

SUMMARY
Lettuce downy mildew (Bremia lactucae) is a rapidly adapting oomycete pathogen affecting commercial lettuce cultivation. Oomycetes are known to use a diverse arsenal of secreted proteins (effectors) to manipulate their hosts. Two classes of effector are known to be translocated by the host: the RXLRs and Crinklers. To gain insight into the repertoire of effectors used by B. lactucae to manipulate its host, we performed massively parallel sequencing of cDNA derived from B. lactucae spores and infected lettuce (Lactuca sativa) seedlings. From over 2.3 million 454 GS FLX reads, 59 618 contigs were assembled representing both plant and pathogen transcripts. Of these, 19 663 contigs were determined to be of B. lactucae origin as they matched pathogen genome sequences (SOLiD) that were obtained from >270 million reads of spore‐derived genomic DNA. After correction of cDNA sequencing errors with SOLiD data, translation into protein models and filtering, 16 372 protein models remained, 1023 of which were predicted to be secreted. This secretome included elicitins, necrosis and ethylene‐inducing peptide 1‐like proteins, glucanase inhibitors and lectins, and was enriched in cysteine‐rich proteins. Candidate host‐translocated effectors included 78 protein models with RXLR effector features. In addition, we found indications for an unknown number of Crinkler‐like sequences. Similarity clustering of secreted proteins revealed additional effector candidates. We provide a first look at the transcriptome of B. lactucae and its encoded effector arsenal.
INTRODUCTION
Oomycete plant pathogens cause devastating diseases on a wide variety of crops. These organisms resemble fungi, but are more closely related to brown algae. Well‐known oomycete pathogens are the obligate biotrophic downy mildews and the hemibiotrophic Phytophthora species, including P. infestans, the causative agent of the Irish potato famine, and P. ramorum, which causes sudden oak death. The downy mildews have a narrow host range, for instance Hyaloperonospora arabidopsidis grows only on living Arabidopsis thaliana plants, Plasmopara viticola is an important grape pathogen and Bremia lactucae is the most important pathogen of lettuce (Lactuca sativa). The control of B. lactucae is an increasingly difficult task as fungicides have been phased out because of environmental concerns, and fungicide resistance is becoming more widespread (Brown et al., 2004). Genetically controlled resistance to B. lactucae is present in most commercial lettuce varieties, but is quickly overcome by new rapidly evolving B. lactucae races.
The B. lactucae life cycle starts from a spore landing on the plant's epidermis, followed by penetration, hyphal colonization of host tissue and sporulation, leading to the release of large numbers of spores. At all stages, B. lactucae is in contact with its host, so that it can interfere with defence and manipulate host processes to obtain nutrients. In contrast with many oomycetes, B. lactucae infection usually starts with the direct germination of asexual spores, without a zoospore intermediate stage. B. lactucae also does not rely on entry via stomata, but, instead, usually penetrates directly through the cuticula into an epidermal cell. A primary vesicle and, later, a secondary vesicle are then formed in the epidermal cell, after which hyphae grow in the intercellular space of the mesophyll tissue. Haustoria are formed in most mesophyll and epidermal cells encountered by the hyphae (reviewed in Lebeda et al., 2008). The B. lactucae–lettuce interaction is a classic example of a gene‐for‐gene interaction, in which single dominant avirulence genes in B. lactucae are genetically recognized by dominant resistance genes in lettuce (reviewed by Michelmore and Wong, 2008). Knowledge on the molecular biology and basis of B. lactucae pathogenicity, however, is mostly lacking.
Studies of oomycete–plant interactions at the molecular level are now revealing more and more details on the molecular toolbox used by these remarkable pathogens. The genomes of the sequenced oomycete species Albugo candida (Kemen et al., 2011), A. laibachii (Links et al., 2011), H. arabidopsidis (Baxter et al., 2010), P. infestans (Haas et al., 2009), P. sojae (Tyler et al., 2006), P. ramorum (Tyler et al., 2006) Pseudoperonospora cubensis (Tian et al., 2011) and Pythium ultimum (Lévesque et al., 2010) represent a treasure trove of information on the effector repertoires secreted by these pathogens to manipulate their hosts. Various types of effectors are predicted to act in the apoplast and, in addition, two classes of oomycete effector translocate into the host cell (reviewed in Stassen and Van den Ackerveken, 2011). Effectors from one of the host‐translocated classes are referred to as RXLR effectors, after the RXLR amino acid motif contained by the first characterized effectors of this class (Rehmany et al., 2005), although variations in this motif also permit host translocation (Kale et al., 2010). Examples of variants of RXLR motifs include the QXLR motif found in host‐translocating effectors of the oomycete pathogen of cucumber, Ps. cubensis (Tian et al., 2011), and the GXLR motif found in the P. infestans effector SNE1 (Kelley et al., 2010). Furthermore, ATR5 of H. arabidopsidis is recognized inside host cells and has homology to the RXLR motif‐containing effectors, but does not contain an RXLR motif (Bailey et al., 2011). A second class of host‐translocated effectors are the Crinklers, which contain two conserved amino acid motifs preceding a modular C‐terminal section. In a number of Crinklers, the domains that form the modules in this C‐terminal section induce cell death when expressed in Nicotiana benthamiana (Haas et al., 2009). Two Crinklers from P. sojae are even thought to be indispensable for successful infection of soybean (Liu et al., 2011). Although the effector repertoire is generally highly divergent between species, features such as motifs associated with host‐translocated effectors allow for the identification of potential effectors encoded by gene models or mRNA sequences.
Access to the catalogue of the effectors of a species allows for the cloning and investigation of individual effector genes. This has led to the identification of various activities of effectors on host processes. Examples include the disruption of host cell wall–membrane adhesion (Bouwmeester et al., 2011), suppression of defensive protease activity (Kaschani et al., 2010; Song et al., 2009; Tian et al., 2007) and suppression of effector‐triggered immunity (e.g. Kelley et al., 2010; Liu et al., 2011). In addition, the first cytoplasmic target and mode of action of a host‐translocated effector has been identified; host E3 ligase CMPG1 is stabilized by P. infestans effector AVR3a (Bos et al., 2010), revealing more details about the mechanisms by which oomycetes establish a successful infection of their hosts.
In this article, we investigate the potential effector arsenal of B. lactucae by cDNA sequencing and de novo assembly of transcript sequences. Transcripts obtained from B. lactucae‐infected lettuce were compared with short reads of B. lactucae genomic DNA (gDNA) to select for B. lactucae transcripts. From the predicted B. lactucae secretome, we identified homologues of known effector families as well as new groups of potential effector proteins.
RESULTS
Bremia lactucae transcriptome sequencing
Two sources of RNA were used for B. lactucae transcriptome sequencing: RNA isolated from asexual B. lactucae conidiospores and RNA isolated from B. lactucae‐infected lettuce leaves. Many effector genes are known to be differentially expressed during different stages of infection, with transcripts of early‐stage effectors being present in spores, and transcripts of later stage (hyphal growth) effectors being expressed in B. lactucae growing in planta. To increase the number of different infection stages and, as a result, transcript diversity, lettuce seedlings were inoculated twice with a 3‐day interval (see time line in Fig. 1A). Lettuce seedlings were inoculated with conidiospores (Fig. 1B) that germinate on the leaf surface, penetrate through epidermal cells and then continue to grow intercellular hyphae in the mesophyll, forming haustoria in plant cells (Fig. 1C). Plants were kept under low relative humidity and, as a result, when material was harvested for RNA isolation (day 7), no B. lactucae sporulation was observed, although abundant growth of intercellular hyphae and the formation of haustoria were visible (Fig. 1D).
Figure 1.
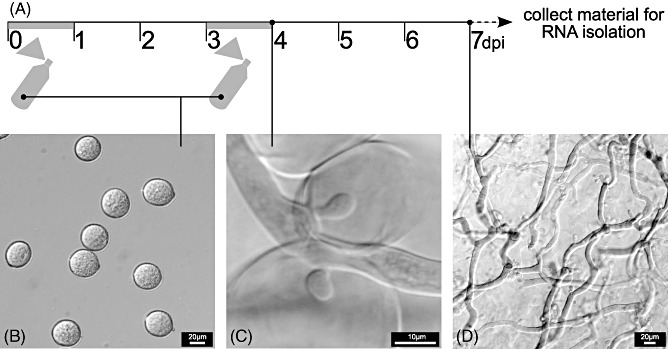
Preparation of Bremia lactucae‐infected lettuce leaves for transcriptome analysis. (A) Time line of sampling. Light grey bars represent periods in which infected lettuce was kept at high humidity. (B) Spores were spray inoculated at time point 0 and at a second time point at 3 days post‐inoculation (dpi). (C) Bremia lactucae establishes an infection and forms hyphae between plant cells, creating haustoria in adjacent cells. (D) Leaf material heavily colonized with B. lactucae hyphae and haustoria is harvested for RNA isolation at 7 dpi.
Transcript sequencing was performed using 454 sequencing technology on cDNA of spores and B. lactucae‐infected cotyledons generated by either oligo dT priming and 5′ enrichment, or by random priming and normalization. An overview of the number of obtained sequences per approach is given in Table 1. Normalization selectively removes sequences present in relatively high abundance, shifting the odds of a randomly sequenced transcript representing a rare transcript towards that of sampling of a high‐copy transcript. It is important to sample transcripts that are present in low abundance, as effectors may be produced very locally and can therefore be present at low abundance in the overall sample. We generated random‐primed cDNA to more evenly spread coverage over the entire length of the transcripts. Pools 3 and 4 (Table 1) are random primed and normalized, and were used to generate the majority of reads (>90%). We also sequenced the 450–650‐bp fragments from both the 3′ and 5′ ends, as the expected ∼200‐bp read size of the 454 sequencing technology (GS FLX) used would otherwise probably miss the 3′ ends of transcripts. In addition, we sequenced the 5′‐enriched non‐normalized pools to provide stronger coverage of the 5′ ends of transcripts that are most important when looking for N‐terminal signal peptides of secreted proteins.
Table 1.
Materials and method of preparation of cDNA for transcriptome sequencing. The contribution of each pool to the number of reads and number of bases is indicated.
| Pool | Material | Strategy | 5′ reads* | Total 5′ nucleotides | 3′ reads | Total 3′ nucleotides |
|---|---|---|---|---|---|---|
| 1 | Spores | 5′ sequencing | 86 352 | 22 212 388 | – | – |
| 2 | Interaction | 5′ sequencing | 115 413 | 30 335 206 | – | – |
| 3 | Interaction | Random primed + normalized | 701 499 | 182 312 369 | – | – |
| 4 | Mixed | Random primed + normalized | 637 709 | 165 624 293 | – | – |
| 3+4† | Mixed | Random primed + normalized | – | – | 1 013 411 | 262 824 074 |
The DNA code in the adapter that links a read to a pool could not be resolved for 19 422 reads that are not included in this table.
The DNA code was not sequenced in 3′ reads.
A total of 523 477 146 bases from 2 349 338 cDNA reads (91.28% of all available reads) was assembled into 59 618 contigs. The total length of contig consensus sequences was 36 217 753 bases and the average coverage of these sequences was 14.6‐fold. The average length of contig consensus sequences was 607.5 bases. Further statistics relating to the assembled sequences can be found in Table 2.
Table 2.
Assembly process statistics regarding the number of input reads, reads after filtering and remaining singletons. The number and total length of contigs are given for all and large (>500 nucleotide) contigs.
| Transcriptome assembly | |
|---|---|
| Reads | |
| Total number of reads | 2 573 806 |
| Number of reads after filtering | 2 349 338 (91.28%) |
| Number of bases in reads after filtering | 523 477 146 (90.65%) |
| Singletons remaining after assembly | 82 194 (3.19%) |
| Contigs | |
| Contigs | 59 618 |
| Bases in consensus sequences | 36 217 753 |
| Large contigs (>500 bp) | 26 358 (44.21%) |
| Bases in large contigs | 26 543 633 (73.29%) |
| Average size of large contigs | 1 007 |
| N50 size of large contigs | 1 087 |
The assembled contigs are derived from both B. lactucae and lettuce, as most transcript sequences were derived from RNA of infected plants. To determine which transcripts are derived from B. lactucae and to correct 454 sequencing errors, we generated additional sequence information by SOLiD sequencing of B. lactucae gDNA. Conidiospores were used for gDNA isolation as they are the only life stage of B. lactucae that can be well separated from its lettuce host. We sequenced gDNA rather than mRNA from spores, as the transcript pool in spores most probably lacks many transcripts that are involved in the interaction with the host plant. The B. lactucae gDNA SOLiD sequencing data obtained consist of 173 428 926 reads of ∼50 bp.
By mapping B. lactucae gDNA reads to the assembled 454 reads, contigs representing B. lactucae transcripts can be differentiated from lettuce‐derived or poorly assembled sequences. Transcripts with high average gDNA read coverage probably represent B. lactucae transcriptome sequences, whereas low or no coverage indicates a lettuce or contaminant origin. A total of 20 925 of 59 618 contigs, or 35.2%, had at least one SOLiD gDNA read mapped to the sequence. The average gDNA read coverage per contig peaked at 37–40‐fold coverage, as shown in Fig. 2. The shoulder seen at approximately half this coverage (18–20‐fold coverage) in Fig. 2 may represent alleles that have been assembled as separate transcripts. We defined the B. lactucae transcriptome set as the 19 663 contigs with a 10‐fold or higher average SOLiD‐sequenced gDNA read coverage. The vast majority of excluded sequences had no gDNA read coverage and, of those that did, most had an average gDNA read coverage of less than one‐fold (Fig. 2). The average length of the sequences in our B. lactucae transcriptome set of 19 663 contigs was 736.7 bp, up from 607.5 bp in the overall set, probably as a result of the exclusion of short contaminant sequences. Of all the assembled spore‐derived reads (pool 1), over 93% were found in consensus sequences that were classified as B. lactucae. For the non‐normalized, interaction‐derived reads (pool 2), we found 20% of reads in B. lactucae contigs, whereas, from normalized pools (pools 3 and 4), 47% matched to B. lactucae. This suggests that the normalization procedure increased the proportion of B. lactucae transcript sequences in the interaction cDNA. To assess whether lettuce transcripts were successfully excluded, we compared our assembled sequences with a collection of 226 050 expressed sequence tags (ESTs) from Lactuca species (The Compositae Genome Project; http://cgpdb.ucdavis.edu). For the sequences excluded from the B. lactucae transcriptome, we found matching ESTs for ∼70% of our sequences. More importantly, we found only 151 sequences (<1%) in our B. lactucae set with Lactuca EST matches. However, comparison of these transcripts with the National Center for Biotechnology Information (NCBI) nonredundant (NR) protein sequence collection revealed that 89 of these most closely matched P. infestans proteins, suggesting that they are of oomycete origin, and are therefore correctly assigned to B. lactucae. We conclude that the B. lactucae set contains few contaminant sequences, as only a very minor number of possible lettuce sequences were detected.
Figure 2.
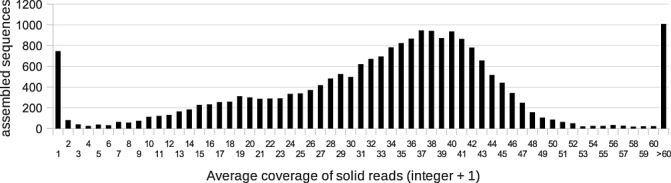
Number of consensus sequences with indicated average genomic DNA (gDNA) read coverage. Read coverages are assigned into groups by taking the integer + 1 of the actual average read coverage.
We also used data on the differences between the 454 transcripts and the mapped SOLiD reads to mitigate problems arising from insertions and deletions of small numbers of bases. We corrected 3048 of the 454 transcripts with SOLiD data. To assess whether sequences were improved, we compared nucleotide sequences before and after correction with the protein sequences in the NCBI NR protein database by blastx. Errors in the sequence that lead to frame shifts yield two short hits with the same database sequence, rather than one single hit. We found 619 cases in which the corrected sequences had fewer hits to the same database sequences than the uncorrected version, and 152 cases in which there were more hits. This indicates that the incorporation of the information from the SOLiD mapping improved the assembly quality, although, project‐wide, most contigs remained unchanged.
Translation of the 19 663 B. lactucae sequences resulted in 16 372 protein sequences of at least 20 amino acids and starting with a methionine. The majority of coding sequences ended with a stop codon, whereas about one‐third lacked a stop codon and apparently lacked 3′ sequences. To obtain an indication of the coverage of the transcriptome, we compared the protein models with a set of 248 hidden Markov models (HMMs) of core eukaryotic genes used for genome completeness surveys (http://korflab.ucdavis.edu/Datasets/genome_completeness/). The HMMs are based on extremely highly conserved proteins thought to be present in all eukaryotes in small numbers of paralogues (Parra et al., 2007). For 79% of all HMM models of conserved eukaryotic genes, matches were found to our B. lactucae transcript sequences. The same approach, using the P. infestans and H. arabidopsidis protein models based on their complete genomes, yielded 95% and 96% coverage, respectively. The B. lactucae transcripts represent the genes expressed during infection, and may therefore not represent all genes that are present in the genome. With a match of 79%, the B. lactucae transcriptome is well represented.
The protein sequences corresponding to a set of 119 core orthologous genes were used to determine the place of B. lactucae in oomycete phylogeny by comparison with six pathogenic oomycete species and four nonpathogenic stramenochromes for which predicted proteomes are available. As depicted in Fig. 3, the stramenochromes branch off first, after which Saprolegnia parasitica and then Py. ultimum branch off in the evolutionary tree. We then find B. lactucae, followed by H. arabidopsidis and, finally, three Phytophthora species. We can therefore compare B. lactucae with the earlier branching necrotrophic oomycete Py. ultimum and the later branching biotrophic downy mildew H. arabidopsidis and hemibiotrophic Phytophthora species.
Figure 3.
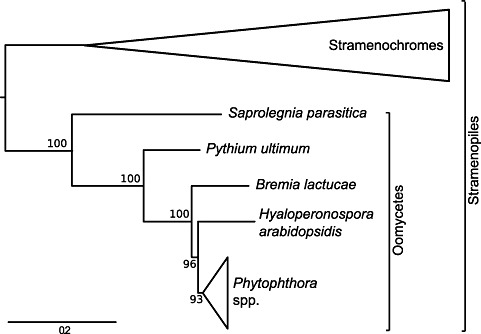
Phylogeny of Bremia lactucae and related stramenochromes determined from a multiple sequence alignment of 119 concatenated orthologous sequences. The scale bar represents 0.2 substitutions per amino acid. Numbers represent the bootstrap support for the given branch.
To define the B. lactucae secretome, we determined which protein models contained a signal peptide but no transmembrane helix predictions. Of the 16 372 models considered, 1023 were predicted to be secreted. The size of the secretome is close to that predicted for the downy mildew H. arabidopsidis (1016 predicted secreted proteins) and Py. ultimum (1123 secreted proteins), as shown in Fig. 4. The 1023 candidate secreted B. lactucae proteins were further analysed for protein domains and other features related to effectors.
Figure 4.
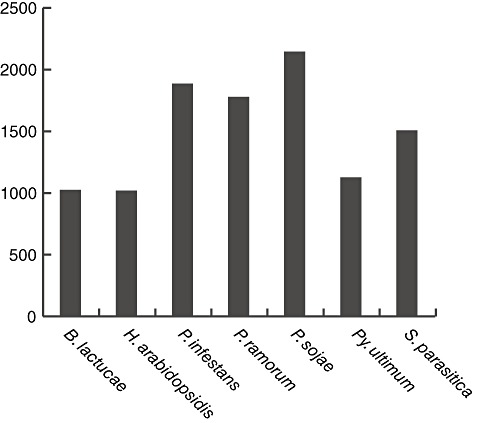
The size of the secretome encoded in the genomes of different oomycetes, as determined from the presence of signal peptides and the absence of transmembrane helices in the protein models of each species.
Domain composition of the B. lactucae secretome
An initial approach to catalogue the secretome was to determine which Pfam domains, families and repeats were present. We obtained a total of 348 Pfam annotations for 295 of the 1023 secretome protein models. Prevalent domains in the B. lactucae secretome (Table 3) included various domains that may be linked to pathogenicity. The most prevalent domain in the B. lactucae secretome was the elicitin domain. Elicitins are small highly conserved secreted proteins characterized by a domain that contains six cysteine residues important for structure. Of the 12 B. lactucae protein models with elicitin domain matches, nine contained at least six cysteines in the predicted mature peptide. Phytophthora elicitins trigger a hypersensitive response in host plants, an effect that is linked to sterol binding and transporting activity for which a surface‐exposed polar residue is important (Hirasawa et al., 2004; 1997, 1998; Pleškováet al., 2011; Ricci et al., 1989; Vauthrin et al., 1999). Elicitins may play a role in the uptake of sterols by oomycetes that do not synthesize their own, as the presence of elicitins appears to be correlated with the loss of the sterol biosynthetic pathway (Gaulin et al., 2010; Jiang et al., 2006; Vauthrin et al., 1999). Elicitins are found in higher numbers in the hemibiotroph P. infestans and the necrotroph Py. ultimum than in H. arabidopsidis and B. lactucae (Table 3), suggesting that they may be selected against in biotrophs that adapt a more stealthy infection strategy. All of the B. lactucae elicitins are α‐elicitins (Nespoulous et al., 1992), which generally provoke a less marked host response than do β‐elicitins.
Table 3.
Number of protein models in Bremia lactucae, Phytophthora infestans, Hyaloperonospora arabidopsidis and Pythium ultimum that are predicted to contain the domains that are most prevalent in the B. lactucae secretome.
| Pfam domain | B. lactucae | H. arabidopsidis | P. infestans | Py. ultimum |
|---|---|---|---|---|
| Elicitin | 12 | 9 | 28 | 37 |
| Trypsin | 11 | 4 | 13 | 14 |
| Jacalin | 7 | 4 | 4 | 0 |
| DnaJ | 6 | 3 | 5 | 5 |
| Cysteine‐rich secretory protein family | 6 | 7 | 14 | 9 |
| Ricin‐B‐lectin | 4 | 2 | 1 | 0 |
Trypsin domains, found in peptidases, are present in B. lactucae in numbers similar to those in P. infestans and Py. ultimum, whereas fewer are found in H. arabidopsidis. The role of oomycete proteases in pathogenesis has not been determined, with only a single report of a P. infestans protease with an intact catalytic triad that is expressed during infection (Raffaele et al., 2010). More common are catalytically inactive trypsin‐like proteins that act as glucanase inhibitors (Rose et al., 2002). The B. lactucae proteins with trypsin matches do not have intact catalytic residues in the active site and could therefore function as glucanase inhibitors. Host glucanases can damage the oomycete cell wall, impeding oomycete growth and releasing glucan‐oligosaccharide elictors (van Loon et al., 2006), and are therefore a potentially useful target for inhibition by an invading oomycete.
Jacalin domains and Ricin‐B‐lectin domains, both found in lectins, are present in larger numbers than in the other oomycetes. A Jacalin domain‐matching oomycete protein has been proposed to act in cell wall degradation (Raffaele et al., 2010), and roles in host attachment have been suggested on the basis of homology to lectins (Ottmann et al., 2009) and lectin‐like activities (Gaulin et al., 2002). Host lectins, however, act in defence and may recognize oomycete‐derived carbohydrates or glycosylation features. Pathogen lectins could therefore also be instrumental in masking these signals, as has been demonstrated for the ECP6 effector of the plant‐pathogenic fungus Cladosporium fulvum, which masks chitin oligosaccharides that normally trigger plant immune responses (de Jonge et al., 2010).
Cysteine‐rich secreted proteins and proteins mediating disulphide bond formation may play an important role in dealing with the protease‐rich conditions in the extracellular environment. Cysteine residues can form disulphide bridges that may contribute to protein stability and protect against proteases. In addition, DnaJ proteins may act as molecular chaperones to further stabilize apoplastic proteins. Many protein families involved in pathogenicity, found in other oomycetes, are small cysteine‐rich proteins. Examples include the aforementioned elicitins, Phytophthora PcFs (Bos et al., 2003; Liu et al., 2005; Orsomando et al., 2001) and H. arabidopsidis PPATs (Bittner‐Eddy et al., 2003) and HaCRs (Cabral et al., 2011). Although there are six proteins matching the Pfam cysteine‐rich secretory domain model, these protein models contain few cysteine residues. Nonetheless, 135 cysteine‐rich proteins (>5% cysteine) are found in the B. lactucae secretome. The majority of these are short (<100 amino acid) peptide predictions and not all predictions include a stop codon. Of all proteins in the secretome, 13.2% are cysteine‐rich, compared with 8.4% of nonsecreted proteins. There is high diversity in the pattern of spacing between cysteine residues in the cysteine‐rich proteins, and no groups of proteins with similar cysteine spacing could be defined.
In terms of protein families in Pfam that have significant hits in the B. lactucae secretome, the NLP [necrosis and ethylene‐inducing peptide 1 (NEP1)‐like protein] family is most prevalent, with eight members. Originally thought to act as cytolytic toxins, the NLPs are found in many oomycetes. Structurally, they were shown to be related to actinoporins, but their resemblance to lectins may indicate an alternative function in attachment to the host (Ottmann et al., 2009). Although the P. sojae NLP PsojNIP and P. infestans NLP PiNPP1.1 induce cell death when expressed in tobacco (Kanneganti et al., 2006; Qutob et al., 2002), the NLPs of the obligate biotroph H. arabidopsidis do not induce necrosis (Baxter et al., 2010; Cabral et al., 2012). The H. arabidopsidis NLPs are predominantly found in a species‐specific subclade within the NLPs. NLP sequences of B. lactucae are more widely distributed in the oomycete NLP gene phylogeny (Fig. S1, see Supporting Information). A cell death‐inducing activity of the B. lactucae NLPs is, however, unlikely as not all residues that are critical for the induction of cell death by NLPs (Ottmann et al., 2009) are conserved in B. lactucae NLPs.
Many contigs, corresponding to protein models with different domains, show differential abundance between B. lactucae spores and infected lettuce leaves. This was deduced by estimating the relative abundance levels from the number of transcript sequence reads from the non‐normalized 5′ cDNA sequencing pools of spore and interaction stages (Table S1, see Supporting Information). It is striking to see that many transcripts encoding secreted proteins are already abundant in spores, e.g. transcripts for several elicitins, trypsins and Jacalin domain proteins. Many other transcripts are more abundant in infected lettuce plants, e.g. an elicitin and several cysteine‐rich proteins. For none of the NLP contigs was a large number of reads or differential abundance observed, suggesting that they are not highly expressed at the B. lactucae stages analysed.
Host‐translocated effectors
To find potential host‐translocated RXLR effectors, a search for the amino acid motif RXLR was performed on the predicted B. lactucae secretome. This search identified 43 potential RXLR effectors, counting only hits with the RXLR located between positions 30 and 60 in proteins of at least 65 amino acids. As variants of the RXLR motif also allow host entry, we expanded our search to include protein models that are similar to known RXLRs. We therefore performed a blast search with all H. arabidopsidis (Baxter et al., 2010) and Phytophthora spp. (Haas et al., 2009) RXLRs against the B. lactucae secretome. To rule out matches based on homology to conserved signal peptide sequences, they were removed from the B. lactucae proteins. In this search, 38 matches were found in the B. lactucae secretome, irrespective of the presence or absence of an RXLR or RXLR‐like motif. In a third approach to find RXLR effectors, we selected the RXLR motif and 20 residues on either side in the sequences that were identified by the motif search, and used these as input for a jackhmmer search. jackhmmer is an iterative search that uses matches to an initial input sequence to construct an alignment profile which is employed to search for additional matches in the target database and to further refine the profile. Eight new candidates were identified in a set of 53 that included 43 input sequences and two sequences that were found by blast. Eleven proteins were predicted by all three methods (Fig. 5). The combined set consists of 78 unique potential RXLR effectors, the details of which are provided in Table 4. We also found protein models by blast comparison that did not have an RXLR motif or variant thereof, but did have an EER motif, albeit slightly more N‐terminal of the location at which the RXLR motif would be expected. For 11 B. lactucae contigs encoding RXLR and RXLR‐like proteins, we observed clear differential expression based on transcript reads between spores and in planta stages (Table S1). Most of the B. lactucae RXLR and RXLR‐like effectors were not found in the genomes of other downy mildews. Bidirectional blast matches (E‐value < 1e‐3 both ways) were found for only seven B. lactucae versus eight H. arabidopsidis RXLRs, and none for Ps. cubensis. In addition, between Ps. cubensis and H. arabidopsidis only three RXLR matches were found.
Figure 5.
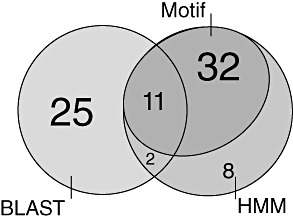
Venn diagram depicting the overlap of the three methods used for the prediction of RXLR effectors. HMM, hidden Markov model.
Table 4.
RXLR and RXLR‐like motifs found in Bremia lactucae secretome protein models identified as potential RXLR effectors based on a motif search, blast comparison with RXLRs of other oomycetes or a jackhmmer search with B. lactucae RXLR motifs and surrounding amino acids as input. All sequence coordinates are given in amino acids. Evidence codes are built up of M (motif search), B (blast comparison) and H (jackhmmer search).
| Contig ID | RXLR start | RXLR | EER start | EER(like) motif* | Stop codon | Evidence† |
|---|---|---|---|---|---|---|
| 16131 | 44 | RRLR | 55 | EER | 363 | MBH |
| 16134 | 41 | RRLR | 50 | EER | 454 | MBH |
| 23155 | 49 | RLLR | 61 | EQEER | – | MBH |
| 27267 | 55 | RALR | 68 | EER | – | MBH |
| 35642 | 42 | RRLR | 60 | EDR | – | MBH |
| 40355 | 50 | RRLR | 64 | DER | 270 | MBH |
| 42290 | 45 | RGLR | 200 | NEK | 333 | MBH |
| 43449 | 35 | RMLR | – | – | MBH | |
| 45396 | 53 | RLLR | – | 130 | MBH | |
| 49490 | 44 | RRLR | 57 | DER | – | MBH |
| 19377 | 49 | RRLR | – | 269 | MB | |
| 22293 | 45 | RFLR | 65 | DEK | – | MB |
| 07991 | 43 | RQLR | 78 | EER | 161 | MH |
| 08247 | 52 | RSLR | – | 103 | MH | |
| 08248 | 52 | RSLR | – | 146 | MH | |
| 08983 | 49 | RRLR | – | – | MH | |
| 16394 | 46 | RRLR | 59 | QNDER | – | MH |
| 18684 | 50 | RRLR | – | 282 | MH | |
| 18840 | 49 | RPLR | 61 | NQEER | – | MH |
| 20700 | 56 | RELR | – | 252 | MH | |
| 21364 | 47 | RALR | 58 | EDK | – | MH |
| 23333 | 35 | RFLR | – | – | MH | |
| 24965 | 57 | RSLR | 62 | DENR | 107 | MH |
| 29191 | 46 | RKLR | – | 75 | MH | |
| 31910 | 45 | RLLR | 54 | DNNEER | 160 | MH |
| 32917‐1 | 47 | RSLR | 60 | NDER | – | MH |
| 33962 | 39 | RILR | – | 74 | MH | |
| 36117 | 38 | RDLR | – | 424 | MH | |
| 36219 | 45 | RHLR | 60 | ENR | 201 | MH |
| 38529 | 40 | RQLR | 91 | EDK | – | MH |
| 39974 | 41 | RQLR | 50 | DER | – | MH |
| 40514 | 44 | RLLR | 56 | EER | 126 | MH |
| 41799 | 42 | RALR | – | 90 | MH | |
| 41935 | 44 | RLLR | 53 | DDNDER | – | MH |
| 43687 | 55 | RSLR | – | 91 | MH | |
| 43968 | 33 | RGLR | – | 185 | MH | |
| 45726 | 42 | RLLR | – | – | MH | |
| 46714 | 45 | RSLR | 82 | NQR | – | MH |
| 48006 | 46 | RALR | 55 | NEDR | 92 | MH |
| 48013 | 55 | RALR | – | 82 | MH | |
| 50216 | 47 | RSLR | 60 | DEER | 102 | MH |
| 53213 | 43 | RGLR | 61 | EER | – | MH |
| 53460 | 47 | RSLR | 60 | DEER | – | MH |
| 22259 | – | 52 | EER | 375 | BH | |
| 29756 | 36 | RSLLQ | 51 | DER | – | BH |
| 56090 | 42 | RSLQ | 55 | EER | – | BH |
| 07727 | 45 | RRLK | 63 | EER | – | B |
| 07952 | – | 65 | NEER | 304 | B | |
| 08155 | 55 | RRLQ | 109 | EEK | 321 | B |
| 09272 | – | 58 | DEER | 346 | B | |
| 09828 | – | 48 | EEK | 652 | B | |
| 11813 | – | 49 | EER | 629 | B | |
| 12298 | 63 | RLKREQ | – | 612 | B | |
| 13165 | 114 | RSLR | 360 | DEK | – | B |
| 14316 | – | 50 | EER | 365 | B | |
| 16501 | 481 | RKLR | – | 485 | B | |
| 17177 | – | – | – | B | ||
| 18477 | 42 | QNLR | – | – | B | |
| 19297 | 57 | KVLR | – | 253 | B | |
| 24419 | – | 54 | DEER | 434 | B | |
| 24503 | – | – | 520 | B | ||
| 28334 | 43 | EKLR | 53 | ENR | 479 | B |
| 29771 | – | – | 263 | B | ||
| 35494 | – | – | – | B | ||
| 35745 | 169 | RSLR | 72 | EER | – | B |
| 48427 | 62 | RLLD | – | – | B | |
| 51693 | – | 49 | EER | – | B | |
| 51725 | 39 | PSLR | – | – | B | |
| 56746 | – | 49 | EER | – | B | |
| 23857 | 42 | GKLR | 55 | DER | 243 | H |
| 23857 | 42 | GKLR | 55 | DER | 243 | H |
| 25695 | 44 | GKLR | 57 | DER | 336 | H |
| 26201 | 58 | RSLR | 63 | DENR | 111 | H |
| 31920 | 44 | GRLR | 57 | DER | 233 | H |
| 38296 | 63 | RRLR | – | – | H | |
| 46806 | 43 | QLRLR | 57 | DER | – | H |
| 50948 | 41 | GRLR | 54 | DER | – | H |
| 59265 | 49 | RLLR | 62 | EE | – | H |
Determined as two or more D,E,N and/or Q residues followed by a R or K residue: [DENQ]{2,}[RK].
Evidence codes are as follows: M, motif; B, blast; H, jackhmmer.
Using the collection of Crinkler effectors of Phytophthora species (Haas et al., 2009), we performed similarity searches to identify these effectors in the B. lactucae secretome. We found six candidates, three of which contained an LXFLA amino acid motif that was similar to the motifs reported in Phytophthora species. Interestingly, gDNA read coverage was higher than expected for four candidates: two containing the LXFLA motif (six‐ and three‐fold) and two without this sequence (22‐ and 40‐fold). This suggests that more gene copies or pseudogenes may be present in the genome than are detected in our transcriptome set, or that multiple genes have been assembled into a single sequence. The sequences containing an LXFLA motif are not complete, suggesting problems with assembly. We therefore used the HMM models of Crinkler domains (Haas et al., 2009) to screen all predicted peptides for the potential presence of Crinkler domains. All peptides were included as many of the potential Phytophthora Crinklers do not have a signal peptide and signal peptides may have been assembled separately. We found a total of 75 sequences that had homology to one or more of the reported Crinkler domains. In total, we found matches to 14 different C‐terminal Crinkler domains in B. lactucae (Table 5), again with higher than expected gDNA coverage. Interestingly, we also found traces of D2, DC and DBF domains, which are also found in cell death‐inducing P. infestans Crinklers (Haas et al., 2009).
Table 5.
Matches to hidden Markov models of Crinkler domains (as Haas et al., 2009) in the Bremia lactucae transcriptome and the predicted copy numbers thereof.
| Domain | Peptide predictions | Predicted copy number* |
|---|---|---|
| N‐terminal | ||
| LFLAK | 15 | 64 |
| DWL | 13 | 101 |
| C‐terminal | ||
| D2 | 11 | 11 |
| DAB | 2 | 4 |
| DBE | 4 | 22 |
| DBF | 3 | 3 |
| DC | 2 | 1 |
| DDB | 3 | 2 |
| DDC | 2 | 1 |
| DFA | 2 | 2 |
| DFB | 8 | 111 |
| DFC | 9 | 68 |
| DN17 | 1 | 1 |
| DN7 | 1 | 1 |
| DX9 | 1 | 1 |
| DXX | 15 | 135 |
The predicted copy number is determined by dividing the observed average genomic DNA coverage of the assembled transcripts in which the indicated domains are encoded by the average genomic coverage of all assembled B. lactucae transcripts.
Expanded sequence families in B. lactucae
The clustering of B. lactucae secretome sequences with those of six other oomycete species and four stramenopile species was performed on the basis of a comparison of all sequences with each other. A cluster of closely related genes may indicate a recently expanded family of genes that plays an important role in pathogenesis and adaptation to the host. Clustering revealed 85 groups of sequences containing multiple B. lactucae members. We focused on eight clusters with three or more B. lactucae members that contained significantly more B. lactucae members than would be expected on the basis of the presence of members of six other oomycetes and four other stramenopiles.
Two clusters could be categorized on the basis of Pfam domain hits. The first is a B. lactucae‐unique cluster containing a three‐member subset of the Pfam elicitin domain‐matching sequences. The second cluster, with 11 B. lactucae members and two H. arabidopsidis, five P. infestans, 10 P. sojae and 18 P. ramorum members, contains all seven Jacalin domain‐containing proteins. The members of this cluster have blast hits in the NCBI NR protein database to P. infestans hypothetical conserved proteins, but also to NPP1, an NLP, and putative adhesins. The sequences are distinct from the sequences with a Pfam NLP prediction, as those cluster together in another cluster, with eight B. lactucae, nine H. arabidopsidis, 25 P. infestans, 40 P. sojae and 56 P. ramorum members.
Of the remaining six clusters, five consist of three B. lactucae sequences each, and one cluster consists of four B. lactucae sequences. All but one cluster could be linked to identified RXLR effector candidates. The four‐member cluster most closely represents the ‘classic’ RXLR effector, with an RXLR motif in all members and a DER motif in one of them. The four clusters of three sequences share similar sequence features that are more apparent when these clusters are aligned (Fig. 6). No significant similarity to sequences in the NCBI NR protein database was found for sequences in these four clusters. All members of the C1 cluster had already been identified as potential RXLR effectors, as well as a single member of each of two other clusters (C4 contig18477 and C3 contig28334). One member of C1, contig49490, has an intact RXLR motif that aligns with similar features in sequences in all sequence clusters. The presence of these sequences in groups of related sequences, with features similar to those found in known RXLR effectors, suggests that these proteins may be host‐translocated effectors. The cluster that could not be linked to RXLR effectors has no RXLR‐like motif (not shown) and contains no members that were identified as potential RXLR effectors, although all members contain an EER motif.
Figure 6.

Alignment of the sequences of four Bremia lactucae‐specific clusters of predicted secreted proteins. The columns in which potential RXLR and dEER‐like motifs can be found are indicated above the alignment.
The clustering of secreted sequences highlights three elicitins specific for B. lactucae. It also shows that the family of jacalin domain proteins occurs in a cluster with varying numbers of family members in other oomycete species, suggesting that they might fulfil a species‐specific role. Finally, it identifies other potential host‐translocated effectors that were not predicted by other methods.
DISCUSSION
The B. lactucae transcriptome sequences that have been generated by next‐generation sequencing methods provide a first look at the toolbox used by B. lactucae to manipulate its host. Potential apoplastic and host‐translocated effectors could be predicted from the assembled transcript sequences. We showed that 79% of a set of conserved eukaryotic genes used to assess genome completion are represented in the B. lactucae transcriptome, underlining a broad sampling of the transcriptome. The transcriptome is far more plastic than the genome, and the remaining 21% of conserved eukaryotic genes may not have been sampled because not all developmental stages of B. lactucae were represented in our material (e.g. sporangiophore formation, but also sexual reproduction). Another possibility is that the transcripts have been sampled, but are not assembled as full‐length transcripts and therefore do not satisfy the cut‐off values for the models of the conserved eukaryotic genes.
Protein predictions were based on the use of homology to known proteins to select the most probable open reading frame (ORF) from the contigs. In 432 cases, this suggested a model that spanned multiple ORFs in a single contig, in which case these ORFs were taken together as a single protein model. Manual inspection revealed that this may be a result of the inclusion of unprocessed introns in the cDNA sample. Although introns are thought to be rare in oomycetes, one study (Shen et al., 2011) found that, of a total of 128 alternative splicing events detected in P. sojae, intron skipping was the most common event, explaining 97 cases.
Sequencing of plant‐pathogenic oomycetes has revealed a variable number of RXLR effectors in different organisms. We identified 78 potential RXLR effectors in the B. lactucae secretome, 43 of which contained a conserved RXLR motif. This is a relatively small number compared with the 134 found in H. arabidopsidis (Baxter et al., 2010) and the 563 found in P. infestans (Haas et al., 2009), but close to the 32 RXLR effectors found in Ps. cubensis (Tian et al., 2011). In addition, 29 potential effectors with a QXLR motif were found in Ps. cubensis. There is evidence that QXLR motif proteins can also translocate into the host (Tian et al., 2011), illustrating that strict conformation to the RXLR motif is not required. In B. lactucae, we identified four clusters of three related B. lactucae sequences that all contain sequence features reminiscent of RXLR motifs.
We reported traces of Crinkler proteins in the B. lactucae transcriptome. These sequences were not completely assembled and had a higher than expected coverage of genomic DNA reads. This may indicate that our sampling strategy is suboptimal for capturing Crinklers; EST sequencing of H. arabidopsidis material collected in a similar manner revealed a single partially assembled Crinkler (Cabral et al., 2011). It may be that, in these obligate biotrophs, Crinklers are expressed in smaller numbers or in a more limited timeframe. Alternatively, key Crinkler‐derived reads may be lost during normalization and assembly because of high sequence similarity.
Sequencing of the transcriptome of B. lactucae has provided a wealth of information about the proteins expressed during infection of the host. The protein models described are an extensive source of potential effectors. Candidate effectors can now be cloned and tested to study their role in the infection process, e.g. as suppressors of host immunity. An important next challenge is to determine how these proteins manipulate host processes. Ultimately, host susceptibility and resistance genes can then be identified from Lactuca resources, allowing for novel strategies in breeding for resistance against B. lactucae.
EXPERIMENTAL PROCEDURES
454 sequencing
Bremia lactucae isolate BL24 was grown on Lactuca sativa cultivar Olof and the enhanced B. lactucae‐susceptible Lactuca sativa cv. Olof × Lactuca saligna recombinant inbred line BIL4.4 (Zhang et al., 2009a, b). Plants were kept at 17 °C under 9 h light per day (100 µE/m2/s). Bremia lactucae spores were spray inoculated at 150 spores/µL suspension on 7‐day‐old soil‐grown lettuce seedlings until run‐off was imminent. Inoculation was repeated at 3 days after the initial inoculation. After each inoculation, plants were kept under high humidity for 24 h and thereafter at low humidity. Trypan blue staining was performed by boiling in staining solution for 5 min, and otherwise as described for H. arabidopsidis (Van Damme et al., 2005). Material was harvested 7 days after the initial inoculation. For spore isolations, inoculated plants were kept at high humidity for 7 days, after which spores were rinsed off leaves in water. Spores were filtered through a double layer of miracloth and then spun down (3650 g) and washed in water three times. Spores and infected leaf material were snap frozen and ground using a mortar and pestle. RNA was isolated from ground material using the Qiagen (Venlo, The Netherlands) plant RNA mini kit employing RLC buffer according to the manufacturer's instructions.
Isolated RNA was sent for preparation for sequencing to Vertis Biotechnologie AG (Freising, Germany). Poly(A) RNA was selected from all samples. Two cDNA synthesis strategies were then performed. In the first, the poly(A) RNA was incubated with calf intestine phosphatase (CIP) and tobacco acid pyrophosphatase (TAP), followed by ligation of an RNA adapter to the 5′‐phosphate of decapped mRNAs. First‐strand cDNA synthesis was then performed with an adapter‐random hexamer primer and M‐MLV‐RNase H reverse transcriptase. The resulting cDNAs were amplified with 21 cycles of long and accurate polymerase chain reaction (LA‐PCR). In the second approach, first‐strand cDNA synthesis was primed with a random hexamer primer, after which 454 adapters A and B were ligated to the 5′ and 3′ ends of the cDNA. The cDNA was finally amplified with PCR using a proof reading enzyme (17 cycles). To normalize samples, one cycle of denaturation and reassociation of the cDNA and subsequent separation of single‐stranded cDNA (normalized cDNA) of double‐stranded RNA by hydroxylapatite chromatography were performed. Normalized cDNA was amplified with eight PCR cycles. For both approaches, cDNA in the size range 450–650 bp was eluted from agarose gel using the Macherey & Nagel (Düren, Germany) NucleoSpin Extract II kit. Sequencing was performed on the 454 GS FLX system, using standard reagents.
SOLiD sequencing
Lettuce leaves were surface sterilized in 4% bleach for 3 min, rinsed with water and placed abaxial side up on wet filter paper. The leaves were then spray inoculated (200 spores/µL) with B. lactucae isolate BL24 and kept at high humidity for 8 days. Spores were snap frozen and lysed by bead beating and incubation in cetyltrimethylammonium bromide (CTAB). DNA was extracted with phenol–chloroform; 7 µg of DNA was sheared into 100–110‐bp mean size fragments, end‐repaired with the End‐It DNA End‐Repair Kit (Epicentre Biotechnology, Madison, WI, USA) according to the manufacturer's instructions, and cleaned by phenol–chloroform extraction. Adapters were ligated as recommended by Applied Biosystems (Carlsbad, CA, USA) and cleaned by phenol–chloroform extraction. Fragments of 150–200 bp were excised from a 2% agarose electrophoresis gel and purified using the QIAquick Gel Extraction Kit (Qiagen). Further steps in sample preparation and sequencing were taken as recommended by Applied Biosystems.
Sequence analysis
The 454 transcriptome sequences were assembled using Roche (Basel, Switzerland) gsAssembler (version 2.0.00) software with an expected coverage of 15‐fold (based on initial assembly coverage levels) and otherwise default settings. SOLiD reads were mapped to the assembled 454 reads by Burrows–Wheeler Aligner (BWA) (Li and Durbin, 2009) (version 0.5.8 r1442; maximum edit distance, 10; first 25 bases as seed; maximally two mismatches in the seed; otherwise default settings for colourspace alignment). Assembled sequences were corrected using insertion/deletion information from the mapping process. Correction effectiveness was determined by comparison with the NCBI NR database by blastx (Altschul et al., 1997) (E‐value = 1e‐3, best hit). Sequences with >10‐fold average SOLiD read coverage were kept as B. lactucae sequences. Proteins were predicted from these sequences by selecting an ORF based on the blastx comparison with the NCBI NR database or, failing matches, selecting the ORF(s) of greatest length. In cases in which blast comparison led to the selection of adjacent ORFs, these ORFs were concatenated into a single model. ORF lengths were determined from methionine to stop unless the ORF was at the 5′ or 3′ end, in which case the length was determined from the 5′ or up to the 3′, respectively, as sequences may be incomplete. Sequences were trimmed to start with a methionine and sequences shorter than 20 amino acids were removed before signal peptide prediction by SignalP (3.0a) (Bendtsen et al., 2004; Nielsen and Krogh, 1998) (first 70 amino acids, eukaryote, default cut‐offs). Transmembrane helices were predicted using TMHMM (2.0) (http://www.cbs.dtu.dk/services/TMHMM/). Secretome criteria were the prediction of a signal peptide by both neural network and HMM prediction methods and no predicted transmembrane helices (unless overlapping at least 10 amino acids of the signal peptide). All blast comparisons were performed with an E‐value cut‐off of 1e‐3. HMM and jackhmmer (version 2.3.2; http://hmmer.org/) searches were performed with an E‐value cut‐off of 1e‐3 and otherwise default settings, except for the genome completeness models, for which bit‐score cut‐offs were provided. For jackhmmer searches using B. lactucae RXLRs, the RXLR motif and the 20 amino acids N‐ and C‐terminal thereof were used as input. Pfam (Finn et al., 2010) searches were performed at the gathering threshold.
To obtain a phylogeny of NLP proteins in B. lactucae and other oomycetes, we predicted the presence of the NLP domain (PF05630, Pfam v24; Finn et al., 2010; http://pfam.sanger.ac.uk/) in their proteomes using hmmer3 (http://hmmer.org/) (gathering cut‐off). To predict full‐length NLP domains, we retrieved the seed alignments from the Pfam database and created a calibrated hmmer2 model. hmmer2 (gathering cut‐off) was used with this model on the hmmer3‐identified candidates. Short domain hits (<180 amino acids) were discarded. The sequences were aligned with mafft (Katoh and Toh, 2008) (L‐INS‐i); the phylogeny was predicted with RAxML (Stamatakis, 2006) (gamma model of site heterogeneity; WAG substitution matrix) and the robustness was assessed with 1000 bootstrap replicates. To obtain an estimation of the positioning of short fragmented predicted NLPs from B. lactucae, we manually added them to the set of predictions, aligned them and retrieved a phylogeny as outlined above.
Sequence clustering and phylogeny
Clusters of similar sequences were generated from a sequence similarity network, the edges of which were determined by all versus all blastp comparison of predicted proteins (for phylogeny) or secretomes (for potential effectors) of different stramenopiles (Armbrust et al., 2004; Baxter et al., 2010; Bowler et al., 2008; Cock et al., 2010; Gobler et al., 2011; Haas et al., 2009; Lévesque et al., 2010; Tyler et al., 2006; Saprolegnia parasitica Sequencing Project, Broad Institute of Harvard and MIT http://www.broadinstitute.org/) (see also Table S2 in Supporting Information). Signal peptides were masked for clustering of the secretomes and short sequences were removed. Only edges that represented a match in which high‐scoring pairs (HSPs) spanned >50% of both target and query sequences, and had >20% effective coverage (fraction of covered amino acids), were taken into account, with the exception that B. lactucae sequences did not need to span 50% of non‐B. lactucae sequences. Bremia lactucae sequences may be truncated and therefore not span 50% of the non‐B. lactucae sequence. The similarity network was then clustered into families using the Markov‐Cluster‐Algorithm (MCL; http://micans.org/mcl/, v09‐308, 1.008, inflation = 3) (van Dongen, 2000; Enright et al., 2002). Clustered sequences were aligned in Jalview (Waterhouse et al., 2009) using the MAFFT web service (Katoh and Toh, 2008). We predicted a reliable species phylogeny utilizing a multi‐protein marker containing 119 concatenated 1 : 1 : 1 families retrieved from the protein clusters determined beforehand. These families were aligned using mafft (v6.713b; maxiterate 1000; localpair) (Katoh and Toh, 2008) and subsequently concatenated. Positions containing >20% gaps, as well as less conserved adjacent positions, were removed until a conserved column with a median of the pairwise BLOSUM62 of ≥0 was reached. The phylogenetic tree was predicted using RAxML (v7.0.4; gamma model of invariant sites, WAG substitution model, fast bootstrap approximation) (Stamatakis, 2006) and the robustness of the topology was assessed by 1000 bootstrap replicates.
Accession numbers
Bremia lactucae sequences longer than 200 nucleotides were deposited in the NCBI Transcriptome Shotgun Assembly sequence database under accession numbers JP948721‐JP965883. All nucleotide data and protein translations are available at http://web.science.uu.nl/pmi/data/bremia/.
Supporting information
Fig. S1 Phylogenetic relationship between NLPs [necrosis and ethylene‐inducing peptide 1 (NEP1)‐like proteins] from various oomycete plant pathogens. Four of the NLPs in the Bremia lactucae secretome provided full‐length NLP domain matches and are included in the main tree, indicated in purple. The positions of fragments of the domains found in the complete B. lactucae transcriptome were determined separately (indicated in the main tree by stars), as they are difficult to locate based on the alignment of the full‐length domain. Bremia lactucae NLPs are spread more widely across the oomycete NLP gene tree than are the Hyaloperonospora arabidopsidis NLPs (indicated in bold as clade or individual common names), and do not fall within H. arabidopsidis or Pythium ultimum subclades. Bremia lactucae NLPs cluster close to the two H. arabidopsidis NLPs outside of the H. arabidopsidis‐specific subclade. NLP names indicated in blue belong to NLPs that have been shown to induce necrosis when expressed in tobacco leaves. Scale bar indicates 0.1 substitutions per amino acid. Stars indicate the location of subtrees that contain short fragments (incomplete assembly) of Bremia NLPs that were assessed separately.
Table S1 Number of reads from spore and infection sequencing pools per contig for those that show a five‐fold or greater difference in the number of reads between spore and infection stages as determined from 5′ end sequences of the non‐normalized sequencing pools. Differential expression was observed for contigs corresponding to protein models of all the indicated categories, except for those encoding for DnaJ domain proteins and NLPs [necrosis and ethylene‐inducing peptide 1 (NEP1)‐like proteins] for which no difference in abundance was found.
Table S2 Versions and sources of genomes used for analysis.
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGEMENTS
This project was supported by the foundation TTI Green Genetics (TTI GG) in collaboration with Wageningen University (Wageniningen, The Netherlands), Enza zaden (Enkhuizen, The Netherlands), Nunhems (Nunhem, The Netherlands), Syngenta (Enkhuizen, The Netherlands), Rijk‐Zwaan (De Lier, The Netherlands) and Vilmorin (La Ménitré, France). We thank Ewart de Bruijn and Patrick van Zon for excellent technical assistance.
REFERENCES
- Altschul, S.F. , Madden, T.L. , Schäffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrust, E.V. , Berges, J.A. , Bowler, C. , Green, B.R. , Martinez, D. , Putnam, N.H. , Zhou, S. , Allen, A.E. , Apt, K.E. , Bechner, M. , Brzezinski, M.A. , Chaal, B.K. , Chiovitti, A. , Davis, A.K. , Demarest, M.S. , Detter, J.C. , Glavina, T. , Goodstein, D. , Hadi, M.Z. , Hellsten, U. , Hildebrand, M. , Jenkins, B.D. , Jurka, J. , Kapitonov, V.V. , Kröger, N. , Lau, W.W.Y. , Lane, T.W. , Larimer, F.W. , Lippmeier, J.C. , Lucas, S. , Medina, M. , Montsant, A. , Obornik, M. , Parker, M.S. , Palenik, B. , Pazour, G.J. , Richardson, P.M. , Rynearson, T.A. , Saito, M.A. , Schwartz, D.C. , Thamatrakoln, K. , Valentin, K. , Vardi, A. , Wilkerson, F.P. and Rokhsar, D.S. (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science, 306, 79–86. [DOI] [PubMed] [Google Scholar]
- Bailey, K. , Cevik, V. , Holton, N.J. , Byrne‐Richardson, J. , Sohn, K.H. , Coates, M. , Woods‐Tör, A. , Aksoy, H.M. , Hughes, L. , Baxter, L. , Jones, J.D.G. , Beynon, J. , Holub, E.B. and Tör, M. (2011) Molecular cloning of ATR5Emoy2 from Hyaloperonospora arabidopsidis, an avirulence determinant that triggers RPP5‐mediated defense in arabidopsis. Mol. Plant–Microbe Interact. 24, 1–59. [DOI] [PubMed] [Google Scholar]
- Baxter, L. , Tripathy, S. , Ishaque, N. , Boot, N. , Cabral, A. , Kemen, E. , Thines, M. , Ah‐Fong, A. , Anderson, R. , Badejoko, W. , Bittner‐Eddy, P. , Boore, J.L. , Chibucos, M.C. , Coates, M. , Dehal, P. , Delehaunty, K. , Dong, S. , Downton, P. , Dumas, B. , Fabro, G. , Fronick, C. , Fuerstenberg, S.I. , Fulton, L. , Gaulin, E. , Govers, F. , Hughes, L. , Humphray, S. , Jiang, R.H.Y. , Judelson, H. , Kamoun, S. , Kyung, K. , Meijer, H. , Minx, P. , Morris, P. , Nelson, J. , Phuntumart, V. , Qutob, D. , Rehmany, A. , Rougon‐Cardoso, A. , Ryden, P. , Torto‐Alalibo, T. , Studholme, D. , Wang, Y. , Win, J. , Wood, J. , Clifton, S.W. , Rogers, J. , Van den Ackerveken, G. , Jones, J.D.G. , McDowell, J.M. , Beynon, J. and Tyler, B.M. (2010) Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science, 330, 1549–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen, J.D. , Nielsen, H. , von Heijne, G. and Brunak, S. (2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. [DOI] [PubMed] [Google Scholar]
- Bittner‐Eddy, P.D. , Allen, R.L. , Rehmany, A.P. , Birch, P. and Beynon, J.L. (2003) Use of suppression subtractive hybridization to identify downy mildew genes expressed during infection of Arabidopsis thaliana . Mol. Plant. Pathol. 4, 501–507. [DOI] [PubMed] [Google Scholar]
- Bos, J.I.B. , Armstrong, M. , Whisson, S.C. , Torto, T.A. , Ochwo, M. , Birch, P.R.J. and Kamoun, S. (2003) Intraspecific comparative genomics to identify avirulence genes from Phytophthora . New Phytol. 159, 63–72. [DOI] [PubMed] [Google Scholar]
- Bos, J.I.B. , Armstrong, M.R. , Gilroy, E.M. , Boevink, P.C. , Hein, I. , Taylor, R.M. , Zhendong, T. , Engelhardt, S. , Vetukuri, R.R. , Harrower, B. , Dixelius, C. , Bryan, G. , Sadanandom, A. , Whisson, S.C. , Kamoun, S. and Birch, P.R.J. (2010) Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc. Natl. Acad. Sci. USA, 107, 9909–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwmeester, K. , de Sain, M. , Weide, R. , Gouget, A. , Klamer, S. , Canut, H. and Govers, F. (2011) The lectin receptor kinase LecRK‐I.9 is a novel Phytophthora resistance component and a potential host target for a RXLR effector. PLoS Pathog. 7, e1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler, C. , Allen, A.E. , Badger, J.H. , Grimwood, J. , Jabbari, K. , Kuo, A. , Maheswari, U. , Martens, C. , Maumus, F. , Otillar, R.P. , Rayko, E. , Salamov, A. , Vandepoele, K. , Beszteri, B. , Gruber, A. , Heijde, M. , Katinka, M. , Mock, T. , Valentin, K. , Verret, F. , Berges, J.A. , Brownlee, C. , Cadoret, J. , Chiovitti, A. , Choi, C.J. , Coesel, S. , De Martino, A. , Detter, J.C. , Durkin, C. , Falciatore, A. , Fournet, J. , Haruta, M. , Huysman, M.J.J. , Jenkins, B.D. , Jiroutova, K. , Jorgensen, R.E. , Joubert, Y. , Kaplan, A. , Kröger, N. , Kroth, P.G. , La Roche, J. , Lindquist, E. , Lommer, M. , Martin‐Jézéquel, V. , Lopez, P.J. , Lucas, S. , Mangogna, M. , McGinnis, K. , Medlin, L.K. , Montsant, A. , Oudot‐Le Secq, M. , Napoli, C. , Obornik, M. , Parker, M.S. , Petit, J. , Porcel, B.M. , Poulsen, N. , Robison, M. , Rychlewski, L. , Rynearson, T.A. , Schmutz, J. , Shapiro, H. , Siaut, M. , Stanley, M. , Sussman, M.R. , Taylor, A.R. , Vardi, A. , von Dassow, P. , Vyverman, W. , Willis, A. , Wyrwicz, L.S. , Rokhsar, D.S. , Weissenbach, J. , Armbrust, E.V. , Green, B.R. , Van de Peer, Y. and Grigoriev, I.V. (2008) The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature, 456, 239–244. [DOI] [PubMed] [Google Scholar]
- Brown, S. , Koike, S.T. , Ochoa, O.E. , Laemmlen, F. and Michelmore, R.W. (2004) Insensitivity to the fungicide fosetyl‐aluminum in California isolates of the lettuce downy mildew pathogen, Bremia lactucae . Plant Dis. 88, 502–508. [DOI] [PubMed] [Google Scholar]
- Cabral, A. , Stassen, J.H.M. , Seidl, M.F. , Bautor, J. , Parker, J.E. and Van den Ackerveken, G. (2011) Identification of Hyaloperonospora arabidopsidis transcript sequences expressed during infection reveals isolate‐specific effectors. PLoS ONE, 6, e19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral, A. , Oome, S. , Sander, N. , Küfner, I. , Nürnberger, T. and Van den Ackerveken, G. (2012) Non‐toxic Nep1‐like proteins of the downy mildew pathogen Hyaloperonospora arabidopsidis; repression of necrosis‐inducing activity by a surface‐exposed region . Mol. Plant-Microbe Interact, in press. Available at 10.1094/MPMI-10-11-0269. [DOI] [PubMed] [Google Scholar]
- Cock, J.M. , Sterck, L. , Rouzé, P. , Scornet, D. , Allen, A.E. , Amoutzias, G. , Anthouard, V. , Artiguenave, F. , Aury, J. , Badger, J.H. , Beszteri, B. , Billiau, K. , Bonnet, E. , Bothwell, J.H. , Bowler, C. , Boyen, C. , Brownlee, C. , Carrano, C.J. , Charrier, B. , Cho, G.Y. , Coelho, S.M. , Collén, J. , Corre, E. , Da Silva, C. , Delage, L. , Delaroque, N. , Dittami, S.M. , Doulbeau, S. , Elias, M. , Farnham, G. , Gachon, C.M.M. , Gschloessl, B. , Heesch, S. , Jabbari, K. , Jubin, C. , Kawai, H. , Kimura, K. , Kloareg, B. , Küpper, F.C. , Lang, D. , Le Bail, A. , Leblanc, C. , Lerouge, P. , Lohr, M. , Lopez, P.J. , Martens, C. , Maumus, F. , Michel, G. , Miranda‐Saavedra, D. , Morales, J. , Moreau, H. , Motomura, T. , Nagasato, C. , Napoli, C.A. , Nelson, D.R. , Nyvall‐Collén, P. , Peters, A.F. , Pommier, C. , Potin, P. , Poulain, J. , Quesneville, H. , Read, B. , Rensing, S.A. , Ritter, A. , Rousvoal, S. , Samanta, M. , Samson, G. , Schroeder, D.C. , Ségurens, B. , Strittmatter, M. , Tonon, T. , Tregear, J.W. , Valentin, K. , von Dassow, P. , Yamagishi, T. , Van de Peer, Y. and Wincker, P. (2010) The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature, 465, 617–621. [DOI] [PubMed] [Google Scholar]
- van Dongen, S.M. (2000) Graph clustering by flow simulation. Universiteit Utrecht (Thesis) 1–167.
- Enright, A.J. , Van Dongen, S. and Ouzounis, C.A. (2002) An efficient algorithm for large‐scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Mistry, J. , Tate, J. , Coggill, P. , Heger, A. , Pollington, J.E. , Gavin, O.L. , Gunasekaran, P. , Ceric, G. , Forslund, K. , Holm, L. , Sonnhammer, E.L.L. , Eddy, S.R. and Bateman, A. (2010) The pfam protein families database. Nucleic Acids Res. 38, D211–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulin, E. , Bottin, A. and Dumas, B. (2010) Sterol biosynthesis in oomycete pathogens. New Phytol. 5, 258–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulin, E. , Jauneau, A. , Villalba, F. , Rickauer, M. , Esquerré‐Tugayé, M. and Bottin, A. (2002) The CBEL glycoprotein of Phytophthora parasitica var‐nicotianae is involved in cell wall deposition and adhesion to cellulosic substrates. J. Cell Sci. 115, 4565–4575. [DOI] [PubMed] [Google Scholar]
- Gobler, C.J. , Berry, D.L. , Dyhrman, S.T. , Wilhelm, S.W. , Salamov, A. , Lobanov, A.V. , Zhang, Y. , Collier, J.L. , Wurch, L.L. , Kustka, A.B. , Dill, B.D. , Shah, M. , VerBerkmoes, N.C. , Kuo, A. , Terry, A. , Pangilinan, J. , Lindquist, E.A. , Lucas, S. , Paulsen, I.T. , Hattenrath‐Lehmann, T.K. , Talmage, S.C. , Walker, E.A. , Koch, F. , Burson, A.M. , Marcoval, M.A. , Tang, Y. , LeCleir, G.R. , Coyne, K.J. , Berg, G.M. , Bertrand, E.M. , Saito, M.A. , Gladyshev, V.N. and Grigoriev, I.V. (2011) Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl. Acad. Sci. USA, 108, 4352–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Kamoun, S. , Zody, M.C. , Jiang, R.H.Y. , Handsaker, R.E. , Cano, L.M. , Grabherr, M. , Kodira, C.D. , Raffaele, S. , Torto‐Alalibo, T. , Bozkurt, T.O. , Ah‐Fong, A.M.V. , Alvarado, L. , Anderson, V.L. , Armstrong, M.R. , Avrova, A. , Baxter, L. , Beynon, J. , Boevink, P.C. , Bollmann, S.R. , Bos, J.I.B. , Bulone, V. , Cai, G. , Cakir, C. , Carrington, J.C. , Chawner, M. , Conti, L. , Costanzo, S. , Ewan, R. , Fahlgren, N. , Fischbach, M.A. , Fugelstad, J. , Gilroy, E.M. , Gnerre, S. , Green, P.J. , Grenville‐Briggs, L.J. , Griffith, J. , Grünwald, N.J. , Horn, K. , Horner, N.R. , Hu, C. , Huitema, E. , Jeong, D. , Jones, A.M.E. , Jones, J.D.G. , Jones, R.W. , Karlsson, E.K. , Kunjeti, S.G. , Lamour, K. , Liu, Z. , Ma, L. , Maclean, D. , Chibucos, M.C. , McDonald, H. , McWalters, J. , Meijer, H.J.G. , Morgan, W. , Morris, P.F. , Munro, C.A. , O'Neill, K. , Ospina‐Giraldo, M. , Pinzón, A. , Pritchard, L. , Ramsahoye, B. , Ren, Q. , Restrepo, S. , Roy, S. , Sadanandom, A. , Savidor, A. , Schornack, S. , Schwartz, D.C. , Schumann, U.D. , Schwessinger, B. , Seyer, L. , Sharpe, T. , Silvar, C. , Song, J. , Studholme, D.J. , Sykes, S. , Thines, M. , van de Vondervoort, P.J.I. , Phuntumart, V. , Wawra, S. , Weide, R. , Win, J. , Young, C. , Zhou, S. , Fry, W. , Meyers, B.C. , van West, P. , Ristaino, J. , Govers, F. , Birch, P.R.J. , Whisson, S.C. , Judelson, H.S. and Nusbaum, C. (2009) Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature, 461, 393–398. [DOI] [PubMed] [Google Scholar]
- Hirasawa, K. , Amano, T. and Shioi, Y. (2004) Lipid‐binding form is a key conformation to induce a programmed cell death initiated in tobacco BY‐2 cells by a proteinaceous elicitor of cryptogein. Physiol. Plant. 121, 196–203. [DOI] [PubMed] [Google Scholar]
- Jiang, R.H. , Tyler, B.M. , Whisson, S.C. , Hardham, A.R. and Govers, F. (2006) Ancient origin of elicitin gene clusters in Phytophthora genomes. Mol. Biol. Evol. 23, 338–351. [DOI] [PubMed] [Google Scholar]
- de Jonge, R. , van Esse, H.P. , Kombrink, A. , Shinya, T. , Desaki, Y. , Bours, R. , van der Krol, S. , Shibuya, N. , Joosten, M.H.A.J. and Thomma, B.P.H.J. (2010) Conserved fungal LysM effector Ecp6 prevents chitin‐triggered immunity in plants. Science, 329, 953–955. [DOI] [PubMed] [Google Scholar]
- Kale, S.D. , Gu, B. , Capelluto, D.G. , Dou, D. , Feldman, E. , Rumore, A. , Arredondo, F.D. , Hanlon, R. , Fudal, I. , Rouxel, T. , Lawrence, C.B. , Shan, W. and Tyler, B.M. (2010) External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell, 142, 284–295. [DOI] [PubMed] [Google Scholar]
- Kanneganti, T.‐D. , Huitema, E. , Cakir, C. and Kamoun, S. (2006) Synergistic interactions of the plant cell death pathways induced by Phytophthora infestans Nepl‐like protein PiNPP1.1 and INF1 elicitin. Mol. Plant–Microbe Interact. 19, 854–863. [DOI] [PubMed] [Google Scholar]
- Kaschani, F. , Shabab, M. , Bozkurt, T. , Shindo, T. , Schornack, S. , Gu, C. , Ilyas, M. , Win, J. , Kamoun, S. and van der Hoorn, R.A.L. (2010) An effector‐targeted protease contributes to defense against Phytophthora infestans and is under diversifying selection in natural hosts. Plant Physiol. 154, 1794–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. and Toh, H. (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9, 286–298. [DOI] [PubMed] [Google Scholar]
- Kelley, B.S. , Lee, S. , Damasceno, C.M.B. , Chakravarthy, S. , Kim, B. , Martin, G.B. and Rose, J.K.C. (2010) A secreted effector protein (SNE1) from Phytophthora infestans is a broadly acting suppressor of programmed cell death. Plant J. 62, 357–366. [DOI] [PubMed] [Google Scholar]
- Kemen, E. , Gardiner, A. , Schultz‐Larsen, T. , Kemen, A.C. , Balmuth, A.L. , Robert‐Seilaniantz, A. , Bailey, K. , Holub, E. , Studholme, D.J. , Maclean, D. and Jones, J.D. (2011) Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana . PLoS Biol. 9, e1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeda, A. , Sedlářová, M. , Petřivalský, M. and Prokopová, J. (2008) Diversity of defence mechanisms in plant–oomycete interactions: a case study of Lactuca spp. and Bremia lactucae . Eur. J. Plant Pathol. 122, 71–89. [Google Scholar]
- Lévesque, C.A. , Brouwer, H. , Cano, L. , Hamilton, J.P. , Holt, C. , Huitema, E. , Raffaele, S. , Robideau, G.P. , Thines, M. , Win, J. , Zerillo, M.M. , Beakes, G.W. , Boore, J.L. , Busam, D. , Dumas, B. , Ferriera, S. , Fuerstenberg, S.I. , Gachon, C.M. , Gaulin, E. , Govers, F. , Grenville‐Briggs, L.J. , Horner, N.R. , Hostetler, J. , Jiang, R.H.Y. , Johnson, J. , Krajaejun, T. , Lin, H. , Meijer, H.J.G. , Moore, B. , Morris, P.F. , Phuntumart, V. , Puiu, D. , Shetty, J. , Stajich, J.E. , Tripathy, S. , Wawra, S. , van West, P. , Whitty, B.R. , Coutinho, P.M. , Henrissat, B. , Martin, F. , Thomas, P.D. , Tyler, B.M. , De Vries, R.P. , Kamoun, S. , Yandell, M. , Tisserat, N. and Buell, C.R. (2010) Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Links, M.G. , Holub, E. , Jiang, R.H. , Sharpe, A.G. , Hegedus, D. , Beynon, E. , Sillito, D. , Clarke, W.E. , Uzuhashi, S. and Borhan, M.H. (2011) De novo sequence assembly of Albugo candida reveals a small genome relative to other biotrophic oomycetes. BMC Genomics, 12, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Ye, W. , Ru, Y. , Yang, X. , Gu, B. , Tao, K. , Lu, S. , Dong, S. , Zheng, X. , Shan, W. , Wang, Y. and Dou, D. (2011) Two host cytoplasmic effectors are required for pathogenesis of Phytophthora sojae by suppression of host defenses. Plant Physiol. 155, 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Bos, J.I.B. , Armstrong, M. , Whisson, S.C. , da Cunha, L. , Torto‐Alalibo, T. , Win, J. , Avrova, A.O. , Wright, F. , Birch, P.R.J. and Kamoun, S. (2005) Patterns of diversifying selection in the phytotoxin‐like scr74 gene family of Phytophthora infestans . Mol. Biol. Evol. 22, 659–672. [DOI] [PubMed] [Google Scholar]
- van Loon, L.C. , Rep, M. and Pieterse, C.M.J. (2006) Significance of inducible defense‐related proteins in infected plants. Annu. Rev. Phytopathol. 44, 135–162. [DOI] [PubMed] [Google Scholar]
- Michelmore, R.W. and Wong, J. (2008) Classical and molecular genetics of Bremia lactucae, cause of lettuce downy mildew. Eur. J. Plant Pathol. 122, 19–30. [Google Scholar]
- Mikes, V. , Milat, M.L. , Ponchet, M. , Ricci, P. and Blein, J.P. (1997) The fungal elicitor cryptogein is a sterol carrier protein. FEBS Lett. 416, 190–192. [DOI] [PubMed] [Google Scholar]
- Mikes, V. , Milat, M.L. , Ponchet, M. , Panabières, F. , Ricci, P. and Blein, J.P. (1998) Elicitins, proteinaceous elicitors of plant defense, are a new class of sterol carrier proteins. Biochem. Biophys. Res. Commun. 245, 133–139. [DOI] [PubMed] [Google Scholar]
- Nespoulous, C. , Huet, J. and Pernollet, J. (1992) Structure–function relationships of α and β elicitins, signal proteins involved in the plant–Phytophthora interaction. Planta, 186, 551–557. [DOI] [PubMed] [Google Scholar]
- Nielsen, H. and Krogh, A. (1998) Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 122–130. [PubMed] [Google Scholar]
- Orsomando, G. , Lorenzi, M. , Raffaelli, N. , Dalla Rizza, M. , Mezzetti, B. and Ruggieri, S. (2001) Phytotoxic protein PcF, purification, characterization, and cDNA sequencing of a novel hydroxyproline‐containing factor secreted by the strawberry pathogen Phytophthora cactorum . J. Biol. Chem. 276, 21 578–21 584. [DOI] [PubMed] [Google Scholar]
- Ottmann, C. , Luberacki, B. , Küfner, I. , Koch, W. , Brunner, F. , Weyand, M. , Mattinen, L. , Pirhonen, M. , Anderluh, G. , Seitz, H.U. , Nürnberger, T. and Oecking, C. (2009) A common toxin fold mediates microbial attack and plant defense. Proc. Natl. Acad. Sci. USA, 106, 10 359–10 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Plešková, V. , Kašparovský, T. , Obořil, M. , Ptáčková, N. , Chaloupková, R. , Ladislav, D. , Damborský, J. and Lochman, J. (2011) Elicitin–membrane interaction is driven by a positive charge on the protein surface: role of Lys13 residue in lipids loading and resistance induction. Plant Physiol. Biochem. 49, 321–328. [DOI] [PubMed] [Google Scholar]
- Qutob, D. , Kamoun, S. and Gijzen, M. (2002) Expression of a Phytophthora sojae necrosis‐inducing protein occurs during transition from biotrophy to necrotrophy. Plant J. 32, 361–373. [DOI] [PubMed] [Google Scholar]
- Raffaele, S. , Win, J. , Cano, L.M. and Kamoun, S. (2010) Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans . BMC Genomics, 11, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmany, A.P. , Gordon, A. , Rose, L.E. , Allen, R.L. , Armstrong, M.R. , Whisson, S.C. , Kamoun, S. , Tyler, B.M. , Birch, P.R.J. and Beynon, J.L. (2005) Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two arabidopsis lines. Plant Cell, 17, 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, P. , Bonnet, P. , Huet, J.C. , Sallantin, M. , Beauvais‐Cante, F. , Bruneteau, M. , Billard, V. , Michel, G. and Pernollet, J.C. (1989) Structure and activity of proteins from pathogenic fungi Phytophthora eliciting necrosis and acquired resistance in tobacco. Eur. J. Biochem. 183, 555–563. [DOI] [PubMed] [Google Scholar]
- Rose, J.K.C. , Ham, K. , Darvill, A.G. and Albersheim, P. (2002) Molecular cloning and characterization of glucanase inhibitor proteins: coevolution of a counterdefense mechanism by plant pathogens. Plant Cell, 14, 1329–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, D. , Ye, W. , Dong, S. , Wang, Y. and Dou, D. (2011) Characterization of intronic structures and alternative splicing in Phytophthora sojae by comparative analysis of expressed sequence tags and genomic sequences. Can. J. Microbiol. 57, 84–90. [DOI] [PubMed] [Google Scholar]
- Song, J. , Win, J. , Tian, M. , Schornack, S. , Kaschani, F. , Ilyas, M. , van Der Hoorn, R.A.L. and Kamoun, S. (2009) Apoplastic effectors secreted by two unrelated eukaryotic plant pathogens target the tomato defense protease Rcr3. Proc. Natl. Acad. Sci. USA, 106, 1654–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2006) RAxML‐VI‐HPC: maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stassen, J.H.M. and Van den Ackerveken, G. (2011) How do oomycete effectors interfere with plant life? Curr. Opin. Plant Biol. 14, 407–414. [DOI] [PubMed] [Google Scholar]
- Tian, M. , Win, J. , Song, J. , van der Hoorn, R. , van der Knaap, E. and Kamoun, S. (2007) A Phytophthora infestans cystatin‐like protein targets a novel tomato papain‐like apoplastic protease. Plant Physiol. 143, 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, M. , Win, J. , Savory, E. , Burkhardt, A. , Held, M. , Brandizzi, F. and Day, B. (2011) 454 genome sequencing of Pseudoperonospora cubensis reveals effector proteins with a putative QXLR translocation motif. Mol. Plant–Microbe Interact. 24, 543–553. [DOI] [PubMed] [Google Scholar]
- Tyler, B.M. , Tripathy, S. , Zhang, X. , Dehal, P. , Jiang, R.H.Y. , Aerts, A. , Arredondo, F.D. , Baxter, L. , Bensasson, D. , Beynon, J.L. , Chapman, J. , Damasceno, C.M.B. , Dorrance, A.E. , Dou, D. , Dickerman, A.W. , Dubchak, I.L. , Garbelotto, M. , Gijzen, M. , Gordon, S.G. , Govers, F. , Grünwald, N.J. , Huang, W. , Ivors, K.L. , Jones, R.W. , Kamoun, S. , Krampis, K. , Lamour, K.H. , Lee, M. , McDonald, W.H. , Medina, M. , Meijer, H.J.G. , Nordberg, E.K. , Maclean, D.J. , Ospina‐Giraldo, M.D. , Morris, P.F. , Phuntumart, V. , Putnam, N.H. , Rash, S. , Rose, J.K.C. , Sakihama, Y. , Salamov, A.A. , Savidor, A. , Scheuring, C.F. , Smith, B.M. , Sobral, B.W.S. , Terry, A. , Torto‐Alalibo, T.A. , Win, J. , Xu, Z. , Zhang, H. , Grigoriev, I.V. , Rokhsar, D.S. and Boore, J.L. (2006) Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science, 313, 1261–1266. [DOI] [PubMed] [Google Scholar]
- Van Damme, M. , Andel, A. , Huibers, R.P. , Panstruga, R. , Weisbeek, P.J. and Van den Ackerveken, G. (2005) Identification of Arabidopsis loci required for susceptibility to the downy mildew pathogen Hyaloperonospora parasitica . Mol. Plant–Microbe Interact. 18, 583–592. [DOI] [PubMed] [Google Scholar]
- Vauthrin, S. , Mikes, V. , Milat, M.L. , Ponchet, M. , Maume, B. , Osman, H. and Blein, J.P. (1999) Elicitins trap and transfer sterols from micelles, liposomes and plant plasma membranes. Biochim. Biophys. Acta, 1419, 335–342. [DOI] [PubMed] [Google Scholar]
- Waterhouse, A.M. , Procter, J.B. , Martin, D.M.A. , Clamp, M. and Barton, G.J. (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics, 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, N.W. , Lindhout, P. , Niks, R.E. and Jeuken, M.J.W. (2009a) Genetic dissection of Lactuca saligna nonhost resistance to downy mildew at various lettuce developmental stages. Plant Pathol. 58, 923–932. [Google Scholar]
- Zhang, N.W. , Pelgrom, K. , Niks, R.E. , Visser, R.G.F. and Jeuken, M.J.W. (2009b) Three combined quantitative trait loci from nonhost Lactuca saligna are sufficient to provide complete resistance of lettuce against Bremia lactucae . Mol. Plant–Microbe Interact. 22, 1160–1168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Phylogenetic relationship between NLPs [necrosis and ethylene‐inducing peptide 1 (NEP1)‐like proteins] from various oomycete plant pathogens. Four of the NLPs in the Bremia lactucae secretome provided full‐length NLP domain matches and are included in the main tree, indicated in purple. The positions of fragments of the domains found in the complete B. lactucae transcriptome were determined separately (indicated in the main tree by stars), as they are difficult to locate based on the alignment of the full‐length domain. Bremia lactucae NLPs are spread more widely across the oomycete NLP gene tree than are the Hyaloperonospora arabidopsidis NLPs (indicated in bold as clade or individual common names), and do not fall within H. arabidopsidis or Pythium ultimum subclades. Bremia lactucae NLPs cluster close to the two H. arabidopsidis NLPs outside of the H. arabidopsidis‐specific subclade. NLP names indicated in blue belong to NLPs that have been shown to induce necrosis when expressed in tobacco leaves. Scale bar indicates 0.1 substitutions per amino acid. Stars indicate the location of subtrees that contain short fragments (incomplete assembly) of Bremia NLPs that were assessed separately.
Table S1 Number of reads from spore and infection sequencing pools per contig for those that show a five‐fold or greater difference in the number of reads between spore and infection stages as determined from 5′ end sequences of the non‐normalized sequencing pools. Differential expression was observed for contigs corresponding to protein models of all the indicated categories, except for those encoding for DnaJ domain proteins and NLPs [necrosis and ethylene‐inducing peptide 1 (NEP1)‐like proteins] for which no difference in abundance was found.
Table S2 Versions and sources of genomes used for analysis.
Supporting info item
Supporting info item
Supporting info item