

SUMMARY
The mammalian sirtuin, SIRT6, is a key tumor suppressor that maintains genome stability and regulates transcription, though how SIRT6 family members control genome stability is unclear. Here, we use multiple genome-wide approaches to demonstrate that the yeast SIRT6 homologs, Hst3 and Hst4, prevent genome instability by tuning levels of both coding and noncoding transcription. While nascent RNAs are elevated in the absence of Hst3 and Hst4, a global impact on steady-state mRNAs is masked by the nuclear exosome, indicating that sirtuins and the exosome provide two levels of regulation to maintain transcription homeostasis. We find that, in the absence of Hst3 and Hst4, increased transcription is associated with excessive DNA-RNA hybrids (R-loops) that appear to lead to new DNA double-strand breaks. Importantly, dissolution of R-loops suppresses the genome instability phenotypes of hst3 hst4 mutants, suggesting that the sirtuins maintain genome stability by acting as a rheostat to prevent promiscuous transcription.
Graphical Abstract
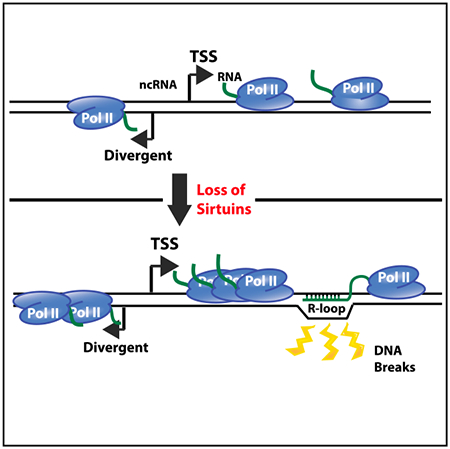
In Brief
Members of the Sirt6 family of histone deacetylases are known to prevent genomic instability and to regulate transcription. Feldman and Peterson find that yeast Sirt6 homologs repress transcription, preventing formation of excess RNA-DNA hybrids. Increased R loops are associated with new DNA double-strand breaks, linking transcriptional regulation to genomic stability.
INTRODUCTION
The epigenetic control of gene expression and genome stability plays a central role in ensuring normal cellular function. Dysregulation has been associated with numerous human malignancies, and chromatin factors have emerged as some of the most frequently affected proteins in cancer (Morgan and Shilatifard, 2015; Shah et al., 2014). SIRT6 is a mammalian member of the Sirtuin family of nicotinamide adenine dinucleotide (NAD+) dependent lysine deacetylases that are conserved across all species (Frye, 2000). SIRT6 functions primarily as a lysine 56 (H3-K56Ac) (Michishita et al., 2009; Yang et al., 2009) and lysine 9 (H3-K9Ac) (Michishita et al., 2008) histone H3 deacetylase at promoters to regulate the expression of genes involved in various pathways, including metabolism, pluripotency, inflammation, and ribosome biogenesis (Etchegaray et al., 2015; Kugel and Mostoslavsky, 2014; Kugel et al., 2016). Global changes in transcription in the absence of SIRT6 have not been reported. Deletion of Sirt6 causes major genomic and metabolic instability (Mostoslavsky et al., 2006), and loss of SIRT6 is sufficient to drive tumorigenesis in mice independent of oncogene activation (Sebastián et al., 2012). Mutations of Sirt6 that affect activity have been identified in human cancers (Kugel et al., 2015), and, strikingly, Sirt6 is deleted in ~60% and ~30% of pancreatic and colorectal cancer cell lines, respectively (Sebastián et al., 2012). Together, the results point to an important role for SIRT6 as a tumor suppressor that regulates transcription and maintains genome stability.
Hst3 and Hst4 are the two SIRT6 homologs in yeast that regulate H3-K56Ac levels (Celic et al., 2006; Maas et al., 2006), with the highest deacetylation activity observed during the S/G2 phase transition (Celic et al., 2006; Maas et al., 2006). Similar to Sirt6, deletion of HST3 and HST4 induces a host of genome instability phenotypes, including spontaneous DNA double-strand breaks, replication fork collapse, increased chromosomal loss, impairment of break-induced replication, and heightened susceptibility to genotoxic agents (Brachmann et al., 1995; Celic et al., 2006; Che et al., 2015). Notably, these phenotypes are alleviated by inactivation of the Asf1 subunit of the Rtt109 histone acetyltransferase (HAT) complex or by a non-acetylatable H3-K56R mutant, suggesting that persistent H3-K56 hyperacetylation promotes genomic instability (Celic et al., 2006, 2008; Maas et al., 2006). A prevailing model proposes that DNA damage is caused by the presence of hyperacetylated nucleosomes due to the lack of Hst3 and Hst4 that either impede replication fork progression or destabilize stalled forks (Celic et al., 2006).
Here, we provide evidence of a functional link between increased transcription driven by loss of the sirtuins and the genomic instability phenotype observed in a hst3 hst4 mutant. Using a combination of native elongating transcript sequencing (NET-seq) (Churchman and Weissman, 2011) and RNA sequencing (RNA-seq) analyses, we show that Hst3 and Hst4 are required to repress transcription of coding and non-coding RNAs. Nascent RNAs are increased throughout coding regions in the absence of Hst3 and Hst4, and we also observe a shift in RNA polymerase II (Pol II) occupancy toward transcription start sites (TSSs). In addition, divergent antisense transcription is increased around the −1 nucleosome, similar to what was observed previously at several promoters (Marquardt et al., 2014). Together, the results are consistent with increased transcription initiation at divergent promoters in a hst3 hst4 mutant, providing an additional mechanism utilized by cells to limit divergent ncRNA abundance. Interestingly, we find that increased nascent RNA is not reflected in the steady-state mRNA pool due to activity of the nuclear exosome. This impact of the exosome was also seen previously in a rtt109Δ mutant that lacked H3-K56Ac. We further use DNA-RNA immunoprecipitation with deep sequencing (DRIP-seq) (Ginno et al., 2012) analyses to identify loci with increased R-loop levels in the absence of Hst3 and Hst4. We show that a subset of regions with increased R-loops are also prone to the formation of DNA double-strand breaks, and we find that overexpression of human RNase-H1 suppresses the sensitivity of a hst3 hst4 mutant to genotoxic stress. Together, the results indicate that the sirtuins function to regulate transcription in order to prevent pervasive R-loop formation and subsequent genomic instability.
RESULTS
Hst3 and Hst4 Repress Nascent RNA Transcription
Deletion of HST3 and HST4 leads to genomic instability (Brachmann et al., 1995; Celic et al., 2006), making such strains susceptible to second-site suppressor mutations. Notably, previous studies showed that the loss of both Hst3 and Hst4 is needed in order to observe measurable phenotypes (Celic et al., 2006), indicating that they perform redundant roles. Therefore, we wanted to establish an alternative approach to characterize the impact of Hst3 and Hst4 on transcription. To this end, the anchor away system (Haruki et al., 2008) was used to conditionally deplete Hst3 from the nucleus in a hst4Δ strain by tagging the C terminus of the HST3 locus with the FKBP12-rapamycin-binding (FRB) domain (hst4Δ HST3-FRB). The parent strain harbors a FK506 binding protein (FKBP12) fused to the C terminus of RPL13A, which is a highly abundant ribosomal protein that shuttles from the nucleus to the cytoplasm during ribosome assembly. A ternary complex between the FRB and FKBP12 domains is formed in the presence of rapamycin and, thus, rapidly depletes Hst3 from the nucleus. In addition, anchor away strains contain a rapamycin resistant tor1-1 allele to ensure rapamycin is not toxic to the wild-type (WT) strain (Haruki et al., 2008).
We first confirmed that depletion of Hst3 in a hst4Δ displays a similar phenotype to a hst3Δ hst4Δ strain by spot dilution assay. The hst4Δ HST3-FRB strain is sensitive to 0.01% methyl metha-nesulfonate (MMS) and 0.1 M hydroxyurea (HU) only in the presence of rapamycin, similar to the hst3Δ hst4Δ strain in the presence of MMS and HU on DMSO (Figure S1A). Consistent with redundant roles for Hst3 and Hst4, the individual HST3-FRB mutant is not sensitive to genotoxic agents in the presence of rapamycin (Figure S1A).
Given that mammalian Sirt6 plays key roles in transcription, we sought to determine the impact of yeast Hst3 and Hst4 on nascent RNA production. We performed NET-seq (Churchman and Weissman, 2011) in WT and the hst4Δ HST3-FRB mutant, using asynchronous cells treated with rapamycin for 3 h. Since we anticipated a potential for global changes in transcription, S. pombe cells were used as a spike-in control for library normalization. Loss of Hst3 and Hst4 led to a global shift in the nascent RNA transcriptome, with an average fold increase of ~1.4 (p < 2 × 10−16, Mann-Whitney U test) (Figure 1A), and approximately a quarter of the genome (1,092 genes) increased by 1.5-fold or greater (false discovery rate [FDR] ≤ 0.1) in the hst4Δ HST3-FRB mutant. Loss of Hst3 and Hst4 has a somewhat larger impact on poorly expressed genes, as the log2 fold change (LFC) between mutant and WT cells is greater for the bottom 25% and 50% of genes transcribed in WT, compared to the top quatriles (p < 2.2 × 10−16, Mann-Whitney U test) (Figure 1B).
Figure 1. Hst3 and Hst4 Globally Repress Nascent Transcription.

(A) Nascent transcript abundance of genes analyzed by NET-seq normalized to S. pombe and adjusted for gene length. Density scatterplots show the log2 mean intensity value for hst4Δ HST3-FRB cells treated with rapamycin for 3 h plotted against WT for two biological replicates. The black line indicates x = y (no change). The red lines indicate ≥ 0.59- or ≤ −0.59-fold change. p value determined by Mann-Whitney U test.
(B) Boxplot comparing the log2 fold change between hst4Δ HST3-FRB and WT mean NET-seq reads for the bottom 25%, bottom middle 25%, top middle 25%, and top 25% of genes expressed in WT. p values determined by Mann-Whitney U test.
(C) Metagene plot of mean NET-seq reads for WT (blue) and hst4 HST3-FRB (green) cells for the bottom 25% of genes expressed in WT (top) and top 25% of genes expressed in WT (bottom). Genes were scaled to 500 bp. Shaded area represents the 95% confidence interval.
(D) Normalized Pol II density forWT (blue) and hst4 HST3-FRB (green) cells. Genes were scaled to 500 bp. NET-seq reads for each gene are normalized by the total number of reads for the region analyzed. Shaded area represents the 95% confidence interval. In A.U., arbitrary units. For all panels, hst4Δ HST3-FRB indicates cells treated for 3 h with rapamycin.
See also Figure S1.
Metagene plots of mean nascent transcript levels, representative genome browser views of NET-seq data, and a heatmap of the log2-fold change between the hst4Δ HST3-FRB mutant and WT confirmed higher levels of transcription throughout genic regions (Figures 1C, S1B, and S1C) in the mutant compared to the WT, especially for genes within the lowest quartile of expression levels (Figure 1C). Genes within the top 25% of the WT expression level also showed increases in nascent RNA, though these increases were greater near the TSS compared to the gene body and transcription termination site (TTS) (Figure 1C, bottom; Figure S1C). In addition, there was an increase in the 5’ to 3’ ratio of RNA transcripts genome wide (p < 2.2 × 10−16, Mann-Whitney U test) (Figure S1D). As further evidence that loss of Hst3 and Hst4 causes a shift in the Pol II distribution toward the TSS, we analyzed the distribution of Pol II after normalizing for differences in overall transcription. In agreement with the 5’ to 3’ ratios (Figure S1D), we observed a shift in Pol II distribution toward the TSS and a corresponding decrease near the TTS (Figure 1D). Taken together, our analyses indicate that Hst3 and Hst4 repress transcription initiation and, furthermore, that the absence of these sirtuins leads to the accumulation of Pol II near the TSS, which may be indicative of increased Pol II pausing.
Hst3 and Hst4 Repress Transcription of Many Non-coding RNAs
Studies in recent years have demonstrated that many eukaryotic promoters are inherently bidirectional (Scruggs et al., 2015; Wei et al., 2011), and transcription termination sequences, RNA degradation complexes, and chromatin modifying factors function to limit the abundance of divergent non-coding RNAs (ncRNAs) relative to mRNAs (Hainer et al., 2015; Huang and Workman, 2013; Marquardt et al., 2014; Wei et al., 2011; Whitehouse et al., 2007). We compared the abundance of divergent antisense nascent transcripts by NET-seq in WT and hst4Δ HST3-FRB mutant cells at tandem genes (2,716) by analyzing antisense reads in the region from −600 to −100 bp from the TSS. We observed a global increase in nascent transcripts up-stream of genes in the absence of Hst3 and Hst4, with an average fold increase of ~1.6 (p < 2 × 10−16, Mann-Whitney U test) (1,051 LFC ≥ 0.59, FDR ≤ 0.1) (Figure 2A). The increase in transcription maps around the −1 and −2 nucleosomes, with little change in the nucleosome depleted region (NDR; Figure 2B). These data are in agreement with previous results that showed increased divergent transcription by northern blot in the absence of Hst3 and Hst4 at several promoters (Marquardt et al., 2014).
Figure 2. Hst3 and Hst4 Repress Divergent Antisense and CUT Transcription.

(A) Nascent antisense transcript abundance upstream of tandem genes(2,716) analyzed by NET-seq and normalized to S. pombe. Density scatterplots show log2 mean intensity values for hst4Δ HST3-FRB plotted against WT fortwo biological replicates. The black line indicates x = y (no change). The red lines indicate ≥ 0.59- or ≤ −0.59-fold change. p value determined by Mann-Whitney U test.
(B) Metagene plot and heatmap displaying the log2 fold changes between hst4Δ HST3-FRB and WT NET-seq antisense reads for tandem genes from −1.0 kb from TSSto 0.2 kb downstream. Black dotted line represents TSS. The average locations ofthe +1, −1, and −2 nucleosomes (Ganguli et al., 2014) are shown for reference.
(C) Nascent transcript abundance of CUTs determined by NET-seq normalized to S. pombe. Density scatterplots show log2 mean intensity value for hst4Δ HST3-FRB plotted against WT. The black line indicates x = y (no change). The red lines indicate ≥ 0.59- or ≤ −0.59-fold change. p value determined by Mann-Whitney U test.
(D) Genome browser view of a representative CUT. NET-seq data (bottom) shown for WT (blue) and hst4Δ HST3-FRB (green).
In addition to divergent antisense transcripts, we investigated the role of Hst3 and Hst4 on cryptic unstable transcript (CUT) levels. CUTs are 5’ capped and polyadenylated ~400 bp transcripts that are rapidly degraded due a high abundance of binding motifs for the Nrd1-Nab1-Sen1 (NNS) termination machinery and subsequent targeting by the nuclear exosome (Arigo et al., 2006; Schulz et al., 2013; Thiebaut et al., 2006). Nascent CUT RNAs are also increased in the hst4 HST3-FRB mutant compared to WT (1.6-fold, p = 6 × 10–13, Mann-Whitney U test) (245 LFC ≥ 0.59, FDR ≤ 0.1) (Figures 2C and 2D). Taken together, our analyses point to an important role for Hst3 and Hst4 in limiting non-coding RNA production.
Steady-State RNA Pool Minimally Affected by Loss of Hst3 and Hst4
Our NET-seq analyses indicated that Hst3 and Hst4 repress the transcription initiation of genes. Therefore, we investigated whether the increase in nascent transcription translated to increased steady-state mRNA levels by analyzing RNA profiles by stranded RNA-seq. Similar to NET-seq analyses, S. pombe cells were used as a spike-in control for library normalization. Unexpectedly, and in contrast to the global increase in nascent transcription, steady-state mRNA levels remained relatively unchanged in the absence of Hst3 and Hst4 (0.981-fold, p = 0.02, Mann-Whitney U test) (Figure 3A). Hst3 and Hst4 negatively regulate the steady-state RNA level of 225 genes (FDR ≤ 0.1, LFC ≥ 0.59, edgeR) and positively regulate 85 genes (FDR ≤ 0.1, LFC ≤ −0.59, edgeR) (Figure S2A). Consistent with what was observed at the nascent RNA level, only very poorly transcribed genes in WT were increased to a greater extent in the mutant compared to the highly transcribed genes (p < 2.2 × 10−16, Mann-Whitney U test) (Figure 3B). Taken together, these results are similar to our previous analyses of the H3-K56 acetyltransferase Rtt109, in which we observed little change in the steady-state mRNA pool despite a global decrease in Pol II occupancy in a rtt109Δ strain (Rege et al., 2015).
Figure 3. Steady-State mRNA Pool Relatively Unchanged in hst4Δ HST3-FRB Mutant.

(A) RNA abundance of genes analyzed by strand-specific RNA-seq normalized to S. pombe and adjusted for gene length. Density scatterplots show log2 mean intensity values for hst4Δ HST3-FRB plotted against WT for two biological replicates. The black line indicates x = y (no change). The red lines indicate ≥ 0.59- or ≤ −0.59-fold change.
(B) (Left) Boxplot comparing the log2 fold changes between hst4Δ HST3-FRB and WT for the bottom 10% or top 10% of genes expressed in WT. p value determined by Mann-Whitney U test. (Right) Genome browser view of WT (blue) and hst4 HST3-FRB (green) RNA-seq reads at a poorly expressed gene in WT (ARG3) and a highly expressed gene (ENO2).
(C) Heatmap of RNA abundance normalized to WT comparing NET-seq and RNA-seq data. k-means clustering was performed.
(D) Cumulative distribution of WT transcript abundance for k-means clusters in (C) for NET-seq data.
See also Figure S2.
The observation that the steady-state mRNA pool remains relatively unchanged even though there is a global increase in nascent RNA production led us to investigate the similarities and differences between the NET-seq and RNA-seq datasets. A k-means clustering approach was used to identify subsets of genes that are differentially regulated at the nascent and steady-state levels (Figure 3C; Table S1; see also Figures S2B and S2C for genome browser views). Group A genes, which are highly transcribed in WT cells (Figure 3D), show small increases at the nascent RNA level, whereas many show an opposite, decreased level in the steady-state RNA pool (Figure 3C). Nascent RNA transcription of the other three groups of genes (groups B–D) was increased in the absence of Hst3 and Hst4, but variable effects are observed at the steady-state level (Figure 3C). With the exception of group B genes, which are the most poorly transcribed genes in WT cells (Figure 3D) and are upregulated in the hst4Δ HST3-FRB mutant by RNA-seq analyses, steady-state mRNA levels are minimally affected (group D) or are decreased (group C) in the hst4Δ HST3-FRB mutant (Figure 3C). The results reveal that many of the increased transcripts observed at the nascent RNA level in the absence of Hst3 and Hst4 are post-transcriptionally regulated and, thus, are not observed in the steady-state RNA pool.
Increased Transcription in the Absence of Hst3 and Hst4 Is Masked by the Nuclear Exosome
In addition to a role in regulating ncRNA transcription (Schneider et al., 2012) and processing small nuclear and nucleolar RNAs (snRNAs and snoRNAs) (Gudipati et al., 2012), the nuclear exosome plays a more general role in the surveillance of nuclear mRNAs (Rege et al., 2015; Schmid et al., 2012). To investigate whether the nuclear exosome might be responsible for masking the impact of Hst3 and Hst4 loss on the steady-state RNA pool, the anchor away system was used to deplete the 3’ to 5’ exonuclease subunit, Rrp6, from the nucleus for 3 h, alone or in combination with the hst4Δ HST3-FRB mutant. Interestingly, growth assays revealed an additive effect of depleting Rrp6 in the absence of both Hst3 and Hst4, as the cells become more sensitive to HU compared to either the RRP6-FRB single mutant or the hst4Δ HST3-FRB double mutant (Figure S3A).
RNA-seq was performed in the hst4Δ HST3-FRB RRP6-FRB triple mutant and RRP6-FRB single mutant, and these datasets were compared to the NET-seq and RNA-seq datasets from the hst4Δ HST3-FRB double mutant, using the same gene groups identified in Figure 3C (Figure 4A; Table S2). Inactivation of the nuclear exosome increased steady-state RNA levels to those more closely resembling what was observed by NET-seq in the absence of Hst3 and Hst4 (Figure 4A). Remarkably, many of the RNAs that increased due to depletion of Hst3 and Hst4 were also increased by the single depletion of the RNA exosome (Figures 4A–C, S3B, and S3C), indicating that Hst3 and Hst4 and the nuclear exosome regulate many of the same target genes. However, there are many genes in groups B–D whose expression is increased to a greater extent in the hst4Δ HST3-FRB RRP6-FRB triple mutant compared to the RRP6-FRB single mutant (Figures 4A, 4C, S3B, and S3C), and there are an additional ~800 genes that are increased ≥ 1.5-fold over WT (FDR ≤ 0.1, edgeR) only in the hst4Δ HST3-FRB RRP6-FRB triple mutant (Figures 4C and S3D). Taken together, the RNA-seq analyses in the absence of the nuclear exosome confirm the observations made by NET-seq. Transcription is elevated in the absence of Hst3 and Hst4, and at many loci the nuclear exosome functions to degrade the increased nascent transcripts. In addition, there are genes that are not targeted by Rrp6 for degradation despite increased NET-seq reads (Figure S3E). In particular, at group C genes, which encode for proteins involved in lipid, sterol, and fatty acid metabolism (Table S1), loss of Rrp6 has little to no effect on steady-state mRNA levels. Thus, indicating there are additional co-transcriptional or post-transcriptional mechanisms for regulating steady-state RNA levels in the hst4Δ HST3-FRB mutant.
Figure 4. Nuclear Exosome Masks Increased Transcription in hst4Δ HST3-FRB Mutant.

(A) Heatmap of RNA abundance for clusters identified in Figure 3G comparing hst4Δ HST3-FRB NET-seq and RNA-seq data with RNA-seq data for hst4Δ HST3-FRB RRP6-FRB and RRP6-FRB mutants. Samples from FRB-tagged strains reflect 3 h of rapamycin treatment. Data are normalized to WT and shown as LFC.
(B) Venn diagram showing genes increased ≥ 0.59 LFC compared to WT (FDR ≤ 0.1) in RRP6-FRB and hst4Δ HST3-FRB RRP6-FRB mutant cells.
(C) Genome browser view of the PTH4 gene regulated by Rrp6 displaying NET-seq data (bottom) for WT (blue) and hst4Δ HST3-FRB (green) as well as RNA-seq data (top) for WT (blue), hst4Δ HST3-FRB (green), RRP6-FRB (gray), and hst4Δ HST3-FRB RRP6-FRB (orange).
Sirtuins Prevent the Accumulation of R Loops That Cause Genomic Instability
The hst3 hst4 mutant is sensitive to genotoxic stress, and loss of Hst3 and Hst4 induces many genomic instability phenotypes (Celic et al., 2006). We hypothesized that increased transcription in the hst4Δ HST3-FRB mutant might be an underlying cause of genomic instability by increasing the prevalence of transcription-associated R-loops. We performed DNA-RNA immunoprecipitation sequencing (DRIP-seq) on WT and hst4Δ HST3-FRB asynchronous cells treated with rapamycin for 3 h to deplete Hst3. We analyzed DRIP-seq reads over open reading frames (ORFs) and compared the signals to RNase-H treated controls. Higher DRIP-seq signals were observed in the mutant compared to WT (214 ≥ 0.59 versus 58 ≤ −0.59, FDR ≤ 0.25) (Figures 5A and 5B), indicating that there is an increased abundance of R-loops in the absence of Hst3 and Hst4. Many of the R-loops identified overlapped with previously mapped R-loops in a rnh1Δ rnh201Δ double mutant (Wahba et al., 2016), and we confirmed the increased presence of R-loops at several genes by DRIP-qPCR (Figure S4A).
Figure 5. Increased R-Loop Abundance in the hst4Δ HST3-FRB Mutant.

(A) Representative genome browser view of normalized DRIP-seq reads in WT (blue) and hst4Δ HST3-FRB (green) cells. NET-seq reads shown below.
(B) Heatmap of DRIP-seq reads summed over ORFs normalized to RNase-H controls for two biological replicates. Shown are genes whose DRIP-seq signal increases ≥ 1.3-fold relative to RNase-H control and the FDR for WT versus hst4Δ HST3-FRB is ≤ 0.25.
(C) Metagene analysis of normalized DRIP-seq reads for genes whose DRIP-seq signal increases ≥ 1.3-fold relativeto RNase-H control. Genes scaled to 500 bp. Shaded area represents standard error.
(D) Cumulative distribution of mean NET-seq reads in WT and hst4 HST3-FRB cells for the genes in (C).
See also Figure S4.
Metagene analysis was performed on genes that had a 1.3-fold or greater DRIP-seq signal in the WT or mutant relative to their respective RNase-H controls (1,105 genes), and as expected, R-loop levels were higher in the hst4Δ HST3-FRB mutant compared to WT (Figure 5B). The increased DRIP-seq signals were found over coding regions, consistent with transcription-associated R-loop formation (Figure 5C). Indeed, nascent transcription levels are increased in the absence of Hst3 and Hst4 at these loci (Figures 5A and 5C). Together, the data suggest that R loops form at a subset of genes with increased nascent transcription in the absence of Hst3 and Hst4.
As a secondary method of identifying R loops genome wide, MACS (model-based analysis of chromatin immunoprecipitation sequencing [ChIP-seq]) (Zhang et al., 2008) was used to identify genomic regions that were significantly enriched for DRIP-seq reads (667 peaks, p < 1 × 10−5) in the hst4Δ HST3-FRB mutant (Figure S4B). Of these, 588 peaks overlapped genes, and there was ~40% overlap with R-loop-enriched genes identified in Figure 5B( Figure S4C). In addition to ORFs, many of the peaks overlap with tRNA, transposable elements, long terminal repeats, and snRNAs and snoRNAs (Figures S4B and S4D), suggesting a possible role for Hst3 and Hst4 in regulating other genomic loci in addition to coding regions.
Recently, it was reported that a small subset of R loops in a sen1Δ rnh1Δ rnh201Δ mutant are hotspots for irreparable DNA damage (Costantino and Koshland, 2018). These persistent sites for R loops hinder DNA repair pathways, leading to large stretches of single-stranded DNA (Costantino and Koshland, 2018), which can function as precursors for gross chromosomal rearrangements (GCRs). To investigate if there are DNA damage sites proximal to DRIP-seq peaks found in the hst4Δ HST3-FRB mutant, we performed Break-seq to identify DNA double-strand breaks (DSBs) (Hoffman et al., 2015) in hst4Δ HST3-FRB and WT cells treated with rapamycin for 3 h. MACS (Zhang et al., 2008) was used to identify genomic regions that were significantly enriched for end-labeled, DSB signals (239 peaks, p < 1 × 10−5) in the hst4Δ HST3-FRB mutant (Figures 6A, 6B, and S5A). Of these, 71 peaks overlapped within a 4-kb region centered around genomic regions that had DRIP-seq peaks in the hst4Δ HST3-FRB mutant (Figures 6A, 6B, and S5A). The large regions of DSBs around R loops (Figure 6B) are similar to what was observed previously for Rad52 ChIP-seq peaks identified in a rnh1Δ rnh201Δ SEN1-AID, consistent with impeded DNA repair. Together, the results identify new sites of DNA damage that are proximal to R loops in the hst4Δ HST3-FRB mutant.
Figure 6. Role of R Loops for Increased DSBs in the hst4Δ HST3-FRB Mutant.

(A) Mean log2 MACS peak signal (p < 1 × 10−5) for two Break-seq biological replicate datasets. Blue boxes are peaks that overlap within 4 kb around genomic regions with DRIP-seq peaks in the hst4Δ HST3-FRB mutant identified by MACS.
(B) Representative genome browser views of two biological replicates of Break-seq data for hst4Δ HST3-FRB and WT using SeqMonk. Genes and DRIP-seq peaks are shown above.
(C) WT anchor away or hst4Δ HST3-FRB strains transformed with empty vector or a vector overexpressing human RNase-H1. Strains were spotted (1/10 dilutions) on 2% glucose media containing either DMSO solvent or 8 μg/mL rapamycin in the presence orabsence of 0.1 M hydroxyurea (HU) and then grown for 3 days at 30°C. Two transformants were spotted for each strain.
See also Figure S5.
To provide functional support for the hypothesis that R loops cause genomic instability in the absence of Hst3 and Hst4, we overexpressed human RNase-H1 in WT and hst4Δ HST3-FRB cells and monitored their sensitivity to genotoxic agents by spot dilution assay. Expression of human RNase-H1 was previously shown to reduce R-loop levels in vivo in yeast (Wahba et al., 2011). Overexpression of RNase-H1 did not affect the growth of WT cells on DMSO or rapamycin, nor in the presence of 0.1 M HU, 0.005% MMS, or 5 μg/mL camptothecin (CPT) (Figures 6C and S5B). Strikingly, RNase-H1 overexpression partially suppressed the genomic instability of the hst4Δ HST3-FRB mutant grown on rapamycin in the presence of HU, MMS, and CPT (Figures 6C and S5B), providing direct evidence that increased R loops, in the absence of Hst3 and Hst4, cause genomic instability.
DISCUSSION
Changes in transcription are tightly regulated to ensure transcription homeostasis, and mis-regulation can have widespread effects on cellular function. The mammalian tumor suppressor SIRT6 functions primarily as a H3-K56 and H3-K9 deacetylase at specific genes (Kugel and Mostoslavsky, 2014), and loss of SIRT6 is sufficient to drive tumorigenesis (Sebastián et al., 2012). The data presented here reveal a link between pervasive, unregulated transcription and the genomic instability phenotypes observed in the absence of the yeast SIRT6 homologs: Hst3 and Hst4. By using a combination of RNA sequencing methodologies (NET-seq and RNA-seq), yeast genetics, DRIP-seq, and Break-seq, we demonstrate that Hst3 and Hst4 are globally required to repress nascent RNA transcription, and transcription-associated R loops and DNA double-strand breaks are elevated in a hst3 hst4 mutant. Furthermore, increased R loops appear to be causative for genomic instability, at least in part, as we observe co-localization of both R loops and DSBs, and overexpression of human RNase H1 can partially alleviate the sensitivity of an hst3 hst4 double mutant to genotoxic stress. Given this partial suppression phenotype, it may be that other pathways are regulated by Hst3 and Hst4 independent of transcriptional regulation. For instance, mammalian Sirt6 also deacetylates nonhistone substrates that impact DNA repair pathways, and it also functions in telomere protection (reviewed in Kugel and Mostoslavsky, 2014). Likewise, several studies have found that increases in transcription are sometimes not sufficient for the formation of R loops (Bayona-Feliu et al., 2017; Salas-Armenteros et al., 2017).
Promoter-proximal nucleosomes flanking active genes are highly acetylated (Rufiange et al., 2007; Yang et al., 2016) and display rapid, replication-independent nucleosome turnover (Dion et al., 2007; Rufiange et al., 2007; Yang et al., 2016). These results suggest that enhanced nucleosome dynamics and histone acetylation are generally beneficial for transcription. Consistent with this view, we find that nascent transcription increases globally in the absence of Hst3 and Hst4, likely due to increased histone acetylation levels, such as H3-K56Ac and/or H3-K9Ac. The increase in nascent transcription is predominantly observed around gene promoters, consistent with enhanced promoter nucleosome dynamics and the creation of a more favorable environment for transcription initiation. While promoter-proximal pausing of Pol II has not been observed in yeast, NET-seq profiles in WT strains show a general accumulation of nascent, coding transcripts near the 5’ end of genes. This position correlates with the location of the +2 nucleosome and the transition point from Pol II transcription initiation to productive elongation, which has been reported to function as a “kinetic” checkpoint (Buratowski, 2009; Rodríguez-Molina et al., 2016). After depletion of Hst3 and Hst4, we observed a greater number of RNA molecules near the 5’ end compared to the 3’ end and the gene body, indicating that many of the polymerases may not transition to productive elongation. This could be a direct effect of increased histone acetylation or indicate that the transition to transcription elongation is influenced by the density of Pol II. Consistent with the latter model, high-density Pol II genes are targets for the nuclear exosome (Rege et al., 2015). Even though termination factors, Nrd1 and Nab3, have been shown to preferentially bind ncRNAs (Schulz et al., 2013), transcription attenuation has been observed for protein coding genes (Colin et al., 2011; Mischo and Proudfoot, 2013). Furthermore, Nrd1 was reported to cross-link to the 5’ end of highly expressed genes (Creamer et al., 2011), providing additional support for the second model.
The significant increase in nascent RNA, combined with minimal changes in steady-state RNA, indicated that post-transcriptional processes are functioning to regulate the increase in nascent transcription. Rapid depletion of the 3’ to 5’ exonuclease subunit of the nuclear RNA exosome, Rrp6, increased steady-state RNA levels, such that expression in the hst4Δ HST3-FRB RRP6-FRB strain more closely resembled nascent transcript abundance observed in the absence of Hst3 and Hst4. Thus, the nuclear exosome appears to repress the increased nascent transcription observed in the hst4Δ HST3-FRB mutant in order to maintain similar steady-state levels of mRNA in the absence of Hst3 and Hst4. We found that rapid depletion of the nuclear exosome, in an otherwise WT strain, increased expression levels of nearly 800 protein-coding genes, and the majority of these genes are also regulated by Hst3 and Hst4. This suggests that histone deacetylation and post-transcriptional exosome activity provides two distinct levels of regulation to maintain transcription homeostasis of a common set of genes. The results are similar to previous studies showing that mRNA synthesis and degradation are dynamically balanced to buffer against changes in either process (Timmers and Tora, 2018). Our results presented here, as well as our previous data for cells lacking the H3-K56 acetyltransferase, Rtt109 (Rege et al., 2015), indicate that a functional relationship might exist between H3-K56Ac, Pol II abundance, and nuclear exosome activity, which may affect the number of polymerases that proceed to productive elongation.
Deletion of HST3 and HST4 induces many genome instability phenotypes (Celic et al., 2006; Che et al., 2015), which can be suppressed by an H3-K56R substitution derivative or by inactivation of the Asf1 subunit of the Rtt109 HAT complex (Celic et al., 2006, 2008; Maas et al., 2006), indicating that H3-K56 hyperacetylation is the cause of the genomic instability. Hst3 and Hst4 remove H3-K56Ac throughout the cell cycle, with the highest activity observed during S phase (Celic et al., 2006; Maas et al., 2006). Chronic γ-H2A phosphorylation is observed in a hst3Δ hst4Δ strain (Celic et al., 2006), and overexpression of a clamp loader protein, RFC1, suppresses hst3 hst4 phenotypes (Celic et al., 2008), indicating that Hst3 and Hst4 prevent DNA damage during replication. The current prevailing model is that prolonged hyperacetylation of H3-K56 in the absence of Hst3 and Hst4 leads to DNA damage by causing the replication machinery to interact with parental nucleosomes ahead of the fork that are H3-K56 acetylated, for example by creating a roadblock that impedes fork progression or affects fork stability (Celic et al., 2006). However, H3-K56Ac nucleosomes are more dynamic than non-H3-K56Ac nucleosomes (Kaplan et al., 2008; Rufiange et al., 2007; Yang et al., 2016), which suggests that they should not impede the replication fork.
We investigated an alternative hypothesis that pervasive, unregulated transcription in a hst3 hst4 mutant poses a risk to the cell by increasing the propensity for transcription-associated R-loop formation. Many of the phenotypes observed in hst3 hst4 mutants are similar to what is observed in cells with unregulated R loops. Previous studies indicate that unregulated R-loops are a source of genomic instability by hindering DNA repair pathways and causing DNA damage during replication (Aguilera and Gómez-González, 2017; Costantino and Koshland, 2018). R-loop abundance, measured by DRIP-seq, is increased over genes that have higher nascent transcription in the absence of Hst3 and Hst4. Furthermore, Break-seq reads are higher in several regions that had DRIP-seq peaks in the hst4Δ HST3-FRB mutant, pointing to the presence of R-loop-induced DNA damage at these sites. Strikingly, overexpression of human RNase-H1 partially suppresses the sensitivity of the hst4Δ HST3-FRB mutant to genotoxic agents, providing direct evidence that R loops contribute in part to the genomic instability of hst3 hst4 mutants. Unregulated R loops in the absence of Hst3 and Hst4 might directly impact replication fork stability, providing an alternative model for how the absence of Sirtuins impacts the replisome.
Our findings suggest an additional chromatin-based mechanism used by cells to regulate transcription and demonstrate that Hst3 and Hst4 function to maintain genome stability by repressing transcription of both coding and non-coding RNAs genome wide. Increased transcription in a hst3 hst4 mutant leads to genomic instability by increasing the abundance of R loops, providing a functional link between pervasive transcription and genomic instability. Given the phenotypic similarities between yeast and mammalian Sirt6 mutants, it seems likely that this functional relationship is evolutionarily conserved.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Craig Peterson (craig.peterson@umassmed.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The S. cerevisiae strains used here are derived from HHY168 (MATα tor1-1 fpr1::NAT RPL13A-2X FKBP12::TRP1 ade 2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL psi+) and HHY221 (MATa tor1-1 fpr1::loxP-LEU2-loxP RPL13A-2 × FKBP12::loxP ade 2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL psi+). Growth conditions are detailed in the Method Details section. The full strain and plasmid list is shown in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG M2 Affinity Gel | Millipore Sigma | Cat#A2220, RRID:AB_10063035 |
| Anti-DNA-RNA Hybrid Antibody, clone S9.6 | Millipore Sigma | Cat#MABE1095 |
| Dynabeads M-280 Sheep Anti-Mouse IgG | Thermo Fisher Scientific | Cat#11201D, RRID: AB_2783640 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Rapamycin | LC Laboratories | Cat#R-5000 |
| Hydroxyurea | US Biological Life Sciences | Cat#127-07-1 |
| Methyl methanesulfonate | Millipore Sigma | Cat#129925 |
| Camptothecin | Millipore Sigma | Cat#CP9911 |
| Taq DNA polymerase | NEB | Cat#M0273S |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080093 |
| Random Primers | Thermo Fisher Scientific | Cat#48190011 |
| RNaseH | Thermo Fisher Scientific | Cat#18021071 |
| AmPure XP | Beckman Coulter | Cat#A63880 |
| RNase-Free DNase Set | QIAGEN | Cat#79254 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher Scientific | Cat#65001 |
| T4 DNA Ligase | New England Biolabs | Cat#M0202 |
| T4 DNA Polymerase | New England Biolabs | Cat#M0203 |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | Cat#M0201 |
| NEB Buffer 2 | New England Biolabs | Cat#B7002 |
| dATP Solution | New England Biolabs | Cat#N0440 |
| Klenow Fragment 3’ to 5’ Exo | New England Biolabs | Cat#M0212 |
| T4 DNA Ligase (Rapid) | Enzymatics | Cat#L6030-HC-L |
| Uracil-DNA Glycosylase (UDG) | New England Biolabs | Cat#M0280 |
| Phusion High Fidelity DNA Polymerase | New England Biolabs | Cat#M0530 |
| Ribo-Zero Gold rRNA Removal Kit | Illumina | Cat#MRZY1324 |
| RQ1 Rnase-Free Dnase | Promega | Cat#M6101 |
| Manganese(II) Chloride Solution | Millipore Sigma | Cat#M1787 |
| 3× FLAG Peptide | Millipore Sigma | Cat#F4799 |
| SUPERase.In Rnase Inhibitor | Thermo Fisher Scientific | Cat#AM2694 |
| cOmplete, EDTA-Free Protease Inhibitor Cocktail | Millipore Sigma | Cat#11873580001 |
| T4 RNA Ligase 2, truncated | New England Biolabs | Cat#M0242 |
| Gel Loading Buffer II | Thermo Fisher Scientific | Cat#8546G |
| 10 bp DNA Ladder | Thermo Fisher Scientific | Cat#10821-015 |
| SYBR Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S11494 |
| GlycoBlue Coprecipitant | Thermo Fisher Scientific | Cat#AM9515 |
| CircLigase ssDNA Ligase | Lucigen | Cat#CL4111K |
| Zymolase 20T | Seikagaku Biobusiness | Cat#120491 |
| HindIII-HF | NEB | Cat#R3104M |
| EcoRI-HF | NEB | Cat#R310M |
| XbaI | NEB | Cat#R0145M |
| SspI-HF | NEB | Cat#R3132M |
| BsrG1-HF | NEB | Cat#R3575L |
| NEB Buffer 2.1 | NEB | Cat#B7202S |
| RNase H | NEB | Cat#M0297L |
| Proteinase K | Millipore Sigma | Cat#P2308 |
| Oligo(dT)12- 18 Primer | Thermo Fisher Scientific | Cat#18418012 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A25742 |
| Apex dNTP Set | Genesee Scientific | Cat #42-403 |
| Biotin-14-dATP | Thermo Fisher Scientific | Cat#19524016 |
| β-agarase | NEB | Cat#M0392L |
| HiFi HotStart Ready Mix | Kapa Biosystems | Cat#KK2601 |
| NuSieve GTG Agarose | Lonza | Cat#50081 |
| Critical Commercial Assays | ||
| NextSeq 500/550 High Output v2 Kit (75 cycles) | Illumina | Cat#FC-404-2005 |
| NextSeq 500/550 Mid Output v2 Kit (150 cycles) | Illumina | Cat#FC-404-2001 |
| QIAGen miRNeasy Mini Kit | QIAGEN | Cat#217004 |
| QIAGen Rneasy Mini Kit | QIAGEN | Cat#74104 |
| RNA clean and concentrator - 5 | Zymo Research | Cat#R1013 |
| Qubit 1X dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q33230 |
| End-It DNA End-Repair Kit | Lucigen | Cat#ER81050 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE124132 |
| Software and Algorithms | ||
| GraphPad Prism v 5.0 | GraphPad Software Inc | N/A |
| R | https://www.r-project.org/ | |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| TopHat2 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml. RRID: SCR_013035 |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/. RRID: SCR_002105 |
| Bedtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2. RRID: SCR_006646 |
| deepTools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ RRID:SCR_016366 |
| PIVOT | Zhu et al., 2018 | http://kim.bio.upenn.edu/software/pivot.shtml |
| HTseq 0.9.1 | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/install.html RRID:SCR_005514 |
| Galaxy web platform | Afgan et al., 2018 | https://usegalaxy.org |
| MACS Peak Caller | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/, RRID:SCR_013291 |
| SeqMonk | http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/, RRID:SCR_001913 | |
| Other | ||
| Whatman nitrocellulose membrane filters | Millipore Sigma | Cat#7184-009 |
| Mixer Mill MM 400 | Retsch | |
| Corning Costar SpinX Centrifuge Tube Filters | Millipore Sigma | Cat#CL8162 |
| Qubit Assay tubes | Thermo Fisher Scientific | Cat#Q32856 |
| Novex TBE-Urea Gels 15% | Thermo Fisher Scientific | Cat#EC6885BOX |
| Novex TBE-Urea Gels 10% | Thermo Fisher Scientific | Cat#EC6875BOX |
METHOD DETAILS
Spot Dilution Plate Assays
For serial dilution spot plate assays, cells were cultured to saturation in 5 mL YPD or SD (-Ieu). Yeast was diluted to an OD600 of 1.0 in sterile dH2O, serially diluted 10-fold four times, and 5 μL of each dilution was spotted onto plates of indicated media. Where used, DMSO was 0.1% vol/vol, rapamycin was 8 μg/mL, methyl methanesulfonate was 0.01% or 0.005% wt/vol, hydroxyurea was 0.1 M, and camptothecin was 5 μg/mL.
NET-seq
Library Construction
NET-seq libraries were produced as described in (Churchman and Weissman, 2011) for 4 WT replicates and 3 hstΔ HST3-FRB replicates. Briefly, overnight cultures from single yeast colonies were diluted to an OD600 = 0.05 in 1 L of YPD. Cells were grown at 30°C until OD600 = 0.25. Rapamycin was added at a final concentration of 8 μg/mL and cells were grown for 3 h (OD600 = 0.7-0.8). We utilized a S. pombe spike-in, which contained a flag-tagged Rpb3 subunit (JY741), to normalize the sequencing libraries. S. pombe cells were mixed with S. cerevisiae cells at a 1:10 ratio, and the cells were harvested as described in (Churchman and Weissman 2011). RNA Pol II IP, RNA purification, and library construction was carried out as described previously (Churchman and Weissman, 2011; Mayer et al., 2015). 3’ end sequencing was performed on an Illumina NextSeq 500 with a read length of 75 bp.
Data Analysis
NET-seq reads were processed and aligned as follows using the Galaxy web platform (Afgan et al., 2018). The adaptor sequence was (ATCTCGTATGCCGTCTTCTGCTTG) removed and the random hexamer sequence was removed from the 5’ end. The 3’ ends of the reads were then trimmed for quality using FASTQ Quality Timmer by sliding window (Blankenberg et al., 2010) with a window size of 10 and a step size of 5. The reads were trimmed until the aggregate score was ≤ 21. Reads were first aligned using Bowtie2 (Langmead and Salzberg, 2012; Langmeadet al., 2009) to a combined FASTA file of S. cerevisiae and S. pombe rRNA, tRNA, and RDN sequences to remove contaminating reads. Reads were then aligned to a combined version of the S. cerevisiae genome (SacCer3, SGD) and the S. pombe genome (ASM294v.2, PomBase) with TopHat2 (Kim et al., 2013), allowing up to three mismatches. The reads were separated by their respective genomes with SAMtools (Li et al., 2009), and only uniquely mapped reads were used for further analyses. Libraries were normalized by scaling the uniquely mapped S. pombe reads to 100,000 reads. This scaling factor was then used to scale the uniquely mapped S. cerevisiae reads. To account for differences between sequencing run depth for various NextSeq runs, the pombe-scaled WT S. cerevisiae read counts were then scaled to 1 M reads, and this additional scaling factor was included to scale the sample reads. Finally, only the 5’ end of the sequencing read, which corresponds to the 3’ end of the nascent RNA was recorded and used for downstream analyses. TSS and TTS annotation was obtained from Xu et al. (2009). Read counts for genes and non-coding regions were obtained by summing normalized base pair reads over the region of interest. For average profiles, BAM files of biological replicates were merged and processed as above, and only genes longer than 500 bp were analyzed. Genes were scaled to 500 bp, and samples were scored in 1 bp bins using the deepTools program (Ramírez et al., 2016). Reads were analyzed as in Harlen et al. (2016). To calculate 5’ to 3’ ratios, the sum of reads from 1-250 bp from the TSS were divided by the sum of reads 250 bp upstream of the TTS to the TTS.
RNA-seq
Library Construction
Strand-specific RNA-seq libraries without polyA selection were prepared similarly to Zhang et al. (2012) for 2 biological replicates. Briefly, overnight cultures from single yeast colonies were diluted to an OD600 = 0.05 in 50 mL of YPD. Cells were grown at 30°C until OD600 = 0.25. Rapamycin was added at a final concentration of 8 μg/mL and cells were grown for 3 h (OD600 = 0.7). We utilized a S. pombe spike-in to normalize the sequencing libraries. S. pombe cells were mixed with S. cerevisiae cells at a 1:6 ratio, and cells were harvested by centrifugation. RNA was purified by hot-phenol extraction and ethanol precipitation. RNA was further purified by RNeasy Miniprep kit and the DNA was digested. RiboZero magnetic beads (Illumina) were then used to remove rRNAs from 3 μg of RNA. Libraries were then prepared as in Zhang et al. (2012), and paired-end sequencing was performed on an Illumina NextSeq 500 with a read length of 75 bp.
Data Analysis
FASTQ files from paired end libraries were collapsed by barcode and the Illumina adaptor sequence was trimmed from the 3’ end. Files were uploaded and analyzed using the Galaxy web platform (Afgan et al., 2018). Reads were first aligned using Bowtie2 (Langmead and Salzberg, 2012; Langmead et al., 2009) to a combined FASTA file of S. cerevisiae and S. pombe rRNA, tRNA, and RDN sequences to remove contaminating reads. Reads were then aligned to a combined version of the S. cerevisiae genome (SacCer3, SGD) and the S. pombe genome (ASM294v.2, PomBase) with TopHat2 (Kim et al., 2013) allowing up to two mismatches. The reads were separated by their respective genomes with SAMtools (Li et al., 2009), and only uniquely mapped reads were used for further analyses. For visualization in USCS genome browser, libraries were normalized by scaling the uniquely mapped S. pombe reads to 100,000 reads. To compare RNA expression between samples, HTseq 0.9.1 (Anders et al., 2015) was used to count the number of reads that aligned to each annotated gene. The annotation file for the S. cerevisiae genome was generated from the Xu et al. dataset (Xu et al., 2009), and the S. pombe annotation file was obtained from PomBase. Normalized S. cerevisiae read counts were generated by using a linear regression model to scale the S. pombe read counts relative to WT biological replicate 1 to determine scaling factors for S. cerevisiae read counts. Differential expression analysis was performed using edgeR (Robinson et al., 2010) or in excel using the R qvalue package.
RT-qPCR
DNase-treated RNA (1 μg) was reverse transcribed using SuperScript III (Invitrogen) and oligo-dT according to the manufacturer protocol. qPCR reactions were performed with PowerUp Syber Green Master Mix (Applied Biosystems). Primer sequences are listed in Supplemental Information. Relative mRNA levels were quantified in duplicate for biological replicates using the highly expressed PDC1 gene for normalization. Significance was determined by Student’s t test.
DRIP-seq
Library Construction
DNA-RNA hybrid immunoprecipitation sequencing (DRIP-seq) was performed using the DNA-RNA hybrid specific S9.6 antibody similarly to Bonnet et al. (2017) for 2 biological replicates. Briefly, overnight cultures from single yeast colonies were diluted to an OD600 = 0.1 in 200 mL of YPD. Cells were grown at 30°C until OD600 ≅ 0.3. Rapamycin was added at a final concentration of 8 mg/mL and cells were grown for 3 h (OD600 ≅ 0.8). Genomic DNA was extracted by spheroplasting and EtOH precipitation, excluding a RNase-A digestion. Purified DNA (50 μg) was digested by a cocktail of restriction enzymes (50 U HindIII, EcorI, XbaI, SspI, BsrGI, NEB) in the presence or absence of 30 U of RNase-H (NEB) overnight at 37°C in a final volume of 110 μL. Digested DNA was further diluted to 500 uL with FA-lysis buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) and the samples were pre-cleared with Dynabeads M-280 Sheep Anti-Mouse IgG (ThermoFisher) for 3 h. DNA was then incubated overnight in the presence of 3 μg of S9.6 antibody (Millipore). Immunoprecipitated DNA fragments were captured using Dynabeads M-280 Sheep Anti-Mouse IgG (ThermoFisher), washed. and eluted according standard ChIP procedures (Bennett et al., 2013). Prior to phenol:chloroform extraction and precipiateion, the eluted DNA was incubated with 200 μg of Proteinase Kfor 1 h at 42°C while shaking at 1000 rpm to ensure complete antibody removal. Libraries were prepared by adaptor ligation and PCR amplification for paired-end sequencing on an Illumina NextSeq 500 with a read length of 75 bp.
Data Analysis
FASTQ files from paired end libraries were collapsed by barcode and the Illumina adaptor sequence was trimmed from the 3’ end. Files were uploaded and analyzed using the Galaxy web platform (Afgan etal., 2018). Reads were aligned to the S. cerevisiae genome (SacCer3, SGD) using Bowtie2 with a maximum fragment length for valid paired-end alignments set to 500 bp. Aligned reads were then filtered for quality and only uniquely mapped paired reads were used for future analys3s. For visualization in USCS genome browser and metagene plots, aligned libraries were merged and normalized by scaling to 1 million reads. RNase-H control IP signal was subtracted from the matched sample. To calculate DRIP-seq signals over genes, BEDtools was used to calculate normalized bp counts across the genome for each replicate individually. Subsequent analysis was performed on median read coverage across 100-bp windows with a 50-bp sliding window. The median read counts were then summed over genes. DRIP-seq enriched genes were determined similarly to Zeller et al. (2016) by comparing DRIP-seq signals to RNase-H treated controls. The fold change in the signal versus a matched RNase-H control was calculated, and a 1.3-fold cutoff in the average of the two replicates for either WT or the hst4Δ HST3-FRB mutant was used to identify genes enriched for R-loops. Differential analysis between WT and the hst4Δ HST3-FRB mutant was performed using the R qvalue package. MACS Peak Caller was used to identify new peaks in a non-biased manner in the hst4Δ HST3-FRB biological replicates using the mean WT file as the input (p value 1×10−5, 500 bp fragment size).
DRIP-qPCR
Cells were grown, genomic DNA was purified, and immunoprecipitated as done for DIP-seq. qPCR reactions were performed with PowerUp Syber Green Master Mix (Applied Biosystems). Primer sequences are listed in Supplemental Information. R-loop levels were determined relative to a matched RNase-H-treated sample for 3 biological replicates. Significance was determined by Student’s t test.
Break-seq
Library Construction
Break-seq libraries were prepared similarly to Hoffman et al. (2015) for 2 biological replicates. Briefly, overnight cultures from single yeast colonies were diluted to an OD600 = 0.1 in 100 mL of YPD. Cells were grown at 30°C until OD600 ≅ 0.32. Rapamycin was added at a final concentration of 8 mg/mLand cells were grown for 3 h (OD600 = 1.0). Cells were harvested and resuspended in 1 mL of 50 mM EDTA. Cells (50 μL) were combined with 50 μL of 1% low melting temperature agarose (Lonza). Following in gel labeling and sonication, libraries were prepared as in Hoffman et al. (2015). Paired-end sequencing was performed on an Illumina NextSeq 500 with a read length of 75 bp.
Data Analysis
FASTQ files from paired end libraries were collapsed by barcode and the Illumina adaptor sequence was trimmed from the 3’ end. Files were uploaded and analyzed using the Galaxy web platform (Afgan et al., 2018). Reads were aligned to the S. cerevisiae genome (SacCer3, SGD) using Bowtie2 with a maximum fragment length for valid paired-end alignments set to 500 bp. Aligned reads were then filtered for quality and only uniquely mapped paired reads were used for future analyses. BAM files were uploaded to SeqMonk and normalized by scaling to 1 million reads. MACS Peak Caller was used to identify new peaks in hst4Δ HST3-FRB biological replicates using the mean WT file as the input (p value 1×10−5, 500 bp fragment size). Genome browser views were obtained by generating probes using a running window with a probe size of 1000 bp and a step size of 500 bp.
QUANTIFICATION AND STATISTICAL ANALYSIS
Software and statistical analysis details can be found in the Method Details section of the STAR Methods, as well as the Key Resources Table.
DATA AND SOFTWARE AVAILABILITY
All data from this study have been deposited in the Gene Expression Omnibus (GEO) under accession GEO: GSE124132.
Supplementary Material
Highlights.
Yeast sirtuins, Hst3 and Hst4, globally repress coding and noncoding transcription
Transcriptional regulation by sirtuins prevents formation of excessive DNA-RNA hybrids
Increased R loops due to sirtuin loss are associated with more DNA breaks
Overexpression of RNaseH1 alleviates genomic instability due to sirtuin loss
ACKNOWLEDGMENTS
We thank members of the Peterson lab and particularly Salih Topal for helpful discussions. We also thank the Stirling Churchman (Harvard Medical School) and William Theurkauf (UMMS) labs for sharing sequencing protocols and helping with troubleshooting. This work was supported by grants from the NIH to C.L.P. (GM049650 and GM122519) and J.L.F. (F32GM119229).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.009.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46 (W1), W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, and Gómez-González B (2017). DNA-RNA hybrids: the risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 24, 439–443. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arigo JT, Eyler DE, Carroll KL, and Corden JL (2006). Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol. Cell 23, 841–851. [DOI] [PubMed] [Google Scholar]
- Bayona-Feliu A, Casas-Lamesa A, Reina O, Bernués J, and Azorín F (2017). Linker histone H1 prevents R-loop accumulation and genome instability in heterochromatin. Nat. Commun. 8, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G, Papamichos-Chronakis M, and Peterson CL (2013). DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat. Commun. 4, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, and Nek-rutenko A; Galaxy Team (2010). Manipulation of FASTQ data with Galaxy. Bioinformatics 26, 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet A, Grosso AR, Elkaoutari A, Coleno E, Presle A, Sridhara SC, Janbon G, Geli V, de Almeida SF, and Palancade B (2017). Introns Protect Eukaryotic Genomes from Transcription-Associated Genetic Instability. Mol. Cell 67, 608–621 . e6. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, and Boeke JD (1995). The SIR2 genefamily, conserved from bacteriato humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9, 2888–2902. [DOI] [PubMed] [Google Scholar]
- Buratowski S (2009). Progression through the RNA polymerase II CTD cycle. Mol. Cell 36, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, and Verreault A (2006). The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 16, 1280–1289. [DOI] [PubMed] [Google Scholar]
- Celic I, Verreault A, and Boeke JD (2008). Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics 179,1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che J, Smith S, Kim YJ, Shim EY, Myung K, and Lee SE (2015). Hyper-Acetylation of Histone H3K56 Limits Break-Induced Replication by Inhibiting Extensive Repair Synthesis. PLoS Genet. 11, e1004990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin J, Libri D, and Porrua O (2011). Cryptictranscription and early termination in the control of gene expression. Genet. Res. Int. 2011, 653494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2018). Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol. Cell 71, 487–497. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creamer TJ, Darby MM, Jamonnak N, Schaughency P, Hao H, Wheelan SJ, and Corden JL (2011). Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet. 7, e1002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, and Rando OJ (2007). Dynamics of replication-independent histone turnover in budding yeast. Science 315, 1405–1408. [DOI] [PubMed] [Google Scholar]
- Etchegaray JP, Chavez L, Huang Y, Ross KN, Choi J, Martinez-Pastor B, Walsh RM, Sommer CA, Lienhard M, Gladden A, et al. (2015). The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat. Cell Biol. 17, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA (2000). Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798. [DOI] [PubMed] [Google Scholar]
- Ganguli D, Chereji RV, Iben JR, Cole HA, and Clark DJ (2014). RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res. 24, 1637–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, and Chédin F (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 45, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati RK,Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, and Libri D (2012). Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol. Cell 48, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer SJ, Gu W, Carone BR, Landry BD, Rando OJ, Mello CC, and Fazzio TG (2015). Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes Dev. 29, 362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Trotta KL, Smith EE, Mosaheb MM, Fuchs SM, and Churchman LS (2016). Comprehensive RNA Polymerase II Interactomes Reveal Distinct and Varied Roles for Each Phospho-CTD Residue. Cell Rep. 15, 2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 37,925–932. [DOI] [PubMed] [Google Scholar]
- Hoffman EA, McCulley A, Haarer B, Arnak R, and Feng W (2015). Break-seq reveals hydroxyurea-induced chromosomefragility asaresult of unscheduled conflict between DNA replication and transcription. Genome Res. 25, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, and Workman JL (2013). Directing transcription to the right way. Cell Res. 23, 1153–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Liu CL, Erkmann JA, Holik J, Grunstein M, Kaufman PD, Friedman N, and Rando OJ (2008). Cell cycle- and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 4, e1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 74, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, and Mostoslavsky R (2014). Chromatin and beyond: the multi-tasking roles for SIRT6. Trends Biochem. Sci. 39, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, Feldman JL, Klein MA, Silberman DM, Sebastián C, Mermel C, Dobersch S, Clark AR, Getz G, Denu JM, and Mostoslavsky R (2015). Identification of and Molecular Basisfor SIRT6 Loss-of-Function Point Mutations in Cancer. Cell Rep. 73, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, Sebastián C, Fitamant J, Ross KN, Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, et al. (2016). SIRT6 Suppresses Pancreatic Cancer through Control of Lin28b. Cell 765, 1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 70, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas NL, Miller KM, DeFazio LG, and Toczyski DP (2006). Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol. Cell 23, 109–119. [DOI] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, and Buratowski S (2014). A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 757, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, and Churchman LS (2015). Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 767, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, and Chua KF (2009). Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, and Proudfoot NJ (2013). Disengaging polymerase: terminating RNA polymerase II transcription in budding yeast. Biochim. Biophys. Acta 7829, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, and Shilatifard A (2015). Chromatin signatures of cancer. Genes Dev. 29, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 724, 315–329. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W7), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rege M, Subramanian V, Zhu C, Hsieh TH, Weiner A, Friedman N, Clauder-Münster S, Steinmetz LM, Rando OJ, Boyer LA, and Peterson CL (2015). Chromatin Dynamics and the RNA Exosome Function in Concert to Regulate Transcriptional Homeostasis. Cell Rep. 73, 1610–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD , McCarthy DJ , and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data Bioinformatics 26, 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Molina JB, Tseng SC, Simonett SP, Taunton J, and Ansari AZ (2016). Engineered Covalent Inactivation of TFIIH-Kinase Reveals an Elongation Checkpoint and Results in Widespread mRNA Stabilization. Mol. Cell 63, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, and Nourani A (2007). Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol. Cell 27, 393–405. [DOI] [PubMed] [Google Scholar]
- Salas-Armenteros I, Pérez-Calero C, Bayona-Feliu A, Tumini E, Luna R, and Aguilera A (2017). Human THO-Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 36, 3532–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Poulsen MB, Olszewski P, Pelechano V, Saguez C, Gupta I, Steinmetz LM, Moore C, and Jensen TH (2012). Rrp6p controls mRNA poly(A) tail length and its decoration with poly(A) binding proteins. Mol. Cell 47, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Kudla G, Wlotzka W,Tuck A, and Tollervey D (2012). Transcriptome-wide analysis of exosome targets. Mol. Cell 48, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, and Cramer P (2013). Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 755, 1075–1087. [DOI] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, and Adelman K (2015). Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 58, 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastián C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O,Truelove J, Guimaraes AR, Toiber D, et al. (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 757, 1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MA, Denton EL, Arrowsmith CH, Lupien M, and Schapira M (2014). Aglobal assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, and Libri D (2006). Transcription termination and nucleardegradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol. Cell 23, 853–864. [DOI] [PubMed] [Google Scholar]
- Timmers HTM, and Tora L (2018).Transcript Buffering:ABalancingAct between mRNA Synthesis and mRNA Degradation. Mol. Cell 72, 10–17. [DOI] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, and Vuica-Ross M (2011). RNase H and multiple RNA biogenesisfactors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 44, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Costantino L, Tan FJ, Zimmer A, and Koshland D (2016). S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 30, 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Pelechano V, Järvelin AI, and Steinmetz LM (2011). Functional consequences of bidirectional promoters. Trends Genet. 27, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I , Rando OJ , Delrow J , and Tsukiyama T (2007). Chromatin remodelling at promoters suppresses antisense transcription Nature 450, 1031–1035. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, and Steinmetz LM (2009). Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zwaans BM, Eckersdorff M, and Lombard DB (2009).The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 8, 2662–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Kwon CS, Choi Y, and Lee D (2016). Both H4K20 mono-methylation and H3K56 acetylation marktranscription-dependent histoneturnover in fission yeast. Biochem. Biophys. Res. Commun. 476, 515–521. [DOI] [PubMed] [Google Scholar]
- Zeller P, Padeken J,van Schendel R, Kalck V,Tijsterman M, and Gasser SM (2016). Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 48, 1385–1395. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, and Zamore PD (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Fisher SA, Dueck H, Middleton S, Khaladkar M, and Kim J (2018). PIVOT: platform for interactive analysis and visualization of transcriptomics data. BMC Bioinformatics 19, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data from this study have been deposited in the Gene Expression Omnibus (GEO) under accession GEO: GSE124132.