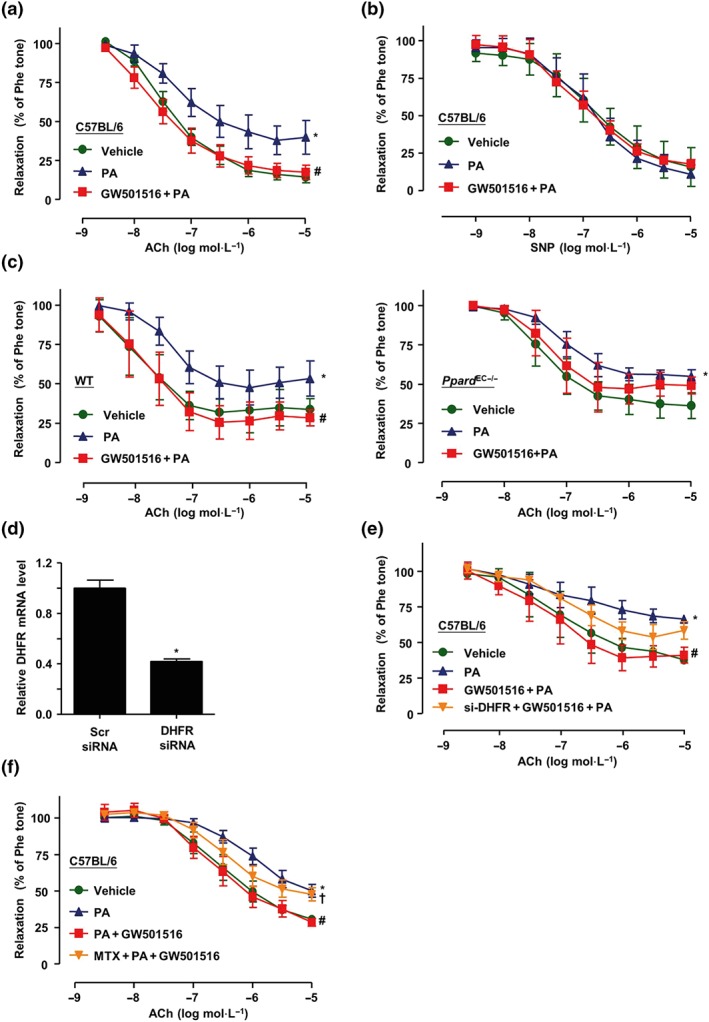
Inhibition of DHFR attenuated the effects of PPARδ on endothelium‐dependent relaxation. C57BL/6J mouse thoracic artery rings were incubated with GW501516 (1 μmol·L−1) and PA (300 μmol·L−1) for 48 hr. (a) ACh‐induced vasodilator responses and (b) SNP‐induced endothelium‐independent dilation were measured (n = 5). (c) Thoracic aortic rings were isolated from Ppard WT littermates or Ppard EC−/− mice, with GW501516 and PA treatment for 48 hr; ACh‐induced vasodilator responses were measured (n = 5). (d) After transfection with DHFR siRNA or Scr siRNA with Lipofectamine RNAi MAX for 24 hr, mouse thoracic aortae RNA was isolated, and DHFR mRNA level was assessed by quantitative RT‐PCR (n = 5). *P < .05 versus Scr siRNA. (e) After being transfected with DHFR siRNA, mouse thoracic aortae were coincubated with GW501516 (1 μmol·L−1) and PA (300 μmol·L−1) for 48 hr; ACh‐induced vasodilator responses were measured (n = 5). (f) Mouse thoracic aortae were preincubated with MTX (2 μmol·L−1, 1 hr) before the exposure to GW501516 (1 μmol·L−1) and PA (300 μmol·L−1) for 48 hr. Then ACh‐induced vasodilation was measured (n = 5). Results are shown as mean ± SEM. *P < .05 versus vehicle; # P < .05 versus PA; † P < .05 versus GW501516 + PA
3.6. Inhibition of PPARδ/DHFR attenuated the effects of GW501516 on NO and superoxide production
Endothelial function is determined by the balance between NO and ROS (Li & Forstermann, 2013). Thus, we investigated the effects of GW501516 on NO and ROS production. In ECs, Ca2+ ionophore A23187 (1 μmol·L−1) induced NO production as reflected by a rise of the DAF‐FM DA fluorescence. Exposure to PA (200 μmol·L−1, 36 hr) significantly reduced NO production, which was improved in the ECs pretreatment with GW501516. However, either GSK0660 or MTX attenuated this effect of GW501516 on NO production. This result was further verified using an EPR spectroscopy (Figure 7a,b).
Figure 7.
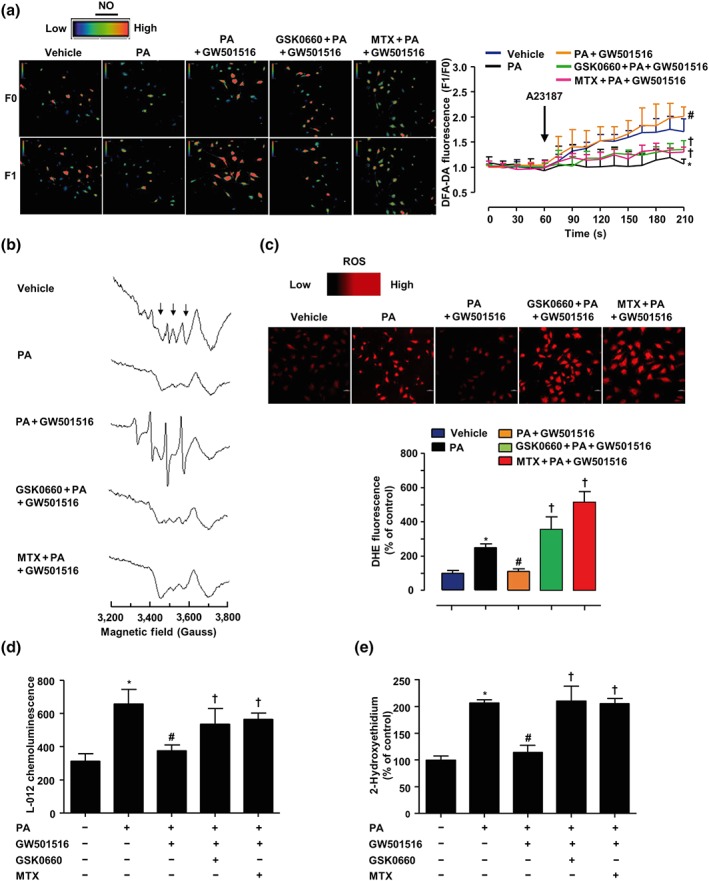
Inhibition of PPARδ/DHFR attenuated the effects of GW501516 on NO and ROS production. BAECs were pretreated with GSK0660 (2 μmol·L−1) or MTX (1 μmol·L−1) for 1 hr, before coincubation with GW501516 (1 μmol·L−1) for 12 hr, and then exposed to PA (200 μmol·L−1) for 12 hr. (a) Representative images of DAF‐FM DA fluorescence signal in ECs in response to A23187 (1 μmol·L−1) under a confocal microscope, analysed by comparing fluorescence intensity before (F0) and after (F1) the addition of A23187. Summarized results showing the levels of NO production in BAECs starting from the addition of A23187 for 210 s. Scale bar: 50 μm (n = 5). (b) NO production was measured with EPR using Fe(DETC)2. (c) Confocal microscopic detection of superoxide with DHE (n = 5). (d) Production of ROS was measured with L‐012 chemiluminescence (n = 5). (e) HPLC analysis of 2‐hydroxyethidium levels (n = 5). *P < .05 versus vehicle; # P < .05 versus PA; † P < .05 versus GW501516 + PA
In contrast, the levels of ROS generation (DHE staining) were increased by PA. This enhanced superoxide generation was abolished by GW501516 pretreatment. In the presence of GSK0660 or MTX, superoxide generation was no longer inhibited by GW501516 (Figure 7c). Since DHE staining is semiquantitative, these ROS results were further confirmed by using luminol derivative L‐012 and DHE‐HPLC profiles (Figure 7d,e). Taken together, these results suggested that the PPARδ/DHFR axis played an essential role in the beneficial effects of GW501516 on endothelial function.
In conclusion, the present study demonstrated that PA and DHA exerted opposite effects on DHFR expression while PPARδ up‐regulation of DHFR increased the generation of BH4 by salvage pathway, reversed eNOS to coupled states under PA stimulation, and decreased superoxide production. Then coupled eNOS increased NO generation and improved EDR (Figure 8).
Figure 8.
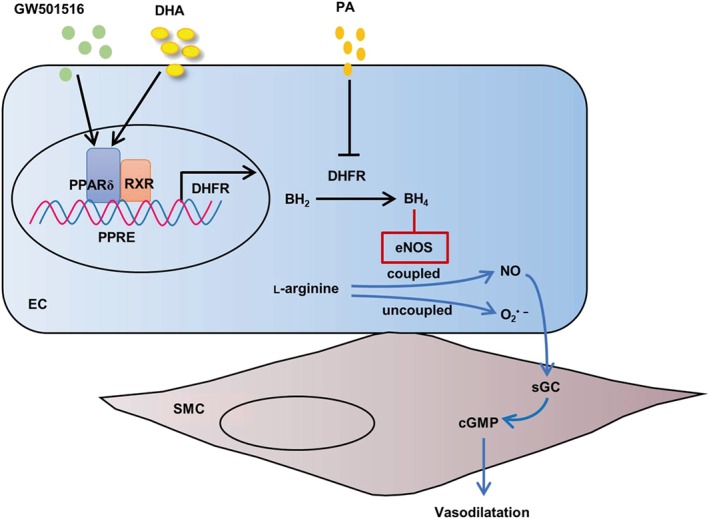
The proposed mechanisms by which PPARδ induces DHFR and protects endothelial function against saturated fatty acids
4. DISCUSSION
In the present study, we demonstrated for the first time that PPARδ activates the expression of the DHFR gene, which is responsible for the BH4 salvage, and maintains eNOS coupling. In addition, we demonstrated that the PPARδ–DHFR axis plays an important role in protecting endothelial‐dependent vasodilation impaired by saturated fatty acid.
Endothelial dysfunction is a hallmark of many metabolic vascular diseases (Vanhoutte, Shimokawa, Feletou, & Tang, 2017). NO is the main endothelium‐dependent vasodilator accounting for vascular relaxation, a common feature among many faces of endothelial functions (Magenta, Greco, Capogrossi, Gaetano, & Martelli, 2014). The pteridine cofactor BH4 is a critical determinant of eNOS activity and can be formed via either de novo biosynthesis or a salvage pathway. GTPCH1 catalyses the conversion of GTP to dihydroneopterin triphosphate. BH4 is generated by further steps catalysed by 6‐pyruvoyltetrahydropterin synthase and sepiapterin reductase (Thony, Auerbach, & Blau, 2000). GTPCH1 appears to be a rate‐limiting enzyme in BH4 biosynthesis (S. Cai et al., 2002). On the other hand, DHFR catalyses the regeneration of BH4 from its oxidized and inactive form, BH2 (Thony et al., 2000; Werner‐Felmayer, Golderer, & Werner, 2002). Sugiyama, Levy, and Michel (2009) showed that knockdown of either DHFR or GTPCH1 attenuated eNOS activity and NO production and these effects were recovered by the BH4 supplement. However, only knockdown of DHFR but not GTPCH1 increased ROS production, implying a relative importance of BH4 salvage/reduction over the de novo synthesis pathway (Sugiyama et al., 2009). It was also suggested that the depletion of BH4 was not sufficient to perturb NO signalling whereas the intracellular level of BH2 and the BH4:BH2 ratio together may determine the redox‐sensitive endothelial responses. In the present study, both in vitro and ex vivo evidence suggested that regulation of DHFR constitutes a novel mechanism for the endothelial protective actions of PPARδ.
It has been reported that DHFR could be regulated on both transcriptional and post‐translational levels. Transcription factors Sp1 and E2F positively modulate DHFR mRNA level (Slansky & Farnham, 1996). MTX is a tight‐binding inhibitor of DHFR (Abali et al., 2008). MicroRNAs such as mir‐24 bind to the 3′‐UTR of the human DHFR gene to interfere with DHFR expression (Mishra et al., 2007). At a post‐translational level, DHFR protein stability can also be modulated by NO via S‐nitrosylation (Z. Cai et al., 2015). Our present study established a novel transcriptional mechanism controlling the expression of DHFR. This conclusion is supported by both loss‐ and gain‐of‐function approaches. First, either PPARδ overexpression or a selective agonist GW501516 induced DHFR expression. Second, such inductive effect was attenuated by the antagonist GSK0660, confirming a PPARδ‐specific action. Third, the cognate binding motifs were identified within the 5′‐flanking region of the human DHFR gene. And the PPARδ‐binding capacities and the functionality of the promoter were corroborated by the results from promoter ChIP and reporter assays. Importantly, the transactivation of DHFR by PPARδ places its transcriptional regulation under a metabolic context linking this BH4 salvage enzyme gene with the lipid‐sensing transcription factors at the interface between endothelia and the fatty acid milieu in the circulation.
Epidemiological evidence suggested that dietary saturated fatty acids such as PA are closely associated with the increased risk for diabetes, hypertension, and coronary heart disease (Cascio, Schiera, & Di Liegro, 2012). In addition to many of its adverse effects on ECs, we further found that PA decreased DHFR expression in EC. The decreased DHFR might instigate eNOS uncoupling and lead to impaired vasorelaxation. Although the exact mechanism by which PA inhibits DHFR expression remains unexplored, it is postulated to be related to the PA‐evoked superoxide production in that several superoxide producing vascular stimuli including angiotensin II and oxidized LDL were reported to decrease DHFR level (Schramm, Matusik, Osmenda, & Guzik, 2012; Yu, Rajapakse, Montani, Yang, & Ming, 2014). Nevertheless, we found that PA‐triggered DHFR suppression as well as endothelial dysfunction could be prevented by GW501516 via a PPARδ activation with the use of a selective antagonist and endothelial‐specific knockout model. Besides directly binding to the promoter to transactivate DHFR gene, activation of PPARδ may also mitigate the suppressive effect of PA via induction of antioxidative target genes including SOD2, catalase, and thioredoxin as we previously reported (Fan et al., 2008). As an unsaturated fatty acid and a naturally occurring agonist for PPARδ, DHA appeared to offset the deleterious effect of PA on DHFR expression (data not shown). Such sharply contrasting effects of PA and DHA on DHFR might aid explaining their specific roles in modifying eNO bioavailability and vascular responses.
In conclusion, these results demonstrated that PPARδ ameliorates endothelial dysfunction via up‐regulation of DHFR, highlighting a new pharmacological action against metabolic vascular diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Z.Z., L.X., and N.W. designed the research. Z.Z., X.X., Y.T., and C.Y. performed the research. Q.Y. and J.L. analysed the data. Z.Z., L.X., and N.W. wrote the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Sequences of primers for qPCR
Table S2. Primers for amplifying the promoter segments
Table S3. Primers for CHIP assay
ACKNOWLEDGEMENTS
This study was supported by grants from the National Natural Science Foundation of China (81830015, 31430045 and 81770497).
Zhang Z, Xie X, Yao Q, et al. PPARδ agonist prevents endothelial dysfunction via induction of dihydrofolate reductase gene and activation of tetrahydrobiopterin salvage pathway. Br J Pharmacol. 2019;176:2945–2961. 10.1111/bph.14745
Contributor Information
Lei Xiao, Email: xiaolei0122@xjtu.edu.cn.
Nanping Wang, Email: npwang@bjmu.edu.cn.
REFERENCES
- Abali, E. E. , Skacel, N. E. , Celikkaya, H. , & Hsieh, Y. C. (2008). Regulation of human dihydrofolate reductase activity and expression. Vitamins and Hormones, 79, 267–292. 10.1016/S0083-6729(08)00409-3 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174(Suppl 1), S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak, Y. , Liao, D. , He, W. , Ong, E. S. , Nelson, M. C. , Olefsky, J. M. , … Evans, R. M. (2002). Effects of peroxisome proliferator‐activated receptor δ on placentation, adiposity, and colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America, 99(1), 303–308. 10.1073/pnas.012610299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall, J. K. , Douglas, G. , McNeill, E. , Channon, K. M. , & Crabtree, M. J. (2014). Tetrahydrobiopterin in cardiovascular health and disease. Antioxidants & Redox Signaling, 20(18), 3040–3077. 10.1089/ars.2013.5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez, P. N. , Bauer, P. M. , Yu, J. , Prendergast, J. S. , He, P. , & Sessa, W. C. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin‐1 using cell‐permeable peptides. Proceedings of the National Academy of Sciences of the United States of America, 102(3), 761–766. 10.1073/pnas.0407224102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant, O. , Foufelle, F. , Scotto, C. , Dauca, M. , & Wahli, W. (1996). Differential expression of peroxisome proliferator‐activated receptors (PPARs): Tissue distribution of PPAR‐alpha, ‐beta, and ‐gamma in the adult rat. Endocrinology, 137(1), 354–366. 10.1210/endo.137.1.8536636 [DOI] [PubMed] [Google Scholar]
- Cai, S. , Alp, N. J. , McDonald, D. , Smith, I. , Kay, J. , Canevari, L. , … Channon, K. M. (2002). GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: Effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovascular Research, 55(4), 838–849. 10.1016/S0008-6363(02)00460-1 [DOI] [PubMed] [Google Scholar]
- Cai, S. , Khoo, J. , & Channon, K. M. (2005). Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovascular Research, 65(4), 823–831. 10.1016/j.cardiores.2004.10.040 [DOI] [PubMed] [Google Scholar]
- Cai, Z. , Lu, Q. , Ding, Y. , Wang, Q. , Xiao, L. , Song, P. , & Zou, M. H. (2015). Endothelial nitric oxide synthase‐derived nitric oxide prevents dihydrofolate reductase degradation via promoting S‐nitrosylation. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(11), 2366–2373. 10.1161/ATVBAHA.115.305796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio, G. , Schiera, G. , & Di Liegro, I. (2012). Dietary fatty acids in metabolic syndrome, diabetes and cardiovascular diseases. Current Diabetes Reviews, 8(1), 2–17. 10.2174/157339912798829241 [DOI] [PubMed] [Google Scholar]
- Crabtree, M. J. , Brixey, R. , Batchelor, H. , Hale, A. B. , & Channon, K. M. (2013). Integrated redox sensor and effector functions for tetrahydrobiopterin‐ and glutathionylation‐dependent endothelial nitric‐oxide synthase uncoupling. The Journal of Biological Chemistry, 288(1), 561–569. 10.1074/jbc.M112.415992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree, M. J. , Tatham, A. L. , Hale, A. B. , Alp, N. J. , & Channon, K. M. (2009). Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric‐oxide synthase coupling: Relative importance of the de novo biopterin synthesis versus salvage pathways. The Journal of Biological Chemistry, 284(41), 28128–28136. 10.1074/jbc.M109.041483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172(14), 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X. , Okon, I. , Liu, Z. , Bedarida, T. , Wang, Q. , Ramprasath, T. , … Zou, M. H. (2017). Ablation of neuropilin 1 in myeloid cells exacerbates high‐fat diet‐induced insulin resistance through Nlrp3 inflammasome in vivo. Diabetes, 66(9), 2424–2435. 10.2337/db17-0132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenborg, E. , & Skogsberg, J. (2013). Peroxisome proliferator‐activated receptor delta and cardiovascular disease. Atherosclerosis, 231(1), 95–106. 10.1016/j.atherosclerosis.2013.08.027 [DOI] [PubMed] [Google Scholar]
- Fan, Y. , Wang, Y. , Tang, Z. , Zhang, H. , Qin, X. , Zhu, Y. , … Wang, N. (2008). Suppression of pro‐inflammatory adhesion molecules by PPAR‐δ in human vascular endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(2), 315–321. 10.1161/ATVBAHA.107.149815 [DOI] [PubMed] [Google Scholar]
- Fang, L. , Zhang, M. , Li, Y. , Liu, Y. , Cui, Q. , & Wang, N. (2016). PPARgene: A database of experimentally verified and computationally predicted PPAR target genes. PPAR Research, 2016, 6042162 10.1155/2016/6042162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton, D. , Ruan, L. , Sood, S. G. , Li, C. , Zhang, Q. , & Venema, R. C. (2008). Agonist‐stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of eNOS phosphorylation at Tyr83. Circulation Research, 102(4), 497–504. 10.1161/circresaha.107.162933 [DOI] [PubMed] [Google Scholar]
- Ghosh, A. , Gao, L. , Thakur, A. , Siu, P. M. , & Lai, C. W. K. (2017). Role of free fatty acids in endothelial dysfunction. Journal of Biomedical Science, 24(1), 50 10.1186/s12929-017-0357-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, T. , Smith, L. A. , Lu, T. , Joyner, M. J. , & Katusic, Z. S. (2011). Activation of peroxisome proliferator‐activated receptor‐δ enhances regenerative capacity of human endothelial progenitor cells by stimulating biosynthesis of tetrahydrobiopterin. Hypertension, 58(2), 287–294. 10.1161/hypertensionaha.111.172189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. , Zhang, Y. Y. , Chen, K. , Hatakeyama, K. , & Keaney, J. F. Jr. (2005). Cytokine‐stimulated GTP cyclohydrolase I expression in endothelial cells requires coordinated activation of nuclear factor‐κB and Stat1/Stat3. Circulation Research, 96(2), 164–171. 10.1161/01.RES.0000153669.24827.DF [DOI] [PubMed] [Google Scholar]
- Karp, N. A. , Meehan, T. F. , Morgan, H. , Mason, J. C. , Blake, A. , Kurbatova, N. , … Brown, S. D. M. (2015). Applying the ARRIVE guidelines to an in vivo database. PLoS Biology, 13, e1002151 10.1371/journal.pbio.1002151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietadisorn, R. , Juni, R. P. , & Moens, A. L. (2012). Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. American Journal of Physiology. Endocrinology and Metabolism, 302(5), E481–E495. 10.1152/ajpendo.00540.2011 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschyov, A. L. , Mollnau, H. , Oelze, M. , Meinertz, T. , Huang, Y. , Harrison, D. G. , & Munzel, T. (2000). Spin trapping of vascular nitric oxide using colloid Fe (II)‐diethyldithiocarbamate. Biochemical and Biophysical Research Communications, 275(2), 672–677. 10.1006/bbrc.2000.3361 [DOI] [PubMed] [Google Scholar]
- Leary, S. , & Golab, G. C. (2013). AVMA guidelines for the euthanasia of animals: 2013 edition. Schaumburg, IL: American Veterinary Medical Association. [Google Scholar]
- Li, H. , & Forstermann, U. (2013). Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Current Opinion in Pharmacology, 13(2), 161–167. 10.1016/j.coph.2013.01.006 [DOI] [PubMed] [Google Scholar]
- Li, H. , Witte, K. , August, M. , Brausch, I. , Godtel‐Armbrust, U. , Habermeier, A. , … Forstermann, U. (2006). Reversal of endothelial nitric oxide synthase uncoupling and up‐regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. Journal of the American College of Cardiology, 47(12), 2536–2544. 10.1016/j.jacc.2006.01.071 [DOI] [PubMed] [Google Scholar]
- Magenta, A. , Greco, S. , Capogrossi, M. C. , Gaetano, C. , & Martelli, F. (2014). Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. BioMed Research International, 2014, 193095 10.1155/2014/193095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga, T. , Kotamraju, S. , Kalivendi, S. V. , Dhanasekaran, A. , Joseph, J. , & Kalyanaraman, B. (2004). Ceramide‐induced intracellular oxidant formation, iron signaling, and apoptosis in endothelial cells: Protective role of endogenous nitric oxide. The Journal of Biological Chemistry, 279(27), 28614–28624. 10.1074/jbc.M400977200 [DOI] [PubMed] [Google Scholar]
- Mishra, P. J. , Humeniuk, R. , Mishra, P. J. , Longo‐Sorbello, G. S. , Banerjee, D. , & Bertino, J. R. (2007). A miR‐24 microRNA binding‐site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proceedings of the National Academy of Sciences of the United States of America, 104(33), 13513–13518. 10.1073/pnas.0706217104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, X. , Xie, X. , Fan, Y. , Tian, J. , Guan, Y. , Wang, X. , … Wang, N. (2008). Peroxisome proliferator‐activated receptor‐δ induces insulin‐induced gene‐1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology, 48(2), 432–441. 10.1002/hep.22334 [DOI] [PubMed] [Google Scholar]
- Quintela, A. M. , Jimenez, R. , Piqueras, L. , Gomez‐Guzman, M. , Haro, J. , Zarzuelo, M. J. , … Duarte, J. (2014). PPARβ activation restores the high glucose‐induced impairment of insulin signalling in endothelial cells. British Journal of Pharmacology, 171(12), 3089–3102. 10.1111/bph.12646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm, A. , Matusik, P. , Osmenda, G. , & Guzik, T. J. (2012). Targeting NADPH oxidases in vascular pharmacology. Vascular Pharmacology, 56(5–6), 216–231. 10.1016/j.vph.2012.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa, M. , & Fleming, I. (2016). The eNOS signalosome and its link to endothelial dysfunction. Pflügers Archiv, 468(7), 1125–1137. 10.1007/s00424-016-1839-0 [DOI] [PubMed] [Google Scholar]
- Slansky, J. E. , & Farnham, P. J. (1996). Transcriptional regulation of the dihydrofolate reductase gene. BioEssays, 18(1), 55–62. 10.1002/bies.950180111 [DOI] [PubMed] [Google Scholar]
- Sugiyama, T. , Levy, B. D. , & Michel, T. (2009). Tetrahydrobiopterin recycling, a key determinant of endothelial nitric‐oxide synthase‐dependent signaling pathways in cultured vascular endothelial cells. The Journal of Biological Chemistry, 284(19), 12691–12700. 10.1074/jbc.M809295200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thony, B. , Auerbach, G. , & Blau, N. (2000). Tetrahydrobiopterin biosynthesis, regeneration and functions. The Biochemical Journal, 347(Pt 1), 1–16. [PMC free article] [PubMed] [Google Scholar]
- Tian, X. Y. , Wong, W. T. , Wang, N. , Lu, Y. , Cheang, W. S. , Liu, J. , … Huang, Y. (2012). PPARdelta activation protects endothelial function in diabetic mice. Diabetes, 61(12), 3285–3293. 10.2337/db12-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte, P. M. , Shimokawa, H. , Feletou, M. , & Tang, E. H. (2017). Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiologica (Oxford, England), 219(1), 22–96. 10.1111/apha.12646 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Cheang, W. S. , Yang, H. , Xiao, L. , Lai, B. , Zhang, M. , … Wang, N. (2015). Nuciferine relaxes rat mesenteric arteries through endothelium‐dependent and ‐independent mechanisms. British Journal of Pharmacology, 172(23), 5609–5618. 10.1111/bph.13021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner‐Felmayer, G. , Golderer, G. , & Werner, E. R. (2002). Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Current Drug Metabolism, 3(2), 159–173. 10.2174/1389200024605073 [DOI] [PubMed] [Google Scholar]
- Widder, J. D. , Chen, W. , Li, L. , Dikalov, S. , Thony, B. , Hatakeyama, K. , & Harrison, D. G. (2007). Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circulation Research, 101(8), 830–838. 10.1161/CIRCRESAHA.107.153809 [DOI] [PubMed] [Google Scholar]
- Xie, X. , Zhang, Z. , Wang, X. , Luo, Z. , Lai, B. , Xiao, L. , & Wang, N. (2018). Stachydrine protects eNOS uncoupling and ameliorates endothelial dysfunction induced by homocysteine. Molecular Medicine, 24(1), 10 10.1186/s10020-018-0010-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Rajapakse, A. G. , Montani, J. P. , Yang, Z. , & Ming, X. F. (2014). p38 mitogen‐activated protein kinase is involved in arginase‐II‐mediated eNOS‐uncoupling in obesity. Cardiovascular Diabetology, 13, 113 10.1186/s12933-014-0113-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzuelo, M. J. , Gomez‐Guzman, M. , Jimenez, R. , Quintela, A. M. , Romero, M. , Sanchez, M. , … Duarte, J. (2013). Effects of peroxisome proliferator‐activated receptor‐β activation in endothelin‐dependent hypertension. Cardiovascular Research, 99(4), 622–631. 10.1093/cvr/cvt152 [DOI] [PubMed] [Google Scholar]
- Zarzuelo, M. J. , Jimenez, R. , Galindo, P. , Sanchez, M. , Nieto, A. , Romero, M. , … Duarte, J. (2011). Antihypertensive effects of peroxisome proliferator‐activated receptor‐β activation in spontaneously hypertensive rats. Hypertension, 58(4), 733–743. 10.1161/HYPERTENSIONAHA.111.174490 [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Vanhoutte, P. M. , & Leung, S. W. (2015). Vascular nitric oxide: Beyond eNOS. Journal of Pharmacological Sciences, 129(2), 83–94. 10.1016/j.jphs.2015.09.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of primers for qPCR
Table S2. Primers for amplifying the promoter segments
Table S3. Primers for CHIP assay