

Abstract
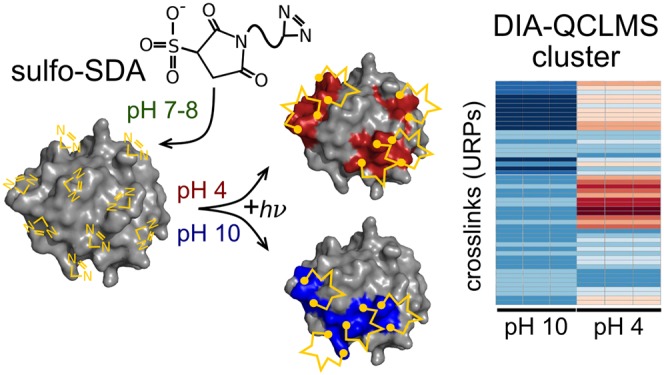
Protein structures respond to changes in their chemical and physical environment. However, studying such conformational changes is notoriously difficult, as many structural biology techniques are also affected by these parameters. Here, the use of photo-crosslinking, coupled with quantitative crosslinking mass spectrometry (QCLMS), offers an opportunity, since the reactivity of photo-crosslinkers is unaffected by changes in environmental parameters. In this study, we introduce a workflow combining photo-crosslinking using sulfosuccinimidyl 4,4′-azipentanoate (sulfo-SDA) with our recently developed data-independent acquisition (DIA)-QCLMS. This novel photo-DIA-QCLMS approach is then used to quantify pH-dependent conformational changes in human serum albumin (HSA) and cytochrome C by monitoring crosslink abundances as a function of pH. Both proteins show pH-dependent conformational changes resulting in acidic and alkaline transitions. 93% and 95% of unique residue pairs (URP) were quantifiable across triplicates for HSA and cytochrome C, respectively. Abundance changes of URPs and hence conformational changes of both proteins were visualized using hierarchical clustering. For HSA we distinguished the N–F and the N–B form from the native conformation. In addition, we observed for cytochrome C acidic and basic conformations. In conclusion, our photo-DIA-QCLMS approach distinguished pH-dependent conformers of both proteins.
The structure of proteins depends on their chemical and physical environment, such as the presence of denaturants, ionic strength, temperature, or pH.1−6 Studying conformational changes as these environmental parameters change is notoriously difficult as many methods of structural biology are themselves affected by the same set of parameters. We set out to investigate whether crosslinking mass spectrometry could be employed in such settings.
The structure of proteins and protein complexes can be revealed through crosslinking mass spectrometry (CLMS).7−13 By forming covalent bonds between the crosslinker and amino acids, proximal amino acid residues in proteins can be detected. Following the proteolytic digestion of a protein, crosslinked peptides can be enriched by strong cation exchange chromatography (SCX)14 or size exclusion chromatography (SEC),15 for example, and identified using liquid chromatography–mass spectrometry (LC-MS). Quantitative crosslinking mass spectrometry reveals structural flexibility and changes in proteins such as protein state changes including activation, protein network, and enzyme activity regulation, complex assembly, or protein–protein interactions.16 However, crosslinking with standard crosslinkers such as bis[sulfosuccinimidyl] suberate (BS3), which contains two NHS groups, is influenced by parameters such as pH and temperature. As such, it is not possible to study conformational changes of proteins across a wide range of pH or temperature values.
As an alternative to NHS-based crosslinkers such as BS,3 photoactivatable crosslinkers can be used in CLMS.17−20 The crosslinking reaction is initiated by UV radiation21,22 and yields a highly reactive carbene intermediate that can react with a variety of groups present in amino acid side chains.23,24 Photo-crosslinking results in more crosslinks than homo-bifunctional NHS-based crosslinkers that are restricted to nucleophilic groups.20 Importantly, since photo-crosslinking chemistry is not influenced by environmental parameters, it may be used to quantify at the residue level conformational changes of proteins resulting from varying conditions, once the crosslinker has been covalently linked to the protein of interest with an inactive diazirine group.
To explore photo-crosslinking as a tool for analyzing pH-dependent conformational changes, we used two model proteins: human serum albumin (HSA) and bovine cytochrome C. HSA and cytochrome C are known to undergo structural changes under different pH conditions.25−28 Human serum albumin is the globular protein in human blood plasma whose main ability is to bind organic and inorganic ligands. Investigation of its denaturation is important for understanding its function as a transporter of physiological metabolites in blood. At least five different pH-dependent conformations have been described for HSA.29 Cytochrome C is a small heme-containing protein found loosely associated with the inner membrane of mitochondria. It is an essential component of the electron transport chain in which it carries electrons between complexes III (coenzyme Q–cytochrome C reductase) and IV (cytochrome C oxidase). Similarly to HSA, cytochrome C undergoes conformational changes depending on pH conditions. Alkaline pH and certain biochemical and biophysical cellular factors induce the so-called “alkaline transition”.30,31 Conformational changes at acidic and neutral pH lead to the interaction of cytochrome C with phospholipids.32,33
Here, we present a workflow combining photo-crosslinking and data-independent acquisition–quantitative crosslinking mass spectrometry (DIA-QCLMS) to study pH-dependent conformational changes and apply it to two model proteins, HSA and cytochrome C. We determine the differential abundance of crosslinked residue pairs in response to different pH conditions. Our study shows that, with use of sulfosuccinimidyl 4,4′-azipentanoate (sulfo-SDA) as the crosslinker, we could pinpoint regions within a protein structure displaying pH-dependent conformational or dynamic changes. Sulfo-SDA is a commonly used hetero-bifunctional crosslinker containing two functional groups: an NHS ester and a diazirine group. First, the NHS ester reacts with the amino acid residues of a protein, followed by the loss of the diazirine group in a second step, induced by UV light exposure.18,34 Relying on established sulfo-SDA analyses of proteins18 and our DIA workflow using Spectronaut,35 we expand the application spectrum of crosslinking mass spectrometry to the wide range of conditions found in life.
Methods
Reagents
Human serum albumin (HSA) and cytochrome C (bovine heart) were purchased individually from Sigma-Aldrich (St. Louis, MO, USA). The crosslinker sulfosuccinimidyl 4,4′-azipentanoate was purchased from Thermo Scientific Pierce (Rockford, IL, USA).
Photo-crosslinking Reaction and Sample Preparation
HSA and cytochrome C were crosslinked separately with sulfo-SDA using a protein-to-crosslinker ratio of 1:0.5 (w/w) (HSA, 15.1 μM:1.5 mM; cytochrome C, 85 μM:1.5 mM). crosslinking was carried out in two stages: first, sulfo-SDA, dissolved in crosslinking buffer (20 mM HEPES–OH, 20 mM NaCl, 5 mM MgCl2, pH 7.8) was added to the target proteins (1 μg/μL total protein concentration) and left to react in the dark for 50 min at room temperature. The sample was then split into seven vials, each adjusted to a different pH, using HCl (18.5%) to lower the pH (pH 4, 5, 6, 7) and NaOH (1 mol/L) to reach basic pH (pH 8, 9, 10). The diazirine group was then photoactivated using ultraviolet light irradiation. A UVP CL-1000L UV crosslinker (UVP, U.K.) at 365 nm was utilized for photoactivation. Samples were spread onto the inside of Eppendorf tube lids to form a thin film, placed on ice at a distance of 5 cm from the lamp, and irradiated for 30 min at 200,000 μJ/cm2. The resulting crosslinked HSA and cytochrome C samples were separated by SDS-PAGE. crosslinked monomer protein gel bands were excised, reduced, alkylated, and digested using trypsin, as previously described.36 Resulting peptides were extracted from gel bands using 80% (v/v) acetonitrile (ACN) and concentrated to a final ACN content of nominally 5% (v/v) using a Vacufuge concentrator (Eppendorf, Germany). Tryptic peptides were desalted using C18–StageTips37 and eluted with 80% (v/v) ACN and 0.1% (v/v) TFA prior to mass spectrometric analysis. Peptides were dried in the Vacufuge concentrator and resuspended in 2% (v/v) ACN and 0.1% (v/v) formic acid (FA) to a final protein concentration of 0.75 μg/μL.
Data Acquisition
LC-MS/MS analysis was performed using a quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer (Orbitrap Fusion Lumos, Thermo Fisher Scientific, California, USA) with a “high/high” acquisition strategy (high resolution on MS1 and MS2). A 1.5 μg amount of peptides was injected for data-dependent acquisition (DDA) and data-independent acquisition (DIA) experiments. The peptide separation was carried out on an EASY-Spray column (50 cm × 75 μm i.d., PepMap C18, 2 μm particles, 100 Å pore size, Thermo Fisher Scientific, Germany). Peptides were separated using a 85 min gradient and analyzed in DDA mode as previously described.38 In short, mobile phase A consisted of water and 0.1% (v/v) formic acid (FA) and mobile phase B consisted of 80% (v/v) ACN and 0.1% (v/v) FA. Peptides were loaded onto the column with 2% buffer B at 0.3 μL/min flow rate and eluted at 0.25 μL/min flow rate with the following gradient: 75 min linear increase from 2 to 37.5% mobile phase B followed by 7 min increase from 37.5 to 47.5%, and 3 min from 47.5 to 95% mobile phase B. Precursor ions were detected in the Orbitrap at 120 K resolution in the m/z range 400–1600. Ions with charge states from 3+ to 7+ were selected for fragmentation by high energy collision dissociation (HCD) and detected in the Orbitrap at 30,000 resolution.39 In DIA mode, precursor ions were acquired using an MS1 master scan (m/z range, 400–1200; maximum injection time, 60 ms; automatic gain control (AGC) target, 4 × 105; detector, Orbitrap; resolution, 60,000), following 66 DIA scans for MS2 within a fragmentation range of m/z 120–1200 using an isolation window width of m/z 12 and a maximum injection time of 50 ms. Ions in the selected m/z window were isolated in the quadrupole, fragmented using HCD (normalized collision energy, 30%), and detected in the Orbitrap at 30K resolution.
Identification of crosslinked Peptides
The raw mass spectrometric data files were processed into peak lists and converted to mgf files using MSconvert (v. 3.0.9576).40 Xi (v. 1.6.731)41 was used for database searches. The database comprised the sequences of HSA (UniProt ID, P02768), cytochrome C (P62894) separately, and the reverse sequence of each of these proteins as decoys. Search parameters were as follows: MS tolerance, 6 ppm; MS/MS tolerance, 10 ppm; enzyme, trypsin; missed cleavages, 3; crosslinker, SDA; fixed modification, carbamidomethylation of cysteine; variable modification, oxidation of methionine and modification by SDA (SDA, SDA-loop, SDA-alkene, SDA-oxid, SDA-hydro) with SDA reaction specificity at lysine, serine, threonine, tyrosine, and N-termini of proteins for the NHS-ester group. Diazirines react with all amino acid residues in proteins.18,20 In a crosslink analysis, the false discovery rate (FDR) can be calculated on different information levels: peptide-spectrum matches (PSMs), peptide pairs, residue pairs (RPs), and protein pairs.42 Here, we considered residue-pair FDR, which was estimated using xiFDR (v. 1.0.22.46)42 following the equation valid for heterobifunctional crosslinkers:43 FDR = (TD – DD)/TT,43 where TT is the number of observed target–target matches, TD the number of observed target–decoy matches, and DD the number of decoy–decoy matches. Filtering was applied to only use crosslink PSMs within proteins. Identification with 5% residue-pair FDR was accepted for quantitation.
Creation of Crosslinked Spectral Library and Quantitation
Quantitation was performed at MS1 and MS2 levels using Spectronaut (version 12.0.20491.13).44,45 The spectral library of crosslinked peptides was introduced as a .csv file using xiDIA-library as previously described.46 In short, xiDIA-library (an open source collaborative initiative available in the GitHub repository https://github.com/Rappsilber-Laboratory/xiDIA-library) was used to extract the top 10 crosslink-containing fragments and the top 10 linear ones by the intensity of b- or y-ion signals in the m/z range of 300–1400. The library was imported as an external library. Protein modifications were defined manually in addition to a default list of modifications in Spectronaut: SDA-loop (82.04 Da), SDA-alkene (68.06 Da), SDA-oxid (98.04 Da), SDA-hydro (100.05 Da), and SDA-N2 (110.05 Da).47,48 MS1 and MS2 filtering was done following the Spectronaut 12 manual with the following deviations: quantitation tab, interference correction unticked; minor (peptide) grouping, by modified sequence; major group top N unticked and minor group top N ticket (maximum, 6; minimum, 1); minor group quantity, mean precursor quantity, decoy method was set to “scrambled”. Normalized data (local normalization49 option) with a q-value of 0.01 (comparable to 1% FDR) were exported from Spectronaut to integrate feature-level quantitation data into residue-level data using a top 3 approach.
For each unique residue pair (URP), pH dependency was assessed by a single-way analysis of variance (ANOVA) test against the null hypothesis that the mean is equal in all groups. After applying the Benjamini–Hochberg multiple testing correction, URPs displaying p-values < 0.05 in the ANOVA test were selected. Using this criterion, 137 of the 742 unique residue pairs (URPs) in the HSA data set were found to display pH-dependent behavior, while, in the cytochrome C data set, 87 of the 300 URPs were selected for further analysis. Once this filtering step was applied, direct comparison between pH series was performed using normalized crosslink abundance (XLnorm), obtained by
| 1 |
where XLpH is the median crosslink abundance of a URP at a given pH, XLmin is the minimum median abundance of the same crosslink across the pH series, and XLmax is its maximum. This results in the normalization of each URP abundance between 0 and 1. The data processing was performed in the statistical language R, and the subsequent hierarchical clustering analysis was performed using the heatmap.2 function.50
Results and Discussion
Spectral Library and Library Quality
We generated a library of fragmentation spectra for data-independent acquisition (DIA) analysis using data-dependent acquisition (DDA). We analyzed two proteins, HSA and cytochrome C, each crosslinked separately in solution using sulfo-SDA (Figure 1a). The sulfo-SDA reaction comprised two steps: first, the NHS-ester functionality was reacted with the proteins at room temperature and pH 7.8. Under these conditions, NHS-esters react efficiently with lysine, serine, threonine, and tyrosine side chains and the N-termini of proteins. The samples were then split into seven aliquots, and the pH was adjusted to pH 4–10 in steps of one pH unit; in the second step, the diazirine functionality was activated by UV light at 365 nm. The carbene radical intermediate generated by diazirine activation efficiently reacts with all amino acid residues.18 Proteins were then subjected to SDS-PAGE and protein monomer bands were excised for trypsin digestion to prevent crosslinks between proteins from entering our analysis. To generate spectral libraries, each pH condition was individually analyzed in triplicates (totaling 21 runs at 2 h each, per protein) by LC-MS using a “high–high” (high-resolution MS1 and MS2) strategy and DDA (Figure 1b). For quantitation, each pH condition was analyzed in triplicates and acquired DIA mode. Protein-specific spectral libraries were then generated using xiDIA-library (Müller et al.46 and Methods) and, in total, at 5% residue-pair FDR, comprised 754 URPs, 1655 precursors, and 22808 fragments for the HSA data set and 305 URPs, 1660 precursors, and 17077 fragments for the cytochrome C data set. In comparison, a previous analysis of sulfo-SDA–crosslinked HSA reported 726 URPs at 5% residue-pair FDR, acquiring 48 runs.20 We selected the top 10 crosslink-containing fragments and the top 10 linear ones by the intensity of b- or y-ion signals for library creation. All URPs from the HSA spectral library were covered by crystallographic protein models, with 662 falling below 25 Å and 92 (12%) above. All 305 URPs form the cytochrome C library resulted in 299 below 25 Å and 6 (2%) above. Importantly, the reference structures were solved at a single pH value while the crosslink data derived from seven different pH values. Both proteins change their conformation in response to pH change,29,33 and we therefore expect some mismatch between our data and the reference structures.
Figure 1.

Label-free DIA-based UV crosslinking quantitation workflow. (a) Sample preparation workflow using sulfo-SDA as the UV-activatable crosslinker. First, the NHS ester group reacts with amino acid residues of the model proteins HSA and cytochrome C to decorate the proteins with diazirine groups. After pH adjustment in the range from 4 to 10, the diazirine group is activated to form links within proteins, induced by UV light exposure. Differential abundances of crosslinks can be monitored using a hierarchical clustering. (b) crosslink quantitation workflow (DIA-QCLMS) using Spectronaut for quantitation. pH-adjusted samples are acquired in DDA mode to create a spectral library, followed by DIA mode sampling to generate quantitation data sets.
Quantitation was performed at MS1 and MS2 levels using Spectronaut (version 12.0.20491.13).44,45 The spectral library of crosslinked peptides was introduced as a .csv file using the “Set up a DIA Analysis from File” wizard in the Analysis tab. Following the automated quantitation of crosslinked peptides, the data set was exported using the Report tab. The identified-to-quantified ratios for the HSA and cytochrome C data sets were 93% (744 out of 797) and 95% (300 out of 315), respectively.
Our raw data, peak files, and results files are accessible in the ProteomeXchange(51) Consortium via the PRIDE52 partner repository.
pH-Induced Changes of HSA Structure
Human serum albumin (HSA) undergoes several conformational changes when experiencing a change in either pH, temperature, salt content in the environment, or the concentration of the protein itself.29 Four isomers of the normal form (N-form, pH 6–7) are known from previous studies.53 Within a pH range between 4.5 and 2.7, HSA transforms into the fast form (F-form), below 2.7 it transforms into the expanded form (E-form), and in the basic region from pH 8 to 10 it takes on the basic transition form (B-form) and the aged form (A-form).29,53 Fluorescence measurements, acidic/base titrations, and nuclear magnetic resonance (NMR) have already been applied to indirectly characterize changes in the N → B transition.1,53−56 Previous studies proposed that the N → B transition of HSA is comparable with the transition caused by the binding of fatty acids (e.g., small rotation of domains I and III relative to domain II29,57,58). Binding sites of HSA are shown in Figure 2. We confirmed our ability to generate distinct structural forms of HSA by changing pH conditions as was previously reported,59 using CD spectroscopy (data not shown).
Figure 2.
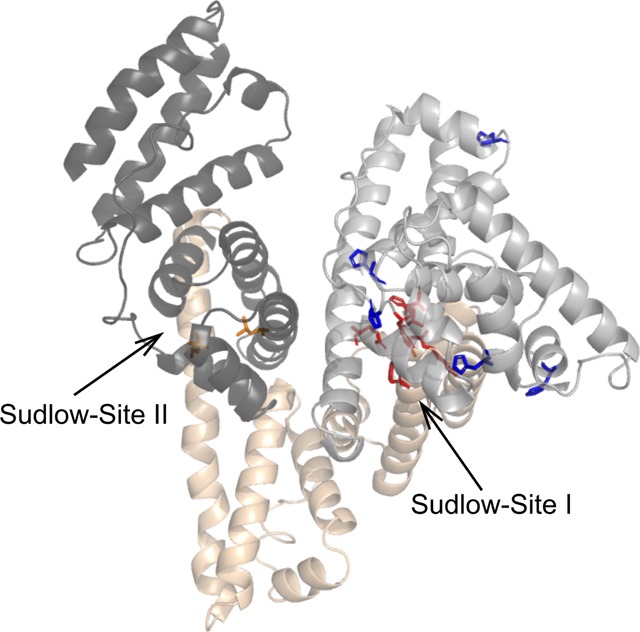
Overview of the domain structure, ligand binding sites, and key residues involved in conformational changes of human serum albumin (HSA) using chain A of the PDB structure 1AO6 (blue, residues referring to the basic transition; red, acidic transition; orange, binding site of diazepam in Sudlow-site II; gray, domain I; sand, domain II; dark gray, domain III).
To investigate the structural differences of HSA in different pH conditions, we crosslinked HSA using sulfo-SDA and quantified the abundance of the individual crosslinks. HSA was crosslinked in different pH conditions, separated by SDS-PAGE, digested in gel using trypsin, and then underwent DIA-LC-MS/MS analysis. Automated quantitation was performed in Spectronaut using our DDA-generated spectral library described above. Normalized data (see Methods) were exported to visualize differences in peak areas of unique residue pairs (URPs) for each pH condition by hierarchical clustering (Figure 3b) based on the changes in normalized median abundance of each URP data series. We applied a cutoff to group the data into the four highest level clusters and to display the pH-dependent abundance of a representative residue pair for each cluster (Figure 3a). The clusters therefore classify URPs based on their pH-dependent relative abundance.
Figure 3.

Hierarchical clustering of normalized median abundance of quantified unique residue pairs (URPs) in HSA. (a) MS1 abundance behavior of representative URPs from human serum albumin (where, for example, 1e+06 represents 1 × 106). The dots show the triplicate MS1 abundance. The line is a smoothed polynomial fit with 95% confidence interval. Representatives were selected on the basis of having features closest to the cluster median. (b) Heat map of median abundances of URPs displaying statistically significant shifts as a function of pH (p < 0.05). Median abundances are normalized between 0 and 1 as described in Methods. Hierarchical clustering was performed by rows: blue, cluster 1; green, cluster 2; red, cluster 3; orange, cluster 4. (c) Visualization of residue pairs corresponding to the four highest level clusters mapped on the structure of human serum albumin (PDB accession code 1AO6). (d) Frequency plot of the euclidean distances corresponding to the URPs within each cluster fitted to a log-normal distribution, highlighting that the crosslink distance distributions of URPs in cluster 4 and cluster 2 do not fit the model.
URPs corresponding to close the distances of domains I–III are mainly sorted into cluster 3, which comprises URPs whose maximum abundance is at neutral pH. This is loosened by a shift to acidic conditions as seen by URPs sorted into cluster 4 with a maximum at pH 4. Cluster 4 shows fewer links between domains I and III compared to cluster 3 (pH 7), and a distance distribution that does not satisfy the model of HSA under neutral conditions, as evidenced by the higher proportion of overlength crosslinks. This is consistent with other characterized motions of the protein such as a separation or rotation of the two domains, possibly to capture or release ligands by entering different compartments. HSA is known as a carrier molecule and hence several binding sites provide interactions with ligands (Figure 2). Sudlow’s side I, in domain IIA, is mostly responsible for interactions with bulky heterocyclic anions, while Sudlow’s side II, located in subdomain IIIA, mainly binds aromatic carboxylates.57,58 Several fatty acid (FA) binding sites (FA1–7) provide the transportation abilities of fatty acids from adipose tissue. Previous studies could show a rotation of domain I relative to domain II due to the binding of FAs to Sudlow’s side I, and movement of Tyr150 to interact with the carboxylate moiety of the lipid. An extensive rearrangement of H-bonds involving Try150, Glu153, Gln196, His242, Arg257, and His288 is the consequence. Additionally, binding of diazepam to Sudlow’s side II is accompanied by a rotation of Leu387 and Leu453 in domain III and consequent side chain movement to encourage drug binding.57,60 Both effects may also be linked to acidic pH conditions and explain the loss of connection between domains I and III at pH 4, compared to the highly crosslinked domains I–III connection at pH7.
URPs in clusters 1–2 have their maximum abundance at basic pH. Cluster 2 is a cluster comprising a small number of residue pairs with a maximum at pH 10 that are mostly located within domain I and between domains I and II. Moreover, the observed distance distribution of cluster 2 deviates from the expected distance distribution of SDA in HSA, consistent with a conformational change of the protein at pH 10. Cluster 1 shows URPs with maxima at pH 9, including crosslinks in domain I and domains II and III. Especially notable is domain I containing His9, His67, His105, and His146, which is heavily crosslinked at pH 8, 9, and 10 (Figure 3c). Previous mutagenesis studies show that His9, His67, His105, His146, and His128 contribute to basic transition.56 Increasing the pH results in deprotonation of residues with a pKa value lower than 9.0, thus triggering the N → B transition.54 crosslinks enriched in basic conditions also fell into domain III, which is in line with previous reports of changes in this domain during the N → B transition.53 The basic transition process was previously described as a structural fluctuation or loosening of human serum albumin including loss of rigidity.57 Overall, our data agree with this as we observed equilibrium states with multiple minima rather than distinct conformation states with just one minimum.
pH-Induced Changes of Cytochrome C Structure
Given its small size, cytochrome C (105 amino acids, 11 kDa) provides an ideal test case for our method of investigating conformational changes in a system of low complexity. The protein was treated as described for HSA. The results of hierarchical clustering are shown in Figure 4b. We applied a cutoff to group the data into the three highest level clusters and to display the pH-dependent abundance of a representative residue pair for each cluster (Figure 4a).
Figure 4.

Hierarchical clustering of normalized median abundance of quantified unique residue pairs (URPs) in cytochrome C. (a) MS1 abundance behavior of representative URPs from cytochrome C (where, for example, 1.5e+06 represents 1.5 × 106). The dots show the triplicate MS1 abundance. The line is a smoothed polynomial fit with 95% confidence interval. Representatives were selected on the basis of having features closest to the cluster median. (b) Heat map of median abundances of URPs displaying statistically significant shifts as a function of pH (p < 0.05). Median abundances are normalized between 0 and 1 as described in Methods. Hierarchical clustering was performed by rows: blue, cluster 1; red, cluster 2; green, cluster 3. (c) Visualization of residue pairs corresponding to the three highest level clusters onto the structure of cytochrome C (PDB accession code 2b4z).
Cluster 1 includes residue pairs which have a maximum at pH 4, cluster 2 at pH 9, and cluster 3 at pH 6–8. The alkaline transition in cytochrome C is described by crosslinks in cluster 2. Links in this cluster are enriched in helix regions 2 (51–55), 3 (62–68), and 4 (72–75) and surrounding Ω-loops, which could indicate flexibility of the protein induced by pH (Figure 4c and Figure 5d). The high crosslinking density in helix regions 2 (51–55), 3 (62–68), and 4 (72–75) and surrounding Ω-loops is in line with previous studies analyzing the alkaline-transition of cytochrome C,31 which show that Met80 is replaced as a ligand of Fe in the heme group with the ε-amino group of a neighboring lysine residue or other surrogate ligands. The change in ligand is thought to increase access of peroxides to the heme center and thus increase the peroxidase activity of cytochrome C.33 The peroxidase activity is critical for translocating cytochrome C from mitochondria into the cytoplasm and nucleolus at the onset of apoptosis.61,62 Additionally, conformational changes induced by a basic pH lead to the interaction of cytochrome C with cardiolipin, which influences homeostasis and stress response in cells.63,64
Figure 5.
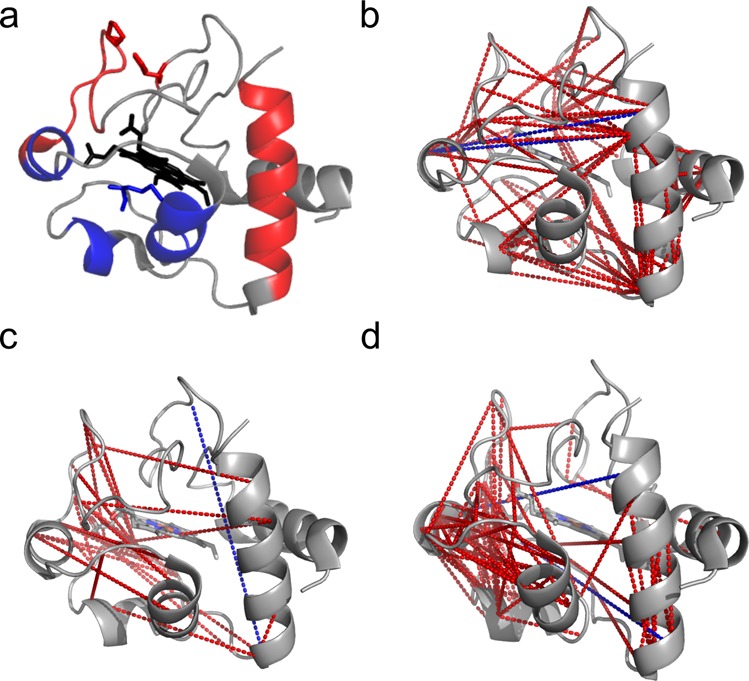
Unique residue pairs matching the PDB (2b4z) crystal structure of cytochrome C. (a) Known residues, helix, and loop regions triggering alkaline and acidic conformational changes in cytochrome C (red, acidic transition; blue, alkaline transition; black, heme group). (b) Residue pairs having maximum abundance at pH ≤ 5, corresponding to the acidic form of cytochrome C (red, links below the distance limit of 25 Å; blue, links longer than 25 Å). (c) Residue pairs having maximum abundance at pH 6–7, corresponding to the neutral form (red, links below the distance limit of 25 Å; blue, links longer than 25 Å). (d) Residue pairs having maximum abundance at pH ≥ 8, corresponding to the alkaline form (red, links below the distance limit of 25 Å; blue, links longer than 25 Å).
In neutral and acidic conditions (clusters 1 and 3), crosslinks are distributed over the entire protein but more frequently between helix regions 5 (89–102) and 3 (62–68) including interconnecting Ω-loops (Figure 4c). crosslinks with high abundance at pH 4 and 5 are combined in Figure 5b to represent the acidic transformation of cytochrome C. The crosslink density is not as localized as for the alkaline transition; nevertheless many crosslinks are concentrated within helix region 5 (89–102), Ω-loop (40–54), and Ω-loop (70–85). Notably, unfolding of δ-loop (40–54) is a known trigger for the acidic transition of cytochrome C.65 The H-bond connection between the imidazole ring of His26 and side chain of Glu44 is disrupted at lower pH. This process induces Met80 substitution by water and thus activates the acidic unfolding pathway of cytochrome C, which would allow crosslinking within the whole protein. Interestingly, the normal form (pH 6–7) shows just a few characteristic crosslinks. This could be linked to the presence of the heme group in the center of the protein, which might sterically interfere with crosslinking (Figure 5c).
Conclusion
In this study, we demonstrate that protein structure can be analyzed under different pH conditions through the use of photo-crosslinking mass spectrometry. Thus, structural changes in proteins can be monitored across a wide range of environmental changes, including pH as shown here, but presumably also temperature, pressure, or concentration. Using standard crosslinkers, this would not be possible as the traditionally employed chemistry is itself influenced by these environmental factors. For standard crosslinkers, changes in crosslink abundance can therefore be linked to a changed structure or to a changed reactivity. These restrictions do not apply to photochemistry, allowing us to probe protein structures here over a pH range from 4 to 10. Although crosslinking involves labeling a protein and thus artifacts cannot be excluded, the overall fold appears to be maintained.66 It will be exciting to see whether our photo-DIA-QCLMS workflow can also be adapted to reveal structural changes induced by environmental parameters within cells.
Acknowledgments
We thank Tobias Baumann for his assistance in acquiring CD spectra. This work was supported by the Wellcome Trust (Grants 103139 and 108504), an Einstein Visiting Fellowship, and the DFG (Grant RA 2365/4-1). The Wellcome Centre for Cell Biology is supported by core funding from the Wellcome Trust (Grant 203149).
The authors declare no competing financial interest.
Notes
Supporting research data containing the mass spectrometry raw files, peak lists, and the result files from xiFDR, xiDIA-library, and Spectronaut used in this study may be accessed from the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD012939 via http://proteomecentral.proteomexchange.org.
References
- Dockal M.; Carter D. C.; Rüker F. Conformational Transitions of the Three Recombinant Domains of Human Serum Albumin Depending on pH. J. Biol. Chem. 2000, 275 (5), 3042–3050. 10.1074/jbc.275.5.3042. [DOI] [PubMed] [Google Scholar]
- Munishkina L. A.; Henriques J.; Uversky V. N.; Fink A. L. Role of Protein-Water Interactions and Electrostatics in Alpha-Synuclein Fibril Formation. Biochemistry 2004, 43 (11), 3289–3300. 10.1021/bi034938r. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Fink A. L. Conformational Constraints for Amyloid Fibrillation: The Importance of Being Unfolded. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1698 (2), 131–153. 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Dzwolak W.; Grudzielanek S.; Smirnovas V.; Ravindra R.; Nicolini C.; Jansen R.; Loksztejn A.; Porowski S.; Winter R. Ethanol-Perturbed Amyloidogenic Self-Assembly of Insulin: Looking for Origins of Amyloid Strains. Biochemistry 2005, 44 (25), 8948–8958. 10.1021/bi050281t. [DOI] [PubMed] [Google Scholar]
- Vaiana S. M.; Manno M.; Emanuele A.; Palma-Vittorelli M. B.; Palma M. U. The Role of Solvent in Protein Folding and in Aggregation. J. Biol. Phys. 2001, 27 (2–3), 133–145. 10.1023/A:1013146530021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foguel D.; Suarez M. C.; Ferrão-Gonzales A. D.; Porto T. C. R.; Palmieri L.; Einsiedler C. M.; Andrade L. R.; Lashuel H. A.; Lansbury P. T.; Kelly J. W.; et al. Dissociation of Amyloid Fibrils of Alpha-Synuclein and Transthyretin by Pressure Reveals Their Reversible Nature and the Formation of Water-Excluded Cavities. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (17), 9831–9836. 10.1073/pnas.1734009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. The Advancement of Chemical Cross-Linking and Mass Spectrometry for Structural Proteomics: From Single Proteins to Protein Interaction Networks. Expert Rev. Proteomics 2014, 11 (6), 733–743. 10.1586/14789450.2014.960852. [DOI] [PubMed] [Google Scholar]
- Liu F.; Heck A. J. R. Interrogating the Architecture of Protein Assemblies and Protein Interaction Networks by Cross-Linking Mass Spectrometry. Curr. Opin. Struct. Biol. 2015, 35, 100–108. 10.1016/j.sbi.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Faini M.; Stengel F.; Aebersold R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci. 2016, 41 (1), 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Schneider M.; Belsom A.; Rappsilber J. Protein Tertiary Structure by Crosslinking/Mass Spectrometry. Trends Biochem. Sci. 2018, 43 (3), 157–169. 10.1016/j.tibs.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J. D.; Bruce J. E. Chemical Cross-Linking with Mass Spectrometry: A Tool for Systems Structural Biology. Curr. Opin. Chem. Biol. 2019, 48, 8–18. 10.1016/j.cbpa.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly F. J.; Rappsilber J. Cross-Linking Mass Spectrometry: Methods and Applications in Structural, Molecular and Systems Biology. Nat. Struct. Mol. Biol. 2018, 25 (11), 1000–1008. 10.1038/s41594-018-0147-0. [DOI] [PubMed] [Google Scholar]
- Yu C.; Huang L. Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Anal. Chem. 2018, 90 (1), 144–165. 10.1021/acs.analchem.7b04431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A.; Jawhari A.; Fischer L.; Buchen C.; Tahir S.; Kamenski T.; Rasmussen M.; Lariviere L.; Bukowski-Wills J.-C.; Nilges M.; et al. Architecture of the RNA Polymerase II-TFIIF Complex Revealed by Cross-Linking and Mass Spectrometry. EMBO J. 2010, 29 (4), 717–726. 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A.; Reischl R.; Walzthoeni T.; Herzog F.; Bohn S.; Förster F.; Aebersold R. Expanding the Chemical Cross-Linking Toolbox by the Use of Multiple Proteases and Enrichment by Size Exclusion Chromatography. Mol. Cell. Proteomics 2012, 11 (3), M111.014126. 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A.; Rappsilber J. Protein Dynamics in Solution by Quantitative Crosslinking/Mass Spectrometry. Trends Biochem. Sci. 2018, 43 (11), 908–920. 10.1016/j.tibs.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin I.; Katritch V.; Sun T.; Xiang Z.; Siu F. Y.; Beyermann M.; Stevens R. C.; Wang L. Genetically Encoded Chemical Probes in Cells Reveal the Binding Path of Urocortin-I to CRF Class B GPCR. Cell 2013, 155 (6), 1258–1269. 10.1016/j.cell.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsom A.; Schneider M.; Fischer L.; Brock O.; Rappsilber J. Serum Albumin Domain Structures in Human Blood Serum by Mass Spectrometry and Computational Biology. Mol. Cell. Proteomics 2016, 15 (3), 1105–1116. 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie N. I.; Makepeace K. A. T.; Petrotchenko E. V.; Borchers C. H. Isotopically-Coded Short-Range Hetero-Bifunctional Photo-Reactive Crosslinkers for Studying Protein Structure. J. Proteomics 2015, 118, 12–20. 10.1016/j.jprot.2014.08.012. [DOI] [PubMed] [Google Scholar]
- Belsom A.; Mudd G.; Giese S.; Auer M.; Rappsilber J. Complementary Benzophenone Cross-Linking/Mass Spectrometry Photochemistry. Anal. Chem. 2017, 89 (10), 5319–5324. 10.1021/acs.analchem.6b04938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J. New Photolabeling and Crosslinking Methods. Annu. Rev. Biochem. 1993, 62, 483–514. 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Bond M. R.; Kohler J. J. Photocrosslinkers Illuminate Interactions in Living Cells. Mol. BioSyst. 2008, 4 (6), 473–480. 10.1039/b803218a. [DOI] [PubMed] [Google Scholar]
- Blencowe A.; Hayes W. Development and Application of Diazirines in Biological and Synthetic Macromolecular Systems. Soft Matter 2005, 1 (3), 178–205. 10.1039/b501989c. [DOI] [PubMed] [Google Scholar]
- Dorman G.; Prestwich G. D. Benzophenone Photophores in Biochemistry. Biochemistry 1994, 33 (19), 5661–5673. 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- Thakur G.; Jiang K.; Lee D.; Prashanthi K.; Kim S.; Thundat T. Investigation of pH-Induced Protein Conformation Changes by Nanomechanical Deflection. Langmuir 2014, 30 (8), 2109–2116. 10.1021/la403981t. [DOI] [PubMed] [Google Scholar]
- Michel L. V.; Ye T.; Bowman S. E. J.; Levin B. D.; Hahn M. A.; Russell B. S.; Elliott S. J.; Bren K. L. Heme Attachment Motif Mobility Tunes Cytochrome c Redox Potential. Biochemistry 2007, 46 (42), 11753–11760. 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan M. G.; Williams M. D.; Bowler B. E. Compressing the Free Energy Range of Substructure Stabilities in Iso-1-Cytochrome c. Protein Sci. 2009, 18 (6), 1155–1164. 10.1002/pro.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döpner S.; Hildebrandt P.; Rosell F. I.; Mauk A. G.; von Walter M.; Buse G.; Soulimane T. The Structural and Functional Role of Lysine Residues in the Binding Domain of Cytochrome c in the Electron Transfer to Cytochrome c Oxidase. Eur. J. Biochem. 1999, 261 (2), 379–391. 10.1046/j.1432-1327.1999.00249.x. [DOI] [PubMed] [Google Scholar]
- Fanali G.; di Masi A.; Trezza V.; Marino M.; Fasano M.; Ascenzi P. Human Serum Albumin: From Bench to Bedside. Mol. Aspects Med. 2012, 33 (3), 209–290. 10.1016/j.mam.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Theorell H.; Åkesson Å. Studies on Cytochrome C. II. The Optical Properties of Pure Cytochrome c and Some of Its Derivatives. J. Am. Chem. Soc. 1941, 63 (7), 1812–1818. 10.1021/ja01852a006. [DOI] [Google Scholar]
- Greenwood C.; Palmer G. Evidence for the Existence of Two Functionally Distinct Forms Cytochrome c Manomer at Alkaline pH. J. Biol. Chem. 1965, 240 (9), 3660–3663. [PubMed] [Google Scholar]
- Hüttemann M.; Pecina P.; Rainbolt M.; Sanderson T. H.; Kagan V. E.; Samavati L.; Doan J. W.; Lee I. The Multiple Functions of Cytochrome c and Their Regulation in Life and Death Decisions of the Mammalian Cell: From Respiration to Apoptosis. Mitochondrion 2011, 11 (3), 369–381. 10.1016/j.mito.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal L.; Tomasina F.; Capdevila D. A.; Demicheli V.; Tórtora V.; Alvarez-Paggi D.; Jemmerson R.; Murgida D. H.; Radi R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 2016, 55 (3), 407–428. 10.1021/acs.biochem.5b01385. [DOI] [PubMed] [Google Scholar]
- Gomes A. F.; Gozzo F. C. Chemical Cross-Linking with a Diazirine Photoactivatable Cross-Linker Investigated by MALDI- and ESI-MS/MS. J. Mass Spectrom. 2010, 45 (8), 892–899. 10.1002/jms.1776. [DOI] [PubMed] [Google Scholar]
- Müller F.; Kolbowski L.; Bernhardt O. M.; Reiter L.; Rappsilber J. Data-Independent Acquisition Improves Quantitative Cross-Linking Mass Spectrometry. Mol. Cell. Proteomics 2019, 18, 786. 10.1074/mcp.TIR118.001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolica A.; Cittaro D.; Borsotti D.; Sennels L.; Ciferri C.; Tarricone C.; Musacchio A.; Rappsilber J. Structural Analysis of Multiprotein Complexes by Cross-Linking, Mass Spectrometry, and Database Searching. Mol. Cell. Proteomics 2007, 6 (12), 2200–2211. 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Mann M.; Ishihama Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using Stage Tips. Nat. Protoc. 2007, 2 (8), 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Müller F.; Fischer L.; Chen Z. A.; Auchynnikava T.; Rappsilber J. On the Reproducibility of Label-Free Quantitative Cross-Linking/Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 405. 10.1007/s13361-017-1837-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbowski L.; Mendes M. L.; Rappsilber J. Optimizing the Parameters Governing the Fragmentation of Cross-Linked Peptides in a Tribrid Mass Spectrometer. Anal. Chem. 2017, 89 (10), 5311–5318. 10.1021/acs.analchem.6b04935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman J. D.; Tabb D. L.; Mallick P. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr. Protoc. Bioinformatics 2014, 46 (1), 13.24.1–13.24.9. 10.1002/0471250953.bi1324s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes M. L.; Fischer L.; Chen Z. A.; Barbon M.; O’Reilly F. J.; Bohlke-Schneider M.; Belsom A.; Dau T.; Combe C. W.; Graham M.; et al. An Integrated Workflow for Cross-Linking/Mass Spectrometry. Bio Rxiv 2018, 10.1101/355396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.; Rappsilber J. Quirks of Error Estimation in Cross-Linking/Mass Spectrometry. Anal. Chem. 2017, 89 (7), 3829–3833. 10.1021/acs.analchem.6b03745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.; Rappsilber J. False Discovery Rate Estimation and Heterobifunctional Cross-Linkers. PLoS One 2018, 13 (5), e0196672 10.1371/journal.pone.0196672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R.; Bernhardt O. M.; Gandhi T.; Miladinović S. M.; Cheng L.-Y.; Messner S.; Ehrenberger T.; Zanotelli V.; Butscheid Y.; Escher C.; et al. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteomics 2015, 14 (5), 1400–1410. 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R.; Bernhardt O. M.; Gandhi T.; Xuan Y.; Sondermann J.; Schmidt M.; Gomez-Varela D.; Reiter L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteomics 2017, 16 (12), 2296–2309. 10.1074/mcp.RA117.000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F.; Kolbowski L.; Bernhardt O. M.; Reiter L.; Rappsilber J. Data-Independent Acquisition Improves Quantitative Cross-Linking Mass Spectrometry. Mol. Cell. Proteomics 2019, 18 (4), 786–795. 10.1074/mcp.TIR118.001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese S. H.; Belsom A.; Rappsilber J. Optimized Fragmentation Regime for Diazirine Photo-Cross-Linked Peptides. Anal. Chem. 2016, 88 (16), 8239–8247. 10.1021/acs.analchem.6b02082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese S. H.; Belsom A.; Rappsilber J. Correction to Optimized Fragmentation Regime for Diazirine Photo-Cross-Linked Peptides. Anal. Chem. 2017, 89 (6), 3802–3803. 10.1021/acs.analchem.7b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callister S. J.; Barry R. C.; Adkins J. N.; Johnson E. T.; Qian W.-J.; Webb-Robertson B.-J. M.; Smith R. D.; Lipton M. S. Normalization Approaches for Removing Systematic Biases Associated with Mass Spectrometry and Label-Free Proteomics. J. Proteome Res. 2006, 5 (2), 277–286. 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G. R.; Bolker B.; Bonebakker L.; Gentleman R.; Liaw W. H. A.; Lumley T.; Maechler M.; Magnusson A.; Moeller S.; Schwartz M.; Venables B.. gplots: Various R Programming Tools for Plotting Data; CRAN, 2016.
- Vizcaíno J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Ríos D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; et al. Proteome X change Provides Globally Coordinated Proteomics Data Submission and Dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; et al. 2016 Update of the PRIDE Database and Its Related Tools. Nucleic Acids Res. 2016, 44 (D1), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz N.; Suárez D. Role of the Protonation State on the Structure and Dynamics of Albumin. J. Chem. Theory Comput. 2016, 12 (4), 1972–1988. 10.1021/acs.jctc.5b01001. [DOI] [PubMed] [Google Scholar]
- Bos O. J.; Labro J. F.; Fischer M. J.; Wilting J.; Janssen L. H. The Molecular Mechanism of the Neutral-to-Base Transition of Human Serum Albumin. Acid/base Titration and Proton Nuclear Magnetic Resonance Studies on a Large Peptic and a Large Tryptic Fragment of Albumin. J. Biol. Chem. 1989, 264 (2), 953–959. [PubMed] [Google Scholar]
- Yamasaki K.; Maruyama T.; Yoshimoto K.; Tsutsumi Y.; Narazaki R.; Fukuhara A.; Kragh-Hansen U.; Otagiri M. Interactive Binding to the Two Principal Ligand Binding Sites of Human Serum Albumin: Effect of the Neutral-to-Base Transition. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1999, 1432 (2), 313–323. 10.1016/S0167-4838(99)00098-9. [DOI] [PubMed] [Google Scholar]
- Yang J.; Ha C.-E.; Bhagavan N. V. Site-Directed Mutagenesis Study of the Role of Histidine Residues in the Neutral-to-Basic Transition of Human Serum Albumin. Biochim. Biophys. Acta, Gen. Subj. 2005, 1724 (1–2), 37–48. 10.1016/j.bbagen.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Ascenzi P.; Fasano M. Allostery in a Monomeric Protein: The Case of Human Serum Albumin. Biophys. Chem. 2010, 148 (1–3), 16–22. 10.1016/j.bpc.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Wenskowsky L.; Schreuder H.; Derdau V.; Matter H.; Volkmar J.; Nazaré M.; Opatz T.; Petry S. Identification and Characterization of a Single High-Affinity Fatty Acid Binding Site in Human Serum Albumin. Angew. Chem., Int. Ed. 2018, 57 (4), 1044–1048. 10.1002/anie.201710437. [DOI] [PubMed] [Google Scholar]
- Kumar Y.; Tayyab S.; Muzammil S. Molten-Globule like Partially Folded States of Human Serum Albumin Induced by Fluoro and Alkyl Alcohols at Low pH. Arch. Biochem. Biophys. 2004, 426 (1), 3–10. 10.1016/j.abb.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Ghuman J.; Zunszain P. A.; Petitpas I.; Bhattacharya A. A.; Otagiri M.; Curry S. Structural Basis of the Drug-Binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353 (1), 38–52. 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- Godoy L. C.; Muñoz-Pinedo C.; Castro L.; Cardaci S.; Schonhoff C. M.; King M.; Tórtora V.; Marín M.; Miao Q.; Jiang J. F.; et al. Disruption of the M80-Fe Ligation Stimulates the Translocation of Cytochrome c to the Cytoplasm and Nucleus in Nonapoptotic Cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (8), 2653–2658. 10.1073/pnas.0809279106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascenzi P.; Coletta M.; Wilson M. T.; Fiorucci L.; Marino M.; Polticelli F.; Sinibaldi F.; Santucci R. Cardiolipin-Cytochrome c Complex: Switching Cytochrome c from an Electron-Transfer Shuttle to a Myoglobin- and a Peroxidase-like Heme-Protein. IUBMB Life 2015, 67 (2), 98–109. 10.1002/iub.1350. [DOI] [PubMed] [Google Scholar]
- Kagan V. E.; Tyurin V. A.; Jiang J.; Tyurina Y. Y.; Ritov V. B.; Amoscato A. A.; Osipov A. N.; Belikova N. A.; Kapralov A. A.; Kini V.; et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1 (4), 223–232. 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Capdevila D. A.; Oviedo Rouco S.; Tomasina F.; Tortora V.; Demicheli V.; Radi R.; Murgida D. H. Active Site Structure and Peroxidase Activity of Oxidatively Modified Cytochrome c Species in Complexes with Cardiolipin. Biochemistry 2015, 54 (51), 7491–7504. 10.1021/acs.biochem.5b00922. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Borsari M.; Loschi L.; Martinelli A.; Sola M. Thermodynamics of the Alkaline Transition of Cytochrome c. Biochemistry 1999, 38 (25), 7900–7907. 10.1021/bi983060e. [DOI] [PubMed] [Google Scholar]
- Rozbeský D.; Rosůlek M.; Kukačka Z.; Chmelík J.; Man P.; Novák P. Impact of Chemical Cross-Linking on Protein Structure and Function. Anal. Chem. 2018, 90 (2), 1104–1113. 10.1021/acs.analchem.7b02863. [DOI] [PubMed] [Google Scholar]